Back to Journals » Breast Cancer: Targets and Therapy » Volume 9
Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells
Authors Cook MT, Liang Y, Besch-Williford C ![]() , Hyder SM
, Hyder SM
Received 17 October 2016
Accepted for publication 17 November 2016
Published 23 December 2016 Volume 2017:9 Pages 9—19
DOI https://doi.org/10.2147/BCTT.S124860
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Matthew T Cook,1,2 Yayun Liang,1,2 Cynthia Besch-Williford,3 Salman M Hyder1,2
1Department of Biomedical Sciences, 2Dalton Cardiovascular Research Center, University of Missouri, 3IDEXX BioResearch, Columbia, MO, USA
Abstract: Most breast cancer-related deaths from triple-negative breast cancer (TNBC) occur following metastasis of cancer cells and development of tumors at secondary sites. Because TNBCs lack the three receptors targeted by current chemotherapeutic regimens, they are typically treated with extremely aggressive and highly toxic non-targeted treatment strategies. Women with TNBC frequently develop metastatic lesions originating from drug-resistant residual cells and have poor prognosis. For this reason, novel therapeutic strategies that are safer and more effective are sought. Luteolin (LU) is a naturally occurring, non-toxic plant compound that has proven effective against several types of cancer. With this in mind, we conducted in vivo and in vitro studies to determine whether LU might suppress metastasis of TNBC. In an in vivo mouse metastasis model, LU suppressed metastasis of human MDA-MB-435 and MDA-MB-231 (4175) LM2 TNBC cells to the lungs. In in vitro assays, LU inhibited cell migration and viability of MDA-MB-435 and MDA-MB-231 (4175) LM2 cells. Further, LU induced apoptosis in MDA-MB-231 (4175) LM2 cells. Relatively low levels (10 µM) of LU significantly inhibited vascular endothelial growth factor (VEGF) secretion in MDA-MB-231 (4175) LM2 cells, suggesting that it has the ability to suppress a potent angiogenic and cell survival factor. In addition, migration of MDA-MB-231 (4175) LM2 cells was inhibited upon exposure to an antibody against the VEGF receptor, KDR, but not by exposure to a VEGF165 antibody. Collectively, these data suggest that the anti-metastatic properties of LU may, in part, be due to its ability to block VEGF production and KDR-mediated activity, thereby inhibiting tumor cell migration. These studies suggest that LU deserves further investigation as a potential treatment option for women with TNBC.
Keywords: luteolin, triple-negative breast cancer, metastasis, cell migration, apoptosis, VEGF
Introduction
Triple-negative breast cancers (TNBCs), which comprise 15–20% of all tumors of the breast, are so named because they fail to express estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor (HER2/neu).1,2 Compared with hormone-responsive breast cancers that are stimulated by the protein–ligand interactions of ER, PR, and/or HER2, the TNBC phenotype is associated with a significantly poorer clinical outcome.1,3,4 Hormone-responsive tumors are commonly treated with adjuvant hormone therapies, e.g., tamoxifen, that act as antagonists to disrupt normal hormone/receptor binding.5 In comparison, women with TNBC by definition do not respond to endocrine therapies and without a reliable prognostic marker rely on toxic chemotherapeutic drug combinations.6 TNBC represents a cancer phenotype that is difficult to treat, and cancers of this type that escape adjuvant treatment and surgery generally emerge as particularly aggressive tumors. Perou et al7 reported that women with basal-like (specifically, triple-negative) breast cancers have an increased risk of tumor recurrence compared with women with other cancer subtypes. TNBC frequently metastasizes to the viscera (i.e., lung, brain, and liver) and, to a lesser extent, bone8–11; in addition, the rate of cancer recurrence 3–5 years post-therapy is greater in women with TNBC than in their non-triple-negative counterparts.10,11 For these reasons, new therapeutic options for TNBC that are less toxic and more effective are needed.
The increased virulence of basal-like breast cancers, which represent ~80% of all triple-negative tumors,12 may be attributed to increased vascular endothelial growth factor (VEGF) secretion. VEGF is a major angiogenic factor and promotes tumor growth and metastasis,13–16 resulting in higher morbidity.17–20 For this reason, VEGF-stimulated migratory and proliferative pathways provide capable adjunctive chemotherapeutic options. Previous reports have established that VEGF, specifically VEGF165, increases migration and invasion potential of T47-D human breast cancer cells.21,22 More recently, our laboratory has shown that VEGF induces mitogenic effects and also acts as a survival factor in TNBC MDA-MB-231 cells mediated through the interaction of VEGF165 and its receptor KDR.23 These reports suggest that indirectly blocking VEGF-induced migration and proliferation may decrease the metastatic potential of TNBC cells and potentially also destroy established metastatic lesions.
In this report, we describe studies aimed at identifying naturally occurring compounds with low toxicity that might be used chemotherapeutically to both treat and prevent the metastasis of TNBC cells. We previously reported on the ability of luteolin (LU; 2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4-chromenone), a non-toxic naturally occurring plant flavonoid, to block progestin-driven VEGF expression and to suppress in vivo tumor progression in two different progestin-associated breast cancer animal models.24,25 Because few clinically accepted standardized chemotherapeutic options to combat metastatic TNBC are available, we tested LU for its ability to disrupt the growth and migration of TNBC cells, as well as to prevent their metastasis. Our findings show that LU effectively suppresses the viability of TNBC cells and blocks their metastasis to the lungs.
Materials and methods
Reagents
LU (2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-4H-1-benzopyran-4-one) was purchased from Tocris (Cat # 2874; Minneapolis, MN, USA) and dissolved in sterile-filtered dimethyl sulfoxide (DMSO; Cat # D2650; Sigma-Aldrich Co., St. Louis, MO, USA). Solutions of LU were prepared weekly, aliquoted for daily use, and stored at −20°C until use. Final DMSO concentration was 0.1% in LU- and vehicle-treated cells. Pierce bicinchoninic acid (BCA) protein reagents were obtained from Fisher Scientific (Pittsburgh, PA, USA). hVEGF165 (AF293NA) and human VEGF receptor (hKDR; AF357) antibodies were purchased from R&D Systems (Minneapolis, MN, USA).
