Back to Journals » Journal of Inflammation Research » Volume 17
Lysophosphatidylcholine 14:0 Alleviates Lipopolysaccharide-Induced Acute Lung Injury via Protecting Alveolar Epithelial Barrier by Activation of Nrf2/HO-1 Pathway
Authors Liu X, Su S ![]() , Xia L, Lei X, Zou S, Zhou L, Yang R, Li K, Lin P
, Xia L, Lei X, Zou S, Zhou L, Yang R, Li K, Lin P ![]() , Li Y
, Li Y
Received 8 September 2024
Accepted for publication 26 November 2024
Published 6 December 2024 Volume 2024:17 Pages 10533—10546
DOI https://doi.org/10.2147/JIR.S495227
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xiling Liu,1,* Shanshan Su,1,* Lijing Xia,1 Xiong Lei,2 Shangpu Zou,1 Liwen Zhou,1 Ruobing Yang,1 Kai Li,3 Pengcheng Lin,1 Yuping Li1
1The Key Laboratory of Interventional Pulmonology of Zhejiang Province, Department of Respiratory and Critical Care Medicine, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325015, People’s Republic of China; 2Emergency Department, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, 325015, People’s Republic of China; 3The First School of Medicine, School of Information and Engineering, Wenzhou Medical University, Wenzhou, 325035, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuping Li, The Key Laboratory of Interventional Pulmonology of Zhejiang Province, Department of Pulmonary and Critical Care Medicine, The First Affiliated Hospital of Wenzhou Medical University, South Baixiang Street, Ouhai District, Wenzhou, Zhejiang Province, 325015, People’s Republic of China, Email [email protected]
Background: Acute lung injury (ALI) is characterized by diffuse alveolar injury and acute non-cardiac pulmonary edema, with high morbidity and mortality. Lysophosphatidylcholine 14:0 (LPC14:0) has anti-inflammatory and anti-oxidative effects in sepsis and bacteremia. We hypothesized that LPC14:0 could be a potential treatment for ALI. Therefore, the effects of LPC14:0 on lung epithelial cells and the underlying mechanism on ALI were investigated.
Methods: Lipopolysaccharide (LPS) was instilled intratracheally in vivo while the Murine Lung Epithelial-12 was stimulated by tert-butyl hydroperoxide (t-BHP) in vitro to induce the ALI model. In vivo, lung injury was evaluated by histopathological changes and pulmonary edema was assessed by wet/dry ratio. Evans blue infiltration in lung tissue, total protein content, total cell counts and inflammatory factors in bronchoalveolar lavage fluid were evaluated for alveolar permeability. In vitro, cell viability and cell death rate were assessed by cell counting kit-8 and Calcein-AM/PI stain respectively. The expression of ZO-1, Occludin, Nrf2, and HO-1 was evaluated by Western blot.
Results: LPC14:0 attenuated the LPS-stimulated lung injury and oxidative stress in vivo, and alleviated the t-BHP-induced cell damage in vitro. Moreover, LPC14:0 significantly inhibited the degradation of the tight junction proteins and activated the Nrf2/HO-1 signaling pathway both in vivo and in vitro. Mechanistically, ML385, the Nrf2 inhibitor, inhibited the protective effects of LPC14:0 on barrier function in vitro.
Conclusion: This study first demonstrated that LPC14:0 mitigated LPS-induced ALI and the destruction of tight junctions, at least in part through up-regulation of the Nrf2/HO-1 pathway.
Keywords: acute lung injury, ALI, alveolar epithelial barrier, lysophosphatidylcholine 14:0, tight junction, Nrf2
Introduction
Acute lung injury (ALI), characterized by diffuse alveolar damage and acute non-cardiac pulmonary edema, which can lead to acute respiratory distress syndrome (ARDS), is a common cause of death in intensive care unit patients.1,2 The current overall management of ALI is mainly focused on supportive care and pharmacological care, such as restrictive fluid strategy,3 prone ventilation,4 mechanical ventilation, and extracorporeal membrane oxygenation.5 Despite the continuous progress in intensive care unit treatment, the mortality rate in ALI is still high.6 Similarly, the morbidity of ALI is also high, especially in the period of COVID-19.1 Although more and more research has explored the pathogenesis of ALI and its possible therapeutic agents, the mechanism remains unclear and there is no precise drug treatment. Therefore, it is urgent to explore a new treatment for ALI.
Existing studies suggest that epithelial cells, as the first line of defense, are vulnerable to reactive oxygen species (ROS) damage.7 In addition, the destruction of alveolar epithelial and resulting alveolar barrier dysfunction are important mechanisms in ALI.5,8 Tight junctions (TJs) are composed of Occludins, zonula occludens (ZO) and claudins proteins, regulating the movement of fluid and ions, which are essential for barrier function.5 Various factors affect TJ proteins, such as infection, cytokines and ROS.9–11 Large amounts of cytokines and ROS are released during the pathophysiology of ALI. Excessive oxidative stress impaired barrier function and down-regulation of TJ proteins.
Lysophosphatidylcholines (LPCs), the major component of oxidized low-density lipoprotein, play vital roles in physiological processes, such as vascular development, reproduction, and myelination. They also play important signaling roles in inflammation and tumor cell invasion.12 Recent studies have found that lower LPC levels are associated with poor outcomes and increased risk of death in rheumatoid arthritis, diabetes mellitus, pulmonary hypertension, and asthma.13,14 Lysophosphatidylcholines14:0 (LPC14:0) <0.24 nmol/mL was associated with gestational diabetes mellitus risk.15 A large amount of evidence proves that LPC has an antioxidant and anti-inflammatory role in sepsis and bacteremia.14,16,17 LPC14:0 was a biological target for community-acquired pneumonia in our previous study.18 It is also reported that LPC14:0 was a biomarker to improve the diagnosis of drug-induced interstitial lung disease.19 However, the potential mechanism is unclear in ALI.
Some researchers found that LPC attenuated ALI by regulating neutrophil motility and the formation of neutrophil extracellular traps.20 Others demonstrated that LPC promoted phagosomal maturation and thus enhanced bactericidal activity by activating the mouse macrophage cell line NF-κB pathway during Salmonella infection.21 Moreover, stearoyl LPC (sLPC) blocked caspase-11 mediated pyroptosis in macrophages,22 while another study suggested sLPC prevented extracellular release of HMGB1 via the G2A/calcium/CaMKKβ/AMPK pathway.23 Nevertheless, the role of LPC on alveolar epithelial barrier dysfunction is unclear. Therefore, we explored whether LPC14:0 protected against lipopolysaccharide (LPS)-induced ALI by improving alveolar epithelial barrier function in this study.
