Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 10
Macrophage activation syndrome: early diagnosis is key
Authors Lerkvaleekul B ![]() , Vilaiyuk S
, Vilaiyuk S ![]()
Received 27 May 2018
Accepted for publication 25 June 2018
Published 31 August 2018 Volume 2018:10 Pages 117—128
DOI https://doi.org/10.2147/OARRR.S151013
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Butsabong Lerkvaleekul, Soamarat Vilaiyuk
Division of Rheumatology, Department of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand
Abstract: Macrophage activation syndrome (MAS) is a life-threatening condition, and it is a subset of hemophagocytic lymphohistiocytosis (HLH). The clinical features include a persistent high-grade fever, hepatosplenomegaly, lymphadenopathy, hemorrhagic manifestations, and a sepsis-like condition. From the clinical features, it is usually difficult to differentiate between a true sepsis, disease flare-ups, or MAS. Although the laboratory abnormalities are similar to those of a disseminated intravascular coagulation, which shows pancytopenia, coagulopathy, hypofibrinogenemia, and an elevated d-dimer test, it can also be a late stage of MAS. Currently, MAS is still underrecognized and usually results in delayed in diagnosis, which leads to high morbidity and mortality. This literature review was conducted in the context of the clinical manifestations and the laboratory abnormalities in MAS, which might provide some clues for an early diagnosis. The best ways for an early recognition and a satisfactory diagnosis were based on the relative changes in the overall parameters from the baseline, together with a thorough and continuous physical examination for these kinds of patients. At present, diagnostic criteria have been proposed for HLH, MAS-associated systemic juvenile idiopathic arthritis, and an MAS-associated systemic lupus erythematosus. Therefore, selecting the proper diagnostic criteria for use is essential because not all of the criteria are suitable for every autoimmune disease.
Keywords: hemophagocytic lymphohistiocytosis, systemic juvenile idiopathic arthritis, systemic lupus erythematosus, Kawasaki disease, autoimmune diseases, early diagnosis
Introduction
Macrophage activation syndrome (MAS) is a life-threatening complication of rheumatic diseases, requiring immediate and appropriate treatment. MAS is a disorder related to hemophagocytic lymphohistiocytosis (HLH), which is divided into primary and secondary HLH. Primary or familial HLH is an inherited disease, whereas secondary HLH is triggered by other diseases, including infections, malignancy, and autoimmune diseases. MAS is a secondary HLH, which is associated with autoimmune diseases.1,2 The most common autoimmune diseases associated with MAS are systemic juvenile idiopathic arthritis (SJIA), followed by systemic lupus erythematosus (SLE), Kawasaki disease (KD), and juvenile dermatomyositis (JDM).3 MAS is caused by an imbalance of the immune system, leading to uninterrupted hyperstimulation of the immune cells. The symptoms of MAS are quite similar to those of many active autoimmune diseases or severe sepsis; therefore, it is quite difficult to make a diagnosis. MAS is still underrecognized, and its treatment is usually delayed, which then leads to high morbidity and mortality. The classical signs and symptoms of patients with MAS are a persistent high-grade fever, hepatosplenomegaly, lymphadenopathy, and hemorrhagic manifestations. Abnormal results of investigation include cytopenia, coagulopathy, and hyperferritinemia. These distinctive features usually occur in the later stages of MAS; this leads to a delay in the diagnosis of the condition, resulting in a worse outcome. Therefore, early recognition of MAS is important and is the key to improving the morbidity and the mortality associated with this condition.
The aim of this current literature review was to identify the initial clinical manifestations and the changes in the laboratory parameters in the early stages of MAS. It was also found that it is important to emphasize the various clues of a suspicion of MAS and to summarize the clinical presentations, the laboratory abnormalities, and the outcomes from the literature review.