Cell lines and culture
The MDA-MB-231 (4175) LM2 breast cancer cell line is a metastatic subpopulation isolated from TNBC MDA-MB-231 cells8 and maintained in DMEM supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich Co.) at 37°C/5% CO2. TNBC MDA-MB-435 cells were obtained from ATCC (Manassas, VA, USA) and maintained at 37°C in phenol red-free DMEM/F12 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS.26 Cells were harvested with 0.05% trypsin–ethylenediaminetetraacetic acid (EDTA) (Thermo Fisher Scientific). For all in vitro experiments, cells were allowed to reach 50–60% confluence, washed in phosphate-buffered saline (PBS), and maintained in DMEM/F12 or DMEM supplemented with 10% FBS for 24 hours prior to treatment. Incubations were carried out in the presence of LU at the various concentrations described later in a medium containing 5% FBS. Control represents cells treated with vehicle alone.
Cell viability assay
Viable cells were quantitated using the previously described sulforhodamine B (SRB) assay,27 a cell protein dye-binding assay. In brief, 1–1.5 × 104 cells in 100 µL 5% FBS DMEM/F12 medium or DMEM were seeded on a 96-well plate and incubated at 37°C overnight in 5% CO2. Cells were subsequently treated for 24 or 48 hours with the indicated concentrations of LU. Treatments were performed in sextuplet. Each experiment was repeated at least twice.
Cell migration assay
Viable MDA-MB-435 or MDA-MB-231 (4175) LM2 cells were counted and plated (4 × 104 cells/well) on an Oris Pro Cell Migration Assay 96-well tissue culture-treated plate (Cat # PROCMA1; Platypus Technologies, Fitchburg, WI, USA) containing a bead of biocompatible gel. The plates were incubated at 37°C for 1 hour to allow the gel to dissolve, leaving a circular zone of detection. Two hours later, once the cells were properly adhered to the plates, they were treated with LU and/or hVEGF165 or hKDR antibodies (or DMSO; vehicle) for 48 hours. Images were taken at 0-, 24-, and 48-hour intervals on an EVOS XL Cell Imaging System (Thermo Fisher Scientific,) utilizing a 4× objective. Migration was analyzed by determining the difference in the area of the detection zone between pre-migration (0 hour) and subsequent time points (closure), using the lasso and measurement tools in Adobe Photoshop CS 5.5 (San Jose, CA, USA) on an inductive pen-enabled Wacom touch screen. Inter-well coefficients of variability given by the manufacturer for the detection zone created by the biocompatible gel were ≤12%.
Apoptosis assay
LU-induced apoptosis of MDA-MB-231 (4175) LM2 cells was measured by fluorescence-activated cell sorting (FACS) analysis using the Annexin V–fluorescein isothiocyanate (FITC) and propidium iodide (PI) binding assay (Cat # K101-100; BioVision, San Francisco, CA, USA). Cells were grown to 50–60% confluence, washed twice, and treated with varying concentrations of LU in 5% FBS/DMEM for an additional 24 hours. Final concentration of DMSO was 0.1% in the medium with 5% FBS. Cells were harvested using Accutase and stained for FACS analysis as per the manufacturer’s protocol.
VEGF enzyme-linked immunosorbent assay (ELISA)
The Quantikine human VEGF ELISA kit (Cat # DVE00) was purchased from R&D Systems. MDA-MB-231 (4175) LM2 cells were grown to 50–60% confluence, washed twice, treated with vehicle or LU at the indicated concentrations, and incubated for 16 hours at 37°C. Supernatant from each sample was collected, and VEGF concentrations were measured by ELISA according to the manufacturer’s protocol. Experiments were performed in quadruplicate, and each sample was analyzed in duplicate on a microplate reader. Inter- and intra-assay coefficients of variance given by the manufacturer for the cell culture supernatant assay were 5–8.5% and 3.5–6.5%, respectively.
BCA protein assay
MDA-MB-231 (4175) LM2 cells were washed twice with PBS, then harvested, pelleted, and resuspended in 300 µL lysis buffer (50 mM Tris/HCl, pH 8, 150 mM NaCl, and 1% Nonidet P-40). Protein concentration was determined using a microplate reader to measure absorbance at 562 nm with a bovine serum albumin standard (Cat # 23209; Thermo Fisher Scientific). Experiments were performed in triplicate, and samples were analyzed in duplicate.
Reverse transcription-polymerase chain reaction (RT-PCR)
MDA-MB-231 (4175) LM2 cells were grown to 60% confluence and treated with 10, 25, or 50 µM LU for 6 hours, after which total messenger RNA (mRNA) was collected using the phenol-based extraction method (RNAzol RT; Molecular Research Center, Cincinnati, OH, USA), as per the manufacturer’s recommendations. Total mRNA was converted to complementary DNA (cDNA) and amplified using the Invitrogen SuperScript III One-Step RT-PCR System (Cat # 12574-026) with the following primers:
VEGF
F 5′ - CTG TCT TGG GTG CAT TGG
R 5′ - CAC CGC CTC GGC TTG TCA CAT
GAPDH
F 5′ - ATG AGA AGT ATG ACA GCC
R 5′- TGA GTC CTT CCA CGA TAC C
PCR products were run on 1.5% agarose gels for 1.5 hours at 100 V and quantified using a Bio-Rad Laboratories imager and software (Hercules, CA, USA). Relative VEGF band intensity was determined by normalization to GAPDH signals.
TNBC lung metastasis xenograft model
All animal studies were approved by the Animal Care and Use Committee at the University of Missouri, Columbia, MO, USA, and the study adhered to the guidelines of the US Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training. Female athymic nu/nu nude mice, 6 weeks old (18–22 g), were purchased from Harlan Sprague Dawley, Inc (Indianapolis, IN, USA). Mice were housed in a laminar airflow cabinet under specific pathogen-free conditions. All facilities were approved by the American Association for Accreditation of Laboratory Animal Care in accordance with current US federal regulations and standards. TNBC cells were harvested by trypsinization and washed twice with DMEM/F12 medium or DMEM. Cell pellets were then suspended in medium and injected intravenously (iv) into the tail vein. MDA-MB-435 and MDA-MB-231 (4175) LM2 cell lines were injected at 2.2 × 106 and 2.0 × 105 cells per 150 µL medium without FBS, respectively (Figure 1). Intraperitoneal (ip) injections of LU began on the fifth day post-inoculation and continued every other day until the end of the study, though the MDA-MB-231 (4175) LM2-inoculated mice received a daily loading dose of LU for the first week. Mice inoculated with MDA-MB-435 cells were given ip injections of 10 or 20 mg/kg LU; those inoculated with MDA-MB-231 (4175) LM2 cells received a higher LU dose of 40 mg/kg due to the highly aggressive nature of the MDA-MB-231 (4175) LM2 TNBC cell line. Control animals were injected with the same volume of vehicle (70% DMSO/30% PBS). Animals were weighed every 2–4 days throughout the study. Lungs were harvested following sacrifice of animals on Days 51 and 42 for mice inoculated with MDA-MB-435 and MDA-MB-231 (4175) LM2 cells, respectively. Lungs were collected and fixed in Bouin’s Fixative Solution (Cat # 1120-32; Ricca Chemical Company, Arlington, TX, USA), as per the manufacturer’s recommendations. Superficial lung colonies were counted using a stereoscopic dissection microscope, and the results from two independent researchers were averaged and then statistically analyzed using SigmaPlot software, version 12.5 (San Jose, CA, USA).