Materials and Methods
Cell Culture
The Murine Lung Epithelial-12 (MLE-12) cells (CRL-2110TM, ATCC, USA) were cultured in DMEM/F-12 medium (C11330500BT, Gibco, USA) with 6% Fetal Bovine Serum (FBS) (Z7185FBS, Zeta life, USA) and 1% penicillin/streptomycin (15070063, Gibco, USA) at 37°C under saturated humidity conditions and 5% CO2.
T-BHP Stimulation and LPC14:0 Treatment
Oxidative stress and barrier dysfunction are the main pathogenic mechanisms of ALI. ROS is a cause of epithelial barrier dysfunctions.24 Based on previous study, t-BHP (B802372, Macklin, China), a chemical induces an oxidative environment, was used to stimulate the MLE-12 to simulate ALI.25–27 When MLE-12 cells grew to 60%-70% confluence, a new medium containing 10 μmol/mL LPC14:0 (855575P, Sigma‒Aldrich, USA) dissolved in 1%BSA (R21512, Shyuanye, China) or the same amount of 1%BSA was added. Then the cells were stimulated with 30 μmol/mL t-BHP for 24 hours after 1 hour.
Animal Experimental Design
Eight-week-old male C57BL/6 mice (n=60, 22–25g) were obtained from Zhejiang Vital River Laboratory Animal Technology Company and maintained in a standard pathogen-free environment at the Wenzhou Medical University’s Laboratory Animal Center with free food and water. The mice were randomly divided into four groups: Control group (1%BSA+PBS), LPS group (1%BSA+LPS), LPC14:0 group (LPC14:0+PBS), and LPS+LPC14:0 group (LPC14:0+LPS). The mice were subcutaneous injection with 1%BSA or LPC14:0 (10mg/kg).18,23 Two hours later, all mice were anesthetized with 0.5% pentobarbital sodium (50 mg/kg), and then mice were challenged intratracheally by PBS or LPS (10mg/kg, dissolved in PBS) (L2880, Sigma‒Aldrich, USA). The LPC14:0 group and LPS+LPC14:0 group were injected subcutaneously with LPC14:0 again 12 hours later and the mice were sacrificed after LPS instilled intratracheally 24 hours later (Figure S1). The animal experiments were approved by the Institutional Animal Care and Use Committee of the first affiliated hospital of Wenzhou Medical University (License number: WYYY-IACUC-AEC-2023-026). Animals were received humane care in compliance with the National Institutes of Health guidelines.
Hematoxylin-Eosin (H&E) Staining and Pathological Scoring
After cardiac perfusion with normal saline, lung tissue was fixed in 4% paraformaldehyde, then dehydrated with an ethanol gradient and embedded in paraffin, sliced (3μm thick), and stained with H&E. A semi-quantitative scoring system was applied to assess lung injury score according to the previous study.28,29 In each group of mice, 6 visual fields were selected and evaluated for semi-quantitative scores in a blinded manner. The scoring system is according to the degree of pulmonary edema, alveolar interstitial inflammation, alveolar and interstitial hemorrhage, and thickness of alveolar wall or hyaline membrane formation. No damage is 0; < 25% injury is 1; 25%-50% injury is 2; 50–75% injury is 3; diffuse injury is 4.
Measurement of Lung Wet/Dry (W/D) Ratio and the Determination of Bronchoalveolar Lavage Fluid (BALF)
The lung W/D ratio, as an indicator of pulmonary edema, reflects the increased lung permeability and the severity of lung injury. Lung tissues were collected, selected from the same pulmonary lobe, washed with normal saline, blotted with filter paper, immediately weighed for wet weight (W), and then placed in an oven at 65°C for 72 hours to obtain the dry weight (D). The ratio W/D obtained by dividing wet weight by dry weight was used to evaluate the degree of pulmonary edema.
Mice were sacrificed after LPS stimulation for 24 hours, then 1.0 mL of cold PBS intratracheal injected into the lung bulged and lavage three times to obtain BALF. After centrifugation at 4°C, 3000rpm for 20 min, the supernatant was collected. The protein concentration of untreated BLAF was determined using the BCA Protein Assay Kit (23225, Thermo scientific, USA). Then, the lower layer of cells were dissolved with red blood cell lysate, washed three times with cold PBS, and then centrifuged again until there were no red blood cells. The cells were suspended with PBS and counted the total number of cells in PBS.
Enzyme Linked Immunosorbent Assay (ELISA)
Determination of cytokines in BALF use ELISA. BALF was obtained from each sample separately as above. IL-1β (BP-E20533), IL-6 (BP-E20012), IL-18 (BP-E200001) and TNF-α (BP-E20220A) protein levels were determined using ELISA kits (Biotech Boyun, China).
Dihydroethidium (DHE) Staining for Determination of ROS
Fresh lung tissue was frozen and cut into 8 μm thick sections (3 sections per mouse), stained with a 1:200 dilution of oxidized fluorescent dye DHE (S0063, Beyotime, China) for 30min, rinsed three times in PBS, incubated by DAPI for 10 min. Pictures were photographed with a fluorescence microscope (80i, Nikon, Japan), and fluorescence images were quantified using Image J software.
Measurement of Superoxide Dismutase (SOD), Malondialdehyde (MDA) Content in Lung Tissue
Lung tissues were washed with PBS and then homogenised. After centrifugation, the supernatants were collected for an MDA assay (BYS0001, Biotech Boyun, China) in accordance with the manufacturer’s protocol. The SOD level was assessed by the SOD assay (PC0027957, Biotech Boyun, China).
Myeloperoxidase (MPO) Activity
MPO activity, closely associated with tissue neutrophil content, is a marker for the assessment of neutrophil infiltration in tissues.30,31 Lung tissue was homogenized and collected as above mentioned, and the MPO activity was measured with the MPO colorimetric activity assay kit (BYS0012, Biotech Boyun, China) according to the manufacturer’s protocol.
Lung Evans Blue Index
To test the permeability of lung, Evans blue dye extravasation assay was performed as previously described.32 In brief, 1% Evans Blue (6 mL/kg) (HY-B1102, MCE, USA) was injected into mice via the tail vein. After 30 minutes, mice were anesthetized and the right ventricle was perfused with 20 mL normal saline for 2 minutes. Then, the lung tissues were excised and photographed. Lung tissues were weighed and homogenized with formamide (1mL/100mg) (HY-Y0842, MCE, USA) at 60°C for 24 hours followed by centrifugation at 12000 rpm for 20 minutes. The concentration of Evans Blue in supernatant was determined by spectrophotometry at 620 nm.
