Back to Journals » Journal of Inflammation Research » Volume 18
Macrophage Metabolic Reprogramming in Inflammatory Bowel Diseases: From Pathogenesis to Therapy
Authors Zhang YF ![]() , Shi TT, Lin YL, Zhu YT, Lin S, Fang TY
, Shi TT, Lin YL, Zhu YT, Lin S, Fang TY
Received 14 April 2025
Accepted for publication 14 August 2025
Published 27 August 2025 Volume 2025:18 Pages 11821—11839
DOI https://doi.org/10.2147/JIR.S534447
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Nadia Andrea Andreani
Yi-Fang Zhang,1 Ting Ting Shi,1 Yi-Ling Lin,2 Yu-Ting Zhu,3 Shu Lin,4,5 Tai-Yong Fang1
1Department of Gastroenterology, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian, People’s Republic of China; 2Department of Gastroenterology, Anxi Maternal and Child Health Hospital, Quanzhou, Fujian, People’s Republic of China; 3School of Medicine and School of Biomedical Sciences, Huaqiao University, Quanzhou, 362000, People’s Republic of China; 4Centre of Neurological and Metabolic Research, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian, People’s Republic of China; 5Group of Neuroendocrinology, Garvan Institute of Medical Research, Sydney, NSW, Australia
Correspondence: Shu Lin, Centre of Neurological and Metabolic Research, The Second Affiliated Hospital of Fujian Medical University, No. 34 North Zhongshan Road, Quanzhou, Fujian, 362000, People’s Republic of China, Tel +86 15659068071, Email [email protected] Tai-Yong Fang, Department of Gastroenterology, The Second Affiliated Hospital of Fujian Medical University, No. 34 North Zhongshan Road, Quanzhou, Fujian, 362000, People’s Republic of China, Tel +86 13805992641, Email [email protected]
Abstract: Inflammatory bowel disease (IBD), encompassing two subtypes, ulcerative colitis, and Crohn’s disease, is a chronic, non-specific gastrointestinal disorder with a complex etiology stemming from various factors. The incidence of IBD has been steadily rising in the past few years, causing great physical and mental strain on patients. Traditional IBD therapeutic drugs include anti-inflammatory drugs, immunosuppressants, and biologics; however, they may have serious adverse effects. This has fueled active clinical research exploring new targets for IBD treatment, focusing on the unique metabolic pathways and functions of macrophages. Macrophage immune metabolism plays a crucial role in IBD; however, the mechanism is unclear. This review discussed the role and potential mechanisms of macrophage metabolic reprogramming in IBD and the link between macrophages and ferroptosis. While these findings from preclinical models suggest novel therapeutic avenues for IBD, such as targeting macrophage metabolic reprogramming and hypothetical approaches like ferroptosis modulation, their clinical applicability remains speculative; rigorous disease-specific validation is imperative.
Keywords: macrophage, metabolic reprogramming, inflammatory bowel diseases, pathogenesis, therapy
Introduction
Inflammatory bowel disease (IBD) is a chronic, non-specific gastrointestinal disorder encompassing two subtypes: Crohn’s disease (CD) and ulcerative colitis (UC). CD is characterized by transmural, progressive, and destructive chronic inflammation that can attack any segment of the gastrointestinal tract,1 while UC typically presents with superficial mucosal inflammation originating in the rectum and extends proximally.2 The rising prevalence of IBD in recent years poses a considerable burden on the quality of life and socioeconomic status of affected individuals.3 Although the exact etiology of IBD remains elusive, its pathogenesis involves genetic susceptibility, environmental influences, internal immunity, and the microbiota.4 Current IBD management relies on immunosuppressants, biologics, and small-molecule inhibitors. While these therapies can induce remission, significant limitations persist—including non-response in 30–40% of patients, risk of serious infections, and loss of efficacy over time.5 Under dual biologic therapy (DBT), for instance, the clinical response rate in refractory Crohn’s disease (CD) patients is only 50%, highlighting the therapeutic challenges in this patient population. This underscores an urgent need for novel therapeutic strategies targeting alternative pathways.6
Macrophages are immune cells derived from bone marrow progenitors and are present in various tissues.7,8 Macrophages and dendritic cells, collectively referred to as antigen-presenting cells (APCs), play a crucial role in maintaining homeostasis within the intestinal lamina propria.9 These cells are essential for immune surveillance, tolerance induction, and the regulation of inflammatory responses, ensuring the balance between immune activation and suppression in the gut environment. Macrophages undergo polarization into distinct functional phenotypes (M1/M2) from a resting state (M0) in response to various cytokines, growth factors, or other microenvironmental stimuli, a dynamic and reversible process.10 The intestine is enriched with macrophages of different phenotypes and functions, which synergistically maintain a healthy intestinal flora and tolerance to dietary antigens.8,11,12 One of the critical drivers of macrophage polarization is metabolic reprogramming in a variety of inflammatory diseases, including IBD.13,14 Macrophages undergo adaptive modifications in their metabolic pathways (such as glycolysis, oxidative phosphorylation, and fatty acid oxidation) during differentiation and activation to satisfy functional requirements under diverse physiological or pathological conditions, a process termed metabolic reprogramming.15 This process is termed metabolic reprogramming. Studies have shown that key metabolic regulators such as PPAR-γ and HIF can induce the polarization direction of macrophages to repair the intestinal mucosal barrier.16 Moreover, the metabolic process of macrophages also influences the polarization direction through transcriptional and post-transcriptional events. For example, N6-methyladenosine (m6A) modification has been proposed as a potential regulatory mechanism during macrophage metabolic reprogramming, though its role in IBD remains hypothetical and requires validation.17 However, critical knowledge gaps persist, such as regulatory nodes controlling metabolic switching (eg, the ROS-hexokinase/pyruvate kinase M2 isoform axis); microbiota-metabolite-epigenetic crosstalk; metabolic heterogeneity driving functional macrophage subsets. Unraveling these mechanisms is vital for developing precise metabolic interventions in inflammatory bowel disease (IBD). While recent reviews have comprehensively covered macrophage metabolism in IBD,16,18 they lack in-depth analysis of ferroptosis as a pathogenic hub and nanoparticle-based therapeutic targeting. This review uniquely integrates macrophage metabolic reprogramming with ferroptosis-driven immunopathology and critically evaluates emerging nanotherapies, bridging mechanistic insights to clinical translation gaps.
Ferroptosis is a distinct form of regulated cell death, distinguished by disturbed iron metabolism, lipid peroxidation, and glutathione (GSH) depletion.19 Ferroptosis is emerging as a key pathogenic driver in IBD macrophages. Unlike other death pathways, ferroptosis directly links iron dysregulation, oxidative stress, and chronic inflammation, amplifying tissue damage and fibrosis. Its unique mechanism and therapeutic tractability justify focused discussion in this review. An intrinsic link exists between ferroptosis and macrophage polarization, suggesting that ferroptosis can regulate macrophage polarization status. Therefore, elucidating this complex relationship between ferroptosis and macrophages will lay the groundwork for innovative targeted therapies. While preclinical data suggest a mechanistic link, ferroptosis-targeted therapies in IBD remain speculative and warrant rigorous disease-specific investigation. This article reviews and discusses the role and possible mechanisms of macrophage metabolic reprogramming in IBD and the connection between macrophages and ferroptosis. Moreover, the findings from this study may lead to the development of targeted therapies for IBD using macrophage metabolic reprogramming.
Discussion
IBD and Macrophage
IBD
IBD comprises a group of chronic inflammatory conditions affecting the gastrointestinal tract. Recent studies show that the development of IBD is intricately linked to the interaction of genetic factors, the gut microbiota, and the host immune system. Regarding genetic factors, genome-wide association studies have identified several genomic regions associated with the risk of IBD. Notably, the nucleotide-binding oligomerization domain-containing 2 (NOD2), autophagy-related 16-like 1 gene (ATG16L1), transforming growth factor β1 (TGF-β1), interleukin 23 receptor (Il23R) and toll-like receptor 4 (TLR4) are crucial in the progression of IBD fibrosis.20–24 Additionally, 28 genes shared between CD and UC have been identified.25 The immune system regulates the interaction between the host and the gut microbiota. Macrophages help to maintain intestinal homeostasis, and different subtypes are recruited to relevant sites of inflammation at different stages of disease.26 CD14+ macrophages, a subset of intestinal macrophages, are increased in Crohn’s disease (CD) patients. These cells exacerbate inflammation by initiating Th17/Th1 responses through the excessive secretion of IL-23 and TNF-α.27 Th17 cells play a critical role in the pathogenesis of inflammatory bowel disease (IBD), with an increased proportion observed in the blood of IBD patients.28 Th17 cells secrete IL-17 and IL-21, and excessive levels of these cytokines can disrupt the intestinal epithelial barrier.29 Reportedly, patients with IBD exhibit reduced bacterial diversity and abundance in their gut microbiota,30 particularly obligate anaerobes from the Bacteroidetes and Firmicutes phyla.31,32 Certain bacterial groups, such as Fusobacterium nucleatum and Escherichia coli, significantly influence the development and progression of colorectal cancer.33,34
Macrophage
Macrophages derived from blood monocytes exhibit remarkable plasticity, with phagocytosis and pathogen clearance as their primary function.35 However, recent studies using transgenic mouse models and cell tracking techniques revealed myb-independent hematopoiesis in early embryonic and adult stages.
These studies suggest that some tissue-resident macrophages originate from progenitor cells in the the yolk sac or fetal liver rather than from circulating blood monocytes.36 They include Kupffer cells in the liver, Langerhans cells in the epidermis, and microglia in the central nervous system. These macrophages are integral to various tissues, performing a range of supportive functions in the intestine, such as regulating immune responses, maintaining mucosal barrier integrity, regulating smooth muscle contraction, and protecting vascular function.37,38 Intestinal macrophages have a high phagocytic capacity and are responsible for preserving the integrity of the intestinal mucosal barrier by removing apoptotic and senescent epithelial cells. They promote epithelial integrity by secreting tissue-remodeling metalloproteinases and stimulatory factors that promote epithelial stem cell renewal, such as prostaglandin E2, hepatocyte growth factor, and Wnt ligands.38 The regulation of gastrointestinal motility is controlled by interactions between muscular layer macrophages and intestinal neurons. Studies have found that macrophages secrete bone morphogenetic protein 2, while enteric neurons release cytokine CSF1, together regulating intestinal activity and influencing gastrointestinal motility.39
Macrophages are traditionally classified into M1 and M2 based on the stimuli obtained in vitro. Lipopolysaccharide (LPS) stimulates the polarization of macrophages toward the M1 phenotype, while interleukin-4 (IL-4) induces polarization toward the M2 phenotype.40 Classically activated M1 macrophages exhibit pro-inflammatory properties and secrete various pro-inflammatory cytokines, including IL-6, TNF-α, IL-1, and IL-12. In contrast, alternatively activated M2 macrophages possess anti-inflammatory and pro-tumorigenic functions.41–43 Classically activated M1 macrophages have pro-inflammatory effects, whereas alternatively activated M2 macrophages have anti-inflammatory effects.41–43 M2 macrophages can be further classified into four subtypes: M2a, M2b, M2c, and M2d.44 The biomarkers of M1 and M2 macrophages also differ significantly. M1 macrophages are characterized by the expression of CD80, CD86, CD64, and CD32, while M2 macrophages predominantly express CD163 and CD206.45,46
Metabolic Reprogramming of Macrophages
Recent research on cellular metabolism underscores the critical of metabolic reprogramming in macrophage activation. Macrophage function is influenced by various metabolic pathways, including glycolysis, the tricarboxylic acid cycle (TCA), the pentose phosphate pathway (PPP), and lipid, amino acid, and macrophage micronutrient metabolism. They coordinate immune responses based on altered nutrient metabolism and polarize into M1 or M2 types, each with different metabolic and energy requirements. Therefore, we will discuss how disruptions in intracellular nutrient metabolism affect macrophage polarization and function in IBD.
