Back to Journals » OncoTargets and Therapy » Volume 12
Luteolin suppresses tumor proliferation through inducing apoptosis and autophagy via MAPK activation in glioma
Authors You Y ![]() , Wang R, Shao N, Zhi F
, Wang R, Shao N, Zhi F ![]() , Yang Y
, Yang Y
Received 17 October 2018
Accepted for publication 4 February 2019
Published 28 March 2019 Volume 2019:12 Pages 2383—2396
DOI https://doi.org/10.2147/OTT.S191158
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Yijie You,1–3 Rong Wang,2 Naiyuan Shao,1–3 Feng Zhi,1,2 Yilin Yang1–3
1Department of Neurosurgery, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China; 2Modern Medical Research Center, The Third Affiliated Hospital of Soochow University, Changzhou, Jiangsu, China; 3Department of Neurosurgery, The First People’s Hospital of Changzhou, Changzhou, Jiangsu, China
Purpose: Glioma is a malignant tumor that originates in the brain and spine and is difficult to be completely removed. Though glioma patients receive active treatment, the survival rate is still poor. Therefore, it is urgent to discover a new medicine to treat glioma patients in order to improve the survival rate. In this study, we explored the anticancer effect and the potential mechanism of luteolin on glioma in vitro.
Materials and methods: Cell viability was determined by Cell Counting Kit-8 (CCK-8) assay. Fluorescent microscopy and flow cytometry analysis were used to determine the cellular apoptosis. Western blot analysis was performed to explore the changes in protein expression. Quantitative reverse transcription-PCR (qRT-PCR) analysis was utilized to evaluate the expression level of the tumor suppressor miR-124-3p.
Results: CCK-8 assays indicated that luteolin significantly inhibited glioma cell proliferation in a time- and dose-dependent manner. Fluorescent microscopy and flow cytometry analysis confirmed that luteolin induced glioma cell apoptosis. Western blot analysis showed that luteolin induced cellular apoptosis in glioma cells via MAPK activation (JNK, ERK, and p38). Luteolin stimulated the death receptor (FADD) to regulate the apoptosis proteins (Caspase-8, Caspase-3, and PARP). Luteolin increased the expression levels of LC3B II/I and downregulated the level of p62 that promotes cell autophagy. Finally, qRT-PCR confirmed that luteolin upregulated the expression levels of miR-124-3p.
Conclusion: These findings illustrate that luteolin may be a potential drug for glioma treatment.
Keywords: glioma, luteolin, apoptosis, autophagy, miR-124-3p
Introduction
Glioma, the most common malignant neoplasm of the brain, accounts for 81% of brain cancers.1,2 Conventional therapies, including surgical resection, chemotherapy, and radiotherapy, are still unsuccessful for completely curing glioma.3 However, the researchers for the current study found no additional effective drugs to treat glioblastoma, except temozolomide (TMZ).4 Therefore, to improve therapeutic efficacy, the demand for development and introduction of new medicines and treatments is imminent.
In recent decades, because of the antitumor effects in brain cancers, the chemical constituents of traditional Chinese medicines have attracted great attention in the academic field.5 Luteolin has considerable pharmacological value, including antitumor, antioxidative, neuroprotective, and anti-inflammatory effects.6–9 The anticancer effect of luteolin has been investigated in various cancers.10–15 For example, luteolin inhibited the proliferation of human cholangiocarcinoma cells by inducing apoptosis and interrupting the JNK/STAT3 pathway.10 It also induced apoptosis in Hela cells through suppressing the expression of human papilloma virus E6 and E7 oncogenes and increasing the expression of death receptors by regulating the Fas/FasL, DR5/TRAIL, and FADD.11 In the field of glioma, Souza et al reported that flavonoids mixture could inhibit cell proliferation in glioma.16 Wang et al indicated that luteolin inhibited glioma cell proliferation through endoplasmic reticulum stress and mitochondrial pathway.17 Anson et al mentioned that luteolin decreased cell proliferation by inhibiting the epidermal growth factor signaling pathway in glioma.19 Though some efforts have been made on luteolin in glioma treatment, these in vitro results were mainly based on the glioma cell line U87, which was recently reported by Allen that this cell line was lack of representativeness of glioma.20 miRNAs are small non-coding RNAs that negatively regulate mRNA translation.21 miRNAs are reported to be involved in luteolin’s anticancer activity.18,22 Chakrabarti and Ray discovered that luteolin induced apoptosis in glioblastoma by regulating miR-7-1-3p expression.18 Yang et al found that miR-6809-5p mediated luteolin-induced anticancer effects by targeting flotillin 1 in hepatoma.23 In our previous studies, we have identified several miRNAs as tumor suppressors in glioma, including miR-124-3p,24 miR-137,25 miR-181b-5p,26 and miR-454-3p,21 and we would like to examine whether these four miRNAs are involved in luteolin’s anticancer activity in glioma. In the present study, the glioma cell lines LN229 and U251 were chosen. Luteolin was found to inhibit glioma cell proliferation, induce cellular apoptosis, and promote cellular autophagy through the activation of death receptor and MAPK signaling pathways and miR-124-3p upregulation. Our results suggested the potential of luteolin as a novel therapeutic drug for glioma.
Materials and methods
Chemicals and materials
Luteolin was purchased from Aynor Medicine Technology Co. Ltd (Xi’an, China) at a purity of 98%. TMZ and 3-methyadenine (3-MA) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Dimethyl sulfoxide (DMSO) was obtained from MP Biomedicals LLC (Santa Ana, CA, USA). Trypsin-EDTA 0.25% (1×), FBS, and DMEM were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The Hoechst 33258 kit and Cell Counting Kit-8 (CCK-8) kit were obtained from the Beyotime Institute of Biotechnology (Shanghai, China). Antibodies, including β-actin, P-JNK, JNK, PARP, FADD, P-p38, p38, LC3B, p62, Caspase-3, cleaved Caspase-3, Caspase-8, and cleaved Caspase-8, were purchased from Cell Signaling Technology (Danvers, MA, USA). The antibodies of P-ERK, and ERK were purchased from Beyotime Institute of Biotechnology. The horseradish peroxidase-conjugated secondary biotinylated antibodies were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). All other reagents were of standard biochemical quality unless otherwise stated.
Cell culture
The human glioma cell line U251 was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The human cell line HEB and glioma cell line LN229 were obtained from Nanjing Beierbo Co. Ltd (Nanjing, China). The cells were cultured in DMEM containing 10% FBS. All the cells were maintained at 37°C with 5% CO2 in a humidified atmosphere. Finally, the cells were planted at an appropriate density and in a good condition before the experiments.
CCK-8 assay
Cell survival was measured by CCK-8 assays. Cells in a good condition were digested with trypsin and plated in a 96-well plate with 8×103 cells per well and then incubated overnight to allow attachment and growth. Luteolin or TMZ was dissolved in DMSO at indicated concentrations and then mixed with 10% FBS-DMEM to prepare solutions with different concentrations. The cells were operated under different treatments for 24 and 48 hours. Twenty microliters of CCK-8 solution was added to each well (200 μL) and incubated for 1 hour at 37°C. Afterward, ELx800 Absorbance Microplate Reader (BioTek, Winooski, VT, USA) was utilized to measure the OD at 490 nm.