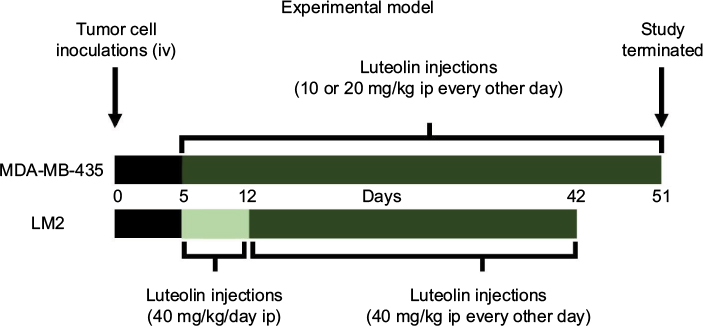 | Figure 1 LU treatment protocol and experimental design. Notes: Female nude mice were inoculated (iv) via tail vein with MDA-MB-435 or MDA-MB-231 (4175) LM2 (LM2) TNBC cells on Day 0. LU (10, 20, or 40 mg/kg), or vehicle alone, was injected (ip) every 48 hours until termination of study (Day 51 or 42). Please note that animals administered LM2 cells received initial loading dose of LU in the form of daily injections for the first week and then LU treatment was continued every 48 hours. At the termination of the study, animals were sacrificed and the lungs excised for analysis. Abbreviations: ip, intraperitoneally; iv, intravenously; LU, luteolin; TNBC, triple-negative breast cancer. |
Statistical analysis
Statistical significance was tested using t-test or one-way analysis of variance (ANOVA) followed by a Newman–Keuls multiple comparison test to determine the difference in mean between groups. If normality failed, the data were tested using a nonparametric one-way ANOVA on ranks (Kruskal–Wallis), followed by the Newman–Keuls nonparametric multiple comparison test or Dunn’s method in the presence of an uneven n. The Student’s t-test was used to report the difference between two group mean; when normality failed, the Mann–Whitney rank sum test was used. Animal weight comparisons were analyzed using a two-way repeated measures ANOVA. Data were reported as mean ± standard error of the mean (SEM). For all comparisons, P ≤ 0.05 was regarded as statistically significant, and analyses were performed using SigmaPlot 12.5 software.
Results
LU inhibits metastasis of human TNBC cells in mouse models
To determine the effectiveness of LU as an anti-metastatic compound that might be used to combat breast cancer, we utilized a xenograft metastasis model that mimics secondary-site colonization (Figure 1). Mice were inoculated with MDA-MB-435 cells. A dose of 20 mg/kg LU significantly reduced the number of MDA-MB-435-derived lung colonies to 5.3 ± 0.5, compared with 14.1 ± 1.6 superficial lung colonies formed in vehicle-treated control animals. The lower dose of 10 mg/kg LU reduced the mean number of metastatic colonies (8.4 ± 0.9), though this did not reach significance (Figure 2A). No significant difference in animal weights was observed between vehicle-treated control animals and animals receiving LU throughout the study (Figure 2B).
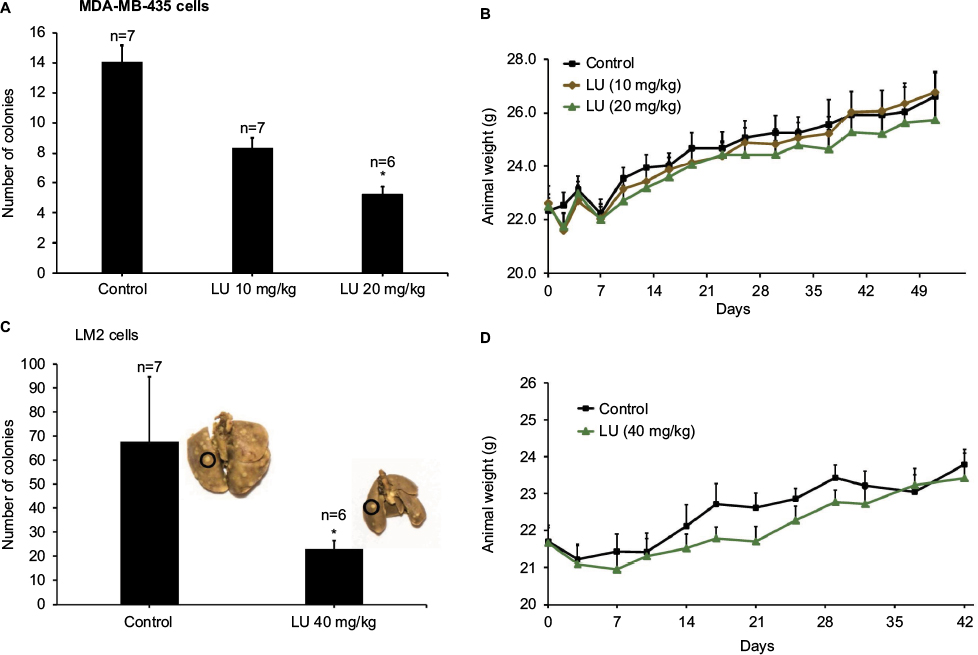 | Figure 2 LU suppresses metastasis of TNBC cells to the lungs. Notes: (A) Female nude mice were inoculated with MDA-MB-435 cells (2.2 × 106) via tail vein on Day 0. Treatment with LU (10 or 20 mg/kg ip) or vehicle alone began 5 days post-inoculation. LU was injected ip every other day until termination of the study. Bar graph represents mean number of lung colonies ± SEM. *Significantly different compared with control group (P < 0.05, ANOVA on ranks followed by Dunn’s method). (B) Animals were weighed every 3–4 days throughout the experiment shown in (A). No significant differences between treatment groups were observed throughout the study using the two-way repeated measures ANOVA. (C) Female nude mice were inoculated with MDA-MB-231 (4175) LM2 cells (2.0 × 105) via tail vein on Day 0 and subsequently treated with LU (40 mg/kg ip) or vehicle (control). Inserts are representative pictures from each group; colonies appear as off-white specks on the lungs (an example is circled). Bar graph represents mean number of lung colonies ± SEM. *Significantly different compared with controls (P < 0.05, Mann–Whitney rank sum test). (D) Animals were weighed every 3–4 days throughout the experiment shown in (C). No significant weight differences were observed between vehicle-treated animals and those administered LU using the two-way repeated measures ANOVA. Abbreviations: ANOVA, analysis of variance; ip, intraperitoneally; LU, luteolin; SEM, standard error of the mean; TNBC, triple-negative breast cancer. |
Since LU reduced metastasis in the MDA-MB-435 model, we sought to determine whether this effect was cell specific by inoculating mice with a particularly aggressive TNBC cell line (4175 LM2) that is an MDA-MB-231 variant with a molecular signature specific to lung metastasis.8 Based on the observations by Minn et al,28 we reduced the number of cells used for inoculation by 10-fold and increased the LU dosage to one approaching the maximum reported in the literature. Inoculation with MDA-MB-231 (4175) LM2 cells increased the mean number of lung colonies in control animals by approximately 5-fold compared with MDA-MB-435 cells (67.6 ± 27.1 colonies vs 14.1 ± 1.6 colonies, respectively), a finding that was highly significant (Mann–Whitney rank sum test, P < 0.001). Administration of LU (40 mg/kg) significantly reduced the number of lung colonies formed by MDA-MB-231 (4175) LM2 cells to 22.8 ± 3.6 (Figure 2C). As in the MDA-MB-435 study, LU had no significant effect on animal weights (Figure 2D).