Glucose Metabolism
Hypoxia is a key pathogenic mechanism of inflammatory tissues. In chronic inflammatory patho-immune environments, hypoxia and inflammation are often interdependent. Growing evidence suggests a link between IBD and mucosal hypoxia.47 Under hypoxic conditions, degradation of hypoxia-inducible factor 1α (HIF-1α) is inhibited,48 and expression of key glycolytic proteins is increased, causing macrophage metabolism to shift from oxidative phosphorylation (OXPHOS) to glycolysis.18 This metabolic reprogramming is a characteristic feature of M1 macrophages. Increased glycolysis enables rapid adaptation to the hypoxic tissue microenvironment to meet energy demands.49 During IBD, PKM2, a key glycolysis enzyme, translocates to the nucleus as a dimer, binds to HIF-1α, and stimulates aerobic glycolysis.50 Shikonin alleviates colitis in mice by inhibiting PKM2 dimerization and tetramerization in macrophages, hindering aerobic glycolytic reprogramming.51
HIF-1α is a pivotal regulator of inflammatory cell metabolism. It plays a crucial role in the response to hypoxia and promotes the expression of inflammatory genes.48 Specific deletion of HIF-1α in bone marrow cells ameliorates acute colitis.52 By integrating metabolomics and transcriptomic data, Jha et al revealed that pro-inflammatory M1 macrophages exhibit disrupted TCA carbon flux at two breakpoints, causing citrate and succinate accumulation.53–55 Succinate inhibits HIF-1α degradation, enhances HIF-1 stability, and induces IL-1α production via HIF-1β.56 Activated macrophages release succinate into the extracellular environment, increasing SUCNR1 expression and promoting IL-1β production, creating a positive feedback loop, and increasing SUCNR1 levels.14 SUCNR1 expression is elevates in the intestinal tissues of patients with CD, and its activation can trigger and progress inflammatory responses and fibrosis.57 Inhibition of citrate efflux leads to histone deacetylation, inactivating HIF-1α target genes, which causes metabolic inhibition of aerobic glycolysis and inflammatory responses.58
While inflammatory hypoxia typically promotes HIF-1α-driven glycolysis and M1 polarization, recent studies reveal context-dependent duality in its function. In colitis, HIF-1α activation via roxadustat (ROX) or HIF-overexpressing MSC-derived extracellular vesicles (EVs) promotes M2 polarization and mucosal healing.59,60 This apparent paradox may stem from two key factors. Firstly, HIF-1α initially exacerbates inflammatory responses in the early stages of disease (eg, via succinate/IL-1β), which subsequently facilitates tissue repair by inducing anti-inflammatory mediators such as IL-10. Secondly, microenvironmental signals, including pro-inflammatory priming factors like TNF-α, synergize with HIF-1α to optimize the cargo of extracellular vesicles (EVs), thereby promoting M2 reprogramming (Figure 1).
 |
Figure 1 Macrophage glucose metabolism in IBD. During inflammatory bowel disease (IBD), macrophages shift from oxidative phosphorylation to glycolytic metabolism. In the mitochondria, the tricarboxylic acid cycle presents two break points, resulting in the accumulation of citrate and succinate. Citrate can be transported to the cytoplasm via the SLC25a1 transporter on the mitochondrial membrane to produce reactive oxygen species (ROS), nitric oxide, and Prostaglandins. Succinate activates HIF-1α. Succinate can bind to SUCNR1 on the cell membrane and increase IL-1β. |
Lipid Metabolism
Studies indicate that lipid metabolism is critical for regulating immune cells, influencing inflammation and host defense. The peroxisome proliferator-activated receptor (PPAR) family includes ligand-activated transcription factors, with PPAR-γ mainly active in adipose tissue and macrophages.61,62 PPAR-γ serves as a pivotal immunometabolic regulator in the pathogenesis of IBD, yet the dichotomy of its functions and therapeutic translation remains contentious. Canonical perspectives posit that PPAR-γ activation exerts potent anti-inflammatory effects by suppressing NF-κB signaling and production of pro-inflammatory cytokines (eg, TNF-α, IL-6, IL-1β).63 PPAR-γ drives macrophage polarization toward an anti-inflammatory M2 phenotype, enhancing efferocytic clearance, promoting IL-10 secretion, and facilitating tissue repair through metabolic reprogramming toward fatty acid oxidation.64 In macrophages, PPAR-γ can inhibit pro-inflammatory molecule expression and regulate macrophage migration.65 Studies have reported that the differentiation of M2a macrophages is dependent on the ligand-activated transcription factor PPARγ.66 PPAR-γ activation promotes macrophage polarization toward M2 phenotype by increasing STAT6 phosphorylation.67 Animal studies demonstrate that in dextran sulfate sodium (DSS)-induced colitis, PPAR-γ gene-deficient mice exhibit exacerbated colonic injury compared to wild-type controls.68 PPAR-γ agonists have been shown to alleviate colonic inflammation induced by DSS.69 Macrophage-specific PPAR-γ knockout reduces the number of regulatory T (Treg) cells, increasing the susceptibility of mice to IBD and exacerbating disease severity.70 However, significant paradoxical aspects exist in its biological functions. On one hand, mice with intestinal epithelium-specific PPAR-γ deletion exhibit markedly increased tumor incidence in carcinogenesis models, supporting its tumor-suppressive role.71 On the other hand, early studies suggest that PPAR-γ activation may potentially promote colorectal tumorigenesis, sparking controversy regarding its function in IBD-associated colorectal cancer.72 Clinical evidence demonstrates that the expression level of PPAR-γ in the rectal tissue is significantly reduced in patients with active UC, whereas it is increased in patients with UC in remission.73 Downregulation of PPAR-γ is observed in both active and inactive stages of ulcerative colitis (UC) and Crohn’s disease (CD), suggesting it may represent a chronic pathological feature of inflammatory bowel disease (IBD) rather than being solely associated with inflammatory activity.74 Mechanistically, PPARγ enhances glucose uptake by upregulating Glucose transporter type 1(GLUT1) expression, thereby providing substrates for lipid synthesis and playing a pivotal role in regulating glucose and lipid metabolism. PPAR-γ also suppresses the expression of HIF-1α—a key regulator of glycolysis—while promoting arginase-1 expression.75 Arginase depletes its substrate arginine, consequently reducing nitric oxide (NO) production. This NO reduction mitigates oxidative stress and inflammatory responses, thereby facilitating the transition of macrophages from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 state.68 Furthermore, PPAR-γ promotes the expression of mitochondrial fatty acid transport proteins. The carnitine palmitoyltransferase (CPT) system comprises two isozymes—CPT1 and CPT2—which collectively constitute the carnitine shuttle and function synergistically in FAO. CPT1 transports activated fatty acids into mitochondria, whereas CPT2 regenerates oxidizable fatty acyl-CoA within the mitochondrial matrix. PPAR-γ agonists (eg, troglitazone) significantly upregulate mRNA levels of carnitine palmitoyltransferase I (CPT-1).76 Nomura et al observed high expression of M2 macrophage markers, production of anti-inflammatory factors, and impaired FAO in a mouse model with macrophage-specific CPT2 deficiency.77 In human and murine macrophages, the inflammatory factors IFN-γ and TNFα reduced PPAR-γ expression and activity, affecting lipid metabolic homeostasis and altering metabolism and inflammatory disease progression.78 Research has shown that arachidonic acid inhibits the polarization of M2 macrophages. However, its metabolite, prostaglandin E2 (PGE2), promotes M2 polarization and enhances OXPHOS by inhibiting PPAR-γ.79 A study found that PPAR-γ expression in both inflamed and non-inflamed regions of UC was lower than that in the control group and CD group.80 This reduction may be attributed to impaired TLR4 and PPAR-γ signaling. In TLR4-SNP mice treated with DSS, the capacity for PPARγ activation and M2a macrophage marker expression was found to be impaired.81 During IBD, inflammatory factors such as IFN-γ and TNFα down-regulate PPAR-γ expression, resulting in reduced FAO, impaired polarization, and increased FAS in M2 macrophages.
While PPAR-γ primarily governs FAO, macrophage lipogenesis is coordinately regulated by sterol regulatory element binding proteins (SREBPs), which control de novo fatty acid synthesis.82 Sterol regulatory element-binding proteins (SREBPs), master transcriptional regulators of cellular fat metabolism, promote lipid accumulation. This dual regulatory axis—PPAR-γ-driven FAO and SREBP-mediated synthesis—collectively shapes lipid metabolism during macrophage polarization. SREBP-1a is responsible for regulating two key enzymes involved in lipid synthesis: acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS). In M1 macrophages, the activation of SREBP-1a leads to enhanced fatty acid and cholesterol synthesis, thereby supporting the energy demands associated with inflammation and immune responses. Srebp1-a, which is abundantly expressed in macrophages, plays a positive regulatory role in the inflammatory response. Reportedly, lipopolysaccharide (LPS) induction in macrophages upregulates Srebp1-a expression. Bone marrow-derived macrophages lacking SREBP-1a exhibit reduced lipogenesis and phagocytosis.83 Additionally, mice lacking SREBP-1a show defects in innate immune responses.84 The activation of SREBP1 is closely associated with the activation of M2 macrophages. IL-4 stimulation leads to the activation of SREBP1, resulting in increased fatty acid synthesis.85 In patients with ulcerative colitis (UC), SREBP2 is significantly elevated, and SREBP2 can promote intestinal mucosal healing by regulating cholesterol synthesis86 (Figure 2).
