Back to Journals » OncoTargets and Therapy » Volume 12
m6A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways
Authors Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, Liu H, Deng Q, Wu X, Lan P, Deng Y
Received 11 January 2019
Accepted for publication 15 April 2019
Published 4 June 2019 Volume 2019:12 Pages 4391—4402
DOI https://doi.org/10.2147/OTT.S201052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Ru Deng,1,* Yikan Cheng,2,* Shubiao Ye,3,4,* Jianwei Zhang,1 Runqing Huang,3 Peisi Li,5 Huashan Liu,3 Qiling Deng,3 Xianrui Wu,3 Ping Lan,3 Yanhong Deng1,*
1Department of Medical Oncology, The Sixth Affiliated Hospital, Guangdong Provincial Key Laboratory of Colorectal and Pelvic Floor Diseases, Guangzhou, Guangdong, People’s Republic of China; 2Department of Radiation Oncology, The Sixth Affiliated Hospital, Guangdong Provincial Key Laboratory of Colorectal and Pelvic Floor Diseases, Sun Yat-sen University, Guangzhou, Guangdong, People’s Republic of China; 3Department of Colorectal Surgery, The Sixth Affiliated Hospital, Guangdong Provincial Key Laboratory of Colorectal and Pelvic Floor Diseases, Sun Yat-sen University, Guangzhou, Guangdong, People’s Republic of China; 4Guangdong Provincial Key Laboratory of Colorectal and Pelvic Floor Diseases, The Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou, Guangdong, People’s Republic of China; 5School of Medicine, Sun Yat-sen University, Guangzhou, Guangdong, People’s Republic of China
*These authors contributed equally to this work
Purpose: Although many biological processes are involved in the modification of N6-methyladenosine (m6A), the exact role of m6A in the development of malignant tumors remains unclear. Methyltransferase 3 (METTL3) is a major RNA N6-methyladenosine methyltransferase. We aimed to explore the role of METTL3 in colorectal cancer (CRC) carcinogenesis and disease progression.
Methods: In this study, immunohistochemistry was performed with a tissue microarray. qRT-PCR and Western blots were used to evaluate the expression of METTL3 in CRC cells. The effect of METTL3 on cell proliferation, migration and invasion of CRC cells was examined by IncuCyte Live Cell Analysis System and transwell assay, respectively.
Results: The results suggested that positive expression of METTL3 was significantly associated with longer survival time (P=0.011). We next demonstrated that overexpression of METTL3 could inhibit proliferation, migration and invasion in CRC cells, while downregulation of METTL3 shows the opposite result. Furthermore, downregulation of METTL3 resulted in activation of p-p38 and p-ERK. Moreover, the inhibitors of p38 or ERK kinase could significantly reverse the effect of migration and invasion, which was induced by knockdown of METTL3.
Conclusion: We concluded that METTL3 played a tumor-suppressive role in CRC cell proliferation, migration and invasion through p38/ERK pathways, which indicated that METTL3 might be a novel marker for CRC carcinogenesis, progression and survival.
Keywords: METTL3, colorectal cancer, N6-methyladenosine, migration
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide.1 The survival outcomes in CRC remain poor, due in part, to the presence of distant metastases.2 Thus, investigating the molecular mechanism of CRC progression and metastasis is crucial in the development of future therapeutic advances. Accumulating evidence has revealed the multistep process in CRC carcinogenesis that involves complicated and dynamic interplay between host genetics, epigenetics and transcriptomic alterations.3,4
The importance of DNA methylation plays in the proliferation and metastasis of CRC is well-established.5 However, the role of N6-methyadenosine (m6A) in the modification of RNA remains unclear. m6A is recognized as the most common internal modification in mammalian mRNA,6 which is a dynamic reversible process and plays a crucial role in epigenetic regulation.7,8 METTL3 and METTL14 were identified as m6A “writers” that form a heterodimeric complex with the support of WTAP in the catalysis of m6A methylation.9,10 By contrast, FTO and ALKBH5 are referred to as m6A “erasers” that were shown to regulate cancer stem cell properties.11,12 METTL3 was originally identified as a methyltransferase that acted as one component of the methyltransferase complex.13 Furthermore, METTL3 was shown to be essential in cancer cell survival by modifying p53-mediated signaling14 and epigenetically silences tumor-suppressor genes and serves as an oncogene in promoting progression in hepatocellular carcinoma.15 By contrast, METTL3 was reported as a tumor suppressor in kidney cancer.16 Therefore, it is of great significance to explore the functional role and potential mechanism of METTL3 in CRC. Herein, we investigated the function of METTL3 in CRC disease progression. This might reveal the potential for future drug development in the setting of CRC.