Epidemiology
The actual incidence of MAS in rheumatic diseases is still unknown, due to its underrecognition. Most of the studies have diagnosed MAS according to the HLH-2004 criteria4 or the criteria proposed by Ravelli et al.5 The estimated prevalence of MAS in SJIA was ~10%, and it increased up to 40% in subclinical MAS, which showed the evidence of MAS occurrence only in the bone marrow.6 The prevalence of MAS in SLE varied from 0.9% to 4.6% and it increased up to 9.4% in those patients with a hepatic dysfunction.7 The occurrence of hemophagocytosis was not related to the severity of SLE.8 The prevalence of MAS in KD was less frequent than in SJIA and SLE, which was estimated to be ~1.1%.9 The mortality rate varied depending on the centers and the underlying diseases of the patients. Previous reports showed that the mortality rate in SJIA varied between 8% and 23%,10–13 whereas the mortality rate in SLE was ~5%–35%.14–17 The mortality rate was also higher in adults, which was around 50%.18
Pathogenesis
The pathogenesis of MAS is still unknown because the clinical features of MAS are quite similar to familial HLH (FHLH) and there are possibly some related pathogeneses between these two conditions. The presence of abnormalities in the cytolytic pathway and decreased activities of natural killer (NK) cells have been shown in MAS-associated SJIA in many studies.19,20 These abnormalities occur by altering the protein variants of the various genes, including PRF1, MUNC13-14, STX11, STXBP2, LYST, and RAB27A, which are involved in the granule-mediated cytotoxic pathway.21–24 Heterozygous mutations in some of these genes might be associated with the development of MAS, especially if triggered by infections.25 Moreover, hyperinflammation, especially in SJIA, results in high levels of interleukin (IL)-6. This has been shown to be a contributing factor for the transient decreased cytotoxic activities of NK cells, as well as its influence on granule exocytosis.20 The main mechanisms of MAS may be triggered by the high activities of the autoimmune diseases or the infectious agents, resulting in a prolonged immune activation, predominantly by the cytotoxic T cells and the macrophages. Any defect in terminating the immune responses leads to hypercytokinemia or to a cytokine storm. These cytokines include interferon gamma (IFNγ), tumor necrosis factor alpha (TNFα), IL-2, IL-1, IL-6, IL-18, as well as the macrophage colony stimulating factor (M-CSF).26–30 A previous study also demonstrated marked elevation of both IFNγ and an IFNγ-induced chemokine, chemokine (C-X-C motif) ligand 9 (CXCL9), in SJIA patients with MAS and other secondary HLH patients, whereas their levels were normal in both active SJIA without MAS and inactive SJIA.31
Some of the previous studies demonstrated cytokine changes during MAS occurrence and this has led to the suspicion of an MAS-associated SJIA. A decreased ratio of IL-18/IFNγ raised the suspicions of an MAS development in SJIA as well.30 In addition, Shimizu et al32 reported that SJIA patients with high elevation of their IL-18 levels, especially with >47,750 pg/mL, were at a high risk of developing MAS and that monitoring of the serum IL-18 levels was beneficial in predicting MAS development.
Clinical and laboratory features: how to recognize MAS early
Diagnostic criteria involving clinical and laboratory data are reported for defining MAS. However, in the reviewers’ opinion, these criteria have some limitations in making an early diagnosis. The relative changes in the clinical and laboratory features from the baseline are more useful for providing an early diagnosis, due to the different baseline characteristics and the dynamic changes over time in the concomitant underlying diseases. Generally, almost all of the SJIA patients with MAS have a fever. The typical patterns of fever in MAS change from an intermittent fever to a nonremitting fever and these changes help to discriminate the patients between underlying disease flare-ups and MAS. Hepatosplenomegaly and lymphadenopathy are both present in those patients with an active SJIA and MAS, but these abnormalities deteriorate when MAS occurs. However, in a study by Kostik et al,33 these clinical features showed high sensitivity, with poor specificity.
Approximately 35% of the patients with MAS developed a central nervous system (CNS) dysfunction, including seizures, as well as alterations in their mental status, although not necessarily at the same time. They also showed a certain amount of irritability, with lethargy, comas, and headaches.10 These CNS involvements, as found in MAS, were more prominent than in active SJIA; however, they were not regarded as an early presentation for an onset of MAS.34 Hemorrhagic manifestations were often seen in a full-blown MAS, which ranged from an easy bruising to a mucosal bleeding, a gastrointestinal bleeding, and a disseminated intravascular coagulation. These findings were also presented in later stages of MAS, and they were found in 20% of the MAS patients.10 Severe cases of MAS requiring an admission to an intensive care unit had multiple-organ failures, and they often had heart, lung, and kidney involvements, leading to a fatal outcome. When MAS started to occur, all of the clinical features were not totally presented. Therefore, it was difficult to make a diagnosis. The clinical features were usually presented after MAS had started for a while. Thus, using only the clinical features was not a practical way to get an early diagnosis of MAS, although they were sometimes useful for predicting the outcome.