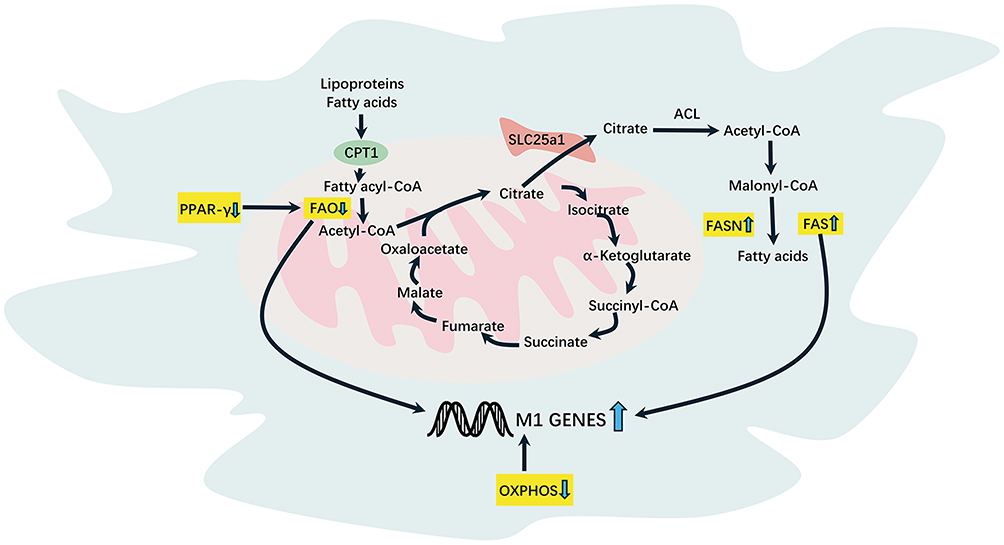 |
Figure 2 Macrophage lipid metabolism in IBD. Downregulation of PPAR-γ expression leads to reduced fatty acid oxidation during IBD. Citrate can be transported to the cytoplasm via the SLC25a1 transporter on the mitochondrial membrane to generate Fatty acids in response to FASN, resulting in increased fatty acid synthase. |
Amino Acid Metabolism
Amino acids are pivotal regulators of various metabolic processes, especially in immune and inflammatory responses. The relationship between specific amino acids and IBD has recently garnered significant attention. Glutamine levels are significantly reduced in the serum of patients with IBD and DSS-induced colitis mice. Additionally, the absence of ATF4 reduces intestinal glutamine uptake.87–89 Some researchers proposed that glutamine is a significant factor in activating HIF-1 signaling and mTOR1C in LPS-induced M1 macrophages.56,90,91 Conversely, some studies indicated that glutamine metabolism favors M2 polarization. As a key regulator of macrophage phenotype, glutamine metabolism responds to different environmental stimuli.92 For instance, glutamine promotes macrophage M2 polarization by inhibiting SIRT5-mediated pyruvate dehydrogenase (PDH) desuccinylation.93 On the other hand, succinate, produced through glutamine-dependent anaerobic glycolysis or the γ-aminobutyric acid pathway, promotes M1-type macrophage polarization.94 Ala-Gln inhibits Th17 cell production, reduces inflammation, and decreases chemokine concentrations in intestinal tissues, thereby attenuating DSS-induced colitis-induced weight loss and colonic edema.95 A high α-KG/succinate ratio stimulates M2 macrophage activation whereas a low ratio favors M1 macrophage activation.96 This ratio regulates M1 polarization through PHD-dependent IKKb proline hydroxylation.97 SENP1-SIRT3 signaling deacetylates glutamate dehydrogenase 1, which promotes the hydrolysis of glutamine to α-KG, leading to macrophage M2 polarization.98
Arginine (Arg) is widely utilized in metabolism and supplemented in various disease states.99 In macrophages, Arg serves as a precursor of two crucial metabolic pathways. In M1 macrophage, it is converted to nitric oxide (NO) and citrulline by inducible NO synthase (iNOS).100 Endogenous NO restricts aconitase 2 and PDH, resulting in M1 macrophage entry into glycolysis.101 In M2 macrophages, Arg is converted to ornithine and urea by arginase-1 (Arg-1).102 A clinical study confirmed that L-Arg levels positively correlate with the severity of UC, with elevated levels observed in patients with severe colitis.103 Reportedly, patients with UC had reduced cellular uptake, increased NOS2 consumption, and decreased ARG1 expression, leading to decreased L-arginine in the tissues.104 Additionally, L-arginine supplementation attenuated the damage and inflammatory response in DSS-induced colitis.105 The active transport of L-arginine is mediated by a family of cationic amino acid transporter proteins, including SLC7A1 and SLC7A2.106 Deletion of SLC7A2 increased inflammatory factors in the colonic tissues and exacerbated colonic tumor development.107
Tryptophan (Trp) plays a role in various pathophysiological processes, mainly through metabolites that affect inflammatory responses, oxidative stress, immune responses, and intestinal homeostasis.108 M1 macrophages use Trp as a precursor for the synthesis of NO via the nitrate pathway.
The NO produced has antimicrobial defense and contributes to microbial killing in inflammatory sites. Emerging evidence links Trp metabolism disorders to IBD development. During IBD, macrophages typically exhibit increased Trp uptake to satisfy their needs in immunomodulation and antimicrobial activity. Patients with IBDs have reduced serum Trp levels and increased indoleamine 2,3-dioxygenase 1 (IDO1) expression in the colon.109,110 Ido1−/− mice suffered from more severe colitis induced by 2,4,6-trinitrobenzene sulfonic acid (TNBS).111 However, recent studies have shown no significant differences in KYN/TRP levels between patients with UC and CD.112 L-trp significantly regulates serotonin levels in the colon and alleviates inflammatory responses in a DSS-induced colitis mouse model.113 New research suggests that supplementing with xanthurenic acid (XANA) and kynurenic acid (KYNA) can alleviate symptoms of colitis.114
α-aminobutyric acid (AABA) inhibits M1 macrophage polarization and function, reducing the severity of colitis by promoting OXPHOS metabolism as well as glutamine and Arg while inhibiting glycolysis.115 Glycine metabolism inhibits M1 macrophage polarization. Tsune et al found that glycine attenuates TNBS and DSS-induced colitis.116,117 Hydroxyproline significantly attenuated symptoms and inflammatory responses in DSS-induced colitis mice, reducing inflammatory cytokines levels and inhibiting activation of NF-κB and STAT3 signaling pathways.118 Plasma levels of D-alanine (D-Ala) were low in DSS-induced colitis mice, UC mouse models, and mice with active intestinal inflammation.119 D-Ala inhibits the differentiation of M1 macrophages into M2.119
Characteristics of amino acid metabolism in macrophages are presented in Table 1.
 |
Table 1 Amino Acid Metabolism and Macrophage Polarization |
Macrophage Micronutrient Metabolism
Patients with IBD frequently experience malnutrition during active and remission phases, characterized by micronutrient deficiencies such as B12, folate, iron, and vitamin D, essential for various physiological processes.120 Selenium is a trace element that plays a critical role in the antioxidant defense system of cells in the form of selenoproteins. Reduced selenoprotein P (SEPP1) function significantly increased M2 macrophages. In a DSS-induced colitis model, SEPP1 deficiency exacerbates severe inflammation and intestinal damage.121 SELENOW is a small selenoprotein that affects glucose and Arg metabolism in macrophages during inflammation. Its absence during inflammation affects macrophage metabolism.122 Macrophage selenoprotein expression is closely related to 15-hydroxyprostaglandin dehydrogenase (15-PGDH). Selenoproteins in macrophages alleviate colitis in mice by inhibiting pro-inflammatory mediators.123 These findings suggest that selenium and its related proteins are crucial in modulating the immune system and inflammatory response, particularly in colitis models exhibiting significant protective effects.
Vitamin A is a lipid-soluble vitamin essential for preserving intestinal homeostasis, influencing immune function, and supporting epithelial barrier function. Its deficiency impairs macrophage function and increases susceptibility to induced colitis pathology. Vitamin A supplementation protects by reducing inflammation and apoptosis through the inhibition of NLRP3 inflammasome and caspase-1 activity.124,125 Conversely, vitamin D is an immunomodulator. The active form 1,25(OH)2D has immunosuppressive effects on various immune cells.126 Deficient levels of vitamin D heighten the risk of developing IBD, and supplementation significantly improves the prognosis of IBD in humans.127 Wang et al found that 1,25-dihydroxyvitamin D directly inhibits M1 polarization.128
Niacin, a vitamin B complex, ameliorates UC by activating the D-prostanoid receptor 1. This activation reduces the infiltration of inflammation-associated cells and increases the number of anti-inflammatory cells.129 Despite vitamins’s role in regulating macrophage immunity, evidence of the effects of vitamins on macrophage immune metabolism in IBD remains limited. A deeper understanding of these modulators of immunometabolism is required to develop new therapeutic strategies to modulate macrophage function and prevent IBD.
Roles of micronutrients on macrophages are presented in Table 2.
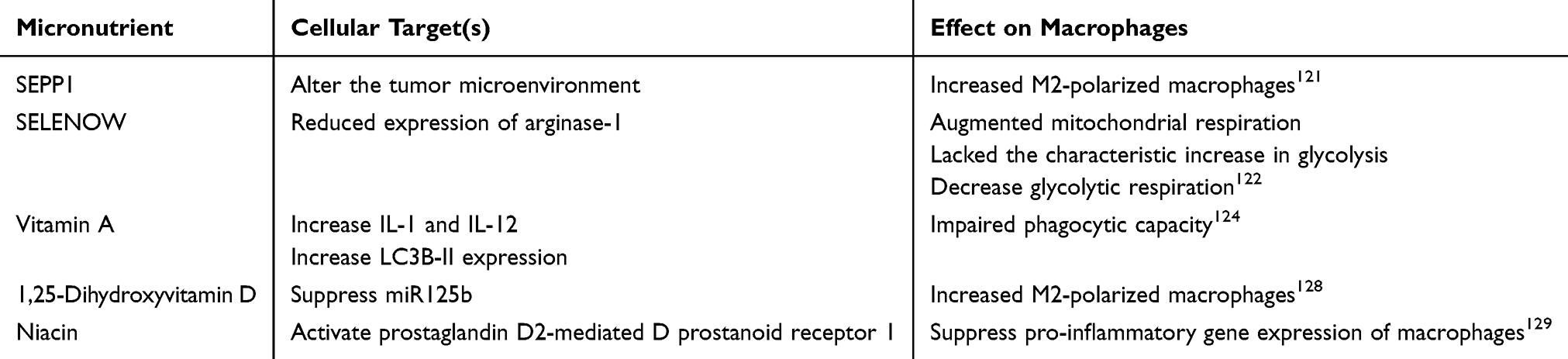 |
Table 2 Role of Micronutrients on Macrophages |
Macrophages and Ferroptosis
Iron Metabolism and Macrophage Polarization
Iron deficiency anemia is prevalent systemic complication of IBD.130,131 Iron is crucial for various physiological processes, encompassing cell proliferation, metabolism, and energy production. Its regulation is closely linked to macrophage polarization. Studies have shown that excess iron ions direct macrophages toward inflammation-activated polarization, increasing the expression of M1 markers such as IL-1β, TNF-α, and iNOS.132–134 M1 macrophages exhibited significantly higher ferritin H levels and lower transferrin receptor 1 than M2 macrophages.135 Iron overload increases reactive oxygen species (ROS) and hepcidin levels, promoting M1 macrophage polarization by inducing p53 acetylation and 4E-BP1 phosphorylation.133,136 During iron overload in macrophages, elevated hepcidin expression enhances STAT3 phosphorylation while inhibiting STAT6 expression, further driving M1 macrophage polarization.137–139 In M1 and colonic macrophages from mice with UC, a positive correlation was observed between Fe2+ accumulation and the production of inflammatory factors.140 Handa et al revealed that dietary iron overload triggered M1 activation in hepatic macrophages of mice, resulting in significant up-regulation of inflammation-related genes, and significant downregulation of M2 pathway-related genes.134 Treatment of IBD-associated anemia often involves oral or parenteral iron supplementation. Although oral iron may improve iron deficiency anemia, it may trigger oxidative stress, causing bowel inflammation and mucosal injury.141,142 Dietary iron has been shown to increase colonic iron levels and lipid peroxidation, promoting colitis development. This was achieved by activating the IL-6/IL-11-STAT3 signaling pathway, further promoting colitis and tumor progression.141 Future studies may delve into the mechanisms regulating iron metabolism in macrophages to identify new therapeutic targets to prevent excess iron-induced inflammation. Additionally, efforts should focus on finding safer and more effective iron supplementation strategies to reduce oral iron-induced oxidative stress and intestinal damage.
The Role of Macrophages in Ferroptosis
Ferroptosis is an iron-dependent, non-apoptotic cell death characterized by iron accumulation, excessive reactive oxygen species (ROS) production, and high lipid peroxidation levels. Impaired iron metabolism underlies ferroptosis and influences the development and progression of various diseases.143–145 Iron ions as Fe3+ bind to transferrin (Tf) and are internalized through Tf receptor 1 (TfR) located in the cell membrane, facilitating their transport from the extracellular environment into the cell. Within the cell, they are reduced to Fe2+.146,147 Fe2+ is stored in the labile iron pool (LIP) or ferritin (FT). FT is the primary intracellular iron storage protein, composed of FT heavy and light chains. Alternatively, Fe²+ can be exported from the cell via Ferroportin1 (FPN).148,149 FPN is an important iron transport protein that mainly facilitates the outward transport of non-heme iron absorbed from the intestine and stored iron, maintaining iron homeostasis in the body by regulating iron flow. It plays a significant role in the process of ferroptosis. Decreased FPN activity leads to iron accumulation, triggering iron ferroptosis. Excessive Fe3+ and Fe2+ induce the Fenton reaction, generating ROS. ROS damage lipids, proteins, and DNA, ultimately causing ferroptosis. Therefore, maintaining cellular iron homeostasis is essential to prevent oxidative damage, cytotoxicity, and cell death. Polyunsaturated fatty acids (PUFA) are converted to PUFA-CoA by long-chain acyl-CoA synthase-4 (ACSL4), binding to membrane phospholipids (PLs) via lysophosphatidylcholine acyltransferase 3. Subsequently, under the influence of lipoxygenases, non-toxic PL-PE-OH converts to toxic PL-PE-OHO, a reaction reversible by GSH peroxidase 4 (Gpx4). Excessive lipid peroxidation damages cell membrane PLs and alters the nature of the cell membrane lipid bilayer, ultimately accelerating ferroptosis.