Material and methods
CRC specimens and cell-lines
The samples were collected from patients who underwent surgery at the Taizhou Hospital in Zhejiang province from 2008 to 2011 with a follow-up information till 2017. All clinical samples were collected with written informed consent from patients, and the ethical approval was granted from the Committees for Ethical Review of Shanghai Outdo Biotech Company. The human CRC cell lines (HCT-116, DLD-1, KM-12 and HCT-8) were obtained from the American Type Culture Collection (ATCC) and maintained in RPMI-1640 culture medium that was supplemented with 10% FBSand working strength penicillin-streptomycin Solution as antibiotics (all from Gibco, New York, NY, USA) at 37ºC in an atmosphere of 5% CO2 in air. The final concentration of p-38 (SB203580, Selleckchem, Shanghai, People's Republic of China) and ERK (SCH772984, MCE) inhibitor was both 2uM.
Establishment of stable knockdown and overexpressed cells
The open-reading frame (ORF) of METTL3 was amplified from the cDNA of HCT-116 cells and inserted into the Psin vector. CRC cells were stably transduced with METTL3-overexpressing lentiviral and Psin control vectors. For the METTL3 knockdown studies, CRC cells were stably transfected with PLKO control vectors and transduced with METTL3 knockdown lentiviral vectors (Table S1). Lentiviruses were packaged by transfecting the 3-plasmid transfer vector: psPAX2: pMD2.G=6: 3: 1.5 ug into 293 Ta cells. The released virus in the liquid media was harvested at 48 hrs and 72 hrs post-transduction and concentrated by ultracentrifugation. To establish the stable knockdown and overexpressing cells, both HCT-116 and DLD1 cells were transduced with the lentivirus at a multiplicity of infection (MOI) of 10 with 5 μg/mL polybrene (Sigma, Saint Louis, MO, USA). The stable expressing cells were selected by treating with puromycin for 2 weeks.
Immunohistochemistry and western blotting
The slide was incubated in 0.3% hydrogen peroxide for 30 mins to block endogenous peroxidase activity. Tissues were permeabilized with 0.5% Triton X-100 for 30 mins and incubated with working strength citrate buffer (pH 6.0) for 15 mins to retrieve the antigen of interest. After three washes in PBS, the slides were blocked in 5% normal goat serum. Samples were then cross-reacted with anti-METTL3 antibody (Abcam, ab195352; Cambridge, UK) that was diluted 1:100 and incubated with the sample for 2 hrs at room temperature. A secondary horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (H+L) was incubated with the slides for 30 mins. In addition, nuclei were counterstained with DAPI. Stained sections were visualized using a Leica fluorescence microscope.
Cell monolayers were lysed by incubating with RIPA buffer (Beyotime, Shanghai, People's Republic of China) that was supplemented with a protease inhibitor cocktail (Sigma) on ice for 30 mins. Lysates were centrifuged at 12,000 g for 20 mins, and supernatants were harvested and saved. The protein concentrations were determined using a BCA Protein Assay Kit (Boster, Pleasanton, CA, USA). Proteins were separated by SDS-PAGE using an 8–10% PAGEand transferred to nitrocellulose membranes (BioRad, Hercules, CA, USA).