Most of the studies supported subtle laboratory alterations and monitoring of the laboratory changes, along with an early recognition of the clinical presentations for MAS onset; these were crucial for an early diagnosis of MAS.5,33,35,36 The laboratory parameters, including platelet count, as well as the aspartate and alanine aminotransferases, together with the ferritin, lactate dehydrogenase, triglyceride, and d-dimer levels, showed >50% changes between a pre-MAS visit and MAS onset.10 These simple and broad laboratory tests are sensitive screening tools for an MAS diagnosis. Additionally, the relative decreases in platelet count appeared to be the most valuable indication for differentiating between MAS onset and a disease flare-up.7,36 The study by Kostik et al33 showed that a combination of >3 of the following laboratory variables were reliable indicators for an early diagnosis: these were decreased platelet and white blood cell counts; decreased levels of albumin and fibrinogen; increased levels of ferritin, aspartate aminotransferase, and lactate dehydrogenase; and the presence of proteinuria. These indications provided the highest sensitivity and specificity necessary for an early diagnosis of MAS-associated SJIA.33 Anemia was also seen in those patients with a disease flare-up or MAS onset, but the percentages of change in the hemoglobin levels were smaller when comparing them with other parameters.10 A drop in the erythrocyte sedimentation rate (ESR), or a disproportion between the ESR and the C-reactive protein (CRP) levels, raised a suspicion of MAS. However, this finding occurred in the latter stages of MAS, concomitant with hypofibrinogenemia, causing a drop in the ESR.
A marked increase of the serum ferritin levels was also an important diagnostic biomarker for MAS, especially when it was >5,000–10,000 ng/mL. Serum ferritin measurements were used to screen patients who were at a risk of developing MAS.37 However, the absolute values of the ferritin levels as a one-time measurement were not associated with disease severity or outcome. The ferritin levels varied between the patients in the survival and the nonsurvival groups. Serial measurements of ferritin levels in an individual patient were more informative for assessing a treatment response and in predicting a prognosis.38 Measurements of the soluble IL-2 receptor alpha chain and the soluble CD163 levels, which represent an activation of the T cells and the macrophages, respectively, reflected a subclinical form of MAS, and these were reported to be helpful in monitoring the disease activities; these are all promising biomarkers.39 However, these investigations are not available in routine clinical practice. In addition, there were no cutoff levels for identifying those patients at a risk of developing MAS. The evidence of hemophagocytosis upon examination of their bone marrow, together with their lymph nodes, their spleen, or any other organs, was also a characteristic feature of MAS. Approximately 60% of the SJIA patients demonstrated hemophagocytosis upon bone marrow aspiration and ~30% of these patients showed hemophagocytosis on biopsy of their lymph nodes and/or their liver.10 Therefore, when it was undetectable on bone marrow aspiration, or on biopsy of their lymph nodes or liver, it did not exclude a diagnosis of MAS, especially the early stage of MAS. The abnormalities of laboratory data in MAS were related to the pathophysiology. Anemia, thrombocytopenia, and leukopenia result from hemophagocytosis. Elevated liver enzymes could be explained by the infiltration of histiocytes and lymphocytes into the liver.40 A markedly elevated ferritin level may result from hypercytokinemia and active production of ferritin by macrophages.41 High triglycerides are caused by decreased lipoprotein lipase activity, which is initiated by TNFα. Hypofibrinogenemia is a consequence of increased plasminogen activator secretion by macrophages, resulting in the conversion of plasminogen to plasmin. Moreover, hypofibrinogenemia leads to a drop in ESR.40
In the research of the literature, there were several studies that reported on the clinical manifestations and the laboratory data in all of the various MAS conditions. These were associated with the various autoimmune diseases. In the opinions of the reviewers, the following points are important clues for an early diagnosis of MAS. Nearly all patients with MAS have a fever, although they may have different underlying autoimmune conditions. MAS-associated SJIA tends to have more patients with hepatosplenomegaly and lymphadenopathy than does MAS-associated SLE. This is explained by the abnormal features that are common presentations in those patients with active SJIA, more so than in those with an active SLE. The MAS-associated KD patients always present hepatosplenomegaly, whereas this is an uncommon presentation in patients with active KD. In fact, cervical lymphadenopathy is one of the clinical criteria that is found in patients with typical presentations of KD. There are only limited data describing the different characteristics of lymph nodes in the KD condition, with or without an MAS condition. The CNS manifestations in SLE patients, including seizures, comas, alterations of consciousness, and headaches, are found in both active disease and MAS-associated SLE; therefore, it is difficult to differentiate between these two conditions. However, this is quite different in those patients with SJIA and KD, in that a CNS involvement is not being presented in the manifestations of the active disease in these two conditions. Therefore, MAS is suspected in those patients with an underlying SJIA and KD, who perhaps have a CNS involvement.