Another critical feature of ferroptosis is glutathione depletion and reduced Gpx4 levels. GSH, an important endogenous antioxidant, scavenges ROS, maintaining intracellular redox balance and reducing oxidative damage, with cysteine being the rate-limiting substrate for its synthesis. Cystine/glutamate antiporter (System Xc−), comprising SLC7A11 and SLC3A2 proteins, transports cysteine into cells for conversion to cysteine.
In essence, a close connection exists between ferroptosis and macrophages, which is crucial for iron homeostasis maintenance and immunomodulation. Macrophages regulate iron homeostasis by recognizing and adhering to senescent red blood cells through surface receptors and transporting them intracellularly via vesicles. This process is facilitated by heme oxygenase-1 (HO-1), generating free iron, carbon monoxide, and biliverdin.150 HO-1 can decrease intracellular iron storage and suppress the inflammatory response of macrophages by inhibiting M1 polarization.151 In cases of intracellular iron overload in macrophages, the Fenton reaction occurs, generating large amounts of ROS and ultimately leading to ferroptosis. Therefore, comprehensively studying the interactions between macrophages and ferroptosis is crucial to unraveling the molecular mechanisms underlying the immune response and iron homeostasis. This exploration is important for identifying new therapeutic targets for related diseases (Figure 3).
 |
Figure 3 Macrophage ferroptosis in IBD. Fe3+ binds to transferrin and is transported to the cell through TfR1, where it is reduced to Fe2+, which is partially stored in the labile iron pool. Excessive intracellular accumulation of Fe3+ and Fe2+ can cause the Fenton reaction, which produces ROS and eventually leads to ferroptosis. |
The Characteration of Ferroptosis in Macrophages Polarization
Impaired iron metabolism can trigger ferroptosis and macrophage polarization. Iron overload disrupts the balance between M1/M2 macrophage polarization and promotes M1 macrophage polarization.134 RSL3 is an inducer of iron apoptosis, inhibiting iron-related cell death and controlling inflammatory progression by increasing Nrf2 expression.152 NRF2, an antioxidant transcription factor, upregulates GPX4 expression. Under inflammatory conditions, NRF2 expression is reduced in ferroptotic macrophages, leading to increased ROS levels and exacerbating cell death.153 Additionally, ROS can activate Chk2, promoting M1 macrophage polarization.154 GPX4 plays a role in inhibiting iron-related cell death. Gpx4-deficient macrophages can be induced to undergo ferroptosis in response to IL-4 stimulation.155 M2 macrophages co-activate the ERK-cPLA2-ACSL4 pathway to induce ferroptosis and further exacerbate mucosal damage in UC.156 In DSS-induced UC, macrophages undergo iron-related cell death and can be activated by β-caryophyllene to activate the type 2 cannabinoid receptor, which inhibits iron-induced cell death.157 Regulating iron-related cell death in macrophages has important implications for inflammatory responses and tissue damage. A more comprehensive understanding of the mechanisms of macrophage ferroptosis in IBD pathogenesis could facilitate the development of novel therapeutic agents and strategies.
Macrophage Metabolic Reprogramming in the Treatment of IBD
Traditional Therapeutic Drugs
In the 20th century, significant advances were made in IBD treatment. Initially, treatment measures for colitis were mainly bed rest and colonic irrigation due to the limited understanding of the disease at that time.158 As research progressed, more effective treatment options emerged, such as salicylates, corticosteroids, immunosuppressive drugs, and biologics. Mild-to-moderate UC is commonly treated with 5-aminosalicylic acid to alleviate inflammation. For left-sided UC, combined enema therapy is often recommended.159 However, some patients may experience non-responsiveness or intolerance to 5-ASA. Acute 5-ASA intolerance syndrome can increase the risk of colectomy and further worsen symptoms.160–162 Combining 5-ASA with Fer-1 treats UC and improves the colonic mucosal barrier by promoting M2 macrophage polarization.156 Corticosteroids effectively alleviate acute moderate-to-severe IBD symptoms by suppressing the immune response and decreasing intestinal permeability to improve the patient’s condition. Short-term corticosteroid use in high doses is linked to adverse effects, such as hyperglycemia and hypertension, making it unsuitable for maintenance therapy.163 Traditional immunosuppressive agents include mercaptopurine, methotrexate, and 6-mercaptopurine, which are primarily utilized in treating corticosteroid-refractory or corticosteroid-dependent IBD cases. These drugs effectively retard the progression of IBD by modulating immune system activity, offering patients a long-term treatment option.164 When patients fail to respond adequately to conventional therapies, biologics such as TNF antibodies (infliximab), anti-α4β7 integrin monoclonal antibodies (vedolizumab), and Janus kinase (JAK) inhibitors (upadacitinib) can be considered as alternative treatments. However, approximately 30% of patients do not respond to biologics during initial treatment, and 50% experience reduced efficacy or non-responsiveness with long-term use.165 Prolonged use of biologics may lead to the development of anti-drug antibodies or increase the risk of cancer.166 Research in the 20th century on IBD treatment has made significant progress through continuous drug innovation and progressive refinement of therapeutic strategies. However, the efficacy and safety of current drugs remain significant challenges. Therefore, the development of new, safer targeted therapies and drug delivery systems is urgently needed.
Current Research Status on Macrophage Immuno-Metabolic Therapy
Approaches targeting macrophages for treating IBD include pharmacotherapy and drug delivery systems. In pharmacological treatment, current research focuses on modulating macrophage function and phenotype to promote inflammation abatement and tissue repair.
New drugs can alleviate inflammation by inhibiting inflammatory signaling pathways, modulating macrophage polarization, and inducing anti-inflammatory cell phenotypes. For example, isosteviol sodium attenuates DSS-induced chronic colitis by inhibiting NF-κB/P65-regulated macrophage polarization.167 D-mannose, a monosaccharide, significantly inhibits macrophage production of IL-1β, reduces lactate and succinate levels in macrophages, and prevents the activation of HIF-1α, protecting mice from UC.168 Shikonin, a PKM2 inhibitor, binds to the PKM2 protein in macrophages, thereby ameliorating colitis symptoms in mice.51 Bergenin binds to PPAR-γ, promotes PPAR-γ nuclear translocation and transcriptional activity, inhibits macrophage activation, and ameliorates colitis in mice.169 Dioscin has an anti-inflammatory effect and can reduce UC symptoms in DSS-induced mice. The mechanism is suggested to regulate the signal transduction between mTOR, HIF-1α, and PPAR-γ, thereby affecting macrophage polarization.170 Notably, these interventions are primarily supported by murine studies; human IBD validation is ongoing.
Another approach is altering drug delivery to increase drug concentrations within macrophages, improving therapeutic efficacy and reducing systemic side effects. For example, nanoparticles or targeting agents can increase drug delivery efficiency within macrophages. This approach has achieved certain success in treating tumor-associated macrophages and holds potential for IBD treatment. Turmeric-derived nanovesicles (TNVs) significantly attenuate DSS-induced UC pathological injury by a mechanism that promotes M2 macrophage polarization. Additionally, TNVs target sites of inflammation and macrophages.171 Thalidomide, a drug for IBD treatment, mainly inhibits angiogenic activation and has anti-inflammatory properties.172 However, its use is restricted due to high systemic adverse effects. Meng et al developed polydopamine-coated thalidomide nanocrystals, which alleviated colitis symptoms in mice by increasing Thalidomide concentration in inflamed intestinal tissue and inducing M2 macrophage polarization.173
Critical Evaluation of Key Studies and Translational Challenges
While the studies discussed herein provide valuable insights into macrophage metabolic reprogramming in IBD, it is crucial to acknowledge their strengths and limitations, as well as the reasons for inconsistent findings across different models. Mouse models of colitis, such as those induced by dextran sulfate sodium (DSS), 2,4,6-trinitrobenzene sulfonic acid (TNBS), or genetic deficiencies (eg, IL-10 knockout), have been instrumental in elucidating the pathogenic mechanisms of IBD. These models offer the advantage of controlled genetic and environmental conditions, enabling the dissection of specific pathways, such as the roles of HIF-1α, PPAR-γ, and ferroptosis in macrophage function.51,52,60,68,77,156 However, a key limitation is that these models do not fully recapitulate the complex etiology and chronicity of human IBD, which involves polygenic susceptibility (eg, APC gene mutations), environmental triggers, and dynamic host-microbiome interactions.4,30 For instance, the DSS model primarily induces epithelial damage and acute inflammation, but human IBD is characterized by chronic relapse and persistent microbial stimulation.174–176 Moreover, while certain genetic manipulations in mice (eg, TRPA1 deletion) exacerbate tumorigenesis, human data show that low TRPA1 expression in colorectal cancer is associated with poor survival, highlighting fundamental differences in molecular pathology.174 Additionally, the microbiome in mouse models is more malleable and responds markedly to interventions (eg, Blautia hominis LYH1 or Alistipes finegoldii OMVs alleviating inflammation), whereas human IBD features complex, persistent dysbiosis with species-specific adaptations (eg, Malassezia’s oxygen-dependent fitness in the human gut) that are not replicated in mice.177–179 Consequently, therapeutic interventions that show efficacy in mouse models, such as targeting PKM2 or PPAR-γ, may not always translate to human patients.51,68,169
While Pan et al provided an excellent dissection of microbiota-metabolite crosstalk in macrophage glycolysis, and Bittencourt et al broadly surveyed immune cell metabolism, our work advances the field through two critical contributions.16,18 Firstly, establishing ferroptosis as an amplifier of macrophage-driven inflammation in IBD, a mechanistic axis underdeveloped in prior syntheses. Secondly, delivering a critical appraisal of nanotherapy that transcends preclinical data summaries by explicitly addressing scalability challenges and IBD-specific delivery barriers. Crucially, our framework resolves longstanding controversies, including HIF-1α’s dual roles in inflammation/tissue repair and PPARγ’s pro-tumorigenic risks, thereby identifying actionable therapeutic niches.