Cell Viability Assay
To observe the effect of t-BHP and LPC14:0 on cell viability in MLE-12 cells, the cells were inoculated in 96-well plates at a concentration of 1×104 cells/well for 24 hours, and then replaced with fresh medium with different concentration of t-BHP (10, 20, 30, 40, 50, 60) μmol/mL for 24 hours to find the appropriate concentration to model ALI. The cells were also divided into 9 groups respectively treated by 0, 1, 5, 10, 15, 20, 25, 30, 35 μmol/mL LPC14:0 to detect the effect of LPC14:0 on cell viability. As a consequence, the cells in another 96-well plate were pretreated by 0, 1, 5, 10, 15, 20, 25, 30, 35 μmol/mL LPC14:0 for 1 hour, and then were added appropriate concentration of t-BHP for 24 hours. The cell viability was measured using the Cell Counting kit-8 (CCK-8) (GK10001, GLPBIO, USA). Two hours after CCK-8 was added, cell viability was calculated by measuring the absorbent value of 450 nm with a microplate reader.
Calcein-AM/Propidium Iodide (PI) Staining
The treated MLE-12 cells were analyzed with a Calcein/PI cell activity and cytotoxicity assay kit (No. C2015M, Beyotime, China) according to the manufacturer’s instructions. In short, after different stimulation, MLE-12 cells were stained with Calcein-AM/PI at 37°C for 30 minutes in the dark. Subsequently, the fluorescence images were acquired by using the Operetta CLSTM (PerkinElmer, Germany). Image J was used to analyze the average fluorescence intensity.
Immunofluorescence Staining
MLE-12 cells were seeded in 96-well culture plate and treated for 24 hours. The plates were fixed with 4% paraformaldehyde for 30 minutes and then blocked by 1% bovine serum albumin for 30 minutes at room temperature. Next, the cells were then incubated with primary antibodies against ZO-1 (1:200) (21773-1-AP, Proteintech, China) at 4°C overnight. After washing, the cells were incubated with rabbit secondary antibody (1:500, Alexa Fluor® 488 goat anti-rabbit IgG, Beyotime, China) for 60 minutes at room temperature in dark. Finally, the cells were counterstained with DAPI for 10 minutes, and observed under the Operetta CLSTM (PerkinElmer, Germany).
Western Blot Analysis
Total proteins were extracted from lung tissues and MLE-12 cells with RIPA lysis buffer, and the protein concentrations were quantified by BCA assay. Proteins were separated through 10–12% denaturing polyacrylamide gels and then transferred to PVDF membranes. After blocking with the protein free rapid blocking buffer (PS108P, Epizyme, China), the membranes were incubated with the following primary antibodies: Nrf2 (ab76026, Proteintech, China), HO-1 (AB1889491, Abcam, USA) and ZO-1 (21,773-1-AP, Proteintech, China), Occludin (A2601, Abclonal, China) and GAPDH (E-AB-40337, Elabscience, China) overnight at 4°C. After that, the membranes were incubated with HRP-conjugated anti-rabbit secondary antibodies (BL003A, Biosharp, China) for 1 hour at room temperature, and then visualized by the imaging system (iBright™ CL1500 Instrument, Thermo Fisher Scientific, USA). The immunoreactive bands were evaluated using Image J software.
Molecular Docking (MD)
Molecular docking analysis was performed using AutoDockTools (version 1.5.7) to determine the interaction between Nrf2 and LPC14:0. The 2D structure of LPC14:0 was obtained from PubChem database (https://pubchem.ncbi.nlm.nih.gov/) and converted into 3D structure in OpenBabel (version 2.4.1). The x-ray structure of Nrf2 (PDB ID: 8XGK) was obtained from the Protein Data Bank database (http://wwwl.rcsb.org). Before docking, energy minimization was established using default parameters and processed with PyMoL (version 2.6.0). The molecular structure of Nrf2 and LPC14:0 was used for MD analysis. The final molecular images were viewed in 3D by using PyMoL.
Statistical Analysis
All experiments were independently repeated three times. All data was using one-way analysis of variance (ANOVA), followed by the Tukey postmortem test, and expressed as mean ± standard deviation (SD). All statistical analysis was performed using GraphPad Prism 9.0 software, and P < 0.05 was considered to be statistically significant.
Results
LPC14:0 Treatment Alleviated LPS-Induced ALI in vivo
We first evaluated the effect of LPC14:0 on ALI in mice. HE staining revealed that there were extensive alveolar wall thickening, a large number of inflammatory cell infiltration, and lung tissue hemorrhage compared LPS group to control group (Figure 1A). LPC14:0 treatment markedly alleviated those changes above caused by LPS (Figure 1A). The lung injury score in LPS+LPC14:0 group also decreased significantly compared to LPS group (Figure 1B). LPS group was characterized by increased alveolar permeability, increased total cell count and protein content of BALF after pulmonary edema, and increased lung W/D ratio. LPC14:0 treatment reversed these changes (Figure 1C-E). Furthermore, LPS induced significant increases in TNF-α, IL-1β, IL-6, and IL-18 levels of BALF, whereas LPC14:0 treatment inhibited the secretion of those inflammatory factors (Figure 1F-I).