Hoechst 33258 staining
A Hoechst staining kit was used to detect changes in cell nucleus apoptosis. Clean cover slips were steeped in alcohol (75%) for 30 minutes, washed three times with sterile PBS, and then incubated with cells for one night in a six-well plate. Cells were treated with luteolin at different concentrations for 24 hours. After removing the culture media, 0.5 mL of stationary liquid was added to each well for 10 minutes. The fixing solution was discarded, and PBS was used to wash the cells for 3 minutes; the process was repeated several times. The cells were stained with Hoechst 33258 (0.5 mL) for 5 minutes in the dark. After discarding Hoechst 33258 and washing the cells twice with PBS, one drop of mounting medium was added onto each of the cover slips. Then, the cells were observed under an IX71 fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Flow cytometry assay
Flow cytometry was performed to detect apoptosis. The cells were treated with different concentrations of luteolin for 24 hours and then collected. Next, the cells were washed entirely with PBS, separated by trypsin digestion, and transferred into a new centrifuge tube. After centrifugal settling, 1×105 cells were collected and then stained with 5 μL of Annexin V-fluorescein isothiocyanate and 10 μL of propidium iodide (Beyotime Institute of Biotechnology) for 20 minutes in the dark. The cells were immediately analyzed by a Guava EasyCyte 6HT-2L flow cytometer (EMD Millipore, Billerica, MA, USA).
RNA extraction and quantitative reverse transcription-PCR (qRT-PCR)
Twenty-four hours after treatment as described above, total RNA was extracted from cultured cells by TRIzol Reagent (Thermo Fisher Scientific) according to the guide, and RT-PCR reaction was performed immediately. Levels of mature miR-124-3p, miR-137, miR-181b-5p, and miR-454-3p were quantified using TaqMan microRNA probes (Thermo Fisher Scientific) according to the instructions. Basically, 2 μg of total RNA was transformed into cDNA by using AMV reverse transcriptase (TaKaRa, Shiga, Japan) and a stem-loop primer (Thermo Fisher Scientific). The mixture was incubated at 16°C for 15 minutes, 42°C for 60 minutes, and 85°C for 5 minutes to form a library of miRNA cDNAs. qRT-PCR was performed using a TaqMan PCR kit on a 7500 Sequence Detection System (Thermo Fisher Scientific). All the sequences were repeated three times. In addition, U6 small nuclear RNA was used as a control. The relative amount of miRNA to internal control U6 transcript was calculated using the equation, ΔCt=CtmiRNA−CtU6. Synthetic miR-124-3p mimics (124M), miR-124-3p inhibitor (124I), and negative control (NC) were purchased from GenePharma (Shanghai, China). The transfections were carried out with Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s specifications.
Western blot analysis
After different treatments for 24 hours, the cells were digested with trypsin, and single cell suspensions were obtained and washed three times with PBS. A whole-cell Lysis Assay Kit (KeyGEN Biotec, Nanjing, China) was utilized to extract the total protein. The protein concentration was determined by the bicinchoninic acid protein assays (Cell Signaling Technology). Then, the denatured protein samples were separated through SDS-PAGE. Next, the proteins were transferred onto polyvinylidene difluoride (PVDF) flat microporous membranes and blocked for 1 hour with 5% skimmed milk powder. Then, the PVDF membranes were incubated overnight at 4°C with the following primary antibodies: ERK (1:1,000), P-ERK (1:1,000), JNK (1:1,000), P-JNK (1:1,000), p38 (1:1,000), P-p38 (1:1,000), FADD (1:1,000), Caspase-8 (1:1,000), cleaved Caspase-8 (1:1,000), Caspase-3 (1:1,000), cleaved Caspase-3 (1:1,000), PARP (1:1,000), cleaved PARP (1:1,000), LC3B (1:1,000), and β-actin (1:1,000) according to the manufacturer’s instructions. After washing three times with 1× PBST, the membranes were incubated with the diluted secondary antibodies for 1 hour. After being washed three times again, the blots were detected using an enhanced chemiluminescence solution (Thermo Fisher Scientific). The films were processed using a medical X-ray film processor (Taixing Suxing Company, Jiangsu, China). The OD values of each protein were analyzed using the NIH imaging program (ImageJ).
Statistical analysis
The results were generated from at least three repetitions of independent experiments. Data were expressed as the mean ± SD and analyzed with GraphPad Prism 5.0 software. One-way ANOVA and the two-tailed Student’s t-test were employed to compare different groups. P<0.05 values were considered statistically significant.
Results
Luteolin inhibited cell proliferation
Luteolin is a flavonoid mainly extracted from the honeysuckle plant; the chemical structure is shown in Figure 1A. After treatments with different concentrations of luteolin for 24 and 48 hours, the cell viability was examined via CCK-8 assay. Under the same experimental conditions, we treated LN229, U251, and HEB cell lines with different concentrations of luteolin. The 50% growth inhibition (GI50) values of luteolin on LN229 cells at 24 and 48 hours were 18.2 and 12.9 μM, respectively (Figure 1B). The GI50 values of luteolin on U251 cells at 24 and 48 hours were 47.5 and 36.72 μM, respectively (Figure 1C). Meanwhile, the result showed that luteolin-treated HEB cells as normal glial cells had no massive death below 40 μM of luteolin, and the GI50 values of luteolin on HEB cells at 24 and 48 hours were 57.96 and 53.81 μM, respectively (Figure 1D). TMZ, the first-line treatment for glioblastoma, was selected as a positive control. As shown in Figure 1E, luteolin was more effective against glioma and less cytotoxic in normal cells than TMZ at the same concentration (30 μM). The results showed that luteolin treatment apparently slowed down the glioma cell proliferation rate in a concentration- and time-dependent manner, and thus, luteolin may be a safe and therapeutic agent against glioma.
