Back to Journals » International Journal of Nanomedicine » Volume 18
Macrophages as Promising Carriers for Nanoparticle Delivery in Anticancer Therapy
Authors Wróblewska A, Szczygieł A ![]() , Szermer-Olearnik B
, Szermer-Olearnik B ![]() , Pajtasz-Piasecka E
, Pajtasz-Piasecka E
Received 13 May 2023
Accepted for publication 19 July 2023
Published 8 August 2023 Volume 2023:18 Pages 4521—4539
DOI https://doi.org/10.2147/IJN.S421173
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lijie Grace Zhang
Anna Wróblewska, Agnieszka Szczygieł, Bożena Szermer-Olearnik, Elżbieta Pajtasz-Piasecka
Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wrocław, Poland
Correspondence: Anna Wróblewska, Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Weigla 12, Wrocław, 53-114, Poland, Tel +48 71 3709928, Email [email protected]
Abstract: Macrophages play a critical role in the immune response due to their ability to recognize and remove pathogens, as well as present antigens, which are involved in inflammation, but they are also one of the most abundant immune cell populations present in the tumor microenvironment. In recent years, macrophages have become promising cellular carriers for drug and nanoparticle delivery to the tumor microenvironment, mainly due to their natural properties such as biocompatibility, degradability, lack of immunogenicity, long half-life in circulation, crossing biological barriers, and the possibility of migration and accumulation at a site of inflammation such as a tumor. For the effectiveness of this therapeutic strategy, known as “Trojan horse”, it is important that the nanoparticles engulfed by macrophages do not affect their proper functioning. In our review, we discussed how the size, shape, chemical and mechanical properties of nanoparticles influence their internalization by macrophages. In addition, we described the promising research utilizing macrophages, their cell membranes and macrophage-derived exosomes as drug carriers in anticancer therapy. As a prospect of the wider use of this therapeutic strategy, we postulate its future application in boron delivery to the tumor environment in boron neutron capture therapy.
Keywords: macrophages, cellular carriers, anticancer therapy, tumor microenvironment, boron neutron capture therapy
Introduction
Cancer is one of the leading causes of death worldwide right behind cardiovascular disease. Globally, there were 19.3 million new cancer cases and 10 million cancer deaths in 2020. Moreover, the International Agency for Research on Cancer predicts the global cancer burden will reach 28.4 million cases in 2040, an increase of 47% from 2020.1 Therefore, there is a global need to improve existing anticancer therapies and develop new ones that reduce mortality. One promising area of research is the use of immune cells in cancer therapy. Innate immune cells are a diverse group consisting of effector cells (natural killers) and professional antigen-presenting cells (monocytes, macrophages, dendritic cells) considered to be helpful components involved in a new form of immunotherapy. An example of this application are vaccines based on dendritic cells, which have great potential for use in the treatment of cancer, both in monotherapy and in combination therapy.2
In recent years, the use of immune cells as carriers for drug delivery to the tumor microenvironment has become a hopeful therapeutic strategy, due to their ability to engulf foreign particles, specific tissue tropism, flexibility and low cytotoxicity.3 Research into cell-based delivery systems includes red blood cells, neutrophils, monocytes, T cells, natural killer cells, stem cells and macrophages. But safety and efficacy in clinical trials are mainly tested on red blood cells, T cells, natural killer cells and mesenchymal stem cells.4 However, macrophages deserve special attention because of some specific advantages, namely their natural biocompatibility, degradability, non-immunogenicity, long circulation half-life, high load capacity, ability to cross biological barriers, as well as migrate and accumulate at sites of inflammation and tumors.5 Macrophages can deliver anticancer drugs and can be applied as carriers of nanoenzymes such as nano-formulated catalase6 or catalase and polyethyleneimine-poly(ethylene glycol) complex for delivery to the region of the brain affected by Parkinson’s disease.7 Moreover, they can serve as carriers for a nanoparticle formulation of indinavir that induces significant inhibition of human immunodeficiency virus type 1 (HIV-1) infection.8
It is still unclear how nanoparticle surfaces interact with the biological environment to influence recognition and internalization by phagocytes. The search for correlations between the physicochemical characteristics of nanoparticles and their uptake mechanism by macrophages would be a significant step in solving issues related to the development of nanomedicine.9 An important role in the speed and efficiency of nanoparticle internalization by macrophages is played by their size, shape, surface chemistry and mechanical properties. Although macrophages as phagocytic cells show many features that enable them to engulf various types of nanoparticles and migrate to the tumor environment, they have some limitations, such as heterogeneity, limited payload, phenotypic changes, and interaction with non-target tissue. Extensive efforts to overcome these limitations may help in the clinical translation of macrophage-based drug carriers.4 Nevertheless, little is known about the mechanisms of such kind of strategy, as well as the connection between experimental research and clinical translation. Hence, in this review, we will assess the current knowledge on the use of macrophages, their membranes and macrophage-derived exosomes as nanomedicine carriers and the perspective of their favor in clinical settings. In addition, we propose the use of macrophages as carriers of boron-rich compounds in boron neutron capture therapy (BNCT), which is targeted radiotherapy for patients with cancers located in hard-to-reach areas and unresponsive to conventional treatment.
The Classification and Polarization of Macrophages
General Characteristics
Macrophages represent a large subpopulation of myeloid cells and as such, they are derived from bone-marrow precursors and parent monocytes in the peripheral blood in postnatal mammals. Circulating monocytes have a half-life of 1 to 3 days, but when they are localized in tissues and mature into tissue-specific macrophages, their half-life varies from months to years depending on the type of tissue. They are distinguished by sizes ranging from 5 to 50 μm and an irregular shape.10 Macrophages populate all tissues and are responsible for tissue homeostasis, development and repair. More importantly, macrophages play a critical role in the immune response due to their ability to recognize and remove pathogens, as well as process and present antigens, which are involved in inflammation.11 The acquisition of appropriate functions is possible due to the phenotypic plasticity of the macrophages in response to stimuli from the environment.12,13
The polarization and differentiation of macrophages into specific cell subpopulations depend on physiological conditions and pathological states such as cancer, cardiovascular diseases, metabolic disorders, or neurological diseases. The nature of macrophages is associated with the type of incoming signals involved in changes in surface markers, gene expression profile, metabolic properties, and cytokine production giving rise to various biological functions (Figure 1).14,15 The main function of classically activated macrophages (M1) is to remove pathogens, dead cells and foreign bodies, but they also play a crucial role in anticancer response.16,17 M1 macrophages can be induced by products of activated T-helper 1 (Th1) lymphocytes, such as interferon γ (IFN-γ), alone or in combination with bacterial lipopolysaccharide (LPS) or tumor necrosis factor α (TNF-α).18 They can secrete pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, IL-12, IL-23 and TNF-α, C-C motif chemokine ligands (CCL5), but also chemokines such as C-X-C motif chemokine ligands (CXCL9, CXCL10, CXCL11, CXCL16) as well as reactive oxygen species (ROS) and nitric oxide synthase (NOS).19–21 These cells express on their surface major histocompatibility complex II (MHC II), but also other specific markers: TLR-2 (toll-like receptor 2), TLR-4, inducible nitric oxide synthase (iNOS), costimulatory molecules CD80 and CD86, and opsonic receptors such as FcɣRIII (CD16), which are helpful in the presentation of necrotic antigens.19,21,22 M1 gene expression is regulated by transcription factors, such as NF-κB, STAT1, STAT5, IRF3, and IRF5.23 Macrophage cellular metabolism varies according to their activation. The M1 phenotype is mainly characterized by glycolysis, fatty acid synthesis, flux through the pentose phosphate pathway, and shortened tricarboxylic acid (TCA) cycle.24
 |
Figure 1 Classification of macrophages depending on environmental stimulation. There are two main types: M1 and M2 macrophages, among which we can distinguish: M2a, M2b, M2c, and M2d subcategories. Each type of macrophage is characterized by a different cytokine and chemokine production profile, surface receptors, and biological functions. The transition between M1 and M2 cells under the influence of stimulation from the environment is called repolarization. M2 subpopulations occur as a result of specific stimuli and differ in their cytokine production profile and function. M2a cells are classified as macrophages involved in wound healing and are activated by IL-4 or IL-13, and express high levels of CD206, Arg1, Ym1 and FIZZ1. They secrete IL-10, CCL17, CCL18, and CCL22, but also pro-fibrotic factors such as fibronectin, insulin-like growth factor (IGF), and TGF-β, used in tissue repairing and remodeling processes, and also involved in cell growth, and endocytosis. M2b cells are induced by IC, LPS, TLR ligands or agonists of IL-1 receptors. The M2b macrophages secrete both pro- and anti-inflammatory cytokines such as IL-1β, IL-6, IL-10, TNF-α and chemokine CCL1, which regulate the strength of the immune response and inflammatory reactions. M2c macrophages also called inactivated macrophages, generated by IL-10, TGF-β and GCs, play an important role in the phagocytosis of apoptotic and necrotic cells (efferocytosis), and release IL-10, TGF-β, CCL16, CCL18 and CXCL13. The last subtype, M2d macrophages are common in the tumor microenvironment (TME) and are activated by the TLR ligands, A2 adenosine receptor (A2R) agonists, and IL-6. They secrete IL-10, TGF-β and vascular endothelial growth factors (VEGF), which promote tumor progression and angiogenesis.17,19,25–27 |