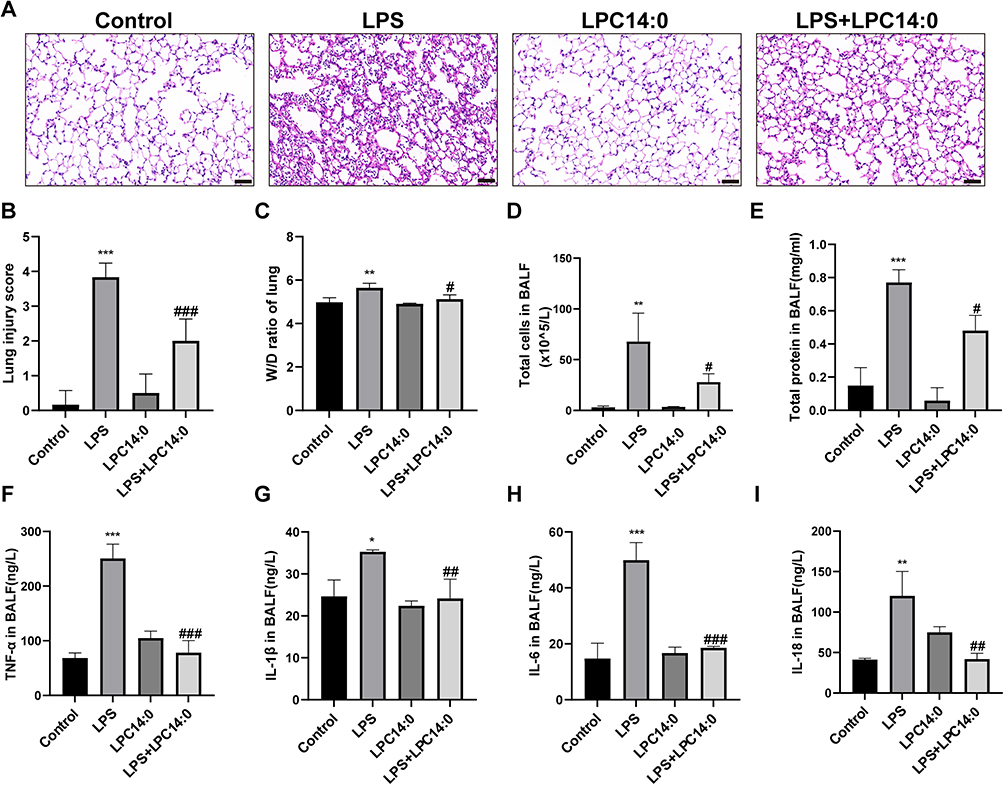 |
Figure 1 LPC14:0 attenuated LPS-induced ALI. (A) Representative images of mice lung sections stained with Hematoxylin and eosin (H&E) (n = 3). Scale bar: 50μm. (B) Lung injury score. (C) Mean values of W/D ratio measured from the ratio of mouse wet lung weight to dry lung weight. (D) Total cell count in BALF. (E) Levels of total protein in BALF. (F-I) Levels of TNF-α, IL-1β, IL-6, and IL-18 in BALF. All experiments were performed three times independently, and the results were described as mean ± SD. n = 3, * vs control group, # vs LPS group, #/*p < 0.05, ##/ **p < 0.01, ###/ ***p < 0.001. |
LPC14:0 Treatment Inhibited LPS-Induced Oxidative Stress and Activated Nrf2/HO-1 Signaling Pathway in vivo
Next, we used DHE staining to assess oxidative stress. As shown in Figure 2A-B, DHE fluorescence intensity was significantly increased in the LPS group, which was decreased in LPC14:0 treated mice. The levels of MPO and MDA in lung tissues of LPC14:0 treated mice were significantly lower than those in LPS-induced mice (Figure 2C-D), while the level of SOD was higher than those in LPS-instilled (Figure 2E). The proteins including Nrf2 and HO-1 were analyzed through Western blotting. As a result, Nrf2 and HO-1 expression were significantly up-regulated in LPC14:0 treatment mice compared with LPS-stimulated mice (Figure 2F-H). These results suggested that LPC14:0 had an antioxidant effect in LPS-induced ALI mice.
 |
Figure 2 LPC14:0 inhibited LPS-induced oxidative stress and activated Nrf2/HO-1 pathway. (A) ROS levels in lung tissues were detected by DHE. Representative DHE fluorescence images of mouse lung tissue (n = 3). Scale bar: 50μm. (B) Quantitative determination of DHE fluorescence intensity. (C) MPO activity in lung tissues of mice. (D-E) The levels of MDA and SOD in lung tissues of mice. (F-H) Western blot analysis for Nrf2 and HO-1 protein expression. All experiments were performed three times independently, and the results were described as mean ± SD. n = 3, * vs control group, # vs LPS group, #/* p < 0.05, ** p < 0.01, ###/ *** p < 0.001. |
LPC14:0 Alleviated Barrier Dysfunction in vivo
In addition, we investigated barrier function and tight junction protein further. The blue color of lung tissue was the result of Evans Blue leakage (Figure 3A). Combined with Evans Blue quantitative analysis (Figure 3B), the infiltration of Evans Blue in lung tissue was significantly increased by LPS induced, whereas LPC14:0 treatment reduced the permeability, indicating that LPC14:0 alleviates alveolar epithelial barrier dysfunction. After LPS induction, the level of the TJ proteins, ZO-1 and Occludin, in lung tissue decreased drastically whereas LPC14:0 treatment recovered the TJ proteins (Figure 3C-E). All these results suggested that LPC14:0 alleviated the barrier function impairment in LPS-induced ALI.
 |
Figure 3 LPC14:0 affected alveolar epithelial barrier function in LPS-induced ALI. (A) Permeability of Evans Blue into mouse lung tissue. (B) Quantitative analysis of Evans Blue in mouse lung tissue. (C-E) Western blot analysis of ZO-1 and Occludin proteins expression. All experiments were performed three times independently, and the results were described as mean ± SD. n = 3, * vs control group, # vs LPS group, #/* p < 0.05, ** p < 0.01, *** p < 0.001. |
LPC14:0 Inhibited t-BHP-Induced Cell Damage in vitro
To further clarify the mechanism of the protective effect of LPC14:0, an in vitro study was performed in MLE-12 cells based on the results above in vivo. The cell viability of MLE-12 cells assessed by CCK-8 assay decreased with the increase of t-BHP concentration, while LPC14:0 had no toxic effect on cells at a concentration of 0–25 μmol/mL. Therefore, we selected 30 μmol/mL t-BHP as the stimulus for cell damage and then pretreated with 0–35 μmol/mL LPC14:0 for 1 hour. The results showed that LPC14:0 at a concentration of 10–25 μmol/mL inhibited the cell damage caused by 30 μmol/mL t-BHP (Figure 4A). Therefore, LPC14:0 (10 μmol/mL) and t-BHP (30 μmol/mL) were selected for subsequent experiments.
 |
Figure 4 LPC14:0 inhibited t-BHP-induced cell damage in MLE-12. (A) CCK-8 cell viability test selected the concentration of t-BHP to stimulate MLE-12 and confirmed that the optimal protective concentration of LPC14:0. (B) The cell death was detected by Calcein-AM/PI staining (n = 3). Scale bar: 100μm. (C) The dead cells (PI positive) were counted. All experiments were performed three times independently, and the results were described as mean ± SD. n = 3, * vs control group, # vs t-BHP group, ##/ ** p < 0.01, ###/ *** p < 0.001. |
Calcein-AM/PI staining showed that the percentage of PI-positive cells was increased after t-BHP exposure, whereas the LPC14:0 pretreatment reversed these changes (Figure 4B-C). These results indicated that LPC14:0 effectively inhibited the death of MLE-12 cells caused by t-BHP.