LU inhibits in vitro TNBC cell migration
Using MDA-MB-435 and MDA-MB-231 (4175) LM2 cells, we examined the effects of LU on cell migration. Cells were grown to 50–60% confluence, harvested, and seeded into 96-well migration assay plates and then subjected to migration assays in the presence or absence of LU. When MDA-435 cells were exposed to very low doses of LU for either 24 or 48 hours, their migration capacity (represented by % closure values) was reduced (Figure 3A). After 24 hours, 5 µM LU significantly suppressed migration compared with controls (40.5 ± 4.7 vs 13.2 ± 6.8% closure). Treatment of cells for 48 hours with a higher (10 µM) concentration of LU resulted in a significant reduction in migration (54.2 ± 5.8 vs 15.9 ± 5.5% closure). At both time points, migration was further suppressed in a dose-dependent manner; a concentration of 50 µM LU completely abolished the migration of MDA-MB-435 cells.
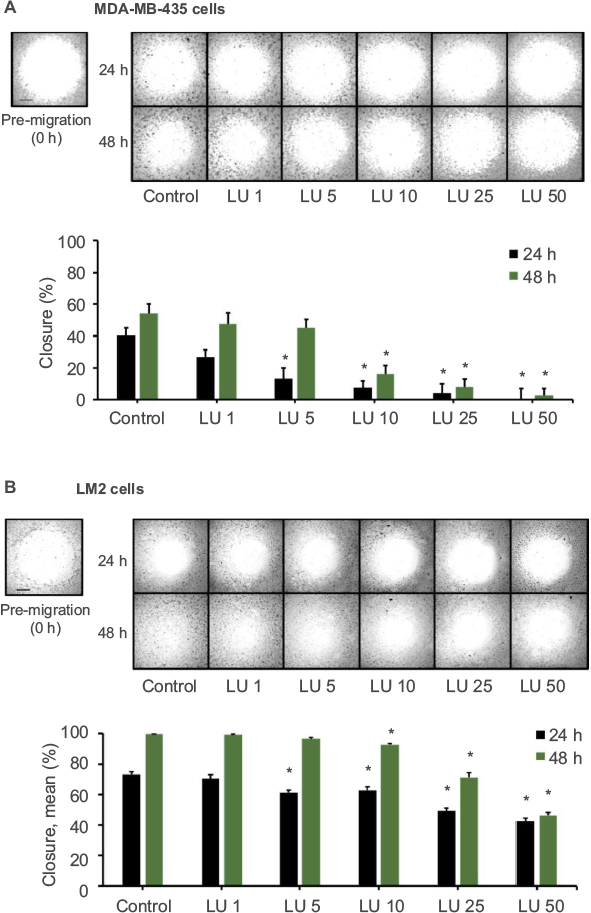 | Figure 3 LU inhibits migration of TNBC cells in vitro. Notes: MDA-MB-435 cells (A) or MDA-MB-231 (4175) LM2 cells (B) (4 × 104 cells/well) were plated in a 96-well migration assay plate and subjected to cell migration assays in the presence of vehicle control or varying concentrations of LU (1–50 µM) for 48 hours. Pictures were taken upon treatment (0 hour; pre-migration) and after 24 and 48 hours. Representative images (top) demonstrate LU-mediated inhibition of migration over 2 days. Bar graph (bottom) quantitates inhibition of cell migration; data represent mean ± SEM (n = 8). (A) *Significantly different compared with controls (P < 0.05, ANOVA). (B) *Significantly different compared with controls (24 hours, P < 0.05; ANOVA; 48 hours, P < 0.05; ANOVA on ranks). Scale bar, 500 µm. Abbreviations: ANOVA, analysis of variance; h, hours; LU, luteolin; SEM, standard error of the mean; TNBC, triple-negative breast cancer. |
Low doses of LU also significantly reduced the migration of MDA-MB-231 (4175) LM2 cells following 24 hours of exposure (Figure 3B). After 48 hours of exposure to 5 µM LU, MDA-MB-231 (4175) LM2 cells overcame the inhibitory effects of the flavonoid and exhibited similar migration behavior as MDA-MB-435 cells. However, as concentrations of LU increased from 10 to 50 µM, there was a dose-dependent reduction in MDA-MB-231 (4175) LM2 cell migration. Interestingly, we observed no significant difference in % closures following exposure of cells to 50 µM LU for 24 and 48 hours (42.6 ± 2.25 vs 46.5 ± 1.8% closure; Student’s t-test, P = 0.189). This suggests that at high dosage (50 µM), the effects of LU on MDA-MB-231 (4175) LM2 migration are maximized after 24 hours.
LU reduces VEGF secretion in TNBC cells
It is well established that the potent angiogenic factor VEGF165 increases the migration and invasion potential of human breast cancer cells22 and that VEGF has mitogenic effects in MDA-MB-231 cells that are mediated through the interaction of VEGFA and its receptor, KDR.23 Given these previous findings and our data showing aggressive metastasis of MDA-MB-231 (4175) LM2 cells to lungs in xenograft models, we sought to determine whether LU suppresses VEGF production in these cells. MDA-MB-231 (4175) LM2 cells were treated with 10, 50, or 100 µM LU or DMSO for 16 hours, after which VEGF levels were measured in the supernatant using a Quantikine human VEGF ELISA kit, which predominantly binds VEGF165. LU concentrations of 10 and 50 µM significantly reduced levels of secreted VEGF (Figure 4A). VEGF secretion was further reduced when cells were exposed to 100 µM LU.