The membranes were then blocked by 5% milk for 1 hr at room temperature. After four washes in PBST (PBS supplemented with 0.5% Triton X-100), membranes were immunoblotted with HRP-conjugated goat anti-mouse antibody (1:10,000) for 1 hr. The signal strength of revealed protein bands was detected with an enhanced chemiluminescence (ECL) substrate and captured with Image-Lab software (BioRad). Rabbit monoclonal antibody METTL3, P38, p-P38, ERK, p-ERK, JNK, and p-JNK were purchased from Cell Signaling Technology, Inc. (St. Louis, MO, USA). Detection of the relative protein band intensities of GAPDH served as an internal loading control.
Proliferation and colony formation assay
Approximately 5×103 cells were seeded into 96-well flat-bottomed plates, and the real-time proliferation of those seeded cells was detected by the IncuCyte® S3 Live Cell Analysis System (Roche Applied Sciences, Indianapolis, IN, USA). Cell growth was assessed for 4 days.
For the colony formation assay, 5–1,000 cells were seeded into 6-well culture plates and cultured for 14 days. Four percent paraformaldehyde was used to fix cells for 10 mins and crystal violet aqueous solution was used to stain cells (Beyotime) for 15 mins. The plates were photographed, and the colonies were then counted. All experiments were independently set up three times, and the representative results of this assay are shown.
Transwell migration and invasion assays
For the migration assays, approximately 4×104 cells were suspended in 200 μL serum-free RPMI-1640 and added into the upper chamber of a Transwell plate (Corning, New York, NY, USA) with an 8 μm pore size polycarbonate filter. The lower chamber had 500 μL complete culture medium with 10% FBS dispensed. The cells were cultured for 48 hrs, following which cells were fixed in 4% paraformaldehyde and then stained with crystal violate. A cotton swab was used to remove cells on the upper surface of the filter manually. The migrating cells were observed by microscopy, and the average value of the crystal violate intensity was calculated. The invasion assay was performed similarly for a migration assay with the exception of dispensing 5% Matrigel into the upper chamber before seeding cells into the culture system.
RNA isolation and quantitative real-time PCR assays
Total RNA was separated from cultured cells with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RT-PCR used a commercially available Reverse Transcription Kit (Toyobo, Osaka, Japan). qPCR assays were performed by the SYBR Green kit (Toyobo) on the ABI7300 instrument. The assay was performed across three biological replicates.
Statistical analysis
An optimized cutoff of METTL3 expression was generated using R (maxstat package; Torsten Hothorn;
Results
Correlation between the METTL3 expression and clinical features in CRC patients
Immunohistochemistry of METTL3 was conducted in a tissue microarray (Figure 1). 181 patients were enrolled with comprehensive baseline clinical characteristics. 181 CRC patients were divided into METTL3 expression group (n=47) and no METTL3 expression group (n=134). The clinical features of both groups were compared (Table 1), which indicated that the negative expression of METTL3 was significantly correlated with larger tumor size (P=0.031) and higher metastasis rates (P=0.002).