LPC14:0 Reduced Alveolar Epithelial Barrier Damage Caused by t-BHP and Activated Nrf2/HO-1 Signaling Pathway in vitro
Immunofluorescence and Western blot analysis were performed to investigate the effect of LPC14:0 on the alveolar barrier damage induced by t-BHP. The results revealed that the expression of ZO-1 and Occludin was reduced by t-BHP exposure, which was blocked by LPC14:0 pretreatment in MLE-12 (Figure 5A-D). These results implicated that LPC14:0 protected the permeability of the alveolar epithelial barrier induced by t-BHP. As demonstrated in Figure 5E-G, t-BHP slightly decreased the expression of Nrf2 and increased the expression of HO-1 proteins, which were further up-regulated by LPC14:0 treatment.
 |
Figure 5 LPC14:0 reduced alveolar epithelial barrier damage caused by t-BHP and activated Nrf2/HO-1 signaling pathway in vitro. (A) Representative immunofluorescent staining images of ZO-1 expression in MLE-12. Scales bar: 50μm. (B-D) Western blot analysis in ZO-1 and Occludin protein expression. (E-G) Western blot analysis in Nrf2 and HO-1 protein expression. All experiments were performed three times independently, and the results were described as mean ± SD. n = 3, * vs control group, # vs t-BHP group, #/* p < 0.05, ##/ ** p < 0.01, *** p < 0.001. |
LPC14:0 Improved Alveolar Epithelial Barrier Function by Activating Nrf2/HO-1 Signaling Pathway in vitro
We studied the interaction between LPC14:0 and Nrf2 by means of molecular docking. The binding energy of LPC14:0 to Nrf 2 is −8.7 (kcal/mol). The binding mode of LPC14:0 to the Nrf2 protein is shown in Figure 6A while Figure 6B shows that LPC14:0 is able to form hydrogen bonds with VAL-418, VAL-465 and ILE-416 residues in Nrf2 protein. These findings indicate that LPC14:0 may interact with Nrf2 to exert antioxidant effects.
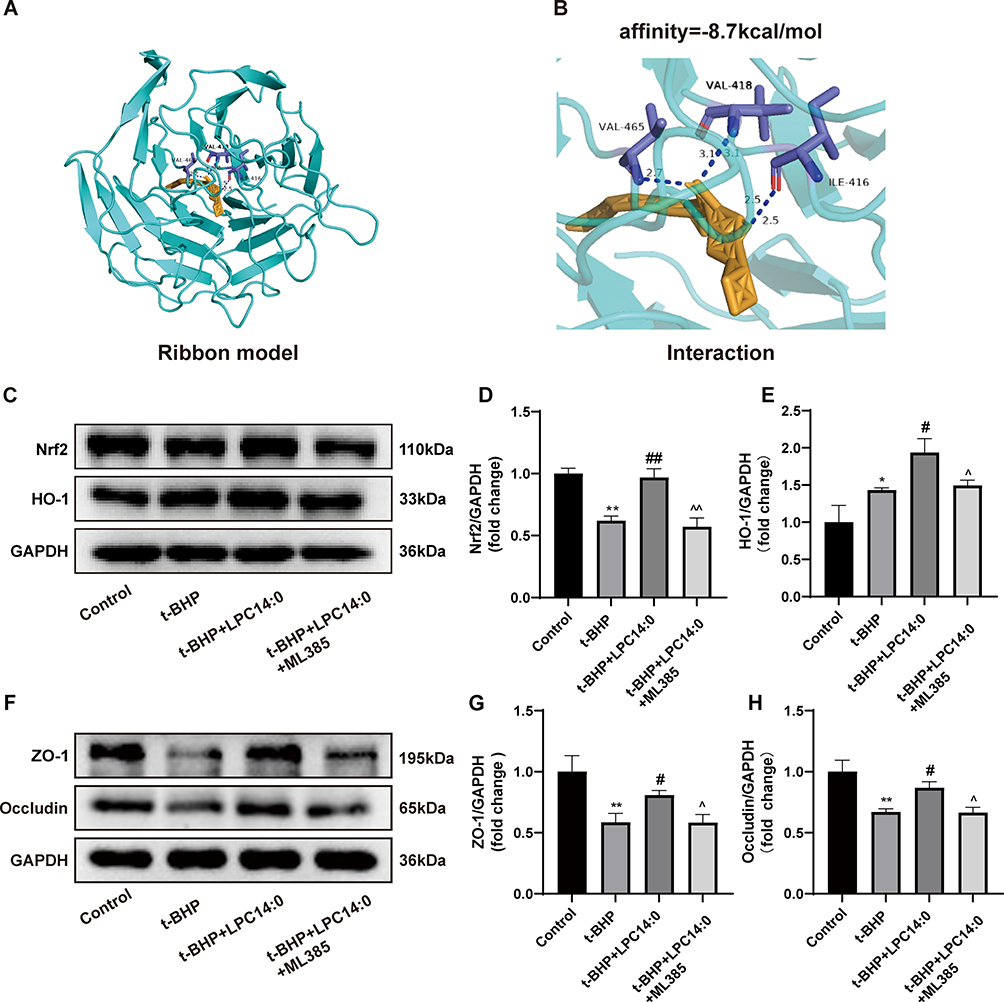 |
Figure 6 LPC14:0 improved alveolar epithelial barrier function by activating Nrf2/HO-1 pathway. (A) The 3D binding model of LPC14:0 and Nrf2 through molecular docking. (B) Local binding between LPC14:0 and Nrf2: three residues (VAL-418,VAL-465 and ILE-416) are involved in the hydrogen bond interaction. (C-E) Western blot analysis in Nrf-2 and HO-1 protein expression. (F-H) Western blot analysis in ZO-1 and Occludin protein expression. All experiments were performed three times independently, and the results were described as mean ± SD. n = 3, * vs control group, # vs t-BHP group, ^ vs t-BHP+LPC14:0 group, #/*/^ p < 0.05, ##/ **/^^ p < 0.01. |
Then, we used a specific Nrf2 inhibitor, ML385, to test whether Nrf2/HO-1 is related to the up-regulation of ZO-1 and Occludin caused by LPC14:0. According to the Western blotting, pretreatment with ML385 significantly inhibited the expression of Nrf2 and HO-1 (Figure 6C-E). In the meantime, we found that the up-regulation of ZO-1 and Occludin caused by LPC14:0 was abolished by ML385 (Figure 6F-H). These results demonstrated that the Nrf2/HO-1 pathway was related to the regulation of TJ proteins by LPC14:0.
Discussion
In this study, we found that LPC14:0 also improved ALI by enhancing tight junction protein expression and enhancing barrier function in addition to its anti-inflammatory and antioxidant effects. We further demonstrated that LPC14:0 alleviated acute lung injury by activating Nrf2/HO-1 pathway to enhance expression of TJ proteins.
Although the pathophysiological mechanism of ALI is complex and not completely clear, it has been found that oxidative stress and barrier dysfunction play major roles in the pathogenesis of ALI. It is reported that LPCs had anti-inflammatory roles in the development of vascular inflammation and atherosclerosis by increasing extracellular superoxide dismutase expression.33 In experimental murine pneumonia models, LPC reduced the bacterial burdens in the lungs and increased the animal survival rate by regulating the production of pro-inflammatory and anti-inflammatory cytokines.34 In the study, we established a mouse model of ALI by intratracheal instillation of LPS, which induced the occurrence of excessive oxidative stress and inflammation response.35,36 LPC14:0 treatment reversed the above pathological changes, such as the increased production of SOD, the decreased production of IL-1β, TNF-α and other inflammatory factors in BALF, which is consistent with previous studies.