Alternatively, activated macrophages (M2, among which we distinguish: M2a, M2b, M2c and M2d subcategories) can be generated by cytokines produced by T-helper 2 (Th2) lymphocytes, such as IL-4 and IL-13, but also IL-10, transforming growth factor β (TGF-β), immune complexes (IC), glucocorticoids (GCs), and LPS.18 M2 macrophages have anti-inflammatory properties and attend to tissue repair and regeneration. They also demonstrate protumor activity due to the production of extracellular matrix components, angiogenic and chemotactic factors that promote tumor formation, progression, and angiogenesis.14,22,28 M2 cells can secrete anti-inflammatory cytokines: IL-4, IL-10, and IL-13. They overexpress non-opsonic receptors such as mannose receptors (CD206, CD209), but also scavenger receptors (CD68, CD163), arginase-1 (Arg1), TGF-β, epidermal growth factor (EGF), Ym1, FIZZ1, indoleamine 2.3-dioxygenase (IDO1) and C-C motif chemokine ligands (CCL1, CCL16, CCL17, CCL18, CCL20, CCL22, CCL24).19,22 M2 gene expression is regulated by transcription factors, such as STAT6, IRF4, JMJD3, PPARδ, and PPARγ.23 However, the metabolism of M2 cells is defined by fatty acid oxidation and the tricarboxylic acid cycle coupled with oxidative phosphorylation.24 It is noteworthy that under the influence of environmental stimulation a transition between M1 and M2 cells (repolarization) has also been described.29
Tumor-Associated Macrophages as a Target in Antitumoral Therapeutic Strategies
Regardless of the above-mentioned subpopulations, macrophages present in tumor tissue are derived from circulating bone marrow monocytes, which are transformed into tumor-associated macrophages (TAMs) under the influence of the tumor microenvironment (TME). In general, these cells are considered to be one of the most abundant immune cell populations present in the tumor microenvironment, reaching up to 50% of the tumor mass.20,30
Initially, TAMs were considered to be exclusively derived from C-C chemokine receptor type 2 positive (CCR2+) monocyte precursors recruited in a CCL2-dependent manner into the tumor microenvironment.31 This concept arose when it was noticed that blocking the CCL2/CCR2 axis causes a substantial reduction in the size of the population of TAMs in tumor tissue.32 One of the main sources of CCR2+ monocytes is located in the bone marrow,33 however, it was revealed that the spleen might also constitute a significant reservoir of these precursors. Cortez-Retamozo et al demonstrated that splenectomy in a mouse lung adenocarcinoma model resulted in a strong reduction of the TAM population.34 Recent evidence suggests that another source of TAM precursors is tissue-resident macrophages, as observed in the pancreatic ductal adenocarcinoma model35 and various lung cancer models.36 These macrophages, as well as circulating monocytes, are recruited to TME upon the influence of various factors, such as inflammation, high levels of lactic acid and several chemokines,37,38 while the main factors causing conversion of these cells towards TAMs are macrophage colony-stimulating factor (M-CSF), VEGF, CCL2 or IL-4.39 It should be added that when the oxygen content in the tissue is reduced (hypoxia) then myeloid-derived suppressor cells (MDSCs) transform into TAMs, which additionally affects the size of this population.40 TAM diversity is associated with different cancer types and stages responsible for tumor inhibition but also progression, metastasis, and resistance to therapy. For this reason, getting to know about the sources and regulatory mechanisms of these macrophages is substantial as these cells play a crucial role in cancer recurrence.41
As mentioned above, macrophages present in tumors differ depending on the prevailing environmental conditions. In the initial stage of tumor growth, M1 cells are the predominant population and show antitumor activity. However, with the further progression of cancer and the presence of cytokines such as IL-4, IL-10 and IL-13, macrophages polarize into M2 cells supporting further tumor growth (Figure 2).42 They are believed to be the main subpopulation of TAMs present in the TME. For this reason, a high number of TAMs in a tumor is correlated with poor overall survival in several cancer types.43
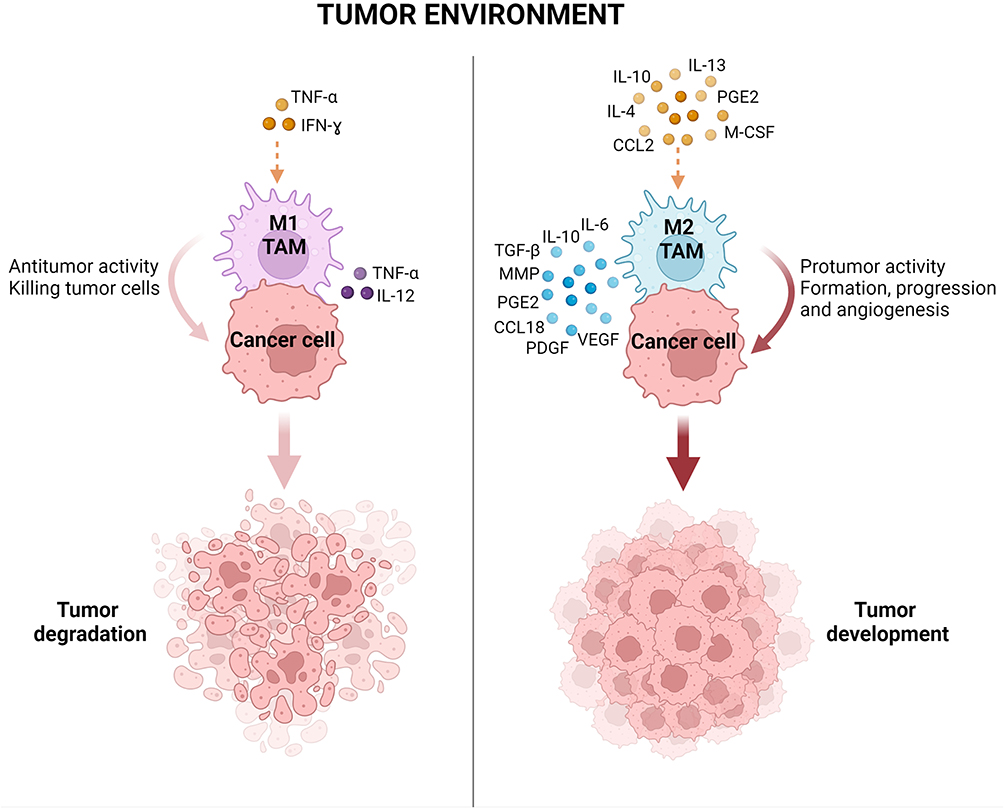 |
Figure 2 Polarization of tumor-associated macrophages to M1 and M2 phenotypes according to stimuli in the tumor microenvironment. The M1 phenotype of macrophages in TAMs is induced by pro-inflammatory cytokines (TNF-α, IFN-ɣ) and these macrophages have antitumor activity promoting cancer cell killing. The M2 phenotype is induced by anti-inflammatory molecules (IL-4, IL-10, IL-13, CCL2, M-CSF, PGE2) and secretes factors with protumor activity leading to tumor formation, progression and angiogenesis. Abbreviations: MMP, matrix metalloproteinases; PDGF, platelet-derived growth factor; PGE2, prostaglandin E2.38,44 |
According to the suppressive properties of most TAMs, they should be considered to play a significant role in suppressing the anticancer response. One of the suppression mechanisms used by TAMs is limiting the availability of amino acids in the environment. By secreting arginase 1 and indoleamine 2.3-dioxygenase into the environment, they contribute to the inhibition of the activation of effector T cells.45,46 In addition, by producing IL-10, TGF-β and prostaglandin E2, and through the interaction of the TAM’s surface ligands with T cell’s programmed death receptor 1 (PD-1) and cytotoxic T cell antigen 4 (CTLA-4) molecules, TAMs adversely affect the functions of cytotoxic CD8+ and CD4+ T cells.47 Considering the dominance of TAMs in the TME, and their ability to produce IL-10, it is believed that these cells, along with MDSCs, constitute the main source of this cytokine in tumor tissue. Moreover, TAMs are responsible for the increased influx of other tumor suppressor cells into this tumor tissue, and through the secretion of chemokines such as CCL2, CCL20, and CCL22, they support further recruitment of regulatory T cells and MDSCs to the TME.48–50
Due to their role, TAMs became a promising target in potential antitumoral therapeutic strategies by blocking the recruitment of TAM precursors into tumor tissue, increasing the ability of TAMs to eliminate cancer cells, as well as TAM depletion by chemotherapeutic agents. Each of these solutions has many advantages and is currently being tested in numerous clinical trials (Table 1). One of such trial is the inhibition of the influx of TAM precursors into tumors. This can be achieved by interrupting crucial recruitment pathways, such as M-CSF/CSF-1R, CCL2/CCR2 and CCL5/CCR5. Blocking the M-CSF-dependent axis may be realized by using small molecule inhibitors or blocking antibodies with potent and selective activity against CSF-R1. A different approach is realized by converting TAMs already present in TME into cells with anticancer potential. However, unlike the previous strategy, this one presumed an increase in the ability of TAMs to eliminate tumor cells via the CD47-SIRPα pathway. This was associated with the observation that the interaction of the tumor CD47 molecule with its signal regulatory protein α (SIRPα) expressed on macrophages leads to the inhibition of the macrophages’ ability to eliminate cancer cells.51 A completely different solution in TAM-targeted anticancer therapy is the selective depletion of these cells from the tumor milieu. This can be achieved with appropriate chemotherapeutics such as clodronate and zoledronate, which belong to the bisphosphonate group. It was reported that besides the direct antitumor effect (cancer cell apoptosis and inhibition of angiogenesis etc.), bisphosphonates indirectly support the effectiveness of therapy by triggering TAMs in the apoptotic pathway.52,53 However, due to the low bioavailability of these drugs and the observed severe side effects, innovative bisphosphonate nano delivery systems are being developed. For example, in a mouse sarcoma model, therapy with lipid-coated calcium zoledronate nanoparticles effectively depleted TAMs, reduced angiogenesis, and prevented the suppression of the antitumor immune response, thus contributing to the inhibition of tumor growth.54
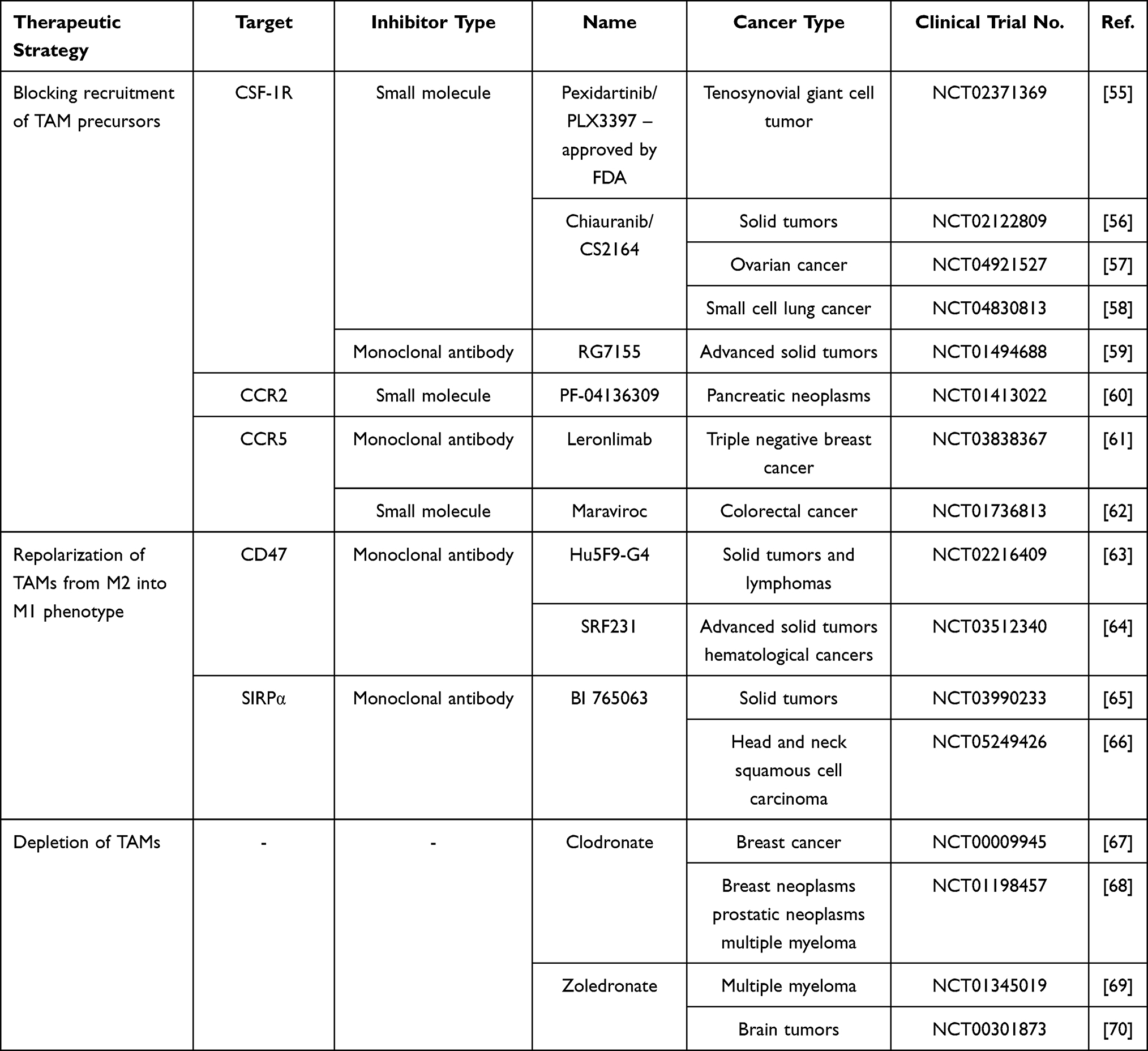 |
Table 1 TAMs as a Target in Potential Antitumoral Therapeutic Strategies |
In summary, TAMs are abundantly present in the TME and have the ability to phagocytosis, and remain mobile in the tumor mass (including hypoxic areas). Thus, TAMs can be considered for the delivery of anticancer drugs due to their better biodistribution in tumor tissue. This application can enhance the therapeutic effect against drug-resistant cells, such as cancer stem cells, which are poorly treated with conventional chemotherapies.71
Macrophages in Anticancer Therapy – Novel Carriers in Nanoparticle Delivery
Nanoscience is a promising field of research based on the design, characterization, and production of materials by controlling their shape and size at the nanometer scale. Their application has become important in nanomedicine, especially in anticancer therapies. The use of nanometer materials such as nanoparticles has pharmacokinetic benefits and demonstrates high efficiency, safety, and targeting properties. However, their toxicity, rapid blood degradation, and accurate characterization pose challenges. Therefore, various nanoparticle delivery systems have been developed, and one promising approach is the use of macrophages as carriers in anticancer therapies.72,73
Mechanism of Nanoparticle Internalization by Macrophages