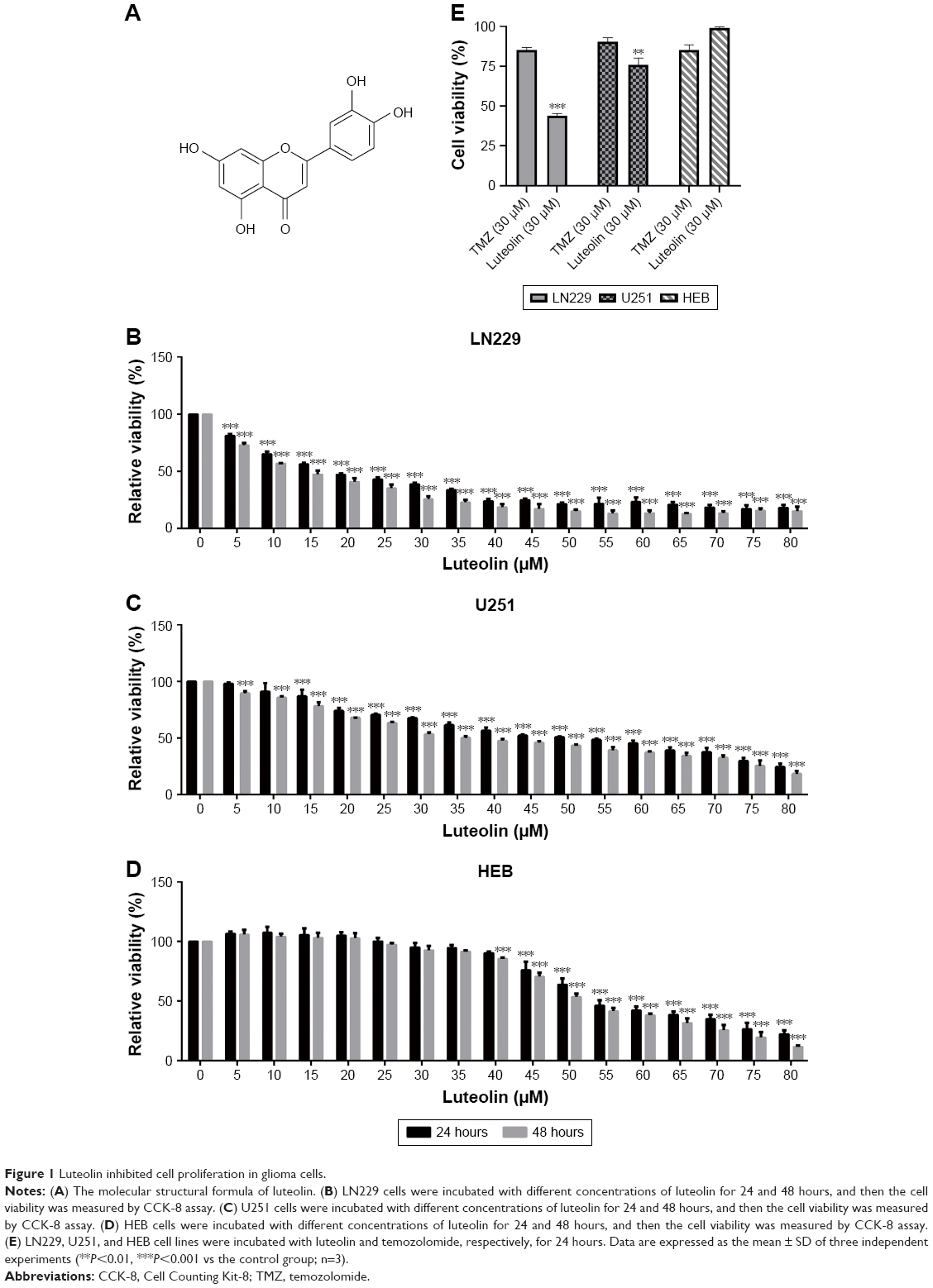 | Figure 1 Luteolin inhibited cell proliferation in glioma cells. |
Luteolin induced cellular apoptosis
To investigate the potential mechanisms of how luteolin markedly suppresses cell proliferation in glioma cells, Hoechst 33258 staining was used to observe the morphological and biochemical features of apoptosis after treating cancer cells with varying levels of luteolin. According to the observation under fluorescence microscopy, karyorrhexis and apoptosis bodies significantly increased compared to the control group. The results indicated that luteolin caused the morphological changes in LN229 cells (Figure 2A). In addition, flow cytometric analysis revealed that luteolin induced apoptosis in LN229 cells: the percentages of early and late apoptotic cells significantly increased after treatment with luteolin for 24 hours. The percentage of early apoptotic cells increased from 3.09% to 21.59% after exposure to luteolin for 24 hours. The percentage of late apoptotic cells also distinctly increased with increased luteolin concentration (Figure 2B). The results obtained for luteolin on U251 cells were similar to those obtained on LN229 cells as shown in Figure 2C and D. However, it is worthy to note that the apoptosis rate induced by luteolin on LN229 cells was more apparent than that on U251 cells.
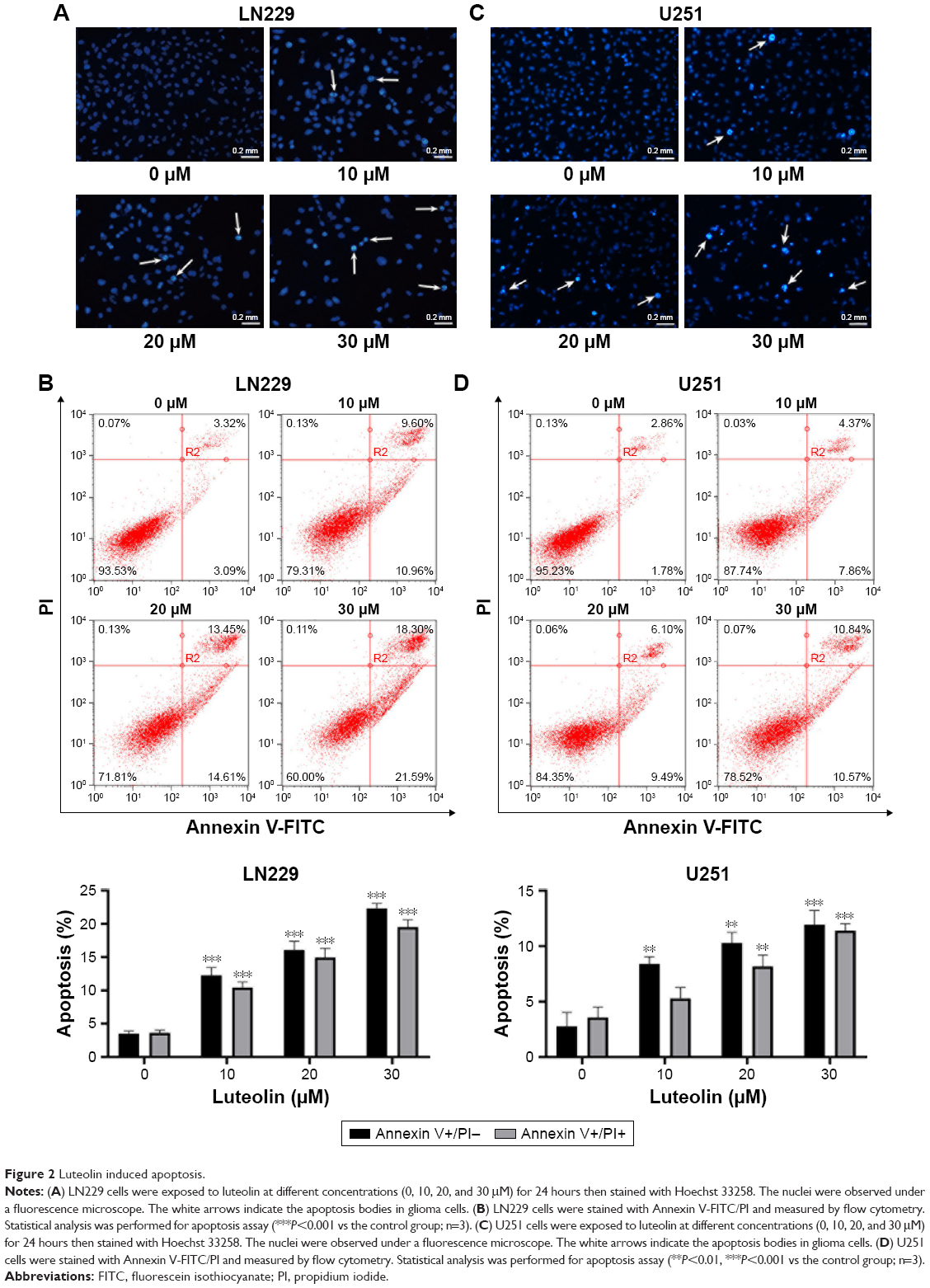 | Figure 2 Luteolin induced apoptosis. |
Luteolin activated the death receptor pathway
The death receptor signaling pathway is an important mechanism involved in cellular apoptosis. We performed Western blot to investigate the changes in some key proteins involved in this pathway in both LN229 and U251 cells. We first tested the expression of some key proteins in LN229 cells treated with luteolin. Our results showed that the expression of FADD was enhanced with increasing luteolin concentration (Figure 3A). There were no obvious changes in the expression levels of PARP in luteolin-treated cells, but cleaved PARP was significantly increased in luteolin-treated cells (Figure 3A). The average densitometric values of FADD, PARP, and cleaved PARP in three independent experiments are shown by the histogram in Figure 3B and C. The expression levels of Caspase-8 had no remarkable change in the luteolin-treated group compared to the control group, while cleaved Caspase-8 was markedly increased after luteolin treatment compared to that in the control group (Figure 3D). Similarly, there was no distinct change in the expression of Caspase-3 in glioma cells after luteolin treatment (Figure 3D), but the expression levels of cleaved Caspase-3 were increased in the cells treated with luteolin (Figure 3D). The average densitometric values of Caspase-8, cleaved Caspase-8, Caspase-3, and cleaved Caspase-3 in three independent experiments are shown in Figure 3E and F. The results were similar in U251 cells as shown in Figure 3G–K.