It has been recognized that alveolar epithelial, the first line of defense in ALI, plays an important role in the survival of patients with ALI.1 Damage to the epithelial barrier facilitates the secretion of chemokines, adhesion molecules, and also impairs the transport of fluid, sodium and chlorine, which maintain a dry airspace by alveolar epithelium. Previous studies have shown the importance of alveolar epithelial cells.31,37 In this study, we found that LPC14:0 treatment markedly reduced lung wet-to-dry ratio and Evans blue index in LPS-induced mice. Our results indicated that LPC14:0 had a protective effect against epithelial damage, which is consistent with the studies in vitro. Additional, LPC14:0 treatment recovered the expression of TJ proteins, such as ZO-1 and Occludin.
TJs are important intercellular junctions that construct epithelial barrier and maintain epithelial polarity.38 The study of alveolar epithelial barrier function is helpful to explore a new method for the treatment of lung injury. In the present study, we found that the expression of ZO-1 and Occludin was reduced by t-BHP exposure in MLE-12, which was blocked by LPC14:0 pretreatment. These results suggested that the protective effect of LPC14:0 on ALI was partly mediated by TJs. Many studies have found that activating Nrf2/HO-1 signaling plays a crucial role in protecting barrier integrity,35,39 thereby inhibiting inflammatory damage. In inflammatory bowel disease, activation of the Nrf2 pathway enhances TJ proteins expression, protects the integrity of the intestinal barrier, and suppresses inflammatory bowel disease.39 Furthermore, the activation of Nrf2-mediated antioxidant stress pathways protects against LPS-induced blood-brain barrier damage.10 HO-1, the downstream of Nrf2, has been found to play a key role in clearing ROS and regulating ROS production, and also reduces the degradation of TJ proteins. Up-regulating HO-1 in LPS-induced ALI can reduce deterioration of TJ proteins.40 To investigate whether LPC14:0 protects barrier integrity by activating Nrf2/HO-1 signaling, we stimulated MLE-12 cells with ML385, the Nrf2-specific inhibitor, for one hour before LPC14:0 treatment and found it could eliminate the effects of LPC14:0 in vitro. The results suggested that LPC14:0 reduced the deterioration of TJ proteins via the Nrf2/HO-1 pathway.
With the development of lipidomics research, increasing evidence proves that lower LPC in plasma correlates with increased mortality risk and poor prognosis.19,41 LPCs have been studied increasingly in a variety of diseases, such as vascular inflammation, cancer, and infectious.42 One study revealed that LPC bound to both G protein-coupled receptor 132 (G2A/GPR132) and G protein-coupled receptor 4 on the cell surface,43 affecting the activation of immune cells. Besides, it has been reported that LPC could induce the activation of the PKA-PI3K-p38 MAPK pathway to promote phagosome maturation, thus effectively controlling the growth of Mycobacterium tuberculosis.44 LPC also induce apoptosis of mouse ovarian granulosa cells and mouse Leydig cells.45,46 LPC14:0, a class of LPCs, was involved in cell signaling through a variety of mechanisms as a bioactive lipid mediator. LPC14:0 plays a protective role against LPS-induced ALI by inhibiting activation of the NLRP3 inflammasome in our previous study.18 In present study, we investigated the effects of LPC14:0 on oxidative stress, inflammation, and TJs in vivo during LPS-stimulated ALI. We first found that LPC14:0 protected LPS-induced ALI by promoting TJ proteins expression in alveolar epithelial cell through the Nrf2/HO-1 pathway in vitro. This result suggested LPC14:0 might be a potential substance for the treatment of ALI.
Our study still has some limitations and shortcomings. For example, many potential upstream or bypass mechanisms regulate the effect of TJs under different situations. We only verified the relationship between Nrf2/HO-1 signaling pathway and TJ protein, but some studies have found that Nrf2/HO-1 alleviated epithelial barrier dysfunction by inhibiting ferroptosis,47 others have found that Nrf2/HO-1 inhibited pyroptosis and improved ALI.48,49 There are many types of programmed cell death, such as apoptosis, autophagy, pyroptosis, and ferroptosis,50 which need to be explored in the TJ completely. In addition, the upstream of Nrf2, such as p-AKT and Sirt1, may also be studied further in the future. So, the deep relationship between the Nrf2/HO-1 pathway and TJ remains to be sufficiently explored. Furthermore, cell research can not completely replace in vivo research. LPC14:0 reducing lung epithelial cell injury by Nrf2/HO-1 pathway needs to be further discussed by using knockout mice and different cell lines. In terms of statistics, various statistical methods may have statistical bias, and we have little data in the present study, in this context, we need more data to support our results in the future.
Conclusion
The study demonstrated that LPC14:0 treatment played a protective role in LPS-induced ALI through anti-inflammatory and antioxidant effects. At the same time, LPC14:0 alleviated the tight junction protein breakage on lung epithelia by activating the Nrf2/HO-1 signaling pathway to improve ALI (Figure 7). In conclusion, we focused on the role of LPC14:0 treatment in alveolar epithelial barrier dysfunction with the involvement of the Nrf2/HO-1 pathway. At the same time, we speculated that LPC14:0 could be used as a new therapeutic agent to alleviate ALI in the future.
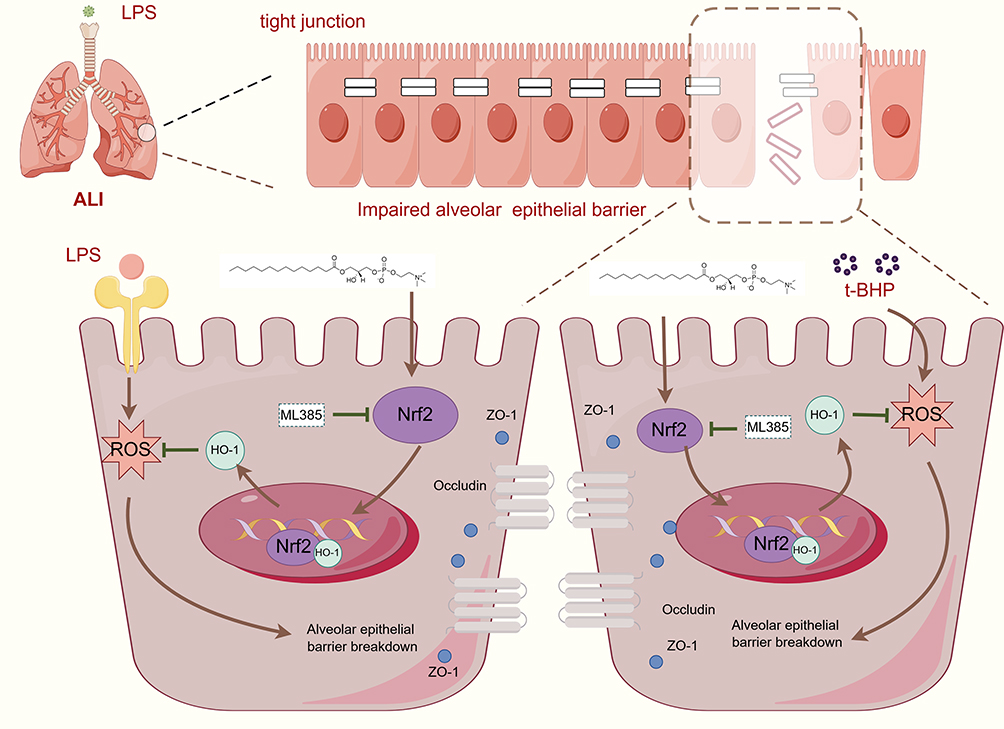 |
Figure 7 Scheme conclude the protective effects of LPC14:0 on LPS-induced ALI via activation of the Nrf2/HO-1 axis. LPC14:0 ameliorates LPS-induced acute lung injury via protection alveolar epithelial barrier by activating the Nrf2/HO-1 pathway. This picture was generated by Figdraw. |
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Wenzhou Collaborative Innovation Center for Technology Enhancement in Diagnosis and Treatment of Severe Respiratory Infections (No.84923003), the Project of Wenzhou Science and Technology Bureau (Y2023109) and the Project of Wenzhou Science and Technology Bureau (Y20210144).