The heterogeneity in HIF-1α function underscores a paradigm shift in macrophage immunometabolism: metabolic regulators are not intrinsically “pro-inflammatory” or “anti-inflammatory” but are dynamic sensors of microenvironmental stress. This explains why therapies like HIF stabilizers succeed in resolution-phase colitis but may fail in acute flares. Consequently, biomarker-driven patient stratification is essential for targeted metabolic therapy. Nanotechnologies offer promise but require optimization for human IBD microbiome and immune landscapes. The paradoxical functions of PPAR-γ in IBD-associated carcinogenesis highlight the context-dependent nature of metabolic regulators, which may vary between acute injury models and chronic human disease.71,72 Furthermore, immune responses in mice are often more tractable; for example, compounds like RTA-408 can readily inhibit T cell proliferation and enhance Treg frequency, and FATS can degrade HIF-1α to suppress M1 macrophage polarization.176,180 However, human IBD involves a more intricate network of immune cells, including diverse innate lymphoid cells (ILCs) whose differentiation and recruitment are multi-factorial, a complexity not fully captured in mouse models.181 Moreover, while ferroptosis has been implicated in macrophage-driven inflammation in mouse models (eg, via Pannexin-1), its relevance in human IBD macrophages remains to be fully established.156 Differences in barrier repair mechanisms also contribute to inconsistencies: mouse models show inducible barrier restoration (eg, via Hyperoside-induced autophagy or Hymenolepis nana ESPs activating tuft cell/IL-13 signaling), but human IBD barrier defects involve sustained epigenetic regulation (eg, IGF2BP2-mediated m6A modification of CBR1, impacting the PI3K/Akt pathway) that is only partially modeled in mice.182–184
Another critical consideration is the inherent differences between murine and human macrophage biology. For example, human intestinal macrophages exhibit distinct developmental pathways and phenotypic markers compared to their murine counterparts.175,184 These differences may account for discrepancies in metabolic responses observed in mouse models versus human studies. Additionally, therapeutic translation faces hurdles: drugs effective in mice (eg, piroxicam analogs inhibiting MEK/ERK/NF-κB or dendritic cell biohybrid modulators) often encounter resistance in human IBD, where biomarkers like IGF2BP2 are needed to predict anti-TNF response, reflecting clinical heterogeneity absent in mouse models.183,185,186 Detection methods also differ, with mouse studies employing techniques like NIR-II imaging for microplastic tracking, while human IBD relies on biomarker assays (eg, MMP-9 detection via VHH-SPR probes).187,188 Furthermore, mouse models typically focus on acute pathology, whereas human IBD involves systemic long-term complications (eg, reproductive endocrine dysregulation) and diverse infectious etiologies (eg, Clostridioides difficile strains with variable toxin profiles) that are not fully represented.189,190 Therefore, future research should prioritize human macrophage studies, including those using organoids, patient-derived cells, and multi-omics approaches, to validate findings from animal models and bridge the translational gap.
Conclusions
Immunometabolic studies have identified significant metabolic differences between M1-type and M2-type macrophages in IBD development and progression. Therefore, targeted modulation of macrophage metabolic reprogramming processes may offer a novel therapeutic approach.191
This review highlighted how disruptions to intracellular nutrient metabolism affect macrophage polarization and function in IBD, and the interactions between ferroptosis and macrophages. Additionally, it summarizes conventional therapeutic agents for IBD and potential drugs targeting macrophage immuno-metabolic reprogramming. Despite recent advances, the mechanisms linking macrophage metabolism to IBD are incompletely defined and require further human validation.192 Therefore, further exploration of the effects of microenvironmental stimuli on macrophage metabolism in IBD is necessary. Mechanisms and effects at the cellular level have been the primary focus of most studies; however, exploration into the clinical importance and application of these findings is lacking. Further studies could establish correlations between these findings and clinical diseases. However, we emphasize that the mechanistic evidence discussed herein primarily derives from preclinical models (eg, murine colitis, in vitro systems), which may not fully recapitulate human IBD pathophysiology. Key hypotheses—such as ferroptosis-driven macrophage inflammation or nanoparticle-based targeting—require rigorous validation in human cohorts. Future studies must prioritize human macrophage multi-omics, longitudinal tissue analyses, and clinical trials to bridge this translational gap.
Abbreviations
IBD, Inflammatory bowel disease; CD, Crohn’s disease; UC, ulcerative colitis; APCs, antigen-presenting cells; GSH, glutathione; NOD2, domain-containing 2; ATG16L1, autophagy-related 16-like 1 gene; TGF-β1, transforming growth factor β1; Il23R, interleukin 23 receptor; TLR4, toll-like receptor 4; LPS, Lipopolysaccharide; IL-4, interleukin-4; TCA, tricarboxylic acid cycle; PPP, the pentose phosphate pathway; HIF-1α, hypoxia-inducible factor 1α; OXPHOS, oxidative phosphorylation; IRG1, immune response gene 1; DSS, dextran sodium sulfate; EVs, Extracellular vesicles; PPAR, peroxisome proliferator-activated receptor; DSS, dextran sulfate sodium; Treg, regulatory T; PGE2, prostaglandin E2; FAO, fatty acid oxidation; CPT2, Carnitine palmitoyltransferase 2; SREBPs, sterol regulatory element binding proteins; ACC, acetyl-CoA carboxylase; FAS, fatty acid synthase; PDH, pyruvate dehydrogenase; Arg, Arginine; NO, nitric oxide; iNOS, inducible NO synthase; Arg-1, arginase-1; Trp, Tryptophan; IDO1, indoleamine 2,3-dioxygenase 1; XANA, xanthurenic acid; KYNA, kynurenic acid; AABA, α-aminobutyric acid; D-Ala, D-alanine; SEPP1, selenoprotein P; 15-PGDH, 15-hydroxyprostaglandin dehydrogenase; ROS, reactive oxygen species; Tf, Transferrin; TfR, Tf receptor 1; LIP, labile iron pool; FT, ferritin; FPN, Ferroportin1; PUFA, Polyunsaturated fatty acids; ACSL4, long-chain acyl-CoA synthase-4; PLs, phospholipids; Gpx4, GSH peroxidase 4; HO-1, heme oxygenase-1; TNVs, Turmeric-derived nanovesicles.
Data Sharing Statement
Data will be made available on request.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Roda G, Chien Ng S, Kotze PG, et al. Crohn’s disease. Nat Rev Dis Primers. 2020;6(1):22. doi:10.1038/s41572-020-0156-2
2. Ordás I, Eckmann L, Talamini M, Baumgart DC, Sandborn WJ. Ulcerative colitis. Lancet. 2012;380(9853):1606–1619. doi:10.1016/s0140-6736(12)60150-0
3. Ramos GP, Papadakis KA. Mechanisms of disease: inflammatory bowel diseases. Mayo Clin Proc. 2019;94(1):155–165. doi:10.1016/j.mayocp.2018.09.013
4. Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3(7):390–407. doi:10.1038/ncpgasthep0528
5. Triantafillidis JK, Zografos CG, Konstadoulakis MM, Papalois AE. Combination treatment of inflammatory bowel disease: present status and future perspectives. World J Gastroenterol. 2024;30(15):2068–2080. doi:10.3748/wjg.v30.i15.2068
6. Yang E, Panaccione N, Whitmire N, et al. Efficacy and safety of simultaneous treatment with two biologic medications in refractory Crohn’s disease. Aliment Pharmacol Ther. 2020;51(11):1031–1038. doi:10.1111/apt.15719
7. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. doi:10.1016/j.immuni.2014.06.013
8. Shapouri-Moghaddam A, Mohammadian S, Vazini H, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi:10.1002/jcp.26429
9. Ogino T, Takeda K. Immunoregulation by antigen-presenting cells in human intestinal lamina propria. Front Immunol. 2023;14:1138971. doi:10.3389/fimmu.2023.1138971
10. Luo M, Zhao F, Cheng H, Su M, Wang Y. Macrophage polarization: an important role in inflammatory diseases. Front Immunol. 2024;15:1352946. doi:10.3389/fimmu.2024.1352946
11. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–995. doi:10.1038/ni.2705
12. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Ann Rev Immunol. 2015;33:643–675. doi:10.1146/annurev-immunol-032414-112220
13. Castegna A, Gissi R, Menga A, et al. Pharmacological targets of metabolism in disease: opportunities from macrophages. Pharmacol Ther. 2020;210:107521. doi:10.1016/j.pharmthera.2020.107521
14. Littlewood-Evans A, Sarret S, Apfel V, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med. 2016;213(9):1655–1662. doi:10.1084/jem.20160061
15. Xu B, Liu Y, Li N, Geng Q. Lactate and lactylation in macrophage metabolic reprogramming: current progress and outstanding issues. Front Immunol. 2024;15:1395786. doi:10.3389/fimmu.2024.1395786
16. Zaiatz Bittencourt V, Jones F, Doherty G, Ryan EJ. Targeting immune cell metabolism in the treatment of inflammatory bowel disease. Inflamm Bowel Dis. 2021;27(10):1684–1693. doi:10.1093/ibd/izab024
17. Wang H, Xu P, Yin K, Wang S. The role of m(6)A modification during macrophage metabolic reprogramming in human diseases and animal models. Front Immunol. 2025;16:1521196. doi:10.3389/fimmu.2025.1521196
18. Pan X, Zhu Q, Pan LL, Sun J. Macrophage immunometabolism in inflammatory bowel diseases: from pathogenesis to therapy. Pharmacol Ther. 2022;238:108176. doi:10.1016/j.pharmthera.2022.108176
19. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
20. Li C, Kuemmerle JF. Mechanisms that mediate the development of fibrosis in patients with Crohn’s disease. Inflamm Bowel Dis. 2014;20(7):1250–1258. doi:10.1097/mib.0000000000000043
21. Jun YK, Kwon SH, Yoon HT, et al. Toll-like receptor 4 regulates intestinal fibrosis via cytokine expression and epithelial-mesenchymal transition. Sci Rep. 2020;10(1):19867. doi:10.1038/s41598-020-76880-y
22. Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nature Genet. 2008;40(8):955–962. doi:10.1038/ng.175
23. Danielpour D, Song K. Cross-talk between IGF-I and TGF-beta signaling pathways. Cytokine Growth Factor Rev. 2006;17(1–2):59–74. doi:10.1016/j.cytogfr.2005.09.007
24. Cleynen I, González JR, Figueroa C, et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: results from the IBDchip European Project. Gut. 2013;62(11):1556–1565. doi:10.1136/gutjnl-2011-300777
25. Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nature Genet. 2011;43(3):246–252. doi:10.1038/ng.764
26. Swirski FK, Nahrendorf M, Etzrodt M, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325(5940):612–616. doi:10.1126/science.1175202