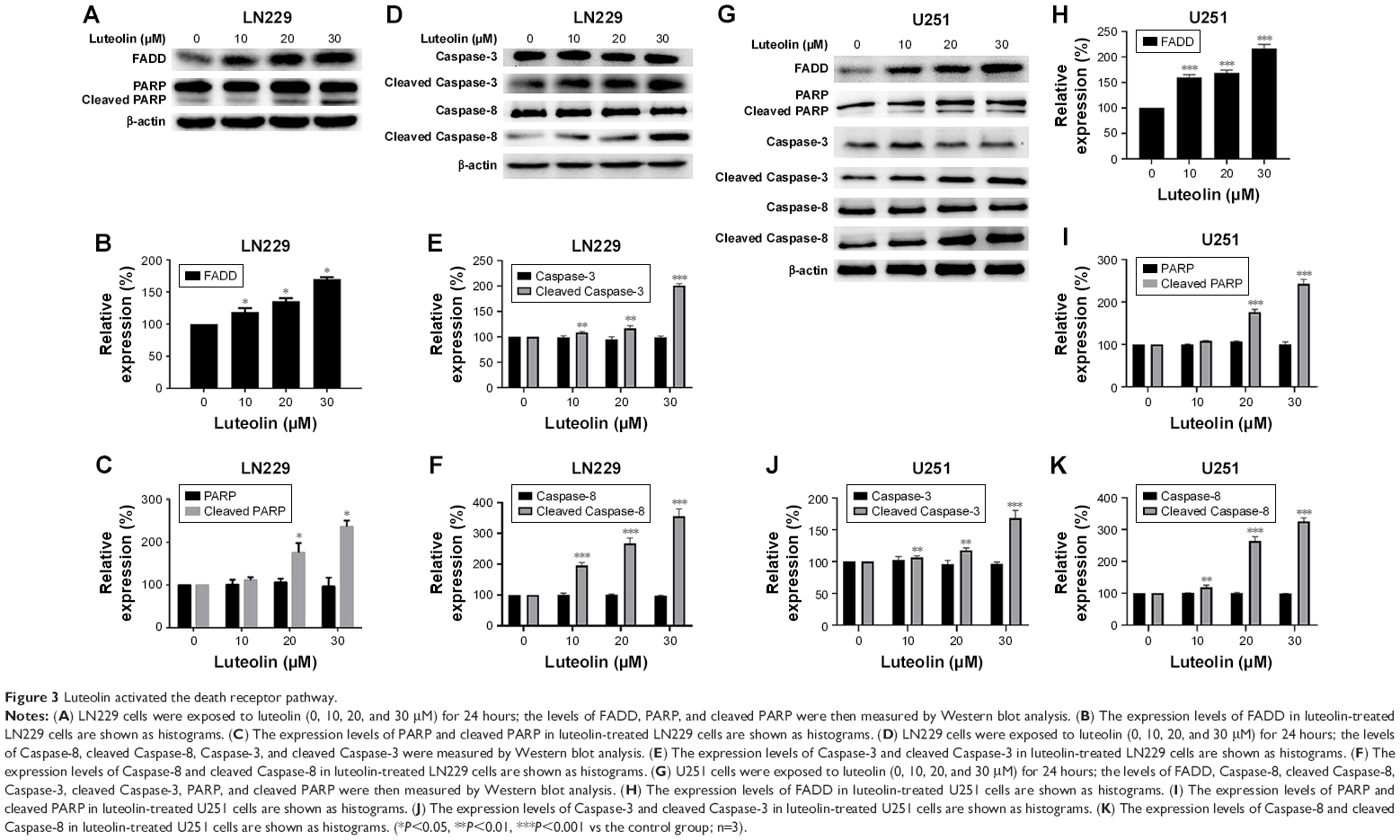 | Figure 3 Luteolin activated the death receptor pathway. |
Luteolin activated the MAPK pathway
We next explored the effect of luteolin on the MAPK pathway in LN229 and U251 cells. We first detected the expression levels of ERK, P-ERK, JNK, P-JNK, p38, P-p38, Bax, and Bcl-2 in LN229 cells. We found that luteolin increased the expression levels of P-ERK, but the expression levels of ERK were unchanged in the luteolin-treated group (Figure 4A). The expression of P-JNK was upregulated in the luteolin-treated group compared to the control group (Figure 4A). Similar to ERK, the expression levels of JNK showed no remarkable changes in the luteolin-treated group (Figure 4A). The expression of P-p38 was markedly increased after luteolin treatment compared to that in the control group, and there were no obvious changes in the expression levels of p38 in luteolin-treated cells (Figure 4A). In addition, we also measured the protein expression levels of Bax and Bcl-2. The level of Bax was significantly upregulated in the luteolin-treated group compared to the control group. Conversely, the expression of Bcl-2 was significantly lower than that in the control group (Figure 4A). The statistical analyses of ERK, P-ERK, JNK, P-JNK, p38, and P-p38 are shown by a bar graph in Figure 4B–D. The ratio of Bax to Bcl-2 increased in a concentration-dependent manner (Figure 4E). The results were similar in U251 cells as shown in Figure 4F–J.