The baseline laboratory data for these autoimmune diseases is also different. The SJIA and KD patients usually express leukocytosis and thrombocytosis, in contrast to the SLE patients, who commonly present leukopenia and thrombocytopenia. In addition, a fever with related cytopenias, in all of the underlying diseases, raise the suspicion of MAS, and further investigations for this condition are recommended. Remarkably, serum ferritin levels are the highest in the MAS-associated SJIA condition, when compared with the MAS-associated SLE and the MAS-associated KD conditions.
From the literature review of the clinical features and the laboratory parameters, in the early stages of MAS in the various autoimmune diseases (Table 1), most of the studies stated that decreased platelet and white blood cell counts, increased serum ferritin levels, as well as increased lactate dehydrogenase and liver enzyme levels. These parameters were all relatively changed in the early stages of MAS. Therefore, serial monitoring of laboratory parameters in these individuals was crucial for an early recognition of MAS in the various autoimmune diseases. These serial monitoring techniques were reported to be better than use of the absolute values in a one-time measurement. The more early the detection and initiation of the treatment for MAS, the better was the disease outcome.
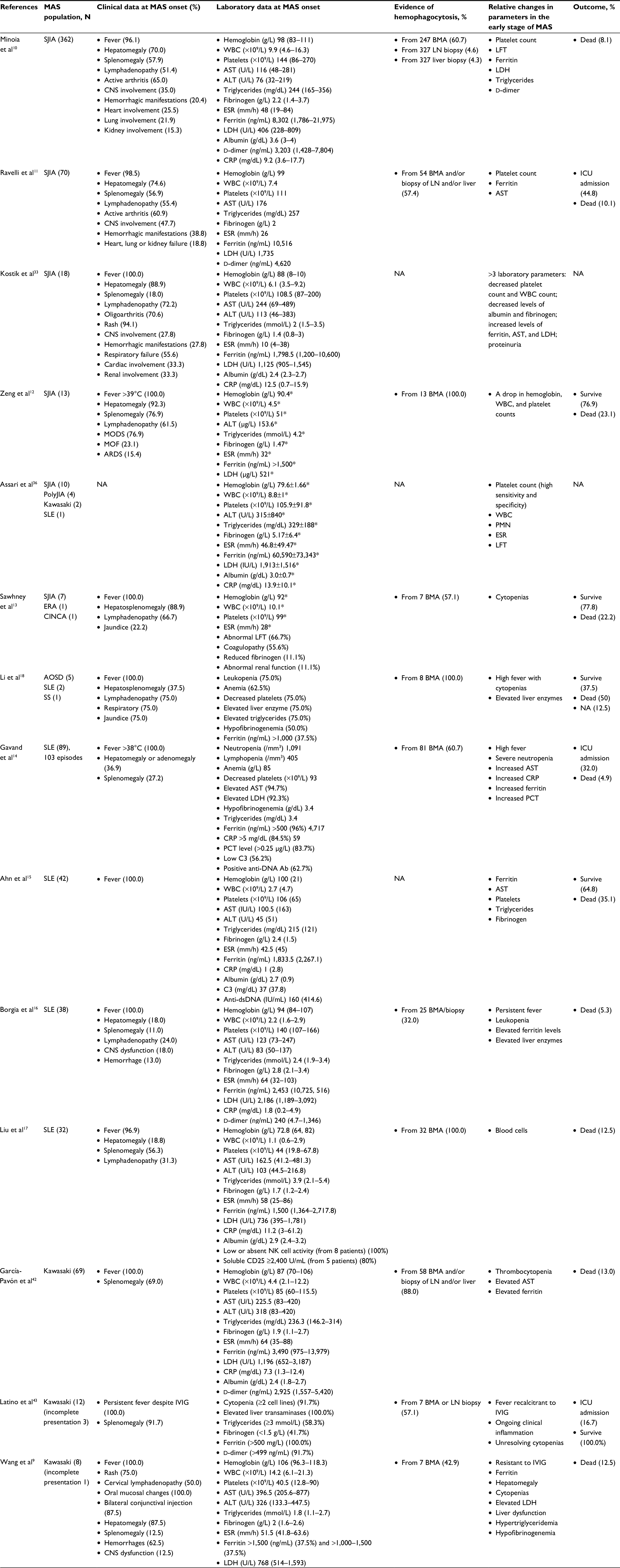 | Table 1 Literature review of the clinical features and the laboratory parameters in the early stages of MAS in the various autoimmune diseases Note: Laboratory data presented as median (interquartile range) and *mean±SD. Abbreviations: Ab, antibody; ALT, alanine aminotransferase; AST, aspartate aminotransferase; APS, anti-phospholipid syndrome; ARDS, acute respiratory distress syndrome; AOSD, adult-onset Still’s disease; BMA, bone marrow aspiration; C3, complement 3; CNS, central nervous system; CINCA, chronic infantile neurological cutaneous articular syndrome; CRP, C-reactive protein; ERA, enthesitis-related arthritis; ESR, erythrocyte sedimentation rate; IVIG, intravenous immunoglobulin; ICU, intensive care unit; LDH, lactate dehydrogenase; LFT, liver function test; LN, lymph node; MAS, macrophage activation syndrome; MCTD, mixed connective tissue disease; MODS, multiple-organ dysfunction syndrome; MOF, multiple-organ failure; NA, not available; PCT, procalcitonin; PMN, polymorphonuclear neutrophils; PolyJIA, polyarticular juvenile idiopathic arthritis; SJIA, systemic juvenile idiopathic arthritis; SLE, systemic lupus erythematosus; SS, Sjögren’s syndrome; WBC, white blood cell count. |
Diagnosis: how to make an early diagnosis
An early diagnosis and prompt initial treatment are both key factors for a favorable outcome. However, the features of MAS are similar to a disease flare-up and a systemic infection, making an early diagnosis challenging. Since MAS is a subset of HLH, many physicians still use the HLH-2004 criteria4 for MAS diagnosis. However, the HLH-2004 criteria are not suitable for patients with an underlying autoimmune disease, especially for those suffering from SJIA. Since the typical laboratory abnormalities in the SJIA patients are leukocytosis and thrombocytosis, using these criteria delays the arrival at an MAS diagnosis. In active SJIA patients, the fibrinogen levels are often elevated due to the inflammatory processes occurring as a nature of the disease. This being so, the cutoff levels of fibrinogen in the HLH-2004 criteria are not to be regarded as proper for the diagnosis of an MAS-associated SJIA condition. In addition, assessments of the NK cell activities and the CD25 levels in the HLH-2004 criteria are not routinely accessible in all centers.