Phagocytosis is a primary mechanism for nanoparticle uptake by macrophages, which is a complex process, consisting of several steps. The first stage is particle internalization initiated by the interaction of specific receptors, called pattern-recognition receptors on the phagocyte with ligands on the surface of the particle. These receptors include mannose receptors, integrins, as well as scavenger receptors. In the next step, the cellular cytoskeleton in the cytosol is rearranged during which actin is polymerized and particles internalized through an actin-based mechanism. Afterward, actin is removed from the phagosome, which then matures to form the phagolysosome through processes that require the coordinated interaction of the actin and tubulin-based cytoskeleton.74 Although this active phagocytosis enables engulf of large particles, but it is limited to a size of 10 microns. Therefore, in some cases, nanoparticles are uptake by pinocytosis, especially when they are fluid and solutes, or by receptor-mediated endocytosis. Pinocytosis can include large volume extracellular clathrin-mediated internalization such as macropinocytosis and circular dorsal ruffles responsible for 0.5–5 µm of particle uptake and smaller volume internalization, such as clathrin and clathrin-independent mechanisms, capable of 20–500 nm internalization. Unlike phagocytosis, there is no uptake into phagocytic vesicles, the cells encapsulate the foreign substance directly into the cytoplasm. Whereas the mechanism of clathrin-mediated endocytosis is based on the formation of clathrin pits in the cell membrane, which envelops and draws particles inside. The membrane fragment is then cleaved with dynamin to form internalized vesicles that enter early endosomes, then mature into late endosomes and are finally transported to other intracellular sites such as lysosomal compartments. However, this process is responsible for the internalization of particle size ranges from 100–350 nm. Therefore, smaller particles from 20–100 nm are internalized via caveolin-mediated endocytosis. During this process, vesicles form by indentation of the membrane and are usually transported to the Golgi apparatus or excreted from the cell, thus avoiding lysosomal internalization, which is crucial for some nanoparticles.9,75,76
Influence of Nanoparticle Properties on Their Internalization by Macrophages
The kinetics of internalization of nanoparticles by macrophages is influenced by their properties, such as size, shape, surface chemistry and mechanical properties. It is not clear how particle size affects their uptake by macrophages.77 Petithory et al demonstrated that silica physisorbed nanoparticles were internalized in two different size-dependent ways. One way was observed for larger nanoparticles (300–450 nm), cells internalized almost all of them. The second was observed for smaller nanoparticles (50–200 nm), where only some of them were internalized by RAW264.7 macrophages, while these cells did not engulf 35 nm nanoparticles even after 12 hours of incubation. The authors suggested that these two ways were associated with phagocytosis and endocytosis, respectively.78 Some research reported that maximum internalization occurs in the size range of 1–3 µm, which corresponds to the size of bacteria.79 There is evidence of a positive correlation between particle size and macrophage uptake. Walkey et al proved that the uptake of gold nanoparticles by J774A.1 macrophages was size-dependent and was higher for the largest ones in serum-independent conditions.80 Similarly, Alqahtani et al showed that the size of gliadin nanoparticles increases their uptake by J774A.1 macrophages.81 Mendes et al demonstrated that the uptake of nanographene oxide by THP-1 cells during short incubation periods was size-dependent. The size of the particles tested for the research ranged from 46–453 nm, with larger flakes being more easily engulfed than smaller ones.82 Nevertheless, the opposite effect was noticed by Chang et al, who proved that nanoparticle size was negatively correlated with their uptake by RAW264.7 macrophages, and smaller ones were more effectively loaded into these cells.83
Taking into consideration that cells are unable to detect nanoparticle size as long as phagocytosis is not complete, then size does not affect the initiation of internalization but the fundamental property governing this process is the shape of the nanoparticles. Champion et al demonstrated that all nanoparticle shapes were able to initiate phagocytosis in at least one orientation, but the shape of the surface at the point of initial contact dictates the type of uptake. One example is the elliptical disk-shaped nanoparticles, which were internalized within a few minutes after the main axis was attached to macrophages. While after attaching a minor axis or a flat size, they were not taken up even after several hours.84 Moreover, Sharma et al demonstrated that the phagocytosis of oblate ellipsoids is significantly greater than that of elongated ellipsoids or spheres for particles up to 690 nm in size.85 Lu et al showed that spheres consisting of complexes of cadmium telluride quantum dots and cystine were phagocytosed more efficiently than rods or needles.86 While Paul et al pointed out that spherical nanoparticles, which were larger than ellipsoid nanoparticles were engulfed five times faster than ellipsoids with smaller surface areas.87 In addition, it was shown that the mechanical properties of the nanoparticles also have a significant impact on the internalization process. Modeling methods revealed that stiff nanoparticles are energetically more prone to complete wrapping than soft ones.88 As the elasticity of the nanoparticles increases, their membrane wrapping gradually becomes less significant. In the case of macrophage phagocytosis and receptor-mediated endocytosis, the elasticity of nanoparticles affects their cellular uptake.89
Surface chemistry also plays a dominant role in the uptake of nanoparticles by macrophages. Hydrophobic nanoparticles are much more susceptible to phagocytosis than their hydrophilic counterparts. The negative charge of the macrophage cell membrane in mammalian cells enables the attachment of cationic nanoparticles to their surface via passive adsorption, such as hydrophobic interactions, hydrogen bonds or electrostatic interactions, which gives them an advantage over anionic ones. Whereas covalent and non-covalent bonds of compounds are possible due to the presence on the cell surface of functional groups derived from proteins, lipids and polysaccharides that form the cell. However, the use of positively charged particles may be limited by their toxicity to cells.90,91 Another property influencing the internalization of nanoparticles is their surface coating. For example, higher coverage of the particles with PEG (polyethylene glycol) reduced their uptake by macrophages.80 Macrophages express surface receptors such as Fc-receptors, mannose, galactose, fibronectin lipoprotein and others, therefore coating nanoparticles with the ligands for these receptors may promote macrophage uptake.92 For example, mannose-functionalized nanoparticles were very efficiently engulfed by macrophages that express the mannose receptor on their surface by receptor-mediated endocytosis.93
To ensure the successful design, modeling, and introduction of new nanoparticles, all of the aforementioned properties must be considered. In addition, they may be helpful during the process of recognition and internalization by macrophages, and as a result, increase the specificity and selectivity of nanoparticle delivery and minimize their cytotoxic effect.
Advantages and Limitations of Using Macrophages as Carriers
The strategy of using macrophages as carriers has many advantages closely related to the natural features of these cells, such as the ability to uptake foreign particles, their presence in many tissues, the ability to modulate the tumor microenvironment, non-immunogenicity and a long half-life.73 In addition, it is a native targeting system due to the properties of macrophages to extravasate through the tight vascular wall into tumors and migrate to their hypoxic regions, such as sites of inflammation, injury, and tumors mediated by various chemoattractants. In the case of a macrophage-encapsulated drug, these properties enable its active accumulation in the tumor in a manner independent of the enhanced permeability and retention (EPR) effect, as well as lower cytotoxicity, and increased efficacy through controlled release and a longer circulating half-life. In contrast, systemically administrated drugs are rapidly eliminated from the bloodstream by phagocytes of the reticuloendothelial system (RES), therefore loading into macrophages may protect them from rapid degradation in the blood, interaction with non-target tissue and minimize the side effects5,94,95 One of the most frequently considered cases is effective drug delivery to brain tumors, which is prevented by the blood-brain barrier (BBB) impermeable to many substances. As a result, many agents do not reach the tumors located in the brain, thus failing to achieve the intended therapeutic effect.96 With the tumor progression (eg in glioma) the integrity of BBB is compromised due to the EPR effect responsible for leakiness of the tumor vasculature and poor lymphatic drainage. However, the increased interstitial fluid pressure inside the tumor and the blood-tumor barrier still impede the delivery of therapeutic agents into the tumor. Only by actively overcoming these barriers, the drug may be successfully delivered to the diseased site.97 However, inflammation arising in tumors can induce overexpression of cell adhesion molecules on the endothelial surface, allowing interaction between macrophages and endothelial cells, facilitating the initial process of macrophage rolling, strong attachment, and transmigration. As a result of this phenomenon, macrophages can cross the blood-brain barrier and enter the parenchyma of the brain from the peripheral circulation.95,98 These cells, constituting the largest population of immune cells infiltrating glioblastoma,99 may become promising carriers for nanoparticle delivery to the brain tumor environment.