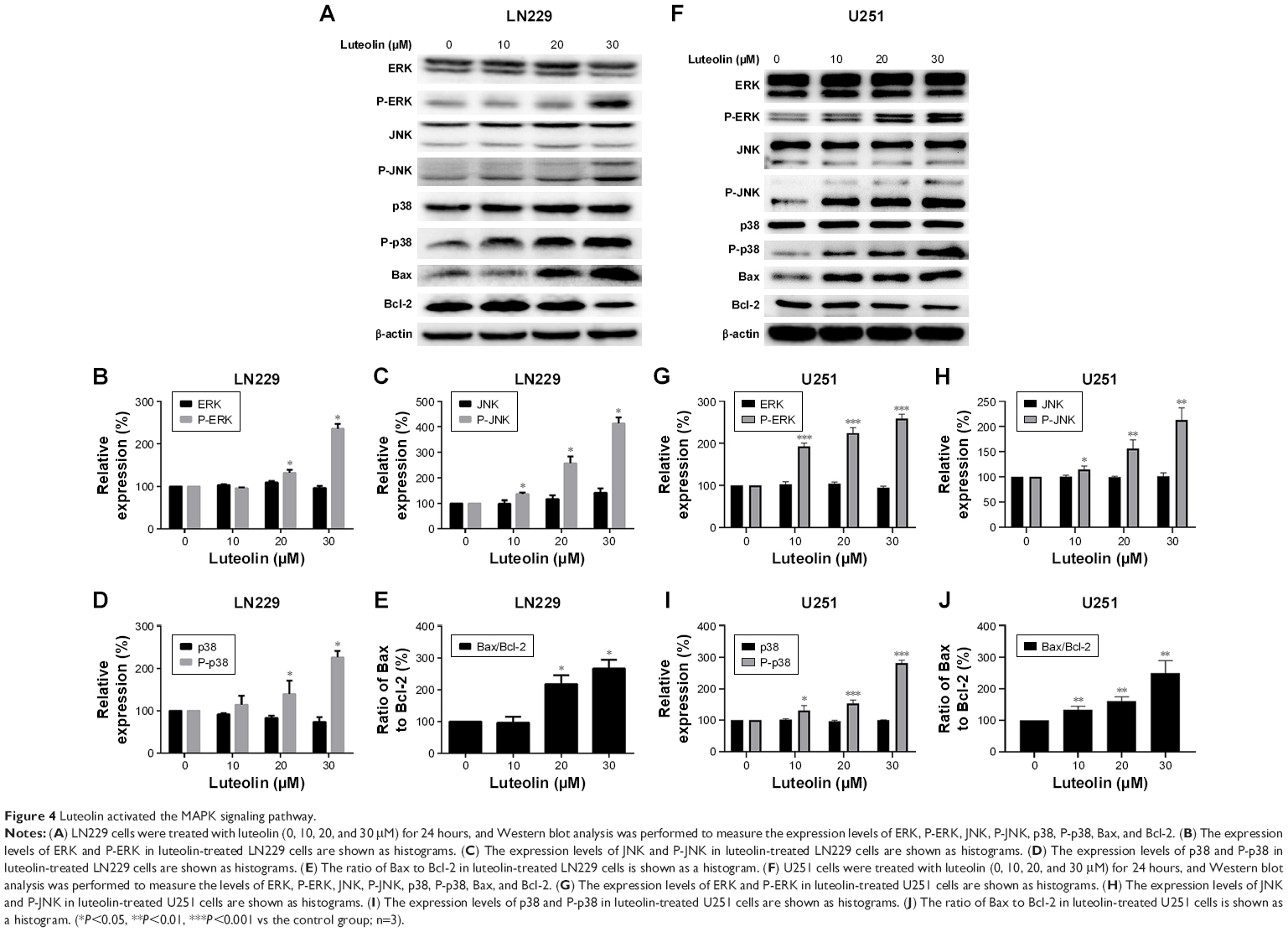 | Figure 4 Luteolin activated the MAPK signaling pathway. |
Luteolin induced cell autophagy
We examined the effect of luteolin on cell autophagy. After the LN229 cells were treated by luteolin, there was no obvious change in the protein expression levels of LC3B I compared to the control group. However, the levels of LC3B II were obviously enhanced with increase in luteolin concentrations (Figure 5A). The ratio of LC3B II to LC3B I increased in a concentration-dependent manner (Figure 5B). LC3 is responsible for delivery of P62 into lysosomes for degradation in the processes of autophagy. We measured the protein expression levels of P62. We found that P62 was expressed at lower levels in luteolin-treated cells than in control cells (Figure 5C). The results were similar in U251 cells (Figure 5D–F). It is worthy to note that luteolin had a relatively stronger effect on inducing autophagy in U251 cells than in LN229 cells. 3-MA, an early-stage inhibitor of the autophagic pathway, was selected to detect the autophagic flux. As shown in Figure 5G, 3-MA significantly inhibited the autophagic flux induced by luteolin in LN229 cells. The statistical analysis for LC3B is shown in Figure 5H and for p62 in Figure 5I. The results were similar in U251 cells as shown in Figure 5J–L.