27. Ogino T, Nishimura J, Barman S, et al. Increased Th17-inducing activity of CD14+ CD163 low myeloid cells in intestinal lamina propria of patients with Crohn’s disease. Gastroenterology. 2013;145(6):1380–91.e1. doi:10.1053/j.gastro.2013.08.049
28. Jiang P, Zheng C, Xiang Y, et al. The involvement of TH17 cells in the pathogenesis of IBD. Cytokine Growth Factor Rev. 2023;69:28–42. doi:10.1016/j.cytogfr.2022.07.005
29. Monteleone I, Sarra M, Pallone F, Monteleone G. Th17-related cytokines in inflammatory bowel diseases: friends or foes? Curr Mol Med. 2012;12(5):592–597. doi:10.2174/156652412800620066
30. Quaglio AEV, Grillo TG, De Oliveira ECS, Di Stasi LC, Sassaki LY. Gut microbiota, inflammatory bowel disease and colorectal cancer. World J Gastroenterol. 2022;28(30):4053–4060. doi:10.3748/wjg.v28.i30.4053
31. Gomaa EZ. Human gut microbiota/microbiome in health and diseases: a review. Antonie van Leeuwenhoek. 2020;113(12):2019–2040. doi:10.1007/s10482-020-01474-7
32. Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018;11(1):1–10. doi:10.1007/s12328-017-0813-5
33. Wang N, Fang JY. Fusobacterium nucleatum, a key pathogenic factor and microbial biomarker for colorectal cancer. Trends Microbiol. 2023;31(2):159–172. doi:10.1016/j.tim.2022.08.010
34. Dougherty MW, Jobin C. Intestinal bacteria and colorectal cancer: etiology and treatment. Gut Microbes. 2023;15(1):2185028. doi:10.1080/19490976.2023.2185028
35. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi:10.1038/nri2448
36. Schulz C, Gomez Perdiguero E, Chorro L, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi:10.1126/science.1219179
37. De Schepper S, Verheijden S, Aguilera-Lizarraga J, et al. Self-maintaining gut macrophages are essential for intestinal homeostasis. Cell. 2018;175(2):400–415.e13. doi:10.1016/j.cell.2018.07.048
38. Bain CC, Schridde A. Origin, differentiation, and function of intestinal macrophages. Front Immunol. 2018;9:2733. doi:10.3389/fimmu.2018.02733
39. Muller PA, Koscsó B, Rajani GM, et al. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell. 2014;158(2):300–313. doi:10.1016/j.cell.2014.04.050
40. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi:10.1172/jci59643
41. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Iimmunol. 2000;164(12):6166–6173. doi:10.4049/jimmunol.164.12.6166
42. Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32(6):463–488. doi:10.1615/critrevimmunol.v32.i6.10
43. Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi:10.1016/j.immuni.2010.05.007
44. Zhang Q, Sioud M. Tumor-associated macrophage subsets: shaping polarization and targeting. Int J Mol Sci. 2023;24(8):7493. doi:10.3390/ijms24087493
45. Cutolo M, Campitiello R, Gotelli E, Soldano S. The role of M1/M2 macrophage polarization in rheumatoid arthritis synovitis. Front Immunol. 2022;13:867260. doi:10.3389/fimmu.2022.867260
46. Tedesco S, Bolego C, Toniolo A, et al. Phenotypic activation and pharmacological outcomes of spontaneously differentiated human monocyte-derived macrophages. Immunobiology. 2015;220(5):545–554. doi:10.1016/j.imbio.2014.12.008
47. Dvornikova KA, Platonova ON, Bystrova EY. Hypoxia and intestinal inflammation: common molecular mechanisms and signaling pathways. Int J Mol Sci. 2023;24(3):2425. doi:10.3390/ijms24032425
48. Corcoran SE, O’Neill LA. HIF1α and metabolic reprogramming in inflammation. J Clin Invest. 2016;126(10):3699–3707. doi:10.1172/jci84431
49. Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab. 2012;15(4):432–437. doi:10.1016/j.cmet.2011.11.013
50. Alves-Filho JC, Pålsson-McDermott EM. Pyruvate Kinase M2: a Potential Target for Regulating Inflammation. Front Immunol. 2016;7:145. doi:10.3389/fimmu.2016.00145
51. Huang B, Wang Q, Jiang L, et al. Shikonin ameliorated mice colitis by inhibiting dimerization and tetramerization of PKM2 in macrophages. Front Pharmacol. 2022;13:926945. doi:10.3389/fphar.2022.926945
52. Bäcker V, Cheung FY, Siveke JT, Fandrey J, Winning S. Knockdown of myeloid cell hypoxia-inducible factor-1α ameliorates the acute pathology in DSS-induced colitis. PLoS One. 2017;12(12):e0190074. doi:10.1371/journal.pone.0190074
53. Jha AK, Huang SC, Sergushichev A, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–430. doi:10.1016/j.immuni.2015.02.005
54. Di Conza G, Ho PC. Metabolic adaptation of macrophages in chronic diseases. Cancer Lett. 2018;414:250–256. doi:10.1016/j.canlet.2017.11.023
55. Michaudel C, Sokol H. The gut microbiota at the service of immunometabolism. Cell Metab. 2020;32(4):514–523. doi:10.1016/j.cmet.2020.09.004
56. Tannahill GM, Curtis AM, Adamik J, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi:10.1038/nature11986
57. Macias-Ceja DC, Ortiz-Masiá D, Salvador P, et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. 2019;12(1):178–187. doi:10.1038/s41385-018-0087-3
58. Li Y, Li YC, Liu XT, et al. Blockage of citrate export prevents TCA cycle fragmentation via Irg1 inactivation. Cell Rep. 2022;38(7):110391. doi:10.1016/j.celrep.2022.110391
59. Gómez-Ferrer M, Amaro-Prellezo E, Dorronsoro A, et al. HIF-overexpression and pro-inflammatory priming in human mesenchymal stromal cells improves the healing properties of extracellular vesicles in experimental Crohn’s disease. Int J Mol Sci. 2021;22(20):11269. doi:10.3390/ijms222011269
60. Kong G, Hua H, Lu Y, et al. Roxadustat ameliorates experimental colitis in mice by regulating macrophage polarization through increasing HIF level. Biochim Biophys Acta Gen Subj. 2024;1868(3):130548. doi:10.1016/j.bbagen.2023.130548
61. Chawla A. Control of macrophage activation and function by PPARs. Circ Res. 2010;106(10):1559–1569. doi:10.1161/circresaha.110.216523
62. Namgaladze D, Brüne B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim Biophys Acta. 2016;1861(11):1796–1807. doi:10.1016/j.bbalip.2016.09.002
63. Caioni G, Viscido A, d’Angelo M, et al. Inflammatory bowel disease: new insights into the interplay between environmental factors and PPARγ. Int J Mol Sci. 2021;22(3):985. doi:10.3390/ijms22030985
64. Toobian D, Ghosh P, Katkar GD. Parsing the role of PPARs in macrophage processes. Front Immunol. 2021;12:783780. doi:10.3389/fimmu.2021.783780
65. Nagy L, Szanto A, Szatmari I, Széles L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol Rev. 2012;92(2):739–789. doi:10.1152/physrev.00004.2011
66. Bouhlel MA, Derudas B, Rigamonti E, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6(2):137–143. doi:10.1016/j.cmet.2007.06.010
67. Xue L, Wu YY. Activation of PPARγ regulates M1/M2 macrophage polarization and attenuates dextran sulfate sodium salt-induced inflammatory bowel disease via the STAT-1/STAT-6 pathway. Kaohsiung J Med Sci. 2025;41(2):e12927. doi:10.1002/kjm2.12927
68. Adachi M, Kurotani R, Morimura K, et al. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55(8):1104–1113. doi:10.1136/gut.2005.081745
69. da Rocha GHO, de Paula-Silva M, Broering MF, et al. Pioglitazone-mediated attenuation of experimental colitis relies on cleaving of Annexin A1 released by macrophages. Front Pharmacol. 2020;11:591561. doi:10.3389/fphar.2020.591561
70. Hontecillas R, Horne WT, Climent M, et al. Immunoregulatory mechanisms of macrophage PPAR-γ in mice with experimental inflammatory bowel disease. Mucosal Immunol. 2011;4(3):304–313. doi:10.1038/mi.2010.75
71. McAlpine CA, Barak Y, Matise I, Cormier RT. Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. Int J Cancer. 2006;119(10):2339–2346. doi:10.1002/ijc.22115
72. Lecarpentier Y, Claes V, Vallée A, Hébert JL. Interactions between PPAR gamma and the canonical Wnt/Beta-Catenin pathway in type 2 diabetes and colon cancer. PPAR Res. 2017;2017:5879090. doi:10.1155/2017/5879090
73. Yamamoto-Furusho JK, Peñaloza-Coronel A, Sánchez-Muñoz F, Barreto-Zuñiga R, Dominguez-Lopez A. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) expression is downregulated in patients with active ulcerative colitis. Inflamm Bowel Dis. 2011;17(2):680–681. doi:10.1002/ibd.21322
74. Fernandes P, MacSharry J, Darby T, et al. Differential expression of key regulators of Toll-like receptors in ulcerative colitis and Crohn’s disease: a role for Tollip and peroxisome proliferator-activated receptor gamma? Clin Exp Immunol. 2016;183(3):358–368. doi:10.1111/cei.12732
75. Yang K, Jiang Q, Wang Z, et al. Mutual inhibitory mechanisms between PPARγ and Hif-1α: implication in pulmonary hypertension. Recept Clin Investig. 2015;2(2):e626. doi:10.14800/rci.626
76. Cabrero A, Cubero M, Llaverías G, et al. Differential effects of peroxisome proliferator-activated receptor activators on the mRNA levels of genes involved in lipid metabolism in primary human monocyte-derived macrophages. Metabolism. 2003;52(5):652–657. doi:10.1053/meta.2003.50100
77. Nomura M, Liu J, Rovira II, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17(3):216–217. doi:10.1038/ni.3366
78. Nagy ZS, Czimmerer Z, Szanto A, Nagy L. Pro-inflammatory cytokines negatively regulate PPARγ mediated gene expression in both human and murine macrophages via multiple mechanisms. Immunobiology. 2013;218(11):1336–1344. doi:10.1016/j.imbio.2013.06.011
79. Xu M, Wang X, Li Y, et al. Arachidonic acid metabolism controls macrophage alternative activation through regulating oxidative phosphorylation in PPARγ dependent manner. Front Immunol. 2021;12:618501. doi:10.3389/fimmu.2021.618501
80. Dubuquoy L, Jansson EA, Deeb S, et al. Impaired expression of peroxisome proliferator-activated receptor gamma in ulcerative colitis. Gastroenterology. 2003;124(5):1265–1276. doi:10.1016/s0016-5085(03)00271-3
81. Vlk AM, Prantner D, Shirey KA, et al. M2a macrophages facilitate resolution of chemically-induced colitis in TLR4-SNP mice. mBio. 2023;14(5):e0120823. doi:10.1128/mbio.01208-23
82. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi:10.1172/jci15593
83. Lee JH, Phelan P, Shin M, et al. SREBP-1a-stimulated lipid synthesis is required for macrophage phagocytosis downstream of TLR4-directed mTORC1. Proc Natl Acad Sci USA. 2018;115(52):E12228–e12234. doi:10.1073/pnas.1813458115
84. Im SS, Yousef L, Blaschitz C, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 2011;13(5):540–549. doi:10.1016/j.cmet.2011.04.001
85. Bidault G, Virtue S, Petkevicius K, et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat Metab. 2021;3(9):1150–1162. doi:10.1038/s42255-021-00440-5
86. Xie B, Zhang A, Li C, et al. Differential analysis of sorting nexin 10 and sterol regulatory element-binding protein 2 expression in inflammatory bowel disease. Immunol Res. 2024;72(6):1417–1423. doi:10.1007/s12026-024-09539-9