Disclosure
The authors declare that they have no conflicts of interest to disclose.
References
1. Bos LDJ, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet. 2022;400(10358):1145–1156. doi:10.1016/S0140-6736(22)01485-4
2. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. 2021;398(10300):622–637. doi:10.1016/S0140-6736(21)00439-6
3. Wiedemann HP, Wheeler AP; National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network. Comparison of two fluid-management strategies in acute lung injury. N Engl J Med. 2006;354(24):2564–2575. doi:10.1056/NEJMoa062200.
4. Guérin C, Reignier J, Richard JC, et al. Prone positioning in severe acute respiratory distress syndrome. N Engl J Med. 2013;368(23):2159–2168. doi:10.1056/NEJMoa1214103
5. Ma M, Rl Z, Ga Z, et al. Acute respiratory distress syndrome. Nature Reviews Disease Primers. 2019;5(1). doi:10.1038/s41572-019-0069-0
6. B G, Jg L, P T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8). doi:10.1001/jama.2016.0291
7. Cheng X, He S, Yuan J, et al. Lipoxin A4 attenuates LPS-induced mouse acute lung injury via Nrf2-mediated E-cadherin expression in airway epithelial cells. Free Radic Biol Med. 2016;93:52–66. doi:10.1016/j.freeradbiomed.2016.01.026
8. D’Agnillo F, Walters KA, Xiao Y, et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci Transl Med. 2021;13(620):eabj7790. doi:10.1126/scitranslmed.abj7790
9. Guttman JA, Finlay BB. Tight junctions as targets of infectious agents. Biochim Biophys Acta. 2009;1788(4):832–841. doi:10.1016/j.bbamem.2008.10.028
10. Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788(4):864–871. doi:10.1016/j.bbamem.2008.08.027
11. Schreibelt G, Kooij G, Reijerkerk A, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21(13):3666–3676. doi:10.1096/fj.07-8329com
12. Birgbauer E, Chun J. New developments in the biological functions of lysophospholipids. Cell Mol Life Sci. 2006;63(23):2695–2701. doi:10.1007/s00018-006-6155-y
13. Liu P, Zhu W, Chen C, et al. The mechanisms of lysophosphatidylcholine in the development of diseases. Life Sci. 2020;247:117443. doi:10.1016/j.lfs.2020.117443
14. Wei J, Liu X, Xiao W, et al. Phospholipid remodeling and its derivatives are associated with COVID-19 severity. J Allergy Clin Immunol. 2023;151(5):1259–1268. doi:10.1016/j.jaci.2022.11.032
15. Liu J, Li J, Li S, et al. Circulating lysophosphatidylcholines in early pregnancy and risk of gestational diabetes in Chinese women. J Clin Endocrinol Metab. 2020;105(4):e982–e993. doi:10.1210/clinem/dgaa058
16. Miró-Canturri A, Ayerbe-Algaba R, Jiménez-Mejías ME, Pachón J, Smani Y. Efficacy of lysophosphatidylcholine as direct treatment in combination with colistin against Acinetobacter baumannii in murine severe infections models. Antibiotics (Basel). 2021;10(2):194. doi:10.3390/antibiotics10020194
17. Parra-Millán R, Jiménez-Mejías ME, Ayerbe-Algaba R, et al. Impact of the immune response modification by lysophosphatidylcholine in the efficacy of antibiotic therapy of experimental models of peritoneal sepsis and pneumonia by Pseudomonas aeruginosa: LPC therapeutic effect in combined therapy. Enfermedades Infecciosas y Microbiología Clínica. 2022;40(1):14–21. doi:10.1016/j.eimc.2020.06.002
18. Nan W, Xiong F, Zheng H, et al. Myristoyl lysophosphatidylcholine is a biomarker and potential therapeutic target for community-acquired pneumonia. Redox Biol. 2022;58:102556. doi:10.1016/j.redox.2022.102556
19. S K, G A, T K, et al. Identification and characterization of lysophosphatidylcholine 14:0 as a biomarker for drug-induced lung disease. Sci Rep. 2022;12(1). doi:10.1038/s41598-022-24406-z
20. Jeong S, Kim B, Byun DJ, et al. Lysophosphatidylcholine alleviates acute lung injury by regulating neutrophil motility and neutrophil extracellular trap formation. Front Cell Dev Biol. 2022;10:941914. doi:10.3389/fcell.2022.941914
21. Lee HJ, Hong WG, Woo Y, et al. Lysophosphatidylcholine enhances bactericidal activity by promoting phagosome maturation via the activation of the NF-κB pathway during salmonella infection in mouse macrophages. MolCells. 2020;43(12):989–1001. doi:10.14348/molcells.2020.0030
22. Li W, Zhang W, Deng M, et al. Stearoyl lysophosphatidylcholine inhibits endotoxin-induced Caspase-11 activation. Shock. 2018;50(3):339–345. doi:10.1097/SHK.0000000000001012
23. Quan H, Bae HB, Hur YH, et al. Stearoyl lysophosphatidylcholine inhibits LPS-induced extracellular release of HMGB1 through the G2A/calcium/CaMKKβ/AMPK pathway. Eur J Pharmacol. 2019;852:125–133. doi:10.1016/j.ejphar.2019.02.038
24. Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, Black SM. ROS signaling in the pathogenesis of Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS). In: Wang YX editor. Pulmonary Vasculature Redox Signaling in Health and Disease. Vol 967. Advances in Experimental Medicine and Biology. Springer International Publishing; 2017:105–137. doi:10.1007/978-3-319-63245-2_8.