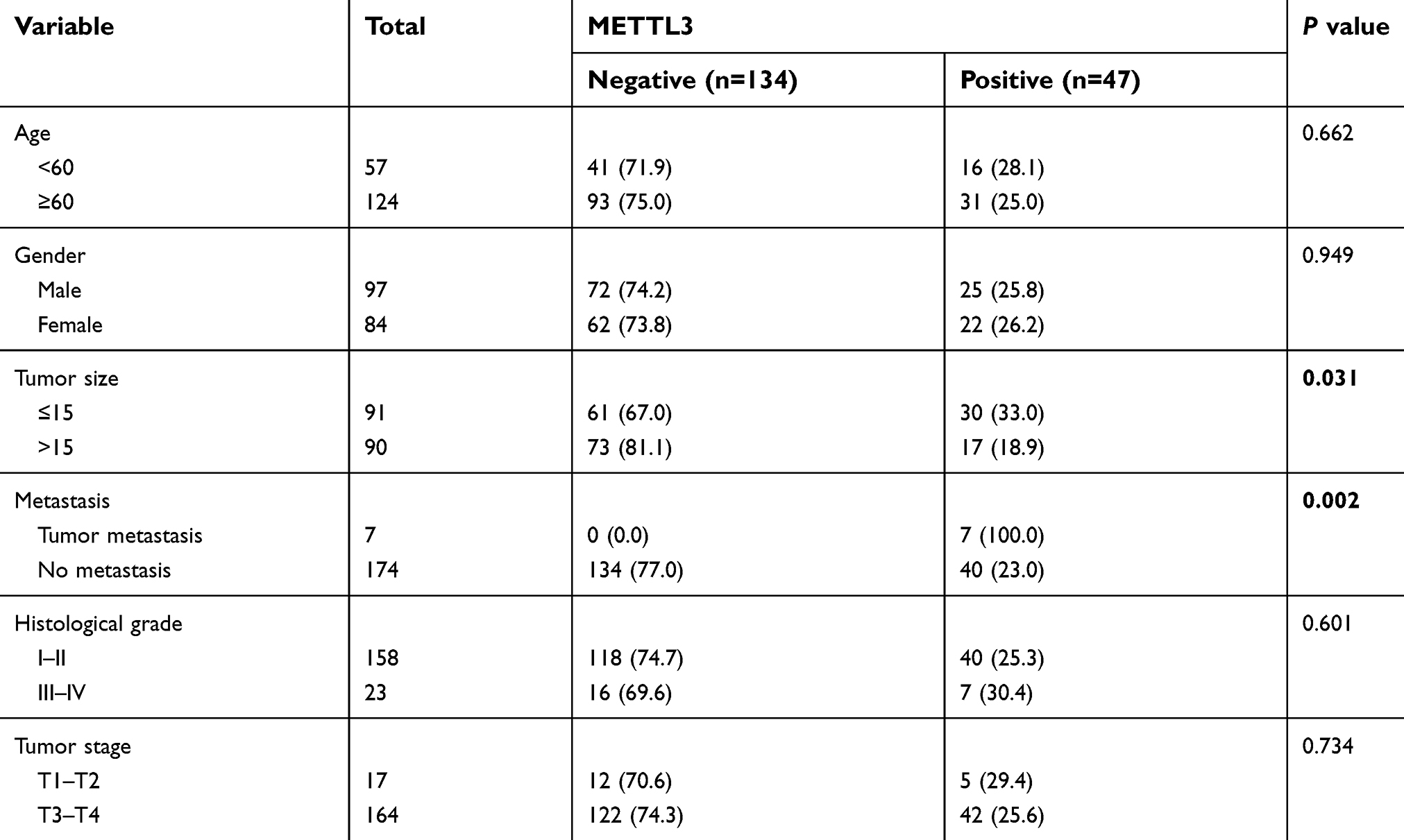 | Table 1 Relationship between methyltransferase 3 (METTL3) expression and the clinicopathological characteristics of the studied patient groups |
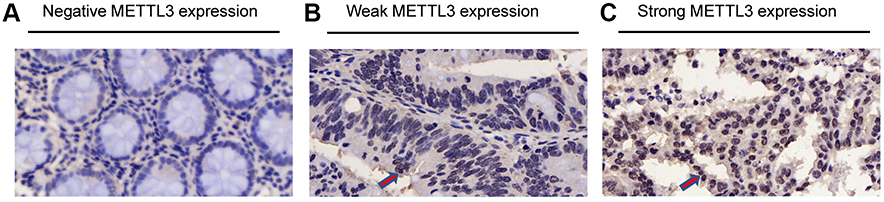 | Figure 1 Differential expression of methyltransferase 3 (METTL3) in microarray. (A) Negative expression of METTL3. (B) Weak expression of METTL3. (C) Strong expression of METTL3. The positive expression of METTL3 is indicated by red arrows. |
Expression of METTL3 is positively correlated with better survival in CRC patients
To further determine the clinical relevance, we assessed the correlation between METTL3 expression and prognosis in CRC patients. Univariate analysis (Figure 2A) indicated that high METTL3 expression was correlated with longer survival time (P=0.011). Analysis of GEO survival data also suggested that CRC patients with high METTL3 expression had lower disease-free survival rates (Figure 2B and C). In addition, tumor size (P=0.006), metastasis (P=0.002), clinical stage (P=0.0001) and tumor classification (P=0.025) were associated with survival by univariate analysis. Moreover, multivariate analysis indicated that tumor location, tumor size, clinical stage, tumor classification and METTL3 expression status were independent prognostic factors (Table 2).