87. Ooi M, Nishiumi S, Yoshie T, et al. GC/MS-based profiling of amino acids and TCA cycle-related molecules in ulcerative colitis. Inflammation Res. 2011;60(9):831–840. doi:10.1007/s00011-011-0340-7
88. Shiomi Y, Nishiumi S, Ooi M, et al. GCMS-based metabolomic study in mice with colitis induced by dextran sulfate sodium. Inflamm Bowel Dis. 2011;17(11):2261–2274. doi:10.1002/ibd.21616
89. Hu X, Deng J, Yu T, et al. ATF4 deficiency promotes intestinal inflammation in mice by reducing uptake of glutamine and expression of antimicrobial peptides. Gastroenterology. 2019;156(4):1098–1111. doi:10.1053/j.gastro.2018.11.033
90. Jiang Q, Qiu Y, Kurland IJ, et al. Glutamine is required for M1-like polarization of macrophages in response to mycobacterium tuberculosis infection. mBio. 2022;13(4):e0127422. doi:10.1128/mbio.01274-22
91. He L, Weber KJ, Schilling JD. Glutamine modulates macrophage lipotoxicity. Nutrients. 2016;8(4):215. doi:10.3390/nu8040215
92. Kieler M, Hofmann M, Schabbauer G. More than just protein building blocks: how amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. 2021;288(12):3694–3714. doi:10.1111/febs.15715
93. Zhu Y, Chen X, Lu Y, et al. Glutamine mitigates murine burn sepsis by supporting macrophage M2 polarization through repressing the SIRT5-mediated desuccinylation of pyruvate dehydrogenase. Burns Trauma. 2022;10:tkac041. doi:10.1093/burnst/tkac041
94. Ren W, Xia Y, Chen S, et al. Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Adv Nutr. 2019;10(2):321–330. doi:10.1093/advances/nmy084
95. Hou YC, Liu JJ, Pai MH, Tsou SS, Yeh SL. Alanyl-glutamine administration suppresses Th17 and reduces inflammatory reaction in dextran sulfate sodium-induced acute colitis. Int Immunopharmacol. 2013;17(1):1–8. doi:10.1016/j.intimp.2013.05.004
96. Liu PS, Wang H, Li X, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985–994. doi:10.1038/ni.3796
97. Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi:10.1016/j.ccr.2004.11.022
98. Zhou W, Hu G, He J, et al. SENP1-Sirt3 signaling promotes α-ketoglutarate production during M2 macrophage polarization. Cell Rep. 2022;39(2):110660. doi:10.1016/j.celrep.2022.110660
99. Morris SM. Arginine: beyond protein. Am J Clin Nutr. 2006;83(2):508s–512s. doi:10.1093/ajcn/83.2.508S
100. Arts RJ, Novakovic B, Ter Horst R, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24(6):807–819. doi:10.1016/j.cmet.2016.10.008
101. Palmieri EM, Holewinski R, McGinity CL, et al. Pyruvate dehydrogenase operates as an intramolecular nitroxyl generator during macrophage metabolic reprogramming. Nat Commun. 2023;14(1):5114. doi:10.1038/s41467-023-40738-4
102. Van den Bossche J, Lamers WH, Koehler ES, et al. Pivotal Advance: arginase-1-independent polyamine production stimulates the expression of IL-4-induced alternatively activated macrophage markers while inhibiting LPS-induced expression of inflammatory genes. J Leukoc Biol. 2012;91(5):685–699. doi:10.1189/jlb.0911453
103. Hong SK, Maltz BE, Coburn LA, et al. Increased serum levels of L-arginine in ulcerative colitis and correlation with disease severity. Inflamm Bowel Dis. 2010;16(1):105–111. doi:10.1002/ibd.21035
104. Coburn LA, Horst SN, Allaman MM, et al. L-arginine availability and metabolism is altered in ulcerative colitis. Inflamm Bowel Dis. 2016;22(8):1847–1858. doi:10.1097/mib.0000000000000790
105. Coburn LA, Gong X, Singh K, et al. L-arginine supplementation improves responses to injury and inflammation in dextran sulfate sodium colitis. PLoS One. 2012;7(3):e33546. doi:10.1371/journal.pone.0033546
106. Closs EI, Boissel JP, Habermeier A, Rotmann A. Structure and function of cationic amino acid transporters (CATs). J Membr Biol. 2006;213(2):67–77. doi:10.1007/s00232-006-0875-7
107. Coburn LA, Singh K, Asim M, et al. Loss of solute carrier family 7 member 2 exacerbates inflammation-associated colon tumorigenesis. Oncogene. 2019;38(7):1067–1079. doi:10.1038/s41388-018-0492-9
108. Xue C, Li G, Zheng Q, et al. Tryptophan metabolism in health and disease. Cell Metab. 2023;35(8):1304–1326. doi:10.1016/j.cmet.2023.06.004
109. Nikolaus S, Schulte B, Al-Massad N, et al. Increased tryptophan metabolism is associated with activity of inflammatory bowel diseases. Gastroenterology. 2017;153(6):1504–1516.e2. doi:10.1053/j.gastro.2017.08.028
110. Sofia MA, Ciorba MA, Meckel K, et al. Tryptophan metabolism through the kynurenine pathway is associated with endoscopic inflammation in ulcerative colitis. Inflamm Bowel Dis. 2018;24(7):1471–1480. doi:10.1093/ibd/izy103
111. Takamatsu M, Hirata A, Ohtaki H, et al. IDO1 plays an immunosuppressive role in 2,4,6-trinitrobenzene sulfate-induced colitis in mice. J Iimmunol. 2013;191(6):3057–3064. doi:10.4049/jimmunol.1203306
112. Manzella CR, Jayawardena D, Pagani W, et al. Serum serotonin differentiates between disease activity states in Crohn’s patients. Inflamm Bowel Dis. 2020;26(10):1607–1618. doi:10.1093/ibd/izaa208
113. Wang B, Sun S, Liu M, et al. Dietary L-tryptophan regulates colonic serotonin homeostasis in mice with dextran sodium sulfate-induced colitis. J Nutr. 2020;150(7):1966–1976. doi:10.1093/jn/nxaa129
114. Michaudel C, Danne C, Agus A, et al. Rewiring the altered tryptophan metabolism as a novel therapeutic strategy in inflammatory bowel diseases. Gut. 2023;72(7):1296–1307. doi:10.1136/gutjnl-2022-327337
115. Li F, Xia Y, Yuan S, et al. α-aminobutyric acid constrains macrophage-associated inflammatory diseases through metabolic reprogramming and epigenetic modification. Int J Mol Sci. 2023;24(13):10444. doi:10.3390/ijms241310444
116. Gan Z, Zhang M, Xie D, et al. Glycinergic signaling in macrophages and its application in macrophage-associated diseases. Front Immunol. 2021;12:762564. doi:10.3389/fimmu.2021.762564
117. Tsune I, Ikejima K, Hirose M, et al. Dietary glycine prevents chemical-induced experimental colitis in the rat. Gastroenterology. 2003;125(3):775–785. doi:10.1016/s0016-5085(03)01067-9
118. Ji Y, Dai Z, Sun S, et al. Hydroxyproline attenuates dextran sulfate sodium-induced colitis in mice: involvment of the NF-κB signaling and oxidative stress. Mol Nutr Food Res. 2018;62(21):e1800494. doi:10.1002/mnfr.201800494
119. Hashimoto H, Takagi T, Asaeda K, et al. D-alanine inhibits murine intestinal inflammation by suppressing IL-12 and IL-23 production in macrophages. J Crohns Colitis. 2024;18(6):908–919. doi:10.1093/ecco-jcc/jjad217
120. Weisshof R, Chermesh I. Micronutrient deficiencies in inflammatory bowel disease. Curr Opin Clin Nutr Metab Care. 2015;18(6):576–581. doi:10.1097/mco.0000000000000226
121. Barrett CW, Reddy VK, Short SP, et al. Selenoprotein P influences colitis-induced tumorigenesis by mediating stemness and oxidative damage. J Clin Invest. 2015;125(7):2646–2660. doi:10.1172/jci76099
122. Misra S, Lee TJ, Sebastian A, et al. Loss of selenoprotein W in murine macrophages alters the hierarchy of selenoprotein expression, redox tone, and mitochondrial functions during inflammation. Redox Biol. 2023;59:102571. doi:10.1016/j.redox.2022.102571
123. Kaushal N, Kudva AK, Patterson AD, et al. Crucial role of macrophage selenoproteins in experimental colitis. J Iimmunol. 2014;193(7):3683–3692. doi:10.4049/jimmunol.1400347
124. Hiraga H, Chinda D, Maeda T, et al. Vitamin A promotes the fusion of autophagolysosomes and prevents excessive inflammasome activation in dextran sulfate sodium-induced colitis. Int J Mol Sci. 2023;24(10):8684. doi:10.3390/ijms24108684
125. Stephensen CB. Vitamin A, infection, and immune function. Annu Rev Nutr. 2001;21:167–192. doi:10.1146/annurev.nutr.21.1.167
126. Prietl B, Treiber G, Pieber TR, Amrein K. Vitamin D and immune function. Nutrients. 2013;5(7):2502–2521. doi:10.3390/nu5072502
127. Meeker S, Seamons A, Maggio-Price L, Paik J. Protective links between vitamin D, inflammatory bowel disease and colon cancer. World J Gastroenterol. 2016;22(3):933–948. doi:10.3748/wjg.v22.i3.933
128. Wang F, Johnson RL, DeSmet ML, Snyder PW, Fairfax KC, Fleet JC. Vitamin D receptor-dependent signaling protects mice from dextran sulfate sodium-induced colitis. Endocrinology. 2017;158(6):1951–1963. doi:10.1210/en.2016-1913
129. Li J, Kong D, Wang Q, et al. Niacin ameliorates ulcerative colitis via prostaglandin D(2)-mediated D prostanoid receptor 1 activation. EMBO Mol Med. 2017;9(5):571–588. doi:10.15252/emmm.201606987
130. Gasché C, Reinisch W, Lochs H, et al. Anemia in Crohn’s disease: importance of inadequate erythropoietin production and iron deficiency. Dig Dis Sci. 1994;39(9):1930–1934. doi:10.1007/bf02088127
131. Stein J, Hartmann F, Dignass AU. Diagnosis and management of iron deficiency anemia in patients with IBD. Nat Rev Gastroenterol Hepatol. 2010;7(11):599–610. doi:10.1038/nrgastro.2010.151
132. Zanganeh S, Hutter G, Spitler R, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nature Nanotechnol. 2016;11(11):986–994. doi:10.1038/nnano.2016.168
133. Zhou Y, Que KT, Zhang Z, et al. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. 2018;7(8):4012–4022. doi:10.1002/cam4.1670
134. Handa P, Thomas S, Morgan-Stevenson V, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol. 2019;105(5):1015–1026. doi:10.1002/jlb.3a0318-108r
135. Corna G, Campana L, Pignatti E, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica. 2010;95(11):1814–1822. doi:10.3324/haematol.2010.023879
136. Fang Z, Wang C, Zhu J, Gou Y. Iron overload promotes hemochromatosis-associated osteoarthritis via the mTORC1-p70S6K/4E-BP1 pathway. Int Immunopharmacol. 2024;131:111848. doi:10.1016/j.intimp.2024.111848
137. Chen X, Dou J, Fu Z, et al. Macrophage M1 polarization mediated via the IL-6/STAT3 pathway contributes to apical periodontitis induced by Porphyromonas gingivalis. J Appl Oral Sci. 2022;30:e20220316. doi:10.1590/1678-7757-2022-0316
138. Dou J, Chen X, Zhang J, et al. Gingivalis induce macrophage polarization by regulating hepcidin expression in chronic apical periodontitis. Int Immunopharmacol. 2024;142(Pt A):113139. doi:10.1016/j.intimp.2024.113139
139. Liu E, Li Z, Zhang Y, Chen K. Hepcidin induces M1 macrophage polarization in monocytes or THP-1 derived macrophages. Iran J Immunol. 2019;16(3):190–199. doi:10.22034/iji.2019.80270
140. Ma H, Shu Q, Li D, et al. Accumulation of intracellular ferrous iron in inflammatory-activated macrophages. Biol Trace Elem Res. 2023;201(5):2303–2310. doi:10.1007/s12011-022-03362-9
141. Chua AC, Klopcic BR, Ho DS, et al. Dietary iron enhances colonic inflammation and IL-6/IL-11-Stat3 signaling promoting colonic tumor development in mice. PLoS One. 2013;8(11):e78850. doi:10.1371/journal.pone.0078850