Despite many advantages, the use of macrophages as carriers has some limitations that pose a challenge in their further development and clinical translation. One potential limitation is the influence of engulfed drugs on the impairment or disruption of innate cellular functions. The particles should not affect the ability of macrophages to migrate and be efficiently delivered and released at the target site. Therefore, it is essential to define the toxic and destructive effects of the internalized drugs and to confirm their safety both for the cellular carrier and the organs.100 The challenge is also the lack of control over the loading quantity of payloads and the limited payload of macrophages, which means that they can only uptake a limited number of particles. Another limitation is the unspecified stability of macrophages loaded with fragile, biodegradable drugs that may undergo intracellular biodegradation or inactivation leading to their premature release. The solution to the above problems may be to encapsulate molecules in protective nanocarriers (eg liposomes) and only then load them into macrophages, or to use only macrophage cell membranes to wrap the particles. In turn, it can also be the attachment of such molecules to the surface of macrophages and the simultaneous prevention of their phagocytosis through chemical conjugation or using a “cellular backpack”.101 Due to the high heterogeneity among macrophages and sensitivity to local environmental changes, the challenge is to control the behavior of macrophages during the transport of particles, ie changes in their phenotype and activation state. In this case, it can be overcome by loading into the “cellular backpack” with factors that maintain the macrophage phenotype in the immunosuppressive environment of the tumor.4,102 Another limitation can be the accumulation of particle-loaded macrophages in the liver, spleen, brain, kidney and even lung, which may result in drug release at an undesirable site and interaction with non-target tissue. Therefore, to avoid interaction with non-target tissues and to increase the homing of cellular carriers to the tumor tissue, modulation of the tumor environment is used. This is achieved by injecting a chemoattractant into target tissues to create a chemokine gradient that increases the influx of cellular carriers expressing receptors for these chemokines. Such strong chemokine signals, resulting in increased recruitment of macrophages, can also be generated by the treatment of the target tissue with chemical or physical stimuli, such as photosensitizer, radiation or hypoxia. It is worth emphasizing that the selection of the optimal route of administration is an important factor in ensuring the specific accumulation of cellular carriers in the tumor tissue because intravenously administrated macrophage carriers tend to accumulate in non-target tissues more than when injected locally. Among the strategies that increase the selectivity of cellular carriers toward the target tissue, are the methods of forced overexpression of chemokine receptors and adhesion molecules using genetic engineering, as well as surface modification of these carriers with targeting ligands or guiding materials.4 Equally important for achieving a therapeutic effect is that macrophages loaded with drugs should migrate to the tumor site in substantial quantities. Therefore, the key challenge may be to scale up the production of macrophages loaded with drugs to deliver a large number of them to the patient’s body. One solution can be the immortalization of macrophages obtained from patients and their propagation so that populations with particular properties can be established.103 However, it is still not known how macrophages should be stored to preserve their biological activity and for how long to avoid their changes affecting undesirable results. Another challenge is to control the immunomodulatory effect of nanoparticles on macrophages. In some cases, the nanoparticles are specifically designed to activate cytokine expression to promote an immune response. However, undesirable changes in cytokine expression can result in immunostimulation, which induces toxic effects on cells, tissues, and even the whole organism, leading in extreme cases to disseminated intravascular coagulation and cytokine storm. The exact mechanisms by which the physicochemical properties of nanoparticles induce the upregulation of inflammatory genes and at which stage of the nanoparticle-macrophage interaction occurs are poorly understood. In most cases, in vitro studies on macrophages correlate well with in vivo results, which allows to establish the dependence between the properties of nanoparticles and the induction of inflammation. The most frequently described changes in the cytokine expression profile are the upregulation of TNF-α, IL-1β, IL-6 and/or IL-8. However, the development of methods to control macrophage immunomodulation by nanoparticles may become crucial for the future introduction of macrophage-based carriers to patient studies.104,105
The above-mentioned limitations pose a challenge to clinical translation, therefore clinical trials using macrophages as a drug delivery system have not yet been reported. However, due to the dynamic progress in research into cellular drug delivery systems and the increase in clinical trials using cells such as red blood cells, T cells, natural killers and stem cells, there is a chance that macrophages will soon be used in the clinic.
“Trojan Horse” Delivery as a Promising Strategy in Anticancer Therapy
As mentioned previously, macrophages, due to their many advantages, are a promising candidate for drug delivery to the tumor microenvironment. Furthermore, it was demonstrated that macrophages successfully accumulate nanoparticles in vacuoles and cytoplasm. The use of macrophages as carriers of therapeutic agents, genes, and nanoparticles for delivery to the TME has been called a “Trojan Horse”.106
The source of macrophages for drug delivery in anticancer therapies can be primary macrophages harvested from animals, such as bone marrow-derived macrophages, peritoneal, and alveolar macrophages, as well as commercially available cells including murine macrophage-like RAW264.7 cells, J774A.1 macrophages or human THP-1 monocytic leukemia cells.86 The procedure for obtaining cellular carriers in humans may consist in separating monocytes from the rest of the cells circulating in the peripheral blood or harvesting stem cells from the bone marrow and differentiating them in culture to obtain macrophages, followed by ex vivo incubation with the nanoparticles for the time necessary for their internalization and reintroduction into the circulation of the obtained cellular carriers (Figure 3).100
 |
Figure 3 A scheme of an exemplary procedure of using macrophages as carriers of nanoparticles in anticancer therapy. Monocytes are isolated from peripheral blood collected from the patient and then differentiated into macrophages using appropriate factors. Subsequently, macrophages are incubated with nanoparticles, eg, anticancer drugs. The macrophages enriched in nanoparticles are re-injected into the patient. |
One of the most widely analyzed pairs is doxorubicin (DOX) with RAW264.7 macrophages. Zhang et al designed doxorubicin-loaded RAW264.7 macrophages with a loading efficiency of chemotherapeutic agents at 16.6 pg per cell. A two-hour exposure resulted in efficient doxorubicin uptake, while prolonged incubation minimally increased their content. In addition, an increased secretion of pro-inflammatory cytokines, such as IL-6, IL-12 and TNF-α, and a decreased production of anti-inflammatory IL-10 was observed, proving that RAW264.7 macrophages have been polarized towards the pro-inflammation M1 phenotype. Moreover, their intravenous injection into a U87MG xenograft model showed tumor accumulation comparable to untreated macrophages.107 Whereas Li et al used RAW264.7 macrophages to load doxorubicin, paclitaxel (PTX), and paclitaxel conjugated N-succinyl-N′-octyl chitosan (SOC), and demonstrated that SOC-PTX-loaded macrophages had the greatest antitumor activity and inhibited the viability of human MCF-7 and MDA-MB-231 breast cancer cells by 80% in vitro. Additionally, SOC-PTX-loaded macrophages resulted in a significant reduction in tumor volume of ~93% in MCF-7 tumor-bearing mice.108 Another team, Fu et al used RAW264.7 macrophages to load doxorubicin, which reduced the viability of mouse 4T1 breast cancer cells in vitro. In addition, in vivo anticancer efficacy was demonstrated by tumor suppression, prolonged life and inhibition of metastasis in 4T1-bearing mice.109 Macrophages can also be used in photothermal therapy (PTT). For example, Qiang et al loaded doxorubicin into nanoparticles composed of reduced graphene oxide, branched polyethylenimine and polyethylene glycol monomethyl ether, and then internalized into RAW264.7 macrophages. Following near-infrared laser (NIR) irradiation, doxorubicin was released as a free drug and demonstrated toxic effects on mouse RM-1 prostate cancer cells in vitro. Additionally, such loaded macrophages efficiently accumulated at the tumor site of RM-1-bearing mice and significantly suppressed tumor growth after NIR irradiation. These results confirm the anticancer effect of chemotherapy combined with photothermotherapy.110 A further example is Nguyen’s research on the use of RAW264.7 macrophages to load liposome-encapsulated doxorubicin and gold nanorods for synergistic photothermal-chemotherapy. The results showed a significantly decreased viability of 4T1 cancer cells following in vitro NIR irradiation. Moreover, loaded macrophages migrated to the tumor and reduced tumor growth in 4T1-bearing mice compared to the other groups after NIR treatment.111
Other types of macrophages have also been tested for use as drug carriers. Huang et al used peritoneal macrophages as delivery carriers for SN38 (7-ethyl-10-hydroxycamptothecin) nanoparticles, which are a highly active metabolite of irinotecan. The macrophages internalized SN38 nanoparticles within 2 hours. Such carriers showed toxic effects after 48 and 72 hours of incubation with human A549 lung cancer cells, as evidenced by the time-dependent increase in the number of apoptotic cells. In addition, macrophage-SN38 nanoparticles accumulated more efficiently at the A549 tumor site compared to normal tissue and inhibited tumor growth.112 Choi et al used liposome-encapsulated doxorubicin loaded into mouse peritoneal macrophages to treat human A549 lung cancer. In vivo studies demonstrated the ability of macrophages to efficiently deliver doxorubicin-loaded liposomes to the tumor site and confirmed their inhibitory effect on A549 tumor growth.113 Pang et al prepared doxorubicin-loaded poly(lactide-co-glycolide) (PLGA) nanoparticles encapsulated in primary M1 macrophages derived from mouse bone marrow cells. Cytotoxicity studies on human U87 glioblastoma cells showed a two-fold lower half-maximal inhibitory concentration (IC50) of DOX-PLGA-loaded macrophages than free doxorubicin and doxorubicin-loaded PLGA nanoparticles. Moreover, the anticancer effect of DOX-PLGA-loaded M1 macrophages was observed in U87-bearing mice by significantly extending the lifespan and the greatest apoptosis of tumor tissue compared to the control and other groups.114 While Madsen et al used rat alveolar macrophages to encapsulate gold-silica nanoshells (AuNS) to treat rat C6 glioblastoma with PTT. After irradiation with an 810 nm laser, in vitro the viability of C6 cells was significantly reduced compared to the control group, and in vivo studies confirmed the antitumor effect of Au-NS-loaded macrophages by reducing tumor volume in C6-bearing rats.115 Choi et al also confirmed that blood monocyte-derived macrophages loaded with gold-silica nanoshells accumulated in intracranial metastatic deposits by crossing the blood-brain barrier in a mouse model of breast cancer metastatic to the brain (MDA-MB-231 BR) when injected into the systemic circulation. Furthermore, these cells surrounded the metastatic cells, delivering loaded nanoparticles less than a cell’s width from the nearest metastatic cell.116