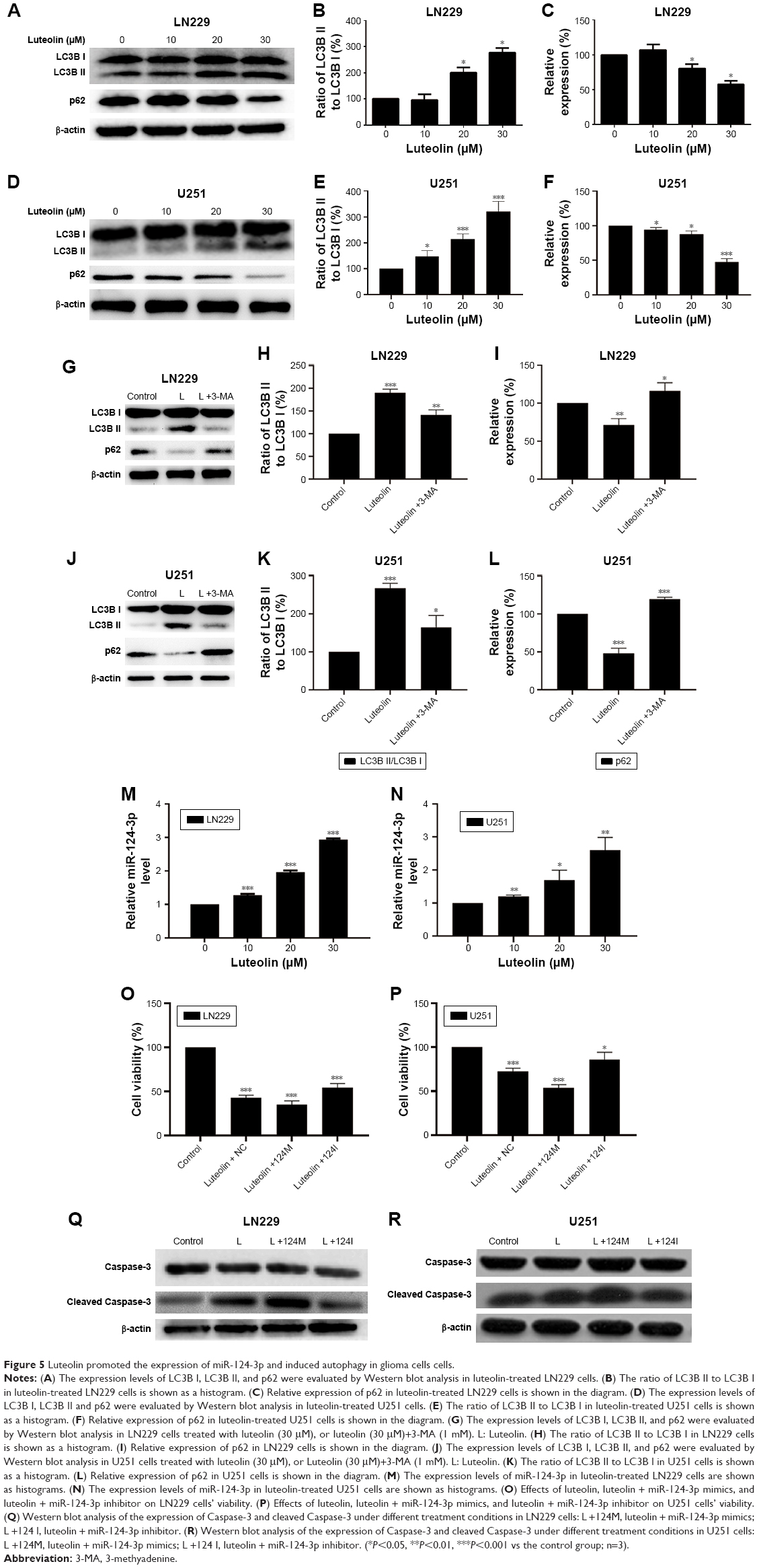 | Figure 5 Luteolin promoted the expression of miR-124-3p and induced autophagy in glioma cells cells. |
Luteolin increased the expression of miR-124-3p
We first detected the expression levels of the four miRNAs after luteolin treatment in LN229 cells. As shown in Figure 5M and N, only the expression levels of miR-124-3p were significantly upregulated in the luteolin-treated group when compared to the control group, while the other three miRNAs were unchanged in both LN229 and U251 cells (data not shown). Next, we investigated the relationship of miR-124-3p with the effect of luteolin on cell proliferation. As shown in Figure 5O, miR-124-3p overexpression could significantly increase cellular cytotoxicity, while miR-124-3p downregulation could decrease cellular cytotoxicity in LN229 cells. It was similar in U251 cells (Figure 5P). Finally, we investigated the relationship of miR-124-3p with the effect of luteolin on apoptosis. As shown in Figure 5Q, miR-124-3p overexpression could significantly increase cleaved Caspase-3, while miR-124-3p downregulation could decrease cleaved Caspase-3 in LN229 cells. It was similar in U251 cells (Figure 5R).
In summary, our results showed that luteolin suppresses tumor proliferation through inducing apoptosis via MAPK and death receptor pathway activation in glioma. We also found that luteolin treatment could induce autophagy and promote the expression of miR-124-3p in glioma cells (Figure 6).
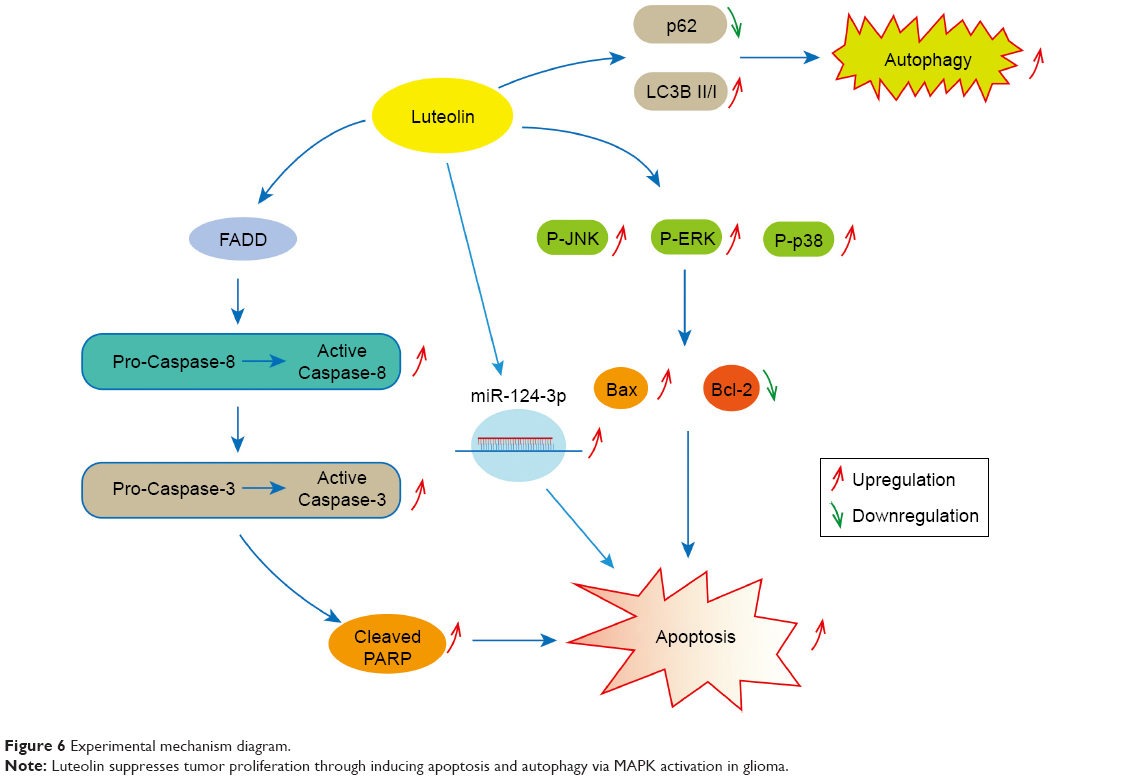 | Figure 6 Experimental mechanism diagram. |
Discussion
Luteolin is a compound of flavones that is used in various traditional Chinese herbal medicines.27 In the present study, we mainly explored the effect of luteolin on human glioma cells. We found that luteolin showed inhibition of glioma cell proliferation and induction of apoptosis. Further studies of the molecular mechanism showed that luteolin could activate MAPK and FADD to induce apoptosis, and could induce autophagy and upregulate the tumor suppressor gene miR-124-3p. These results provide a basis for developing luteolin into a new anti-glioma medicine.