The alternative approaches for MAS diagnosis in SJIA patients are the preliminary diagnostic guidelines for an MAS-complicating SJIA condition,5 as well as the recent 2016 classification criteria for MAS-complicating SJIA condition, as stated by Ravelli et al.1 These two sets of criteria provide more sensitivity and specificity in making a diagnosis of an MAS-associated SJIA condition. Furthermore, Kostik et al33 have proposed the diagnostic criteria for MAS in an active SJIA condition, which demonstrate high sensitivity and specificity for an early diagnosis of an MAS-associated SJIA condition. Moreover, Parodi et al7 propose preliminary diagnostic guidelines for MAS as a complication of juvenile SLE, which also demonstrated a high diagnostic yield.
In the real world, although there are many diagnostic guidelines with high sensitivity and specificity for diagnosing MAS, especially when there are different underlying conditions, the best ways for early recognition and diagnosis were based on the relative changes in the overall parameters from the baseline. Thorough and continuous physical examination in these patients was also stated as being important. These approaches helped the physicians when initiating an early treatment, resulting in improvements for the outcome of the disease.
Management
The mainstay of MAS treatment is glucocorticoid therapy. Most of the physicians start with an intravenous methylprednisolone 30 mg/kg/dose (maximum 1 g) for 1–3 days. If the patients respond well to the high dose of methylprednisolone, this is decreased to 2–3 mg/kg/day in a divided dose. If the clinical status of the patients is stable, the physicians change the dosage to oral prednisolone, thus, preparing for a discharge, as well. For the nonresponders, an additional therapy with cyclosporin A 2–7 mg/kg/day is recommended; for the common side effects from this medication, including hypertension and renal toxicity, the physicians should be cautious.44–46 For patients who are refractory to the high dosages of corticosteroids and cyclosporin A, an HLH-2004 protocol treatment should be considered.4 Other than dexamethasone and cyclosporin A, the HLH-2004 treatment protocol includes etoposide (or VP16) as a treatment regime. As a result, etoposide has been reported as a therapeutic option for MAS. However, etoposide is not recommended to be a first-line therapy for MAS, because of the severe side effects, including severe bone marrow suppression, as well as hepatic and renal toxicity.47 For patients with a refractory MAS, concomitant with hepatic and renal impairment, anti-thymocyte globulin (ATG) is considered to be useful.48 However, there were some data showing that ATG increased the risk of severe infection and mortality.49 Other treatments, including intravenous immunoglobulin, cyclophosphamide, and plasma exchange, have provided inconsistent outcomes. Recently, there have been increased reports of use of biological therapies for MAS. Since IL-1 and IL-6 have major roles in the pathogenesis of SJIA, the IL-1 receptor antagonist, anakinra, has