 | Table 2 Multivariate analysis of overall survival in 181 CRC patients |
 | Figure 2 Correlation between expression of METTL3 and survival. (A) Expression of METTL3 is positively correlated with better survival in CRC patients (log-rank test, P=0.011). (B) Kaplan–Meier survival curves show that patients with high METTL3 expression had reduced disease-free survival time compared with patients with low expression of METTL3 (log-rank test, P=0.018; GSE33113: High METTL3=13; Low METTL3=75; time=months). (C) Patients with high METTL3 expression were associated with poor disease-free survival (log-rank test, P=0.03; GSE39582: high METTL3=371; low METTL3=186; time=months). Abbreviations: CRC, colorectal cancer; METTL3, methyltransferase 3. |
Knockdown of METTL3 promotes proliferation and metastasis in CRC cells
We established a stable METTL3 knockdown models in HCT16 and DLD1 cells with two shRNA (shMETTL3-1 and shMETTL3-2) to investigate the function of METTL3 in CRC cells. Successful knockdown of METTL3 was validated by qRT-PCR and western blot (Figure 3A and B). Knockdown of METTL3 obviously promoted CRC cell proliferation (Figure 3C) and the number of colony formation (Figure 3D). The result of transwell cell migration assay suggested that knockdown of METTL3 significantly increased the migration and invasion abilities of CRC cell (Figure 3E and F). Our results suggest that METTL3 may inhibit CRC cellular growth and metastasis.
 | Figure 3 Knockdown of METTL3 promoted CRC proliferation and migration. (A) Downregulation of METTL3 in HCT116 and DLD1 cell lines was validated with qRT-PCR (*P<0.05). (B) Knockdown of METTL3 in HCT116 and DLD1 cell lines was validated by Western blot assay (*P<0.05). (C) Downregulation of METTL3 significantly promoted CRC cellular proliferation. (D) Downregulation of METTL3 clearly increased the number of colony formation. (E) Downregulation of METTL3 promoted cell migration capacity. (F) Knockdown of METTL3 enhanced cellular invasion ability. *P<0.05. All experiments were independently set up three times. Abbreviations: CRC, colorectal cancer; METTL3, methyltransferase 3. |
Overexpression of METTL3 inhibited CRC cellular growth and metastasis
We then overexpressed METTL3 in HCT8 and KM12 cell lines. Overexpression of METTL3 was demonstrated in both qRT-PCR and western blot analyses (Figure 4A and B). METTL3 overexpression remarkably inhibited CRC cell proliferation and colony formation efficiency (Figure 4C and D). Furthermore, overexpression of METTL3 significantly inhibited CRC cellular migration and invasion abilities (Figure 4E and F). Results from knockdown and overexpression models indicated that METTL3 might serve as a suppressor gene that inhibits CRC cellular proliferation and metastasis.