The apoptotic pathway is divided into the extrinsic death receptor pathway and the intrinsic mitochondrial pathway.28 The extrinsic death receptor pathway involves some related proteins, such as FADD, Caspase-8, and Caspase-3, which can be activated to improve the expression of PARP.29 During drug-induced apoptosis, PARP is cleaved into fragments, and cleaved PARP is usually used as a marker of apoptosis.30 MAPK pathway activates especially the transmitted signals to activate several regulatory molecules to initiate cellular processes.31 For example, activating the MAPK pathway can induce apoptosis, which is an approach utilized in various kinds of chemotherapeutic drugs as an anticancer mechanism.32–34 The MAPK pathway is shared by three main groups of mammalian MAPKs: ERK, JNK, and p38, which are involved in cell apoptosis.35,36 The activation of ERK, JNK, and p38 is most frequently associated with the induction of apoptosis in multiple tumor cells.37,38 In particular, the activated JNK can regulate mitochondrial proteins such as Bcl-2 to trigger a plethora of cellular processes, such as cell proliferation and apoptosis.39 Although Souza et al found that luteolin could inhibit cell proliferation in glioma, the inherent mechanism had not been discussed.16 Wang et al found that luteolin could induce apoptosis in glioma cells through ROS/ER stress and mitochondrial dysfunction.17 Chakrabarti et al found that luteolin could stimulate cell apoptosis via regulating several apoptosis-related proteins and miR-7-1-3p upregulation in glioma.18 Anson et al reported that luteolin decreased the expression of EGFR mRNA to inhibit glioma cell proliferation.19 However, all these studies chose the U87MG cell line, which has been reported to lack representativeness of glioma.20 This issue may weaken the conclusion obtained from their experiments. In our research, two glioma cell lines LN229 and U251 were chosen. Our results indicated that luteolin not only increased the number of apoptotic bodies, but also activated FADD, Caspase-3, Caspase-8, and PARP, all of which participate in the regulation of the extrinsic death receptor pathway. Our results also revealed that luteolin induced the activation and phosphorylation of p38, JNK, and ERK. Based on these results, we believed that luteolin could induce apoptosis to achieve antitumor effect via the death receptor pathway and MAPK activation in human glioblastoma.
Autophagy is a catabolic process whereby damaged organelles, aged proteins, and damaged proteins are sequestered and degraded to maintain cellular homeostasis.40 During autophagy, LC3 is responsible for the delivery of p62 into lysosomes for degradation.41 In recent studies, it was reported that autophagy not only protects cells from cellular metabolic stress, but can also function as a tumor suppressor.42,43 The antitumor function is supported by the observations in other physiological processes.44–47 In the human HCC cell line SMMC-7721, luteolin can inhibit cancer growth through inducing autophagy.44 In addition, luteolin treatment can induce autophagy in NCI-H460 lung carcinoma cells and human osteosarcoma U2OS cells, as well as H9C2 cells.45 In glioma, Chakrabarti et al showed that luteolin decreased the expression of PKCα in the Rapamycin-pretreated glioma cells. They also found that luteolin could inhibit autophagy in RAPA-pretreated glioma cells through regulating some autophagy-related proteins such as Beclin-1, LC3B I, LC3B II, mTOR, and p62.18 However, in our study, we observed a significant increase in the ratio of LC3B II to LC3B I. Conversely, the expression of p62 tapered with increase of luteolin concentration. These results demonstrated that luteolin treatment induced autophagy. Based on the differences between the two studies, we found that Chakrabarti et al used RAPA to pretreat glioma cells, whereas we only used luteolin to treat glioma without any pretreatment. Furthermore, we found that luteolin could increase LC3B II level and decrease p62 level, while the addition of 3-MA could inhibit the induction. This indicated that luteolin may promote autophagosome initiation. Therefore, we believe our study is more convincing with respect to the effect of luteolin on autophagy in glioma cells. Additionally, we found that the cell lines used in the two studies were different. This difference may be an important reason for the different results obtained.
miR-124-3p could promote apoptosis to inhibit cell proliferation, invasion, and bioenergetics by targeting PIM1, which is a key control protein in cell proliferation, cell cycle, apoptosis, and metabolism.48 We found that luteolin distinctly promoted the expression of miR-124-3p. In addition, miR-124-3p mimics significantly enhanced cellular cytotoxicity and apoptosis induced by luteolin, while miR-124-3p inhibitor partially reduced cellular cytotoxicity and apoptosis induced by luteolin. These results established the relationship of miR-124-3p with the effect of luteolin in glioma. We believe that the antitumor effect of luteolin may be associated with regulation of miR-124-3p expression.
Conclusion
Our results indicated that luteolin could suppress cell viability, induce cellular apoptosis and autophagy, and increase the expression of miR-124-3p. These results confirmed that luteolin has an antitumor effect on glioma. Our results will promote further research into the use of luteolin in glioma treatment.
Ethical approval
This article does not contain any study with human participants or animals performed by any of the authors.
Acknowledgments
This study was funded by the National Natural Science Foundation of China (81302197), Changzhou High-Level Medical Talents Training Project (2016CZBJ006), and Changzhou Municipal Commissions of Health and Family Planning Major Scientific and Technological Project (ZD201620).
Disclosure
The authors report no conflicts of interest in this work.
References
Jiang Y, Uhrbom L. On the origin of glioma. Ups J Med Sci. 2012;117(2):113–121. | ||
Ostrom QT, Bauchet L, Davis FG, et al. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16(7):896–913. | ||
de Groot JF. High-grade gliomas. Neuro Oncol. 2015;21:332–344. | ||
Shi L, Sun G. DMC is not better than TMZ on intracranial anti-glioma effects. J Cell Biochem. 2018;119(7):6057–6064. | ||
Zhang FY, Hu Y, Que ZY, et al. Shikonin inhibits the migration and invasion of human glioblastoma cells by targeting phosphorylated β-catenin and phosphorylated PI3K/Akt: a potential mechanism for the anti-glioma efficacy of a traditional Chinese herbal medicine. Int J Mol Sci. 2015;16(10):23823–23848. | ||
Majumdar D, Jung KH, Zhang H, et al. Luteolin nanoparticle in chemoprevention: in vitro and in vivo anticancer activity. Cancer Prev Res (Phila). 2014;7(1):65–73. | ||