142. Mahalhal A, Burkitt MD, Duckworth CA, et al. Long-term iron deficiency and dietary iron excess exacerbate acute dextran sodium sulphate-induced colitis and are associated with significant dysbiosis. Int J Mol Sci. 2021;22(7):3646. doi:10.3390/ijms22073646
143. Lin L, Chen H, Zhao R, Zhu M, Nie G. Nanomedicine targets iron metabolism for cancer therapy. Cancer Sci. 2022;113(3):828–837. doi:10.1111/cas.15250
144. Xu S, He Y, Lin L, Chen P, Chen M, Zhang S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021;12(4):289. doi:10.1038/s41419-021-03559-1
145. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Ann Rev Cancer Biol. 2019;3(1):35–54. doi:10.1146/annurev-cancerbio-030518-055844
146. Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116(4):565–576. doi:10.1016/s0092-8674(04)00130-8
147. Sakajiri T, Yamamura T, Kikuchi T, Yajima H. Computational structure models of apo and diferric transferrin-transferrin receptor complexes. Protein J. 2009;28(9–10):407–414. doi:10.1007/s10930-009-9208-x
148. Arosio P, Elia L, Poli M. Ferritin, cellular iron storage and regulation. IUBMB Life. 2017;69(6):414–422. doi:10.1002/iub.1621
149. Lane DJ, Merlot AM, Huang ML, et al. Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim Biophys Acta. 2015;1853(5):1130–1144. doi:10.1016/j.bbamcr.2015.01.021
150. Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys. 2014;564:83–88. doi:10.1016/j.abb.2014.09.005
151. Tang X, Li Y, Zhao J, et al. Heme oxygenase-1 increases intracellular iron storage and suppresses inflammatory response of macrophages by inhibiting M1 polarization. Metallomics. 2023;15(10):mfad062. doi:10.1093/mtomcs/mfad062
152. Cui Y, Zhang Z, Zhou X, et al. Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after RSL3 stimulation via increasing Nrf2 expression. J Neuroinflammation. 2021;18(1):249. doi:10.1186/s12974-021-02231-x
153. Yang M, Shen Z, Zhang X, et al. Ferroptosis of macrophages facilitates bone loss in apical periodontitis via NRF2/FSP1/ROS pathway. Free Radic Biol Med. 2023;208:334–347. doi:10.1016/j.freeradbiomed.2023.08.020
154. Li C, Deng C, Wang S, et al. A novel role for the ROS-ATM-Chk2 axis mediated metabolic and cell cycle reprogramming in the M1 macrophage polarization. Redox Biol. 2024;70:103059. doi:10.1016/j.redox.2024.103059
155. Piattini F, Matsushita M, Muri J, et al. Differential sensitivity of inflammatory macrophages and alternatively activated macrophages to ferroptosis. Eur J Immunol. 2021;51(10):2417–2429. doi:10.1002/eji.202049114
156. Ye Y, Liu L, Feng Z, et al. The ERK-cPLA2-ACSL4 axis mediating M2 macrophages ferroptosis impedes mucosal healing in ulcerative colitis. Free Radic Biol Med. 2024;214:219–235. doi:10.1016/j.freeradbiomed.2024.02.016
157. Wu YT, Zhong LS, Huang C, et al. β-caryophyllene acts as a ferroptosis inhibitor to ameliorate experimental colitis. Int J Mol Sci. 2022;23(24):16055. doi:10.3390/ijms232416055
158. Actis GC, Pellicano R, Fagoonee S, Ribaldone DG. History of inflammatory bowel diseases. J Clin Med. 2019;8(11):1970. doi:10.3390/jcm8111970
159. Manai F, Zanoletti L, Arfini D, et al. Dimethyl fumarate and intestine: from main suspect to potential ally against gut disorders. Int J Mol Sci. 2023;24(12):9912. doi:10.3390/ijms24129912
160. Mikami Y, Tsunoda J, Suzuki S, Mizushima I, Kiyohara H, Kanai T. Significance of 5-aminosalicylic acid intolerance in the clinical management of ulcerative colitis. Digestion. 2023;104(1):58–65. doi:10.1159/000527452
161. Hibiya S, Matsuyama Y, Fujii T, et al. 5-aminosalicylate-intolerant patients are at increased risk of colectomy for ulcerative colitis. Aliment Pharmacol Ther. 2021;53(1):103–113. doi:10.1111/apt.16120
162. Shimizu H, Arai K, Tang J, Hosoi K, Funayama R. 5-Aminosalicylate intolerance causing exacerbation in pediatric ulcerative colitis. Pediatr Int. 2017;59(5):583–587. doi:10.1111/ped.13235
163. Salice M, Rizzello F, Calabrese C, Calandrini L, Gionchetti P. A current overview of corticosteroid use in active ulcerative colitis. Expert Rev Gastroenterol Hepatol. 2019;13(6):557–561. doi:10.1080/17474124.2019.1604219
164. Crepaldi M, Maniero D, Massano A, et al. Azathioprine monotherapy withdrawal in inflammatory bowel diseases: a retrospective mono-centric study. World J Gastroenterol. 2023;29(27):4334–4343. doi:10.3748/wjg.v29.i27.4334
165. Lu Q, Yang MF, Liang YJ, et al. Immunology of inflammatory bowel disease: molecular mechanisms and therapeutics. J Inflamm Res. 2022;15:1825–1844. doi:10.2147/jir.S353038
166. Chupin A, Perduca V, Meyer A, Bellanger C, Carbonnel F, Dong C. Systematic review with meta-analysis: comparative risk of lymphoma with anti-tumour necrosis factor agents and/or thiopurines in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2020;52(8):1289–1297. doi:10.1111/apt.16050
167. Wang S, Huang J, Tan KS, Deng L, Liu F, Tan W. Isosteviol sodium ameliorates dextran sodium sulfate-induced chronic colitis through the regulation of metabolic profiling, macrophage polarization, and NF-κB pathway. Oxid Med Cell Longev. 2022;2022:4636618. doi:10.1155/2022/4636618
168. Torretta S, Scagliola A, Ricci L, et al. D-mannose suppresses macrophage IL-1β production. Nat Commun. 2020;11(1):6343. doi:10.1038/s41467-020-20164-6
169. Wang K, Li YF, Lv Q, Li XM, Dai Y, Wei ZF. Bergenin, acting as an agonist of PPARγ, ameliorates experimental colitis in mice through improving expression of SIRT1, and therefore inhibiting NF-κB-mediated macrophage activation. Front Pharmacol. 2017;8:981. doi:10.3389/fphar.2017.00981
170. Wu MM, Wang QM, Huang BY, et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol Res. 2021;172:105796. doi:10.1016/j.phrs.2021.105796
171. Gao C, Zhou Y, Chen Z, et al. Turmeric-derived nanovesicles as novel nanobiologics for targeted therapy of ulcerative colitis. Theranostics. 2022;12(12):5596–5614. doi:10.7150/thno.73650
172. Rafiee P, Stein DJ, Nelson VM, Otterson MF, Shaker R, Binion DG. Thalidomide inhibits inflammatory and angiogenic activation of human intestinal microvascular endothelial cells (HIMEC). Am J Physiol Gastrointest Liver Physiol. 2010;298(2):G167–76. doi:10.1152/ajpgi.00385.2009
173. Meng Z, Fu B, Yang Z, et al. Polydopamine-coated thalidomide nanocrystals promote DSS-induced murine colitis recovery through Macrophage M2 polarization together with the synergistic anti-inflammatory and anti-angiogenic effects. Int J Pharm. 2023;630:122376. doi:10.1016/j.ijpharm.2022.122376
174. Dou F, Hu S, Lu D, Gao J. Trpa1 knockout favors colon tumorigenesis in dextran sulfate sodium (DSS)-induced colitis mice. Drug Discov Ther. 2025;19(3):200–207. doi:10.5582/ddt.2025.01022
175. Sandoval AQ, James A, Neufeld KL. Nuclear adenomatous polyposis coli elevates STAT1 and reduces CXCL1,2, and 3 expression and inhibits neutrophil recruitment. Cell Signal. 2025;134:111957. doi:10.1016/j.cellsig.2025.111957
176. Han Y, Xun J, Li T, et al. FATS alleviates ulcerative colitis by inhibiting M1 macrophage polarization and aerobic glycolysis through promoting the ubiquitination-mediated degradation of HIF-1α. Biochem Pharmacol. 2025;240:117053. doi:10.1016/j.bcp.2025.117053
177. Wei J, Liu Y, Li H, et al. Unlocking the power of swine gut bacteria: newly isolated Blautia strain and its metabolites inhibit the replication of Salmonella Typhimurium in macrophages and alleviate DSS-induced colitis in mice. J Anim Sci Biotechnol. 2025;16(1):87. doi:10.1186/s40104-025-01208-7
178. Older EA, Mitchell MK, Campbell A, et al. Human gut commensal Alistipes timonensis modulates the host lipidome and delivers anti-inflammatory outer membrane vesicles to suppress colitis in an Il10-deficient mouse model. Gut Microbes. 2025;17(1):2517380. doi:10.1080/19490976.2025.2517380
179. Cho YJ, Shin SY, Yang J, et al. Live Malassezia strains from the mucosa of patients with ulcerative colitis: pathogenic potential and environmental adaptations. mBio. 2025;16:e0140025. doi:10.1128/mbio.01400-25
180. Dasgupta D, Tripathi A, Griffard-Smith R, Yellapu N, Dhorajiya P, Pyaram K. Therapeutic potential of NRF2 activating drug RTA-408 in suppressing T cell effector responses and inflammatory bowel disease. J Iimmunol. 2025:vkaf117. doi:10.1093/jimmun/vkaf117
181. Yao X, Ma K, Zhu Y, Cao S. Innate lymphoid cells in inflammatory bowel disease. Cells. 2025;14(11):825. doi:10.3390/cells14110825
182. Mou R, Cui X, Wang H, et al. Excretory/secretory products from Hymenolepis nana adult worms alleviate ulcerative colitis in mice via tuft/IL-13 signaling pathway. Parasit Vectors. 2025;18(1):230. doi:10.1186/s13071-025-06893-x
183. Wu W, Zhou Z, Pang C, et al. IGF2BP2 regulates inflammation in ulcerative colitis through N6-methyladenosine-dependent modulation of CBR1. Int Immunopharmacol. 2025;161:115072. doi:10.1016/j.intimp.2025.115072
184. Xia W, Lin H, Zhang J, et al. Hyperoside promotes autophagy of colonic epithelial cells to protect intestinal barrier function in ulcerative colitis. Food Funct. 2025;16(13):5543–5555. doi:10.1039/d5fo00256g
185. Luo JQ, Xu LL, Xiao KX, et al. Enhanced anti-inflammatory efficacy of a new piroxicam analogue through the MEK/ERK/NF-κB pathway in vitro and in vivo. Int Immunopharmacol. 2025;161:115100. doi:10.1016/j.intimp.2025.115100
186. Yi W, Sun F, Qian X, et al. Dendritic cell-based biohybrid immunomodulator alleviates intestinal inflammatory disease. Adv Mater. 2025:e2420113. doi:10.1002/adma.202420113
187. Tian X, Yang Y, Gao W, et al. NIR-II plastic particles for monitoring intestinal motility and microplastic deposition in mice. Anal Chem. 2025;97(25):13474–13479. doi:10.1021/acs.analchem.5c01885
188. Wu D, Yang J, Cong J, et al. A disposable VHH-based SPR fiber probe for sensitive and specific detection of MMP-9 in cancer and inflammatory disease. Biosens Bioelectron. 2025;287:117684. doi:10.1016/j.bios.2025.117684
189. Martell M, Quarnstrom CF, Khoruts A, Vezys V, Staley C, Shmidt E. Chronic intestinal inflammation and microbial dysbiosis are associated with female reproductive outcomes in a mouse model of inflammatory bowel disease. Gastro Hep Adv. 2025;4(7):100670. doi:10.1016/j.gastha.2025.100670
190. DiBenedetto NV, Oberkampf M, Crouzols A, et al. Clostridioides difficile’s virulence requires efficient holin-mediated toxin secretion. iScience. 2025;28(6):112586. doi:10.1016/j.isci.2025.112586
191. Zhuang H, Lv Q, Zhong C, et al. Tiliroside ameliorates ulcerative colitis by restoring the M1/M2 macrophage balance via the HIF-1α/glycolysis pathway. Front Immunol. 2021;12:649463. doi:10.3389/fimmu.2021.649463
192. Singh UP, Singh NP, Murphy EA, et al. Chemokine and cytokine levels in inflammatory bowel disease patients. Cytokine. 2016;77:44–49. doi:10.1016/j.cyto.2015.10.008
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.