been reported to benefit patients with an MAS-associated SJIA condition.50–53 Nevertheless, there are a few reports that anakinra may be a trigger for MAS.54 Careful monitoring of the unexpected complications arising after receiving biologic treatment is recommended for these patients. Another human anti-IL-1β monoclonal antibody, canakinumab, has also been reported to be successful in the treatment of MAS-associated SJIA patients. However, clinical trials demonstrated the development of MAS in SJIA patients treated with canakinumab; nevertheless, the incidence was not statistically different when compared with that in placebo-treated patients. From this study, we can conclude that canakinumab does not change the risk of developing MAS. Furthermore, infections are still the most important trigger in MAS patients because MAS in this study occurred while patients were inactive but had infections.55 The humanized anti-IL-6 receptor monoclonal antibody, tocilizumab, has proven to be beneficial for SJIA, although there are only limited data for the treatment of an MAS-associated SJIA condition. In addition, it has been reported that tocilizumab masked the clinical features of MAS, because tocilizumab blocked the IL-6R and disrupted the IL-6 signaling pathway, which resulted in mild clinical symptoms, but with normal CRP levels.56 A systematic literature review in MAS-associated SJIA with anti-IL-6 or anti-IL-1 therapy demonstrated that ~25% of patients with tocilizumab treatment were afebrile, while all patients undergoing treatment with canakinumab had fever. In addition, MAS patients receiving tocilizumab or canakinumab treatment also had lower ferritin levels when compared to MAS patients without biologic therapy. Therefore, MAS-associated SJIA with biologic treatment might not be able to fulfill the criteria and may lead to a delay in the diagnosis. If clinical manifestations in SJIA patients with biologic treatment are compatible with MAS, physicians should be aware of this condition even though patients are afebrile and have low ferritin levels. The risk of MAS does not decrease in SJIA patients treated with either tocilizumab or canakinumab. This may be explained by the pathophysiology in MAS, which is related to the production of many cytokines. Inhibition of IL-1 or IL-6 might not be able to prevent MAS because other cytokines, for instance, IL-18, might play a major role in developing MAS.57 A summary of the MAS treatment is shown in Table 2.
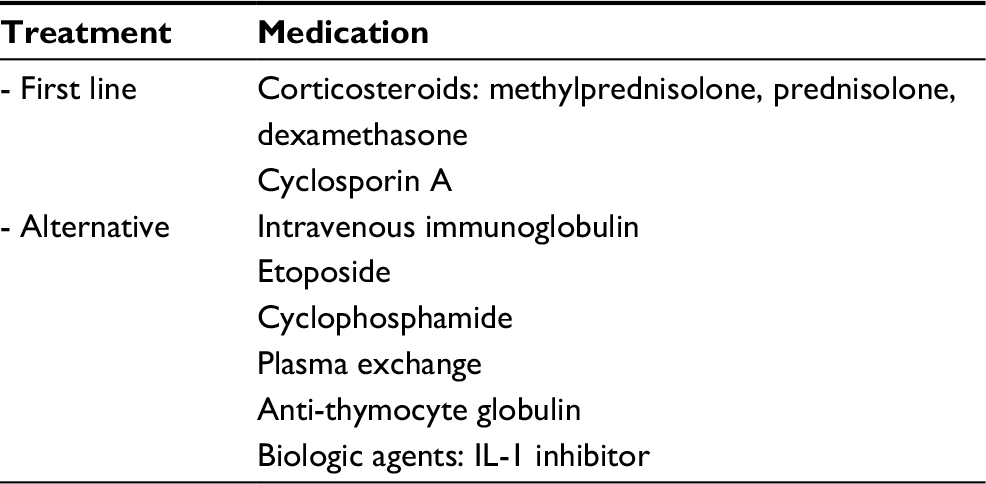 | Table 2 Summary of treatment for macrophage activation syndrome |
Conclusion
An early diagnosis and prompt initial treatment are both key factors for a favorable outcome in MAS. Although the clinical presentations of both MAS and active autoimmune diseases were quite similar, there were some clues from the serial monitoring of laboratory parameters, which helped the physicians in making an early diagnosis of MAS. The relative changes in the laboratory parameters during the early stages of MAS included changes in platelet and white blood cell counts, as well as levels of ferritin, LDH, and liver enzymes. At present, diagnostic criteria have been proposed for HLH and MAS in the various autoimmune diseases. Selecting the proper diagnostic criteria to diagnose MAS is essential, because not all of the criteria are suitable for every autoimmune disease.