In addition, macrophages can be used in anticancer therapies to transfer genetic material. For example, based on the knowledge that calcium integrin binding protein-1 (CIB1) promotes survival and proliferation in triple-negative breast cancer, Wayne et al used small interfering RNA (siRNA) lipoplexes to CIB1 knockdown and load into mouse macrophages IC21. The results showed that macrophages could horizontally transfer siRNA into human MDA-MB-468 breast cancer cells in vitro and caused a dose-dependent reduction in cell viability and decreased tumor sphere growth.117 While Muthana et al prepared macrophages from mononuclear cells isolated from buffy coat to selectively deliver oncolytic adenovirus to hypoxic areas of prostate tumors. For this purpose, macrophages were co-transduced with a hypoxia-regulated E1A/B construct and an E1A-dependent oncolytic adenovirus whose proliferation is restricted to prostate tumor cells using a prostate-specific promoter element. After the migration of macrophages to regions of extreme hypoxia, E1A/B proteins were expressed, activating the replication of the adenovirus that was released and infected adjacent LNCaP prostate cancer cells. As a result, oncolytic adenovirus inhibited tumor growth in LNCaP-bearing mice and reduced pulmonary metastases.118
In anticancer therapies, instead of utilizing whole macrophages, their membrane can also be used to coat drugs and nanoparticles. The application of a macrophage membrane can effectively improve the blood circulation of the drug and increase its accumulation in the tumor due to the complete replication of surface antigens from natural cells to synthetic nanoparticles. For example, Meng et al used RAW264.7 macrophages to prepare a macrophage membrane in order to coat iron oxide (Fe3O4) nanoparticles, which significantly affected tumor regression in MCF-7-bearing mice after PTT.119 Xuan et al developed macrophage cell membrane-camouflaged gold nanoshells for PTT in vivo, which exhibited good biocompatibility, reduced opsonization, extended circulation time and increased accumulation in 4T1 breast tumor, and tumor growth inhibition in mice after NIR irradiation.120 Another approach was described by Bhattacharyya et al, who isolated a TNFα-expressed membrane from human THP-1 monocytes differentiated into macrophages to coat chitosan nanoparticles. These carriers showed a dose-dependent decrease in the viability of human HeLa cervical cancer cells, MCF-7 and MDA-MB-231 breast cancer cells.121 While Xuan et al formed macrophage cell membrane-camouflaged mesoporous silica nanocapsules (MSNCs) loaded with hydrochloride doxorubicin, which accumulated at the site of the 4T1 breast tumor and inhibited tumor growth more efficiently than the components of this complex separately.122 Similarly, in a 4T1 breast cancer model, Cao et al demonstrated effective uptake of emtansine liposomes coated with macrophage membrane and reduced viability of 4T1 cells in vitro and in vivo inhibited lung metastasis in breast cancer.123 However, in other research, albumin nanoparticles coated with RAW264.7 macrophage plasma membranes were loaded with paclitaxel, which demonstrated higher internalization efficiency, cytotoxicity and apoptosis rates than uncoated albumin nanoparticles in B16F10 melanoma cells. Furthermore, such carriers demonstrated sustained blood circulation and selective accumulation at the tumor site in the B16F10 tumor model.124 Other studies on the use of paclitaxel encapsulated in nanoparticles coated with macrophage membrane, which are gradually released in response to pH differences in the tumor microenvironment, were reported by Zhang et al. The carriers obtained were tested on an orthotopic mouse model of breast cancer and showed a favorable tumor-homing ability in systemic circulation and high biocompatibility.125
Another promising approach is the use of macrophage-derived exosomes as drug delivery carriers.126 For example, Kim et al used macrophage-derived exosomes vectorized with aminoethylanisamide-polyethylene glycol (AA-PEG) to deliver paclitaxel for the treatment of lung metastasis. This complex showed a high ability to accumulate in tumor tissue and was taken up by receptor-mediated endocytosis compared to non-vectorized exosomes. Moreover, the antitumor effect of the designed complex was confirmed by inhibiting the growth of metastases and increasing the survival time in mice with lung metastasis.127 While Haney et al proposed the use of macrophage-derived extracellular vesicles for the delivery of doxorubicin and paclitaxel in the treatment of triple-negative breast cancer. In vitro studies proved the efficient accumulation of these carriers in MDA-MB-231 cells and their antiproliferative activity against tumor cells. Additionally in mice with T11 and MDA-MB-231 breast tumors, these extracellular vesicles loaded with chemotherapeutics showed stronger tumor growth suppression than the free drugs.128
All these in vitro and in vivo studies confirm that the use of macrophages, membranes and macrophage-derived exosomes as carriers for the delivery of therapeutic substances to the tumor environment is a promising therapeutic strategy.
Effect of Nanoparticles on the Tumor-Associated Macrophages
Similarly, as in the case of dendritic cells, a low risk of side effects and a high treatment efficacy make macrophage-mediated nanodrug delivery systems a promising strategy combining tumor immunology and nano therapy. The use of nanoparticles is attractive due to the EPR effect, which in the tumor tissue supports the accumulation of nanomedicines at the tumor site.129,130
Recently, it has been discovered that nanoparticles have more advantages in immunological applications by targeting and regulating TAMs. Some nanoparticles can polarize macrophages without reprogramming agents. Polyurethane nanoparticles were found to have surface-dependent immunosuppressive properties, preventing polarization of M1 macrophages by decreasing the production of TNF-α and IL-1β.131 Fuchs et al found that carboxyl-modified polystyrene and amine-modified nanoparticles could inhibit macrophage polarization towards M2 by downregulating the expression of CD200R, CD163 and IL-10.132 The use of nanoparticles, such as silica, carbon, iron oxide, or gold to regulate the polarization states of macrophages has been extensively studied.133 An interesting approach was to use engineered nanoparticles capable of preferentially targeting M2 TAMs by delivering nanoparticles carrying M1 polarizing transcription factors and mRNAs for IRF5. These factors were able to force M2 TAMs to polarize to a pro-inflammatory state and convert to the M1 phenotype.134 Another study where researchers loaded the mannose-modified TAM repolarization agent (IMD-0354) into cationic lipid-based nanoparticles using the presence of mannose receptors on M2 TAMs. This led to the induction of potent antitumor properties of TAMs in Hepa 1-6-bearing mice.135 Chen et al used mannose as a ligand for the targeted delivery of nanoparticles to M2 TAMs by conjugating bivalent mannose receptor ligands to nanoparticles. It was observed that these nanoparticles have a significantly higher level of uptake by M2 macrophages compared to M1 or resting macrophages.136 While albumin-based nanoparticles expressing transferrin receptor binding peptide T12 and mannose were able to repolarize M2 TAMs to M1 and remodel the tumor microenvironment to enable an enhanced antitumor response. In addition, due to the strong expression of albumin-binding receptor SPARC (secreted protein acidic and rich in cysteine) and transferrin receptor by gliomas, and SPARC and mannose by M2 TAMs, these nanoparticles contribute to a dual effect on both gliomas and M2 TAMs.137 Many studies confirm that nanomedicines can be designed to remodel the tumor environment. When considering the use of nanoparticles to facilitate and enhance macrophage-mediated phagocytosis, the following main approaches are of interest: inactivation/elimination of M2 TAMs or reprogramming of M2 TAMs to acquire the pro-inflammatory M1 phenotype.138
Promising Perspectives for Using Macrophages as Boron Carriers in BNCT
Macrophages, due to their natural ability to uptake various types of nanoparticles and deliver them to the tumor microenvironment, may be promising carriers not only for compounds with direct anticancer activity but also for non-medical compounds used in other anticancer therapies. An example of such use is boron neutron capture therapy (BNCT). BNCT is based on the delivery of compounds rich in the boron-10 isotope to cancer cells and irradiation of the tumor site with a neutron beam, causing the breakdown of the boron-10 nucleus. This leads to the formation of lithium nuclei and alpha particles with high linear energy transfer, which destroys the cancer cells where boron was accumulated.139 This therapy is mainly intended for the treatment of patients with tumors located in hard-to-reach areas such as brain tumors, melanomas, or head and neck cancers. The role of macrophages in BNCT would be to deliver boron-rich compounds to the tumor environment, which in the case of brain tumors would be possible due to the ability of these cells to cross the blood-brain barrier.98 Subsequently, after irradiation with a neutron beam, boron-10-containing compounds accumulated in TME would undergo nuclear fission, destroying cancer cells.140
BNCT research has shown that peripheral blood mononuclear cells (PBMCs), including macrophages, delivered boron to cancer cells and were not destroyed after irradiation with a neutron beam. Additionally, these cells demonstrated immunomodulatory activity by increasing the production of the antitumor cytokine IL-12 while reducing the production of IL-10, which negatively affects the antitumor response.141 Other studies have investigated the interaction of macrophages with various boron compounds, which are promising candidates for boron delivery in BNCT. Kozień et al obtained boron carbide nanoparticles functionalized with immunoglobulin G, which showed a higher level of binding to RAW264.7 macrophages than to mouse MC38 colorectal cancer cells.142 Boron nitride nanotubes (BNNTs) are also promising candidates for boron delivery. BNNTs proved no toxicity to RAW264.7 macrophages and did not affect their morphology, as well as the production of the cytokines IL-6, IL-10 and TNF-α by these cells. Macrophages showed the ability to internalize BNNTs, followed by the formation of small aggregates in the perinuclear regions of the cytoplasm.143 Exosomes released by macrophages were used to encapsulate boron-containing carbon dots (BCDs) and demonstrated internalization by U-87-MG glioma cells and distribution around the nucleus in vitro. Moreover, studies in tumor-bearing mice have proved the accumulation of these carriers at the tumor site due to their ability to cross the blood-brain barrier, good ratio of boron in tumor to normal tissue for BNCT, and have shown prolonged survival after neutron irradiation.144 Wu et al prepared boric acid-contained chitosan/alginate/polyvinyl alcohol nanoparticles, which accumulated in SAS oral cancer cells, but also in RAW264.7 macrophages.145 Similarly, Nakagawa proved that the boronophenylalanine–fructose (BPA-F) complex was taken up not only by tumor cells in C6 rat glioblastoma models but also by activated macrophages in the brain. The authors suggested that the inhibition of activated macrophages after irradiation results in a reduction in peritumoral edema.146 Preliminary studies on the use of macrophages and macrophage-derived exosomes to deliver boron-rich compounds to the tumor environment in BNCT appear to be a promising therapeutic approach, but more research is needed on the safety and effectiveness of such treatment.