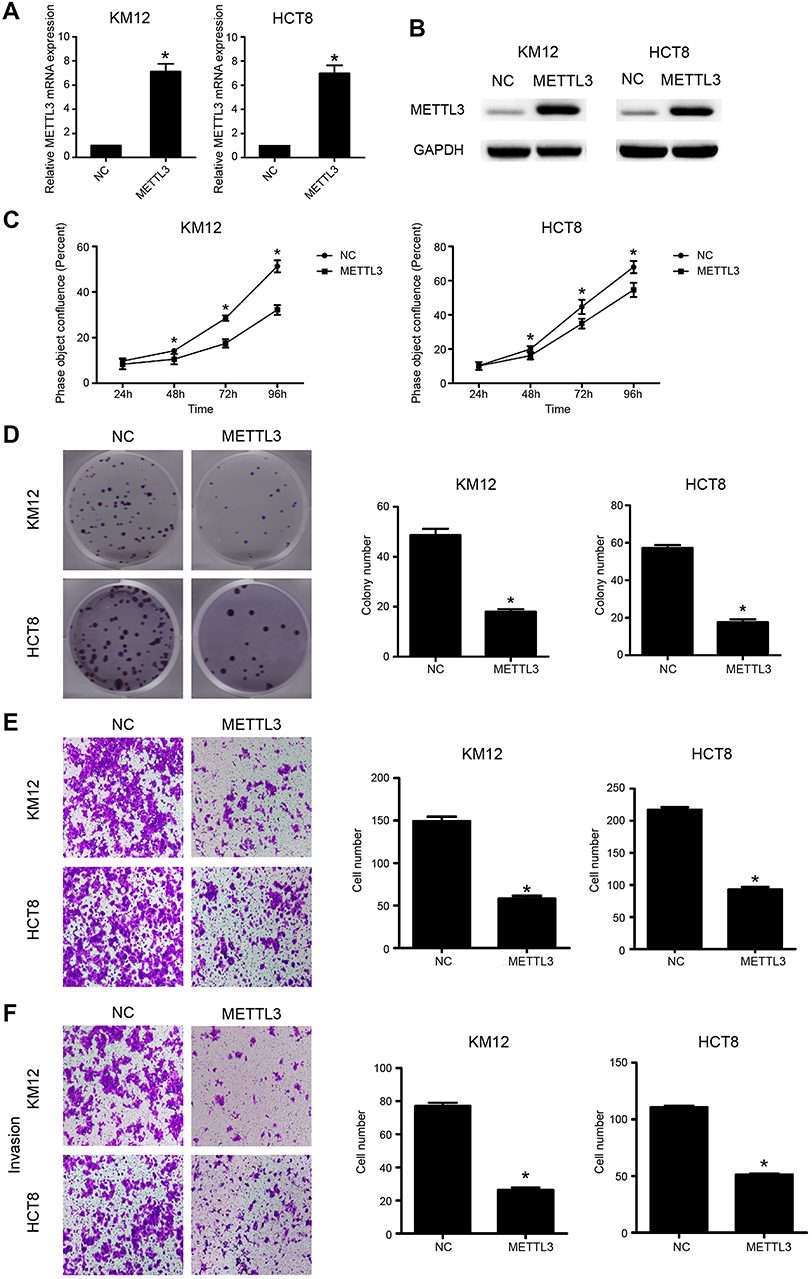 | Figure 4 Overexpression of METTL3 inhibited CRC growth and migration. (A) Overexpression of METTL3 in KM12 and HCT8 cells was validated by qRT-PCR (*P<0.05). (B) Overexpression of METTL3 in KM12 and HCT8 cells was validated by Western blot assay (*P<0.05). (C) Overexpression of METTL3 inhibited cellular proliferation. (D) Overexpression of METTL3 was clearly associated with decreased number of colony formation. (E) Overexpression of METTL3 inhibited cell migration capacity. (F) Overexpression of METTL3 inhibited cell invasion capacity. *P<0.05. All experiments were independently set up three times. Abbreviations: CRC, colorectal cancer; METTL3, methyltransferase 3. |
METTL3 inhibited CRC cell proliferation and migration by modulating the p38/ERK pathways
We further investigated the role of METTL3 in the MAPK signaling pathway. The results demonstrated that p-p38 and p-ERK expression was significantly higher in the METTL3 knockdown models, while lower in the METTL3 overexpression groups. However, the expression of both p-JNK and JNK was unchanged when comparing both groups (Figure 5A). Thus, we tested whether METTL3 affects cell proliferation through the P38 or ERK pathways. In these studies, small-molecule inhibitors of p38 kinase or ERK kinases were, respectively, used to assess the effects of these inhibitors on migration and proliferation (Figure 5B and C). Compared with the control group, the inhibitors of p38 or ERK kinases significantly inhibited the migration, invasion and proliferation abilities that were induced by knockdown of METTL3. Furthermore, we found that the effects of these inhibitors on migration, invasion and proliferation had no significant difference compared with DMSO in HCT116 shNC group (Figure S1A and B). These findings suggested that METTL3 might play a role in inhibiting CRC cell proliferation and migration by modulating the p38/ERK pathways.