25. Pan YN, Jia C, Yu JP, Wu ZW, Xu GC, Huang YX. Fibroblast growth factor 9 reduces TBHP-induced oxidative stress in chondrocytes and diminishes mouse osteoarthritis by activating ERK/Nrf2 signaling pathway. Int Immunopharmacol. 2023;114:109606. doi:10.1016/j.intimp.2022.109606
26. Zhao W, Feng H, Sun W, Liu K, Lu JJ, Chen X. Tert-butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: roles of NOX4 and mitochondrion. Redox Biol. 2017;11:524–534. doi:10.1016/j.redox.2016.12.036
27. Lv H, Liu Q, Wen Z, Feng H, Deng X, Ci X. Xanthohumol ameliorates lipopolysaccharide (LPS)-induced acute lung injury via induction of AMPK/GSK3β-Nrf2 signal axis. Redox Biol. 2017;12:311–324. doi:10.1016/j.redox.2017.03.001
28. Liu J, Yao S, Jia J, et al. Loss of MBD2 ameliorates LPS-induced alveolar epithelial cell apoptosis and ALI in mice via modulating intracellular zinc homeostasis. THE FASEB Journal. 2022;36(2):e22162. doi:10.1096/fj.202100924RR
29. Matute-Bello G, Downey G, Moore BB, et al. An official American thoracic society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44(5):725–738. doi:10.1165/rcmb.2009-0210ST
30. Pulli B, Ali M, Forghani R, et al. Measuring myeloperoxidase activity in biological samples. PLoS One. 2013;8(7):e67976. doi:10.1371/journal.pone.0067976
31. Tang X, Liu J, Yao S, Zheng J, Gong X, Xiao B Ferulic acid alleviates alveolar epithelial barrier dysfunction in sepsis-induced acute lung injury by activating the Nrf2/HO-1 pathway and inhibiting ferroptosis. Pharmaceutical Biology. 2022.
32. Jiang J, Huang K, Xu S, Garcia JGN, Wang C, Cai H. Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. 2020;36:101638. doi:10.1016/j.redox.2020.101638
33. Yamamoto M, Hara H, Adachi T. The expression of extracellular-superoxide dismutase is increased by lysophosphatidylcholine in human monocytic U937 cells. Atherosclerosis. 2002;163(2):223–228. doi:10.1016/S0021-9150(02)00007-2
34. Smani Y, Domínguez-Herrera J, Ibáñez-Martínez J, Pachón J. Therapeutic Efficacy of Lysophosphatidylcholine in severe infections caused by Acinetobacter baumannii. Antimicrob Agents Chemother. 2015;59(7):3920–3924. doi:10.1128/AAC.04986-14
35. Li H, Wang P, Huang F, et al. Astragaloside IV protects blood-brain barrier integrity from LPS-induced disruption via activating Nrf2 antioxidant signaling pathway in mice. Toxicol Appl Pharmacol. 2018;340:58–66. doi:10.1016/j.taap.2017.12.019
36. Carrano A, Hoozemans JJM, van der Vies SM, Rozemuller AJM, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15(5):1167–1178. doi:10.1089/ars.2011.3895
37. Herrero R, Prados L, Ferruelo A, et al. Fas activation alters tight junction proteins in acute lung injury. Thorax. 2019;74(1):69–82. doi:10.1136/thoraxjnl-2018-211535
38. O T, F M. Tight junction structure and function revisited. Trends Cell Biol. 2020;30(10). doi:10.1016/j.tcb.2020.08.004
39. Singh R, Chandrashekharappa S, Bodduluri SR, et al. Enhancement of the gut barrier integrity by a microbial metabolite through the Nrf2 pathway. Nat Commun. 2019;10(1):89. doi:10.1038/s41467-018-07859-7
40. Xie W, Wang H, Wang L, Yao C, Yuan R, Wu Q. Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-induced mice. Lab Invest. 2013;93(9):991–1000. doi:10.1038/labinvest.2013.80
41. Cho WH, Yeo HJ, Yoon SH, et al. Lysophosphatidylcholine as a prognostic marker in community-acquired pneumonia requiring hospitalization: a pilot study. Eur J Clin Microbiol Infect Dis. 2015;34(2):309–315. doi:10.1007/s10096-014-2234-4
42. Knuplez E, Marsche G. An updated review of pro- and anti-inflammatory properties of plasma lysophosphatidylcholines in the vascular system. Int J Mol Sci. 2020;21(12):4501. doi:10.3390/ijms21124501
43. Xu Y. Sphingosylphosphorylcholine and lysophosphatidylcholine: g protein-coupled receptors and receptor-mediated signal transduction. Biochim Biophys Acta. 2002;1582(1–3):81–88. doi:10.1016/s1388-1981(02)00140-3
44. Lee HJ, Ko HJ, Song DK, Jung YJ. Lysophosphatidylcholine promotes phagosome maturation and regulates inflammatory mediator production through the protein Kinase A-Phosphatidylinositol 3 Kinase-p38 mitogen-activated protein kinase signaling pathway during mycobacterium tuberculosis infection in mouse macrophages. Front Immunol. 2018;9:920. doi:10.3389/fimmu.2018.00920
45. Zeng L, Ma B, Yang S, et al. Role of autophagy in lysophosphatidylcholine-induced apoptosis in mouse Leydig cells. Environ Toxicol: Int J. 2022;37(11):2756–2763. doi:10.1002/tox.23634
46. Yang S, Chen J, Ma B, Wang J, Chen J. Role of autophagy in lysophosphatidylcholine-induced apoptosis of mouse ovarian granulosa cells. Int J Mol Sci. 2022;23(3):1479. doi:10.3390/ijms23031479
47. Lou L, Wang M, He J, et al. Urolithin A (UA) attenuates ferroptosis in LPS-induced acute lung injury in mice by upregulating Keap1-Nrf2/HO-1 signaling pathway. Front Pharmacol. 2023;14:1067402. doi:10.3389/fphar.2023.1067402
48. Wang X, Wang W, Zhang R, et al. Melatonin attenuates high glucose‑induced endothelial cell pyroptosis by activating the Nrf2 pathway to inhibit NLRP3 inflammasome activation. Mol Med Rep. 2023;27(3):71. doi:10.3892/mmr.2023.12958
49. Liu L, Shi Q, Wang K, et al. Fibroblast growth factor 10 protects against particulate matter-induced lung injury by inhibiting oxidative stress-mediated pyroptosis via the PI3K/Akt/Nrf2 signaling pathway. Int Immunopharmacol. 2022;113:109398. doi:10.1016/j.intimp.2022.109398
50. Zheng Y, Huang Y, Xu Y, Sang L, Liu X, Li Y. Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discov. 2023;9(1):91. doi:10.1038/s41420-023-01369-2
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.