Acknowledgment
We thank Michael John Howard, Supervisory Proofreader and Administrative Copy Editor, at PRS Proofreading Services, for copy editing and proofreading a draft of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Ravelli A, Minoia F, Davì S, et al. 2016 classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborat. Arthritis Rheumatol. 2016;68(3):566–576. | ||
Grom AA, Passo M. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis. J Pediatr. 1996;129(5):630–632. | ||
Lin CI, Yu HH, Lee JH, et al. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin Rheumatol. 2012;31(8):1223–1230. | ||
Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. | ||
Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146(5):598–604. | ||
Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–1138. | ||
Parodi A, Davì S, Pringe AB, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum. 2009;60(11):3388–3399. | ||
Morales PM, Jimenez BF, Yanes P, Rios D, Godinez R. Marrow B with reactive histiocytosis (RH), hemophagocytosis and storage histiocytes (SH) in systemic lupus erythematosus. Arthritis Rheum. 1992;35(Suppl):S239. | ||
Wang W, Gong F, Zhu W, Fu S, Zhang Q. Macrophage activation syndrome in Kawasaki disease: more common than we thought? Semin Arthritis Rheum. 2015;44(4):405–410. | ||
Minoia F, Davì S, Horne A, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66(11):3160–3169. | ||
Ravelli A, Minoia F, Davì S, et al. Expert consensus on dynamics of laboratory tests for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. RMD Open. 2016;2(1): e000161. | ||
Zeng HS, Xiong XY, Wei YD, Wang HW, Luo XP. Macrophage activation syndrome in 13 children with systemic-onset juvenile idiopathic arthritis. World J Pediatr. 2008;4(2):97–101. | ||
Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child. 2001;85(5):421–426. | ||
Gavand PE, Serio I, Arnaud L, et al. Clinical spectrum and therapeutic management of systemic lupus erythematosus-associated macrophage activation syndrome: a study of 103 episodes in 89 adult patients. Autoimmun Rev. 2017;16(7):743–749. | ||
Ahn SS, Yoo BW, Jung SM, Lee SW, Park YB, Song JJ. In-hospital mortality in febrile lupus patients based on 2016 EULAR/ACR/PRINTO classification criteria for macrophage activation syndrome. Semin Arthritis Rheum. 2017;47(2):216–221. | ||
Borgia RE, Gerstein M, Levy DM, Silverman ED, Hiraki LT, Features HLT. Features, treatment, and outcomes of macrophage activation syndrome in childhood-onset systemic lupus erythematosus. Arthritis Rheumatol. 2018;70(4):616–624. | ||
Liu AC, Yang Y, Li MT, et al. Macrophage activation syndrome in systemic lupus erythematosus: a multicenter, case-control study in China. Clin Rheumatol. 2018;37(1):93–100. | ||
Li X, Qu B, Nie Y, Zhu G, Li W, Mu F. Clinical features of macrophage activation syndrome in the adult northern Chinese population. Lupus. 2014;23(8):785–792. | ||
Grom AA, Villanueva J, Lee S, Goldmuntz EA, Passo MH, Filipovich A. Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr. 2003;142(3):292–296. | ||
Cifaldi L, Prencipe G, Caiello I, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol. 2015;67(11):3037–3046. | ||
Kaufman KM, Linghu B, Szustakowski JD, et al. Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. 2014;66(12):3486–3495. | ||
Vastert SJ, van Wijk R, D’Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology. 2010;49(3):441–449. | ||
Hazen MM, Woodward AL, Hofmann I, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(2):567–570. | ||
Zhang K, Biroschak J, Glass DN, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum. 2008;58(9):2892–2896. | ||
Sepulveda FE, Garrigue A, Maschalidi S, et al. Polygenic mutations in the cytotoxicity pathway increase susceptibility to develop HLH immunopathology in mice. Blood. 2016;127(17):2113–2121. | ||
Henter JI, Elinder G, Söder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991;78(11):2918–2922. | ||
Henter JI, Andersson B, Elinder G, Jakobson A, Lübeck PO, Söder O. Elevated circulating levels of interleukin-1 receptor antagonist but not IL-1 agonists in hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 1996;27(1):21–25. | ||
Schulert GS, Grom AA. Pathogenesis of macrophage activation syndrome and potential for cytokine-directed therapies. Annu Rev Med. 2015;66:145–159. | ||
Maruyama J, Inokuma S. Cytokine profiles of macrophage activation syndrome associated with rheumatic diseases. J Rheumatol. 2010;37(5):967–973. | ||
Put K, Avau A, Brisse E, et al. Cytokines in systemic juvenile idiopathic arthritis and haemophagocytic lymphohistiocytosis: tipping the balance between interleukin-18 and interferon-γ. Rheumatology. 2015;54(8):1507–1517. | ||