Conclusions
In conclusion, the use of macrophages as carriers for nanoparticle delivery is an increasingly promising strategy in anticancer therapy. The properties of these specialized phagocytic cells are the key to the efficient and selective delivery of nanoparticles to tumor tissue, even across the blood-brain barrier. In addition, the abundant presence of TAMs in the tumor microenvironment, their high plasticity, and the possibility of repolarization to the anticancer phenotype can be used to enhance the therapeutic effect. However, in order to achieve the most effective delivery of nanoparticles to the tumor environment using macrophages, it is important to select such nanoparticles whose size, shape, surface chemistry, and mechanical properties will ensure their optimal internalization by these cells, and at the same time will not affect their proper functioning. Although macrophages have many advantages as nanoparticle carriers, their limitations remain crucial. The rapid development of the field of cell-based delivery systems is finding more new solutions to overcome these limitations, giving macrophages an increasing chance of clinical translation. The aim of our review was to collect information that has a significant impact on the study of interactions between macrophages and nanoparticles for use in anticancer therapy. As a further step towards new applications of these carriers, we propose using them to deliver boron-rich compounds in BNCT, which may contribute to the development of new-generation radioimmunotherapy. However, there are still many unknowns about the mechanisms of this type of strategy and further experimental research is needed.
Abbreviations
HIV-1, Human immunodeficiency virus type 1; BNCT, Boron neutron capture therapy; M1, Classically activated macrophages; Th1, T-helper 1 lymphocytes; IFN-γ, Interferon γ; LPS, Lipopolysaccharide; TNF-α, Tumor necrosis factor α; IL, Interleukin; CCL, C-C motif chemokine ligands; CXCL, C-X-C motif chemokine ligands; ROS, Reactive oxygen species; NOS, Nitric oxide synthase; MHC II, Major histocompatibility complex II; TLR, Toll-like receptor; iNOS, Inducible nitric oxide synthase; NF-kB, Nuclear factor kappa-light-chain-enhancer of activated B cells; STAT, Signal transducer and activator of transcription; IRF, Interferon regulatory factor; TCA, Tricarboxylic acid cycle; M2, Alternatively activated macrophages; Th2, T-helper 2 lymphocytes; TGF-β, Transforming growth factor β; IC, Immune complexes; GCs, Glucocorticoids; Arg-1, Arginase-1; EGF, Epidermal growth factor; IDO1, Indoleamine 2,3-dioxygenase; JMJD3, Jumonji domain-containing protein D3; PPAR, Peroxisome proliferator- activated receptor; IGF, Insulin-like growth factor; TME, Tumor microenvironment; A2R, A2 adenosine receptor; VEGF, Vascular endothelial growth factor; TAMs, Tumor-associated macrophages; CCR, C-C chemokine receptor; M-CSF, Macrophage colony-stimulating factor; MDSCs, Myeloid-derived suppressor cells; MMP, Matrix metalloproteinases; PDGF, Platelet-derived growth factor; PGE2, Prostaglandin E2; PD-1, Programmed death receptor 1; CTLA-4, Cytotoxic T cell antigen 4; CSF-1R, Colony stimulating factor 1 receptor; SIRPα, Signal regulatory protein α; PEG, Polyethylene glycol; EPR, Enhanced permeability and retention effect; RES, Reticuloendothelial system; PTX, Paclitaxel; SOC, N-succinyl-N′-octyl chitosan; DOX, Doxorubicin; PTT, Photothermal therapy; NIR, Near-infrared laser; SN38, 7-ethyl-10-hydroxycamptothecin; PLGA, poly(lactide-co-glycolide); IC50, Half-maximal inhibitory concentration; AuNS, Gold-silica nanoshells; CIB1, Calcium integrin binding protein-1; siRNA, Small interfering RNA; Fe3O4, Iron oxide; MSNCs, Mesoporous silica nanocapsules; AA-PEG, Aminoethylanisamide-polyethylene glycol; SPARC, Secreted protein acidic and rich in cysteine; PBMCs, Peripheral blood mononuclear cells; BNNTs, Boron nitride nanotubes; BCDs, Boron-containing carbon dots; BPA-F, Boronophenylalanine–fructose.
Acknowledgments
This work was supported by grants from the National Science Center, Poland (Grant No. 2022/45/N/NZ5/03204 and 2019/33/B/NZ5/02212).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflict of interest.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Yu J, Sun H, Cao W, Song Y, Jiang Z. Research progress on dendritic cell vaccines in cancer immunotherapy. Exp Hematol Oncol. 2022;11(1):3. doi:10.1186/s40164-022-00257-2
3. Soehnlein O, Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10(6):427–439. doi:10.1038/nri2779
4. Chao CJ, Zhang E, Zhao Z. Engineering cells for precision drug delivery: new advances, clinical translation, and emerging strategies. Adv Drug Deliv Rev. 2023;197:114840. doi:10.1016/j.addr.2023.114840
5. Wang H-F, Liu Y, Yang G, Zhao C-X. Macrophage-mediated cancer drug delivery. Mater Today Sustain. 2021;100055:11–12. doi:10.1016/j.mtsust.2020.100055
6. Zhao Y, Haney MJ, Mahajan V, et al. Active targeted macrophage-mediated delivery of catalase to affected brain regions in models of parkinson’s disease. J Nanomed Nanotechnol. 2011;S4:003. doi:10.4172/2157-7439.S4-003
7. Batrakova EV, Li S, Reynolds AD, et al. A macrophage-nanozyme delivery system for Parkinson’s disease. Bioconjug Chem. 2007;18(5):1498–1506. doi:10.1021/bc700184b
8. Dou H, Destache CJ, Morehead JR, et al. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood. 2006;108(8):2827–2835. doi:10.1182/blood-2006-03-012534
9. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle Uptake: the phagocyte problem. Nano Today. 2015;10(4):487–510. doi:10.1016/j.nantod.2015.06.006
10. Serhan CN, Ward PA, Gilroy DW. Fundamentals of Inflammation. Cambridge University Press; 2010. doi:10.1017/CBO9781139195737
11. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887–904. doi:10.1038/nrd.2018.169
12. Kiefer R, Jurisic M, Dahlem C, et al. Targeted delivery of functionalized PLGA nanoparticles to macrophages by complexation with the yeast Saccharomyces cerevisiae. Biotechnol Bioeng. 2020;117(3):776–788. doi:10.1002/bit.27226
13. Visser JG, Van Staden ADP, Smith C. Harnessing macrophages for controlled-release drug delivery: lessons from microbes. Front Pharmacol. 2019;10:22. doi:10.3389/fphar.2019.00022
14. Yao Y, Xu XH, Jin L. Macrophage polarization in physiological and pathological pregnancy. Front Immunol. 2019;10:792. doi:10.3389/fimmu.2019.00792
15. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol. 2019;12(1):76. doi:10.1186/s13045-019-0760-3
16. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi:10.1038/nri3073
17. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. 2019;106(2):345–358. doi:10.1002/JLB.3RU1018-378RR
18. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084. doi:10.3389/fimmu.2019.01084
19. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. doi:10.1016/j.it.2004.09.015
20. Zheng X, Turkowski K, Mora J, et al. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget. 2017;8(29):48436–48452. doi:10.18632/oncotarget.17061
21. Grégoire H, Roncali L, Rousseau A, et al. Targeting tumor associated macrophages to overcome conventional treatment resistance in glioblastoma. Front Pharmacol. 2020;11:368. doi:10.3389/fphar.2020.00368
22. Najafi M, Hashemi Goradel N, Farhood B, et al. Macrophage polarity in cancer: a review. J Cell Biochem. 2019;120(3):2756–2765. doi:10.1002/jcb.27646
23. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014;5:614. doi:10.3389/fimmu.2014.00614
24. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. 2017;8:289. doi:10.3389/fimmu.2017.00289
25. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262(1):153–166. doi:10.1111/imr.12218
26. Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin I, Orekhov AN. Macrophage phenotypic plasticity in atherosclerosis: the associated features and the peculiarities of the expression of inflammatory genes. Int J Cardiol. 2015;184:436–445. doi:10.1016/j.ijcard.2015.03.055
27. Rőszer T. Understanding the mysterious m2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015;2015:816460. doi:10.1155/2015/816460
28. Tarique AA, Logan J, Thomas E, Holt PG, Sly PD, Fantino E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am J Respir Cell Mol Biol. 2015;53(5):676–688. doi:10.1165/rcmb.2015-0012OC
29. Ruytinx P, Proost P, Van Damme J, Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol. 2018;9:1930. doi:10.3389/fimmu.2018.01930
30. Andón FT, Digifico E, Maeda A, et al. Targeting tumor associated macrophages: the new challenge for nanomedicine. Semin Immunol. 2017;34:103–113. doi:10.1016/j.smim.2017.09.004
31. Laviron M, Boissonnas A. Ontogeny of tumor-associated macrophages. Front Immunol. 2019;10:1799. doi:10.3389/fimmu.2019.01799
32. Grossman JG, Nywening TM, Belt BA, et al. Recruitment of CCR2+ tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology. 2018;7(9):e1470729. doi:10.1080/2162402X.2018
33. Kadomoto S, Izumi K, Mizokami A. Roles of CCL2-CCR2 axis in the tumor microenvironment. Int J Mol Sci. 2021;22(16):8530. doi:10.3390/ijms22168530
34. Cortez-Retamozo V, Etzrodt M, Newton A, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012;109(7):2491–2496. doi:10.1073/pnas.1113744109
35. Zhu Y, Herndon JM, Sojka DK, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47(2):323–338.e6. doi:10.1016/j.immuni.2017.07.014
36. Loyher PL, Hamon P, Laviron M, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. 2018;215(10):2536–2553. doi:10.1084/jem.20180534
37. Li X, Liu R, Su X, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019;18(1):177. doi:10.1186/s12943-019-1102-3
38. Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. 2020;10:188. doi:10.3389/fonc.2020.00188
39. Davidov V, Jensen G, Mai S, Chen SH, Pan PY. Analyzing one cell at a TIME: analysis of myeloid cell contributions in the tumor immune microenvironment. Front Immunol. 2020;11:1842. doi:10.3389/fimmu.2020.01842
40. Corzo CA, Condamine T, Lu L, et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–2453. doi:10.1084/jem.20100587
41. Malfitano AM, Pisanti S, Napolitano F, Di Somma S, Martinelli R, Portella G. Tumor-associated macrophage status in cancer treatment. Cancers. 2020;12(7):1987. doi:10.3390/cancers12071987
42. Pan Y, Yu Y, Wang X, Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. 2020;11:583084. doi:10.3389/fimmu.2020.583084
43. Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol. 2018;9:2629. doi:10.3389/fimmu.2018.02629
44. Degboé Y, Poupot R, Poupot M. Repolarization of unbalanced macrophages: unmet medical need in chronic inflammation and cancer. Int J Mol Sci. 2022;23:1496. doi:10.3390/ijms23031496
45. Rodriguez PC, Zea AH, DeSalvo J, et al. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171(3):1232–1239. doi:10.4049/jimmunol.171.3
46. Munn DH, Sharma MD, Baban B, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi:10.1016/j.immuni.2005.03.013
47. Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. 2017;9(3):289–302. doi:10.2217/imt-2016-0135
48. Curiel TJ, Wei S, Dong H, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–567. doi:10.1038/nm863
49. Lesokhin AM, Hohl TM, Kitano S, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72(4):876–886. doi:10.1158/0008-5472.CAN-11-1792
50. Liu J, Zhang N, Li Q, et al. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS One. 2011;6(4):e19495. doi:10.1371/journal.pone.0019495
51. Chao MP, Weissman IL, Majeti R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225–232. doi:10.1016/j.coi.2012.01.010
52. Moreau MF, Guillet C, Massin P, et al. Comparative effects of five bisphosphonates on apoptosis of macrophage cells in vitro. Biochem Pharmacol. 2007;73(5):718–723. doi:10.1016/j.bcp.2006.09.031
53. Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med. 2011;9:177. PMID: 22005011. doi:10.1186/1479-5876-9-177
54. Zang X, Zhang X, Hu H, et al. Targeted delivery of zoledronate to tumor-associated macrophages for cancer immunotherapy. Mol Pharm. 2019;16(5):2249–2258. doi:10.1021/acs.molpharmaceut.9b00261
55. Healey JH, Tap WD, Gelhorn HL, et al. Pexidartinib provides modest pain relief in patients with tenosynovial giant cell tumor: results from ENLIVEN. Clin Orthop Relat Res. 2023;481(1):107–116. doi:10.1097/CORR.0000000000002335
56. Sun Y, Yang L, Hao X, et al. Phase I dose-escalation study of chiauranib, a novel angiogenic, mitotic, and chronic inflammation inhibitor, in patients with advanced solid tumors. J Hematol Oncol. 2019;12(1):9. doi:10.1186/s13045-018-0695-0
57. National Library of Medicine. Clinicaltrials.gov Database. Available from: https://clinicaltrials.gov/ct2/show/NCT04921527.