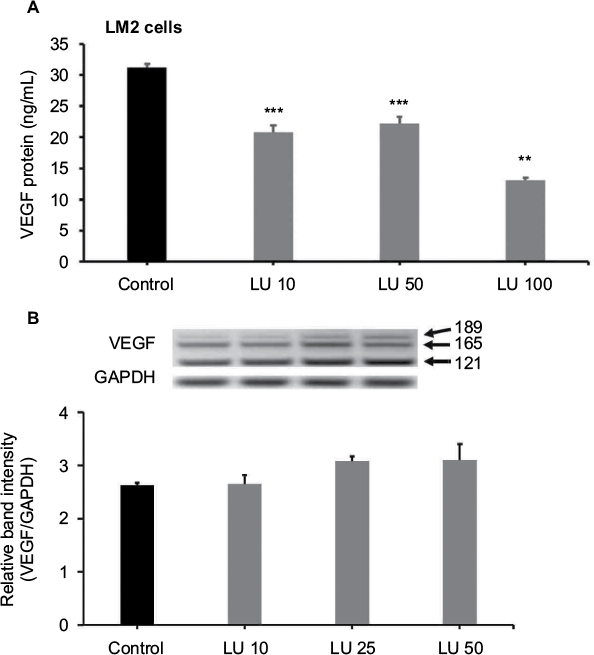 | Figure 4 LU inhibits VEGF secretion but not VEGF mRNA expression in MDA-MB-231 (4175) LM2 cells. Notes: (A) Cells were treated at 37°C for 16 hours with 10, 50, or 100 μM LU or vehicle control. Levels of VEGF in supernatants were measured by ELISA. Data represent mean ± SEM (n = 4). ***Significantly different compared with controls (P < 0.001, ANOVA). **Significantly different compared with controls and either 10 or 50 μM LU (P < 0.001, ANOVA). (B) Cells were treated for 6 hours with 10, 25, or 50 μM LU or vehicle control, after which total mRNA was isolated and RT-PCR for VEGF isoforms performed. Upper panel shows PCR-amplified VEGF bands for VEGF189, VEGF165, and VEGF121, together with a GAPDH band for normalization. Bar graph (bottom) represents mean band intensities (VEGF/GAPDH) ± SEM (n = 3). Results represent values from three independent experiments. No statistically significant differences were observed compared with controls (P = 0.149, ANOVA). Abbreviations: ANOVA, analysis of variance; ELISA, enzyme-linked immunosorbent assay; LU, luteolin; mRNA, messenger RNA; RT-PCR, reverse transcription-polymerase chain reaction; SEM, standard error of the mean; VEGF, vascular endothelial growth factor. |
LU does not reduce VEGF mRNA production by MDA-MB-231 (4175) LM2 cells
In order to gain insight into the mechanism through which LU suppresses VEGF secretion by MDA-MB-231 (4175) LM2 cells, we measured VEGF mRNA levels. MDA-MB-231 (4175) LM2 cells were treated with 10, 25, or 50 µM LU for 6 hours, after which total mRNA was collected, and VEGF mRNA levels were measured by RT-PCR. LU had no effect on total VEGF mRNA levels (Figure 4B), suggesting that it suppresses VEGF secretion post-transcriptionally.
Blocking VEGF receptor-2 but not VEGF165 inhibits migration of MDA-MB-231 (4175) LM2 cells in the presence or absence of LU
VEGF increases the migration and invasion potential of human breast cancer cells.22 We found that 10 µM LU suppresses both VEGF secretion (Figure 4A) and cell migration (Figure 3B) in MDA-MB-231 (4175) LM2 cells. Based on this finding, we used an antibody to inhibit human VEGF165 and ascertained the effect of such treatment on the migration of MDA-MB-231 (4175) LM2 cells. Migration assays of cells treated with 10 µM LU ± VEGF165 antibody (0.5 µg/mL) demonstrated that migration was not inhibited by the VEGF165 antibody (data not shown). VEGF165 actions are potentiated through the VEGF receptor-2 (KDR), which is stimulated by VEGF isoforms.23 Therefore, we incubated MDA-MB-231 (4175) LM2 cells with a human KDR antibody to determine how the receptor is involved in their migration. After 24 and 48 hours, cell migration was inhibited by the KDR antibody and by 10 µM LU (Figure 5). Treating cells with a combination of LU and KDR antibody did not inhibit migration in an additive or a synergistic manner. Taken together, these results suggest that LU may act on KDR or its downstream effectors to inhibit MDA-MB-231 (4175) LM2 cell migration.
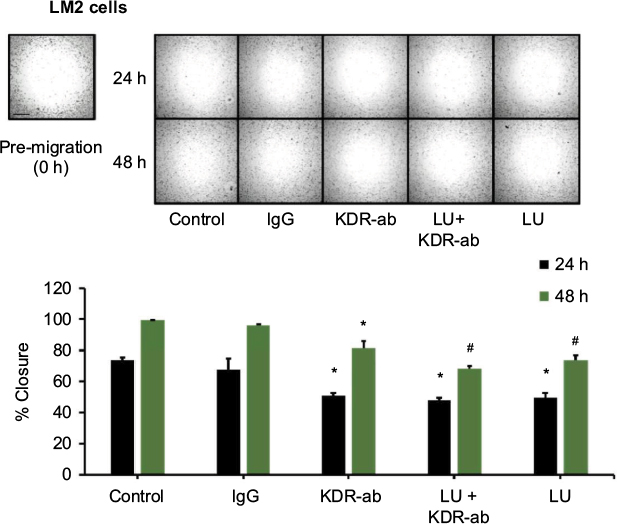 | Figure 5 Blocking VEGF receptor-2 (KDR) inhibits MDA-MB-231 (4175) LM2 cell migration. Notes: Cells (4 × 104/well) were plated in a 96-well migration assay plate and subjected to cell migration assays in the presence of vehicle control, normal IgG, KDR antibody (0.5 µg/mL), 10 µM LU, or 10 µM LU + KDR antibody (0.5 µg/mL). Pictures were taken upon treatment (0 hour; pre-migration) and after 24 and 48 hours. Representative images (top) demonstrate inhibition of migration by KDR antibody, LU, or LU + KDR antibody over 2 days. Bar graph (bottom) quantitates inhibition of cell migration; data represent mean ± SEM (n = 6). *Significantly different compared with respective time controls (P < 0.05, ANOVA). #Significantly different compared to KDR-ab (48 hours, P < 0.05, ANOVA on ranks). Scale bar, 500 µm. KDR, VEGFA receptor. Abbreviations: ANOVA, analysis of variance; IgG, immunoglobulin G; h, hours; LU, luteolin; SEM, standard error of the mean. |
LU reduces viability of TNBC cells
We assessed the effect on cell viability of exposing MDA-MB-435 and MDA-MB-231 (4175) LM2 cells to varying concentrations of LU (0–100 μM) for 24 or 48 hours. The viability of MDA-MB-435 cells was markedly reduced by LU in both a dose- and time-dependent manner (Figure 6A). Following exposure of MDA-MB-435 cells for 24 hours, the IC50 value for LU was between 50 and 75 μM, with minimal or no effect observed at 10 μM. After 48 hours of exposure, the IC50 value fell to ~25 μM. However, MDA-MB-231 (4175) LM2 cells were less sensitive to LU; following 24 hours of exposure, the flavonoid had minimal effect until concentrations exceeded 10 μM (Figure 6B). In addition, after 48 hours of exposure to LU, the IC50 was ~50 μM. Our findings demonstrate that concentrations of LU >10 μM significantly reduce the viability of both MDA-MB-435 and MDA-MB-231 (4175) LM2 cells in a dose- and time-dependent manner.