Bracaglia C, de Graaf K, Pires Marafon D, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017;76(1):166–172. | ||
Shimizu M, Nakagishi Y, Inoue N, et al. Interleukin-18 for predicting the development of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Clin Immunol. 2015;160(2):277–281. | ||
Kostik MM, Dubko MF, Masalova VV, et al. Identification of the best cutoff points and clinical signs specific for early recognition of macrophage activation syndrome in active systemic juvenile idiopathic arthritis. Semin Arthritis Rheum. 2015;44(4):417–422. | ||
Singh S, Chandrakasan S, Ahluwalia J, et al. Macrophage activation syndrome in children with systemic onset juvenile idiopathic arthritis: clinical experience from northwest India. Rheumatol Int. 2012;32(4):881–886. | ||
Davì S, Consolaro A, Guseinova D, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38(4):764–768. | ||
Assari R, Ziaee V, Mirmohammadsadeghi A, Moradinejad MH. Dynamic changes, cut-off points, sensitivity, and specificity of laboratory data to differentiate macrophage activation syndrome from active disease. Dis Markers. 2015;2015:424381–424388. | ||
Emmenegger U, Reimers A, Frey U, et al. Reactive macrophage activation syndrome: a simple screening strategy and its potential in early treatment initiation. Swiss Med Wkly. 2002;132(17–18):230–236. | ||
Vilaiyuk S, Sirachainan N, Wanitkun S, Pirojsakul K, Vaewpanich J. Recurrent macrophage activation syndrome as the primary manifestation in systemic lupus erythematosus and the benefit of serial ferritin measurements: a case-based review. Clin Rheumatol. 2013;32(6):899–904. | ||
Bleesing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(3):965–971. | ||
George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. J Blood Med. 2014;5:69–86. | ||
Cohen LA, Gutierrez L, Weiss A, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood. 2010;116(9):1574–1584. | ||
García-Pavón S, Yamazaki-Nakashimada MA, Báez M, Borjas-Aguilar KL, Murata C. Kawasaki disease complicated with macrophage activation syndrome: a systematic review. J Pediatr Hematol Oncol. 2017;39(6):445–451. | ||
Latino GA, Manlhiot C, Yeung RS, Chahal N, Mccrindle BW. Macrophage activation syndrome in the acute phase of Kawasaki disease. J Pediatr Hematol Oncol. 2010;32(7):527–531. | ||
Mouy R, Stephan JL, Pillet P, Haddad E, Hubert P, Prieur AM. Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr. 1996;129(5):750–754. | ||
Ravelli A, de Benedetti F, Viola S, Martini A. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis successfully treated with cyclosporine. J Pediatr. 1996;128(2):275–278. | ||
Quesnel B, Catteau B, Aznar V, Bauters F, Fenaux P. Successful treatment of juvenile rheumatoid arthritis associated haemophagocytic syndrome by cyclosporin A with transient exacerbation by conventional-dose G-CSF. Br J Haematol. 1997;97(2):508–510. | ||
Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–298. | ||
Coca A, Bundy KW, Marston B, Huggins J, Looney RJ. Macrophage activation syndrome: serological markers and treatment with anti-thymocyte globulin. Clin Immunol. 2009;132(1):10–18. | ||
Mahlaoui N, Ouachée-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622–e628. | ||
Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology. 2011;50(2):417–419. | ||
Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F. Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol. 2011;17(1):23–27. | ||
Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol. 2008;4(11):615–620. | ||
Lurati A, Teruzzi B, Salmaso A, et al. MAS) during anti-IL-1 receptor therapy (anakinra) in a patient affected by systemic onset idiopathic juvenile arthritis (soJIA): a report and review of the literature. Pediatr Rheumatol Online J. 2007;3(2):79–85. | ||
Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63(2):545–555. | ||
Grom AA, Ilowite NT, Pascual V, et al. Rate and clinical presentation of macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis treated with canakinumab. Arthritis Rheumatol. 2016;68(1):218–228. | ||
Shimizu M, Nakagishi Y, Kasai K, et al. Tocilizumab masks the clinical symptoms of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome: the diagnostic significance of interleukin-18 and interleukin-6. Cytokine. 2012;58(2):287–294. | ||
Schulert GS, Minoia F, Bohnsack J, et al. Effect of biologic therapy on clinical and laboratory features of macrophage activation syndrome associated with systemic juvenile idiopathic arthritis. Arthritis Care Res. 2018;70(3):409–419. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.