 | Figure 5 Changes in the p38/ERK pathways. (A) Knockdown of METTL3 resulted in changes in the expression of p38/ERK markers with a gain in p-p38 and p-ERK. By contrast, overexpression of METTL3 promoted losses in p-p38 and p-ERK. *P<0.05. (B) Migration and cellular invasion capacities following METTL3 knockdown could be rescued by using inhibitors of p38 kinase or ERK kinases. (C) Proliferation capacities following METTL3 knockdown could be rescued by using inhibitors of p38 kinase or ERK kinases. All experiments were independently set up three times. Abbreviation: METTL3, methyltransferase 3. |
 | Figure S1 The effects of p38 and ERK kinases inhibitors on migration, invasion and proliferation. (A) The effects of these inhibitors on migration and invasion had no significant difference compared with DMSO in HCT116 shNC group. (B) The effects of these inhibitors on proliferation had no significant difference compared with DMSO in HCT116 shNC group. All experiments were independently set up three times.Abbreviation: DMSO, Dimethyl sulfoxide. |
Discussion
Even though many biological processes are involved in the modification of m6A, its specific role in CRC remains largely obscure. The methyltransferase METTL3 has been reported to participate in tumorigenesis and the development of many cancers.16,17 However, relatively few studies have reported the functional role of METTL3 in CRC carcinogenesis and development.
The present study showed that loss of METTL3 expression was correlated with a larger tumor size, an advanced stage of development and metastatic disease in CRC. Furthermore, positive METTL3 expression was shown to be an independent and favorable prognostic factor in CRC, suggesting that METTL3 expression might affect CRC prognosis. Accordingly, METTL3 can affect CRC cellular functions including proliferation, migration and invasion. More importantly, we identified both p38 and ERK as downstream targets of METTL3. Thus, our findings shed new light on the progression of CRC development.
Our study had indicated that METTL3 acted as a tumor-suppressor gene in CRC, which was consistent with carcinostasis role in renal cancer cell.16 However, Chen et al15 reported that when METTL3 serves as an oncogene, its functional expression could contribute to the progression of hepatocellular cancer development. In their study, it was shown that dysregulation of METTL3 silenced the function of the tumor-suppressor SOCS2 and promoted liver carcinogenesis.15 Moreover, METTL3 can promote breast cancer disease progression by inhibiting the tumor-suppressor let-7g.17 Additionally, METTL3 was also reported to promote bladder cancer progression via AFF4/NF-κB/MYC signaling network.18 The main reason for the dual role of METTL3 in the regulation of cancer might account for the differences in the targeted pathways and cancer heterogeneity.
Functionally, we investigated the potential molecular mechanisms by which METTL3 affects CRC cell proliferation through regulating p38/ERK signaling pathway. It has been reported that the p38/ERK signaling pathway has long been known to be essential in cell proliferation and metastasis in different cancer types.19–21 In this study, p-P38 and p-ERK were activated in the METTL3 knockdown group. Whereas, the expression levels of p-P38 and p-ERK were lower in METTL3 overexpression group. Moreover, blocking p38 and ERK kinase can inhibit the migration and invasion that was induced by knockdown of METTL3. Therefore, this study provided evidence that METTL3 might affect progression in CRC through the p38/ERK signaling pathway. However, it remains unclear whether m6A modification was regulated and which target was modulated. Therefore, further experiments are needed to clarify this fundamental question.