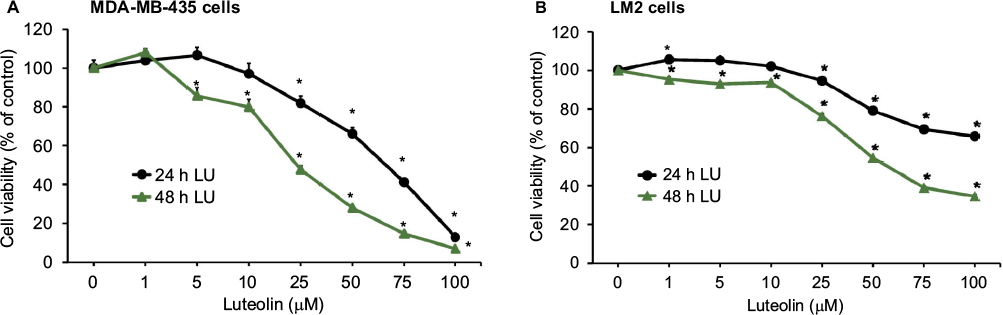 | Figure 6 LU inhibits the viability of TNBC cells in vitro. Notes: MDA-MB-435 cells (4 × 103 cells/well) (A) or MDA-MB-231 (4175) LM2 cells (3 × 103 cells/well) (B) were seeded overnight in a 96-well plate, washed, and then treated with the indicated concentration of LU for either 24 or 48 hours. Cell viability was determined by SRB assay. Data represent mean ± SEM (n = 6). *Significantly different compared with controls (0 µM LU) (P < 0.05, ANOVA). Abbreviations: ANOVA, analysis of variance; h, hours; LU, luteolin; SEM, standard error of the mean; SRB, sulforhodamine B; TNBC, triple-negative breast cancer. |
LU induces apoptosis in MDA-MB-231 (4175) LM2 cells
Finally, using FACS analysis, we determined whether LU induces apoptosis in MDA-MB-231 (4175) LM2 cells. Cells were treated with LU or vehicle for 48 hours, then collected and subjected to FACS analysis. We found that concentrations of LU as low as 10 µM significantly induced apoptosis and that the effect was dose-dependent (Figure 7). These studies show that LU initiates the process of programmed cell death in MDA-MB-231 (4175) LM2 cells even at concentrations too low to influence cell viability.
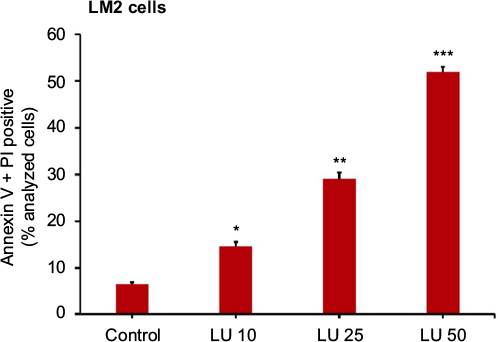 | Figure 7 LU induces apoptosis in MDA-MB-231 (4175) LM2 cells in vitro. Notes: Cells were treated with 10, 25, or 50 μM LU or vehicle control for 24 hours, then stained with AV and PI, and analyzed by FACS. Bars represent mean (AV+ + PI+) ± SEM (n = 4). *Significantly different compared with controls; **significantly different compared with controls and 10 μM LU; and ***significantly different compared with controls, 10 μM LU, and 25 μM LU (P < 0.001, ANOVA). Abbreviations: ANOVA, analysis of variance; AV, Annexin V; FACS, fluorescence-activated cell sorting; LU, luteolin; PI, propidium iodide; SEM, standard error of the mean. |
Discussion
Breast cancer is a deadly disease that afflicts millions of women worldwide. Almost 20% of the detected tumors are of the TNBC type. Because no recognized targeted chemotherapeutic approaches exist for TNBC, patients diagnosed with this type of tumor have a poor prognosis.1–3,29 Many existing treatments for TNBC are highly toxic, and resistant tumors often arise and metastasize despite the use of aggressive chemotherapy regimens.6 Given the urgent need for safer and more effective treatments for TNBC, we embarked on a study of LU, a naturally occurring compound, that might be used to block TNBC tumor growth and metastasis. Such compounds have the benefit of being either nontoxic or of low toxicity and therefore confer significant potential advantages over currently used drugs.
This study determined the effectiveness of LU, a naturally occurring plant flavonoid, to block growth and metastasis of TNBC cells, as well as angiogenesis-associated processes in those cells. Angiogenesis is a vital component for both growth and metastasis of tumor cells, since new blood vessels facilitate the delivery of nutrients to tumors as well as the dissemination of cancer cells to distant sites. We previously reported on the ability of LU to block progestin-driven tumor formation and growth in two different progestin-associated breast cancer animal models.24 In this report, we used a well-established xenograft model of lung metastasis and assessed the ability of LU to reduce the metastatic capacity of TNBC cells using two different TNBC cell lines, MDA-MB-43526 and MDA-MB-231 (4175) LM2.8 Both of these TNBC cell types are known to metastasize to lung, though the latter cell line is particularly aggressive8 and gives rise to significantly more tumors of the lung per animal, which is a testament to its virulence.