Jia Z, Nallasamy P, Liu D, et al. Luteolin protects against vascular inflammation in mice and TNF-alpha-induced monocyte adhesion to endothelial cells via suppressing IKBα/NF-κB signaling pathway. J Nutr Biochem. 2015;26(3):293–302. | ||
Xu J, Wang H, Ding K, et al. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2–ARE pathway. Free Radic Biol Med. 2014;71(6):186–195. | ||
Cao Z, Ding Y, Ke Z, et al. Luteoloside acts as 3C protease inhibitor of enterovirus 71 in vitro. PLoS One. 2016;11(2):e0148693. | ||
Aneknan P, Kukongviriyapan V, Prawan A, Kongpetch S, Sripa B, Senggunprai L. Luteolin arrests cell cycling, induces apoptosis and inhibits the JAK/STAT3 pathway in human cholangiocarcinoma cells. Asian Pac J Cancer Prev. 2014;15(12):5071–5076. | ||
Ham S, Kim KH, Kwon TH, et al. Luteolin induces intrinsic apoptosis via inhibition of E6/E7 oncogenes and activation of extrinsic and intrinsic signaling pathways in HPV-18-associated cells. Oncol Rep. 2014;31(6):2683–2691. | ||
Wu H, Huang M, Liu Y, Shu Y, Liu P. Luteolin induces apoptosis by up-regulating miR-34a in human gastric cancer cells. Technol Cancer Res Treat. 2015;14(6):747–755. | ||
Krifa M, Leloup L, Ghedira K, Mousli M, Chekir-Ghedira L. Luteolin induces apoptosis in Be colorectal cancer cells by downregulating calpain, UHRF1, and DNMT1 expressions. Nutr Cancer. 2014;66(7):1220–1227. | ||
Liping MA, Peng H, Li K, et al. Luteolin exerts an anticancer effect on NCI-H460 human non-small cell lung cancer cells through the induction of SIRT1-mediated apoptosis. Mol Med Rep. 2015;12(3):4196–4202. | ||
Yang MY, Wang CJ, Chen NF, Ho WH, Lu FJ, Tseng TH. Luteolin enhances paclitaxel-induced apoptosis in human breast cancer MDA-MB-231 cells by blocking STAT3. Chem Biol Interact. 2014;213(1):60–68. | ||
Souza PO, Bianchi SE, Figueiró F, et al. Anticancer activity of flavonoids isolated from Achyrocline satureioides in gliomas cell lines. Toxicol In Vitro. 2018;51:23–33. | ||
Wang Q, Wang H, Jia Y, Pan H, Ding H. Luteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastoma. Cancer Chemother Pharmacol. 2017;79(5):1031–1041. | ||
Chakrabarti M, Ray SK. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis. 2016;21(3):312–328. | ||
Anson DM, Wilcox RM, Huseman ED, et al. Luteolin decreases epidermal growth factor receptor-mediated cell proliferation and induces apoptosis in glioblastoma cell lines. Basic Clin Pharmacol Toxicol. 2018;123(6):678–686. | ||
Allen M, Bjerke M, Edlund H, Nelander S, Westermark B. Origin of the U87MG glioma cell line: good news and bad news. Sci Transl Med. 2016;8(354):354re3. | ||
Shao N, Xue L, Wang R, Luo K, Zhi F, Lan Q. miR-454-3p is an exosomal biomarker and functions as a tumor suppressor in glioma. Mol Cancer Ther. 2019;18(2):459–469. | ||
Sun DW, Zhang HD, Mao L, et al. Luteolin inhibits breast cancer development and progression in vitro and in vivo by suppressing Notch signaling and regulating miRNAs. Cell Physiol Biochem. 2015;37(5):1693–1711. | ||
Yang PW, Lu ZY, Pan Q, et al. MicroRNA-6809-5p mediates luteolin-induced anticancer effects against hepatoma by targeting flotillin 1. Phytomedicine. 2019;57:18–29. | ||
Deng D, Wang L, Chen Y, et al. MicroRNA-124-3p regulates cell proliferation, invasion, apoptosis, and bioenergetics by targeting PIM1 in astrocytoma. Cancer Sci. 2016;107(7):899–907. | ||
Deng D, Xue L, Shao N, et al. miR-137 acts as a tumor suppressor in astrocytoma by targeting RASGRF1. Tumour Biol. 2016;37(3):3331–3340. | ||
Zhi F, Wang Q, Deng D, et al. MiR-181b-5p downregulates NOVA1 to suppress proliferation, migration and invasion and promote apoptosis in astrocytoma. PLoS One. 2014;9(10):e109124. | ||
Lin Y, Shi R, Wang X, Shen HM. Luteolin, a flavonoid with potentials for cancer prevention and therapy. Curr Cancer Drug Targets. 2008;8(7):634–646. | ||
Teocchi MA, D’Souza-Li L. Apoptosis through death receptors in temporal lobe epilepsy-associated hippocampal sclerosis. Mediators Inflamm. 2016;2016(6):1–12. | ||
Sartorius U, Schmitz I, Krammer PH. Molecular mechanisms of death-receptor-mediated apoptosis. Chembiochem. 2001;2(1):20–29. | ||
Boulares AH, Yakovlev AG, Ivanova V, et al. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274(33):22932–22940. | ||
Seger R, Krebs EG, Wexler S. The MAPK signaling cascade. FASEB J. 1995;9(9):726–735. | ||
Pang H, Cai L, Yang Y, Chen X, Sui G, Zhao C. Knockdown of osteopontin chemosensitizes MDA-MB-231 cells to cyclophosphamide by enhancing apoptosis through activating p38 MAPK pathway. Cancer Biother Radiopharm. 2011;26(2):165–173. | ||
Chiu SJ, Chao JI, Lee YJ, Hsu TS. Regulation of gamma-H2AX and securin contribute to apoptosis by oxaliplatin via a p38 mitogen-activated protein kinase-dependent pathway in human colorectal cancer cells. Toxicol Lett. 2008;179(2):63–70. | ||
Shin DY, Lee WS, Lu JN, et al. Induction of apoptosis in human colon cancer HCT-116 cells by anthocyanins through suppression of Akt and activation of p38-MAPK. Int J Oncol. 2009;35(6):1499–1504. | ||
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. | ||
Boutros T, Chevet E, Metrakos P. Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol Rev. 2008;60(3):261–310. | ||
Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275(50):39435–39443. | ||
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–1331. | ||
Sui X, Kong N, Ye L, et al. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014;344(2):174–179. | ||
El-Khattouti A, Selimovic D, Haikel Y, Hassan M. Crosstalk between apoptosis and autophagy: molecular mechanisms and therapeutic strategies in cancer. J Cell Death. 2013;6:37–55. | ||
Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008;121(16):2685–2695. | ||
Lamy L, Ngo VN, Emre NCT, et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell. 2013;23(4):435–449. | ||
Luo YH, Wu SB, Wei YH, et al. Cadmium-based quantum dot induced autophagy formation for cell survival via oxidative stress. Chem Res Toxicol. 2013;26(5):662–673. | ||
Cao Z, Zhang H, Cai X, et al. Luteolin promotes cell apoptosis by inducing autophagy in hepatocellular carcinoma. Cell Physiol Biochem. 2017;43(5):1803–1812. | ||
Zhang B, Yu X, Xia H. The flavonoid luteolin enhances doxorubicin-induced autophagy in human osteosarcoma U2OS cells. Int J Clin Exp Med. 2015;8(9):15190. | ||
Yao H, Zhou L, Tang L, et al. Protective effects of luteolin-7-O-glucoside against starvation-induced injury through upregulation of autophagy in H9c2 cells. Biosci Trends. 2017;11(5):557–564. | ||
Park SH, Park HS, Lee JH, et al. Induction of endoplasmic reticulum stress-mediated apoptosis and non-canonical autophagy by luteolin in NCI-H460 lung carcinoma cells. Food Chem Toxicol. 2013;56(2):100–109. | ||
Deng D, Wang L, Chen Y, et al. MicroRNA-124-3p regulates cell proliferation, invasion, apoptosis, and bioenergetics by targeting PIM1 in astrocytoma. Cancer Sci. 2016;107(7):899–907. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.