58. National Library of Medicine. Clinicaltrials.gov Database. Available from: https://clinicaltrials.gov/ct2/show/NCT04830813.
59. Cassier PA, Italiano A, Gomez-Roca C, et al. Long-term clinical activity, safety and patient-reported quality of life for emactuzumab-treated patients with diffuse-type tenosynovial giant-cell tumour. Eur J Cancer. 2020;141:162–170. doi:10.1016/j.ejca.2020.09.038
60. Nywening TM, Wang-Gillam A, Sanford DE, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–662. doi:10.1016/S1470-2045(16)00078-4
61. National Library of Medicine. Clinicaltrials.gov Database. Available online: https://clinicaltrials.gov/ct2/show/NCT03838367.
62. Halama N, Zoernig I, Berthel A, et al. tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell. 2016;29(4):587–601. doi:10.1016/j.ccell.2016.03.005
63. Sikic BI, Lakhani N, Patnaik A, et al. First-in-human, first-in-class phase I trial of the anti-CD47 antibody hu5F9-G4 in patients with advanced cancers. J Clin Oncol. 2019;37(12):946–953. doi:10.1200/JCO.18.02018
64. National Library of Medicine. Clinicaltrials.gov Database. Available from: https://clinicaltrials.gov/ct2/show/NCT03512340.
65. National Library of Medicine. Clinicaltrials.gov Database. Available from: https://clinicaltrials.gov/ct2/show/NCT03990233.
66. National Library of Medicine. Clinicaltrials.gov Database. Available from: https://clinicaltrials.gov/ct2/show/NCT05249426.
67. Paterson AH, Anderson SJ, Lembersky BC, et al. Oral clodronate for adjuvant treatment of operable breast cancer (national surgical adjuvant breast and bowel project protocol B-34): a multicentre, placebo-controlled, randomised trial. Lancet Oncol. 2012;13(7):734–742. doi:10.1016/S1470-2045(12)70226-7
68. National Library of Medicine. Clinicaltrials.gov Database. Available from: https://clinicaltrials.gov/ct2/show/NCT01198457.
69. Terpos E, Raje N, Croucher P, et al. Denosumab compared with zoledronic acid on PFS in multiple myeloma: exploratory results of an international Phase 3 study. Blood Adv. 2021;5(3):725–736. doi:10.1182/bloodadvances.2020002378
70. National Library of Medicine. Clinicaltrials.gov Database. Available from: https://clinicaltrials.gov/ct2/show/NCT00301873.
71. Vinogradov S, Warren G, Wei X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine. 2014;9(5):695–707. doi:10.2217/nnm.14.13
72. Ventola CL. Progress in nanomedicine: approved and investigational nanodrugs. P T. 2017;42(12):742–755.
73. Qi Y, Yan X, Xia T, Liu S. Use of macrophage as a Trojan horse for cancer nanotheranostics. Mater Des. 2021;198:109388. doi:10.1016/j.matdes.2020.109388
74. Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi:10.1146/annurev.immunol.17.1.593
75. Kumari S, Mg S, Mayor S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010;20(3):256–275. doi:10.1038/cr.2010.19
76. Yue H, Wei W, Yue Z, et al. Particle size affects the cellular response in macrophages. Eur J Pharm Sci. 2010;41(5):650–657. doi:10.1016/j.ejps.2010.09.006
77. Adjei IM, Sharma B, Labhasetwar V. Nanoparticles: cellular uptake and cytotoxicity. Adv Exp Med Biol. 2014;811:73–91. doi:10.1007/978-94-017-8739-0_5
78. Petithory T, Pieuchot L, Josien L, Ponche A, Anselme K, Vonna L. Size-dependent internalization efficiency of macrophages from adsorbed nanoparticle-based monolayers. Nanomaterials. 2021;11(8):1963. doi:10.3390/nano11081963
79. Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm Res. 2008;25(8):1815–1821. doi:10.1007/s11095-008-9562-y
80. Walkey C, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134(4):2139–2147. doi:10.1021/ja2084338
81. Alqahtani MS, Syed R, Alshehri M. Size-dependent phagocytic uptake and immunogenicity of gliadin nanoparticles. Polymers. 2020;12(11):2576. doi:10.3390/polym12112576
82. Mendes RG, Mandarino A, Koch B, et al. Size and time dependent internalization of label-free nano-graphene oxide in human macrophages. Nano Res. 2017;10:1980–1995. doi:10.1007/s12274-016-1385-2
83. Chang YN, Guo H, Li J, et al. Adjusting the balance between effective loading and vector migration of macrophage vehicles to deliver nanoparticles. PLoS One. 2013;8(10):e76024. doi:10.1371/journal.pone.0076024
84. Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103(13):4930–4934. doi:10.1073/pnas.0600997103
85. Sharma G, Valenta DT, Altman Y, et al. Polymer particle shape independently influences binding and internalization by macrophages. J Control Release. 2010;147(3):408–412. doi:10.1016/j.jconrel.2010.07.116
86. Lu Z, Qiao Y, Zheng XT, Chan-Park MB, Li CM. Effect of particle shape on phagocytosis of CdTe quantum dot–cystine composites. Med Chem Commun. 2010;1(1):84. doi:10.1039/C0MD00008F
87. Paul D, Achouri S, Yoon YZ, Herre J, Bryant CE, Cicuta P. Phagocytosis dynamics depends on target shape. Biophys J. 2013;105(5):1143–1150. doi:10.1016/j.bpj.2013.07.036
88. Yi X, Shi X, Gao H. Cellular uptake of elastic nanoparticles. Phys Rev Lett. 2011;107(9):098101. doi:10.1103/PhysRevLett.107.098101
89. Hui Y, Yi X, Wibowo D, et al. Nanoparticle elasticity regulates phagocytosis and cancer cell uptake. Sci Adv. 2020;6(16):eaaz4316. doi:10.1126/sciadv.aaz4316
90. Xia T, Kovochich M, Liong M, Zink JI, Nel AE. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano. 2008;2(1):85–96. doi:10.1021/nn700256c
91. Stephan MT, Irvine DJ. Enhancing cell therapies from the outside in: cell surface engineering using synthetic nanomaterials. Nano Today. 2011;6(3):309–325. doi:10.1016/j.nantod.2011.04.001
92. Ahsan F, Rivas IP, Khan MA, Torres Suarez AI. Targeting to macrophages: role of physicochemical properties of particulate carriers-liposomes and microspheres-on the phagocytosis by macrophages. J Control Release. 2002;79(1–3):29–40. doi:10.1016/s0168-3659(01)00549-1
93. Kumar PV, Asthana A, Dutta T, Jain NK. Intracellular macrophage uptake of rifampicin loaded mannosylated dendrimers. J Drug Target. 2006;14(8):546–556. doi:10.1080/10611860600825159
94. Si J, Shao S, Shen Y, Wang K. Macrophages as active nanocarriers for targeted early and adjuvant cancer chemotherapy. Small. 2016;12(37):5108–5119. doi:10.1002/smll.201601282
95. Pang L, Qin J, Han L, et al. Exploiting macrophages as targeted carrier to guide nanoparticles into glioma. Oncotarget. 2016;7(24):37081–37091. doi:10.18632/oncotarget.9464
96. Dymova MA, Taskaev SY, Richter VA, Kuligina EV. Boron neutron capture therapy: current status and future perspectives. Cancer Commun. 2020;40(9):406–421. doi:10.1002/cac2.12089
97. Caro C, Avasthi A, Paez-Muñoz JM, Pernia Leal M, García-Martín ML. Passive targeting of high-grade gliomas via the EPR effect: a closed path for metallic nanoparticles? Biomater Sci. 2021;9(23):7984–7995. doi:10.1039/d1bm01398j
98. Corraliza I. Recruiting specialized macrophages across the borders to restore brain functions. Front Cell Neurosci. 2014;8:262. doi:10.3389/fncel.2014.00262
99. Hussain SF, Yang D, Suki D, et al. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006;8(3):261–279. doi:10.1215/15228517-2006-008
100. Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J Control Release. 2014;190:531–541. doi:10.1016/j.jconrel.2014.03.050
101. Shields CW, Evans MA, Wang LL-W, et al. Cellular backpacks for macrophage immunotherapy. Sci Adv. 2020;6(18):eaaz6579. doi:10.1126/sciadv.aaz6579
102. Liang T, Zhang R, Liu X, et al. Recent advances in macrophage-mediated drug delivery systems. Int J Nanomedicine. 2021;16:2703–2714. doi:10.2147/IJN.S298159
103. Batrakova EV, Gendelman HE, Kabanov AV. Cell-mediated drug delivery. Expert Opin Drug Deliv. 2011;8(4):415–433. doi:10.1517/17425247.2011.559457
104. Song C, Xu J, Gao C, Zhang W, Fang X, Shang Y. Nanomaterials targeting macrophages in sepsis: a promising approach for sepsis management. Front Immunol. 2022;13:1026173. doi:10.3389/fimmu.2022.1026173
105. Dobrovolskaia MA, McNeil SE. Understanding the correlation between in vitro and in vivo immunotoxicity tests for nanomedicines. J Control Release. 2013;172(2):456–466. doi:10.1016/j.jconrel.2013.05.025