In the present studies, LU significantly inhibited metastasis of both MDA-MB-435 and MDA-231 (4175) LM2 TNBC cells with no observed toxicity. To our knowledge, it has never been reported that LU attenuates the ability of TNBC cells to metastasize to the lungs, though it has been suggested in other models that inhibition of metastasis by LU may be potentiated through suppression of cellular migration and neovascularization,30 which are indispensable steps in tumor metastasis.16 Our findings that relatively low concentrations of LU significantly reduced TNBC cell migration suggest that, at low concentrations, LU has the capacity to lessen colonization of metastatic cells at the site of metastasis. Higher concentrations of LU abolished migration in a manner that was dependent upon the particular cell line. The lesser response seen with the more aggressive MDA-MB-231 (4175) LM2 cells suggests that the ability of LU to interfere with migration is most likely an indirect effect, perhaps following LU-mediated suppression of VEGF secretion from tumor cells. Locally produced VEGF is known to act via VEGF receptors to promote tumor cell survival and migration.31–33
VEGF plays an important role in tumor cell migration, survival, and proliferation,14,19 and we found here that LU inhibited VEGF production, but not VEGF mRNA levels, in MDA-MB-231 (4175) LM2 cells (which were observed to be particularly aggressive in their colonization of lung tissue following injection into nude mice). In addition, it has been reported that VEGF165 promotes breast cancer cell migration.22 Consequently, it is reasonable to speculate that suppression of VEGF bioavailability by LU is a plausible mechanism by which LU may modulate cell migration. We found that migration of MDA-MB-231 (4175) LM2 cells was not affected by neutralization of soluble VEGF with an antibody against VEGF165, and no significant difference in migration was observed between cells exposed to both LU and VEGF165 antibody and those exposed to LU alone (data not shown). The anti-migratory effects of LU may be hampered by an inability to block other isoforms of VEGF, such as VEGF121 or VEGF189, or other angiogenic factors such as neuropilin, which continue to activate VEGF receptor-2 (KDR) in the absence of VEGF165. However, our VEGF antibody studies suggest that it is likely that LU acts downstream of the VEGF receptor-2 (KDR) or on the receptor itself. Evidence that LU suppresses VEGF-induced phosphorylation of VEGF receptor-2, resulting in inhibition of prostate tumor growth,34 supports this concept. When we used a KDR-specific antibody to neutralize VEGFR-2, migration of MDA-MB-231 (4175) LM2 cells was significantly inhibited in both the presence and absence of LU. Although our results do not directly demonstrate that LU is inhibiting KDR, the effects of LU on TNBC cell migration suggest at least a partial link between KDR signal propagation and cell migration capacity. Although LU has been demonstrated to have the capacity to inhibit functions of KDR,34 the effect of LU on KDR in breast cancer cells needs to be studied in more detail.
Previous reports indicate that VEGF is potently mitogenic for breast cancer cells and that its actions are mediated through the interaction of VEGF165 and its receptor.23 Furthermore, it has been reported that 10 µM LU is able to block VEGF-induced angiogenesis and proliferation of tumor cells through phosphatidylinositol 3′-kinase (PI3K) inhibition.30 We found that relatively modest concentrations of LU (between 10 and 25 µM) effectively inhibited viability of TNBC cells in a dose-, time-, and cell line-dependent manner. At higher LU concentrations (>10 µM), reduced viability of MDA-MB-231 (4175) LM2 cells appeared to be associated with increased apoptosis. However, we found that a concentration of LU (10 µM) that did not affect MDA-MB-231 (4175) LM2 cell viability after 24 hours was able to induce apoptosis in these cells after 24 hours, the latter of which is in accordance with previous reports in TNBC cells.34,35 The discrepancies between these two findings at low LU concentrations are likely due to the possibility that early effects of LU are not manifested in the SRB cell viability assays but can be observed in the more sensitive FACS analysis. The precise role of VEGF165 in these processes and to what extent its attenuation by LU has on mediating TNBC cell migration, proliferation, and apoptosis remain unclear.
In addition to the findings reported here, little direct evidence shows that LU inhibits the metastasis of TNBC cells. However, in vivo reports have demonstrated potent inhibition of both prostate and colorectal cancer cell metastasis by LU. Zhou et al36 found that LU (5 mg/kg administered 3 times per week ip) upregulated E-cadherin and thereby prevented human PC-3 prostate cancer cells from spontaneously metastasizing to lungs. LU was also shown to suppress Raf and PI3K, thereby diminishing mouse CT-26 colorectal cancer cell metastasis.37 This latter finding is in agreement with Bagli et al30 and Pratheeshkumar et al,34 who reported that low concentrations of LU (10–20 µM) acted as both a potent PI3K inhibitor and an inhibitor of metalloproteinase MMP9. Additional evidence implicates downregulation of PI3K phosphorylation and MMP9 as being responsible for the anti-invasive capacity of LU, perhaps explaining its ability to reduce the invasive potential of various cancer cell lines in vitro.38–42 Because the VEGF-induced PI3K pathway mediates cell survival, migration, and proliferation, we propose that our findings that LU inhibits TNBC cell migration support the notion that it acts via multiple mechanisms likely potentiated through blockade of PI3K phosphorylation and/or MMP-mediated VEGF signaling. While its precise mechanisms of action remain to be determined, we contend that these studies support the further investigation of LU as an anti-metastatic agent that could be used to combat early-stage, as well as late-stage, TNBC, including its metastasis.
Acknowledgments
We would like to thank Dr. Joan Massague from Memorial Sloan-Kettering Cancer Center for the provision of MDA-MB-231 LM2 cell line. The studies reported were supported by a peer-reviewed COR grant from the College of Veterinary Medicine, University of Missouri-Columbia, and by generous support from donors of Ellis Fischel Cancer Center, University of Missouri-Columbia. We would also like to thank Dr. Carolyn Henry for her invaluable support during the completion of this project. SMH is the Zalk Missouri Professor of Tumor Angiogenesis.
Disclosure
The authors report no conflicts of interest in this work.