In summary, the present study has implicated METTL3 as a favorable prognostic factor in CRC patients. In addition, METTL3 regulated the cellular proliferation, migration and invasion capabilities in CRC and discovered that the p38/ERK pathways were involved in the molecular mechanisms of these observed functional behaviors in CRC. Thus, our finding may provide new insights into the role of tumor-suppressor and molecular mechanism of METTL3 by p38/ERK pathways in CRC.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (NSFC; 81703060, 81802441, and 81472249), National Key Research and Development Program of China (2017YFC1308800), Science and Technology Planning Project of Guangdong Province (20160916, 2015B020229001, and 2017A030310644), and Youth Science and Technology Innovation Talent of Guangdong TeZhi Plan (2016TQ03R738).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. van de Velde CJ, Boelens PG, Borras JM, et al. EURECCA colorectal: multidisciplinary management: European consensus conference colon & rectum. Eur J Cancer (Oxford, England: 1990). 2014;50(1):
3. Tang R, Cheng AJ, Wang JY, Wang TC. Close correlation between telomerase expression and adenomatous polyp progression in multistep colorectal carcinogenesis. Cancer Res. 1998;58(18):4052–4054.
4. Pichler M, Stiegelbauer V, Vychytilova-Faltejskova P, et al. Genome-Wide miRNA Analysis Identifies miR-188-3p as a Novel Prognostic Marker and Molecular Factor Involved in Colorectal Carcinogenesis. Clin Cancer Res. 2017;23(5):1323–1333. doi:10.1158/1078-0432.CCR-16-0497
5. Liu X, Chen X, Zeng K, et al. DNA-methylation-mediated silencing of miR-486-5p promotes colorectal cancer proliferation and migration through activation of PLAGL2/IGF2/beta-catenin signal pathways. Cell Death Dis. 2018;9(10):1037. doi:10.1038/s41419-018-1105-9
6. Meyer K, Saletore Y, Zumbo P, Elemento O, Mason C, Jaffrey S. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell. 2012;149(7):1635–1646. doi:10.1016/j.cell.2012.05.003
7. Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. doi:10.1038/nchembio.1432
8. Jia G, Ye F, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genetics Tig. 2013;29(2):108–115. doi:10.1016/j.tig.2012.11.003
9. Ping XL, Sun BF, Wang L, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell research. 2014;24(2):177–189.
10. Schwartz S, Mumbach M, Jovanovic M, et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 2014;8(1):284–296. doi:10.1016/j.celrep.2014.05.048
11. Jia G, Fu Y, Zhao X, et al. Corrigendum: N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. doi:10.1038/nchembio.687
12. Zheng G, Dahl JA, Niu Y, et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. RNA Biol. 2013;49(6):18–29.
13. Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA-a Publ RNA Soc.1997;3(11):1233.
14. Lin S, Choe J, Peng D, Triboulet R, Gregory R. The m 6 A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–345. doi:10.1016/j.molcel.2016.03.021
15. Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–2270. doi:10.1002/hep.29683
16. Li X, Tang J, Huang W, et al. The m6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8(56):96103–96116. doi:10.18632/oncotarget.21726
17. Cai X, Wang X, Cao C, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–19. doi:10.1016/j.canlet.2017.11.018
18. Cheng M, Sheng L, Gao Q, et al. The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. 2019. doi:10.1038/s41388-019-0683-z
19. Walczak K, Turski WA, Rajtar G. Kynurenic acid inhibits colon cancer proliferation in vitro: effects on signaling pathways. Amino Acids. 2014;46(10):2393–2401. doi:10.1007/s00726-014-1790-3
20. Liu ZG, Jiao XY, Chen ZG, Feng K, Luo HH. Estrogen receptorbeta2 regulates interlukin-12 receptorbeta2 expression via p38 mitogen-activated protein kinase signaling and inhibits non-small-cell lung cancer proliferation and invasion. Mol Med Rep. 2015;12(1):248–254. doi:10.3892/mmr.2015.3366
21. Wang J, Guo X, Xie C, Jiang J. KIF15 promotes pancreatic cancer proliferation via the MEK-ERK signalling pathway. Br J Cancer. 2017;117(2):245–255. doi:10.1038/bjc.2017.165
Supplementary materials
 | Table S1 Two oligos for sh-methyltransferase (METTL3) lentivirus construct |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.