106. Choi M-R, Stanton-Maxey KJ, Stanley JK, et al. A cellular trojan horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7(12):3759–3765. doi:10.1021/nl072209h
107. Zhang W, Wang M, Tang W, et al. Nanoparticle-Laden macrophages for tumor-tropic drug delivery. Adv Mater. 2018;30(50):e1805557. doi:10.1002/adma.201805557
108. Li S, Feng S, Ding L, et al. Nanomedicine engulfed by macrophages for targeted tumor therapy. Int J Nanomedicine. 2016;11:4107–4124. doi:10.2147/IJN.S110146
109. Fu J, Wang D, Mei D, et al. Macrophage mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J Control Release. 2015;204:11–19. doi:10.1016/j.jconrel.2015.01.039
110. Qiang L, Cai Z, Jiang W, et al. A novel macrophage-mediated biomimetic delivery system with NIR-triggered release for prostate cancer therapy. J Nanobiotechnology. 2019;17(1):83. doi:10.1186/s12951-019-0513-z
111. Nguyen VD, Min HK, Kim DH, et al. Macrophage-mediated delivery of multifunctional nanotherapeutics for synergistic chemo-photothermal therapy of solid tumors. ACS Appl Mater Interfaces. 2020;12(9):10130–10141. doi:10.1021/acsami.9b23632
112. Huang Z, Sun X, Liu X, Shen Y, Wang K. Macrophages as an active tumour-targeting carrier of SN38-nanoparticles for cancer therapy. J Drug Target. 2018;26(5–6):458–465. doi:10.1080/1061186X.2017.1419359
113. Choi J, Kim HY, Ju EJ, et al. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials. 2012;33(16):4195–4203. doi:10.1016/j.biomaterials.2012.02.022
114. Pang L, Zhu Y, Qin J, Zhao W, Wang J. Primary M1 macrophages as multifunctional carrier combined with PLGA nanoparticle delivering anticancer drug for efficient glioma therapy. Drug Deliv. 2018;25(1):1922–1931. doi:10.1080/10717544.2018.1502839
115. Madsen SJ, Christie C, Hong SJ, et al. Nanoparticle-loaded macrophage-mediated photothermal therapy: potential for glioma treatment. Lasers Med Sci. 2015;30(4):1357–1365. doi:10.1007/s10103-015-1742-5
116. Choi MR, Bardhan R, Stanton-Maxey KJ, et al. Delivery of nanoparticles to brain metastases of breast cancer using a cellular Trojan horse. Cancer Nanotechnol. 2012;3(1–6):47–54. doi:10.1007/s12645-012-0029-9
117. Wayne EC, Long C, Haney MJ, et al. Targeted delivery of siRNA lipoplexes to cancer cells using macrophage transient horizontal gene transfer. Adv Sci. 2019;6(21):1900582. doi:10.1002/advs.201900582
118. Muthana M, Giannoudis A, Scott SD, et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011;71(5):1805–1815. doi:10.1158/0008-5472.CAN-10-2349
119. Meng QF, Rao L, Zan M, et al. Macrophage membrane-coated iron oxide nanoparticles for enhanced photothermal tumor therapy. Nanotechnology. 2018;29(13):134004. doi:10.1088/1361-6528/aaa7c7
120. Xuan M, Shao J, Dai L, Li J, He Q. Macrophage cell membrane camouflaged au nanoshells for in vivo prolonged circulation life and enhanced cancer photothermal therapy. ACS Appl Mater Interfaces. 2016;8(15):9610–9618. doi:10.1021/acsami.6b00853
121. Bhattacharyya S, Ghosh SS. Transmembrane TNFα-expressed macrophage membrane-coated chitosan nanoparticles as cancer therapeutics. ACS Omega. 2020;5(3):1572–1580. doi:10.1021/acsomega.9b03531
122. Xuan M, Shao J, Dai L, He Q, Li J. Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv Healthc Mater. 2015;4(11):1645–1652. doi:10.1002/adhm.201500129
123. Cao H, Dan Z, He X, et al. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano. 2016;10(8):7738–7748. doi:10.1021/acsnano.6b03148
124. Cao X, Tan T, Zhu D, et al. Paclitaxel-loaded macrophage membrane camouflaged albumin nanoparticles for targeted cancer therapy. Int J Nanomedicine. 2020;15:1915–1928. doi:10.2147/IJN.S244849
125. Zhang Y, Cai K, Li C, et al. Macrophage-membrane-coated nanoparticles for tumor-targeted chemotherapy. Nano Lett. 2018;18(3):1908–1915. doi:10.1021/acs.nanolett.7b05263
126. Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32:2002054. doi:10.1002/adma.202002054
127. Kim MS, Haney MJ, Zhao Y, et al. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: in vitro and in vivo evaluations. Nanomedicine. 2018;14(1):195–204. doi:10.1016/j.nano.2017.09.011
128. Haney MJ, Zhao Y, Jin YS, et al. Macrophage-derived extracellular vesicles as drug delivery systems for triple negative breast cancer (TNBC) therapy. J Neuroimmune Pharmacol. 2020;15(3):487–500. doi:10.1007/s11481-019-09884-9
129. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. doi:10.1016/j.addr.2010.03.011
130. Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem. 2010;21(5):797–802. doi:10.1021/bc100070g
131. Huang YJ, Hung KC, Hung HS, Hsu SH. Modulation of macrophage phenotype by biodegradable polyurethane nanoparticles: possible relation between macrophage polarization and immune response of nanoparticles. ACS Appl Mater Interfaces. 2018;10(23):19436–19448. doi:10.1021/acsami.8b04718
132. Fuchs AK, Syrovets T, Haas KA, et al. Carboxyl- and amino-functionalized polystyrene nanoparticles differentially affect the polarization profile of M1 and M2 macrophage subsets. Biomaterials. 2016;85:78–87. doi:10.1016/j.biomaterials.2016.01.064
133. Reichel D, Tripathi M, Perez JM. Biological effects of nanoparticles on macrophage polarization in the tumor microenvironment. Nanotheranostics. 2019;3(1):66–88. doi:10.7150/ntno.30052
134. Zhang F, Parayath NN, Ene CI, et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun. 2019;10(1):3974. doi:10.1038/s41467-019-11911-5
135. Wang T, Zhang J, Hou T, Yin X, Zhang N. Selective targeting of tumor cells and tumor associated macrophages separately by twin-like core-shell nanoparticles for enhanced tumor-localized chemoimmunotherapy. Nanoscale. 2019;11(29):13934–13946. doi:10.1039/C9NR03374B
136. Chen P, Zhang X, Venosa A, et al. A novel bivalent mannosylated targeting ligand displayed on nanoparticles selectively targets anti-inflammatory M2 macrophages. Pharmaceutics. 2020;12(3):243. doi:10.3390/pharmaceutics12030243
137. Zhao P, Wang Y, Kang X, et al. Dual-targeting biomimetic delivery for anti-glioma activity via remodeling the tumor microenvironment and directing macrophage-mediated immunotherapy. Chem Sci. 2018;9(10):2674–2689. doi:10.1039/c7sc04853j
138. Chen S, Lai SWT, Brown CE, Feng M. Harnessing and enhancing macrophage phagocytosis for cancer therapy. Front Immunol. 2021;12:635173. doi:10.3389/fimmu.2021.635173
139. He H, Li J, Jiang P, et al. The basis and advances in clinical application of boron neutron capture therapy. Radiat Oncol. 2021;16(1):216. doi:10.1186/s13014-021-01939-7
140. Wróblewska A, Szermer-Olearnik B, Pajtasz-Piasecka E. Boron-rich nanoparticles as potential carriers in boron-neutron capture therapy. Adv Hyg Exp Med. 2021;75:122–132. doi:10.5604/01.3001.0014.7760
141. Khan AA, Maitz C, Quanyu C, Hawthorne F. BNCT induced immunomodulatory effects contribute to mammary tumor inhibition. PLoS One. 2019;14(9):e0222022. doi:10.1371/journal.pone.0222022
142. Kozień D, Szermer-Olearnik B, Rapak A, et al. Boron-rich boron carbide nanoparticles as a carrier in boron neutron capture therapy: their influence on tumor and immune phagocytic cells. Materials. 2021;14(11):3010. doi:10.3390/ma14113010
143. Rocca A, Marino A, Del Turco S, et al. Pectin-coated boron nitride nanotubes: in vitro cyto-/immune-compatibility on RAW 264.7 macrophages. Biochim Biophys Acta. 2016;1860(4):775–784. doi:10.1016/j.bbagen.2016.01.020
144. Li J, Kong J, Ma S, et al. Exosome‐coated 10 B carbon dots for precise boron neutron capture therapy in a mouse model of glioma in situ. Adv Funct Mater. 2021;31(24):
145. Wu WC, Wang SH, Ou ST, Liu YH, Liu BH, Tseng FG. Electrosprayed chitosan/alginate/polyvinyl alcohol nanoparticles as boric acid carriers for 10Boron neutron capture therapy. Nanomedicine. 2020;15(11):1067–1077. doi:10.2217/nnm-2019-0465
146. Nakagawa N, Akai F, Fukawa N, et al. Early effects of boron neutron capture therapy on rat glioma models. Brain Tumor Pathol. 2007;24(1):7–13. doi:10.1007/s10014-007-0214-4
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.