References
Arvold ND, Taghian AG, Niemierko A, et al. Age, breast cancer subtype approximation, and local recurrence after breast-conserving therapy. J Clin Oncol. 2011;29(29):3885–3891. | ||
Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. | ||
Lund MJ, Butler EN, Hair BY, et al. Age/race differences in HER2 testing and in incidence rates for breast cancer triple subtypes: a population-based study and first report. Cancer. 2010;116(11):2549–2559. | ||
Metzger-Filho O, Sun Z, Viale G, et al. Patterns of recurrence and outcome according to breast cancer subtypes in lymph node–negative disease: results from International Breast Cancer Study Group Trials VIII and IX. J Clin Oncol. 2013;31(25):3083–3090. | ||
Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2(2):101–112. | ||
Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011;16(Suppl 1):1–11. | ||
Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | ||
Minn AJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. | ||
Rodríguez-Pinilla SM, Sarrió D, Honrado E, et al. Prognostic significance of basal-like phenotype and fascin expression in node-negative invasive breast carcinomas. Clin Cancer Res. 2006;12(5):1533–1539. | ||
Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–1281. | ||
Dent R, Hanna WM, Trudeau M, Rawlinson E, Sun P, Narod SA. Pattern of metastatic spread in triple-negative breast cancer. Breast Cancer Res Treat. 2009;115(2):423–428. | ||
Ricardo S, Vieira AF, Gerhard R, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011;64(11):937–946. | ||
Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235(4787):442–447. | ||
Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
O’Reilly MS, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79(2):315–328. | ||
Weidner N. Intratumor microvessel density as a prognostic factor in cancer. Am J Pathol. 1995;147(1):9. | ||
Toi M, Hoshina S, Takayanagi T, Tominaga T. Association of vascular endothelial growth factor expression with tumor angiogenesis and with early relapse in primary breast cancer. Cancer Sci. 1994;85(10):1045–1049. | ||
Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–676. | ||
Gasparini G. Prognostic value of vascular endothelial growth factor in breast cancer. Oncologist. 2000;5(Suppl 1):37–44. | ||
Price JE. Metastasis from human breast cancer cell lines. Breast Cancer Res Treat. 1996;39(1):93–102. | ||
Price DJ, Miralem T, Jiang S, Steinberg R, Avraham H. Role of vascular endothelial growth factor in the stimulation of cellular invasion and signaling of breast cancer cells. Cell Growth Differ. 2001;12(3):129–135. | ||
Liang Y, Hyder SM. Proliferation of endothelial and tumor epithelial cells by progestin-induced vascular endothelial growth factor from human breast cancer cells: paracrine and autocrine effects. Endocrinology. 2005;146(8):3632–3641. | ||
Cook MT, Mafuvadze B, Besch-Williford C, Ellersieck MR, Goyette S, Hyder SM. Luteolin suppresses development of medroxyprogesterone acetate-accelerated 7,12-dimethylbenz(a)anthracene-induced mammary tumors in Sprague-Dawley rats. Oncol Rep. 2016;35(2):825–832. | ||
Cook MT, Liang Y, Besch-Williford C, Goyette S, Mafuvadze B, Hyder SM. Luteolin inhibits progestin-dependent angiogenesis, stem cell-like characteristics, and growth of human breast cancer xenografts. Springerplus. 2015;4:444. | ||
McGee AM, Douglas DL, Liang Y, Hyder SM, Baines CP. The mitochondrial protein C1qbp promotes cell proliferation, migration and resistance to cell death. Cell Cycle. 2011;10(23):4119–4127. | ||
Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13):1107–1112. | ||
Chiu FL, Lin JK. Downregulation of androgen receptor expression by luteolin causes inhibition of cell proliferation and induction of apoptosis in human prostate cancer cells and xenografts. Prostate. 2008;68(1):61–71. | ||
Weigelt B, Peterse JL, Van’t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005;5(8):591–602. | ||
Bagli E, Stefaniotou M, Morbidelli L, et al. Luteolin inhibits vascular endothelial growth factor-induced angiogenesis; inhibition of endothelial cell survival and proliferation by targeting phosphatidylinositol 3′-kinase activity. Cancer Res. 2004;64(21):7936–7946. | ||
Dias S, Hattori K, Zhu Z, et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106(4):511. | ||
Dias S, Choy M, Alitalo K, Rafii S. Vascular endothelial growth factor (VEGF)–C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. 2002;99(6):2179–2184. | ||
Zhang W, Ran S, Sambade M, Huang X, Thorpe PE. A monoclonal antibody that blocks VEGF binding to VEGFR2 (KDR/Flk-1) inhibits vascular expression of Flk-1 and tumor growth in an orthotopic human breast cancer model. Angiogenesis. 2002;5(1–2):35–44. | ||
Pratheeshkumar P, Son Y-O, Budhraja A, et al. Luteolin inhibits human prostate tumor growth by suppressing vascular endothelial growth factor receptor 2-mediated angiogenesis. PLoS One. 2012;7(12):e52279. | ||
Lee E-J, Oh S-Y, Sung M-K. Luteolin exerts anti-tumor activity through the suppression of epidermal growth factor receptor-mediated pathway in MDA-MB-231 ER-negative breast cancer cells. Food Chem Toxicol. 2012;50(11):4136–4143. | ||
Zhou Q, Yan B, Hu X, Li XB, Zhang J, Fang J. Luteolin inhibits invasion of prostate cancer PC3 cells through E-cadherin. Mol Cancer Ther. 2009;8(6):1684–1691. | ||
Kim HY, Jung SK, Byun S, et al. Raf and PI3K are the molecular targets for the anti-metastatic effect of luteolin. Phytother Res. 2013;27(10):1481–1488. | ||
Lee W-J, Wu L-F, Chen W-K, Wang C-J, Tseng T-H. Inhibitory effect of luteolin on hepatocyte growth factor/scatter factor-induced HepG2 cell invasion involving both MAPK/ERKs and PI3K–Akt pathways. Chem Biol Interact. 2006;160(2):123–133. | ||
Cheng W-Y, Chiao M-T, Liang Y-J, Yang Y-C, Shen C-C, Yang C-Y. Luteolin inhibits migration of human glioblastoma U-87 MG and T98G cells through downregulation of Cdc42 expression and PI3K/AKT activity. Mol Biol Rep. 2013;40(9):5315–5326. | ||
Pandurangan A, Dharmalingam P, Sadagopan S, Ganapasam S. Luteolin inhibits matrix metalloproteinase 9 and 2 in azoxymethane-induced colon carcinogenesis. Hum Exp Toxicol. 2014;33(11):1176–1185. | ||
Park S-H, Kim J-H, Lee D-H, et al. Luteolin 8-C-β-fucopyranoside inhibits invasion and suppresses TPA-induced MMP-9 and IL-8 via ERK/AP-1 and ERK/NF-κB signaling in MCF-7 breast cancer cells. Biochimie. 2013;95(11):2082–2090. | ||
Lu J, Li G, He K, et al. Luteolin exerts a marked antitumor effect in cMet-overexpressing patient-derived tumor xenograft models of gastric cancer. J Transl Med. 2015;13:42. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.