Back to Journals » Drug Design, Development and Therapy » Volume 13
Luteolin Attenuates Atherosclerosis Via Modulating Signal Transducer And Activator Of Transcription 3-Mediated Inflammatory Response
Authors Ding X, Zheng L, Yang B ![]() , Wang X, Ying Y
, Wang X, Ying Y
Received 28 February 2019
Accepted for publication 26 September 2019
Published 18 November 2019 Volume 2019:13 Pages 3899—3911
DOI https://doi.org/10.2147/DDDT.S207185
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Tin Wui Wong
Xiaoji Ding,1,* Lulu Zheng,1,* Bo Yang,1,* Xiaodong Wang,2 Yin Ying1
1Department of Pharmacy, Tongde Hospital of Zhejiang Province, Hangzhou 310012, Zhejiang, People’s Republic of China; 2Department of Vascular Surgery, Tongde Hospital of Zhejiang Province, Hangzhou 310012, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yin Ying
Department of Pharmacy, Tongde Hospital of Zhejiang Province, 234 Gucui Road, Hangzhou 310012, Zhejiang, People’s Republic of China
Tel +86-571-89972240
Email [email protected]
Background: Inflammatory factors play a crucial role throughout the development and progression of atherosclerosis, which has been considered as a chronic vascular inflammatory disease. Luteolin, a natural flavonoid which exists in many natural medicinal materials, has anti-inflammatory, anti-fibrotic and other pharmacological effects. Recently, the protective effects of luteolin on the cardiovascular disease have been reported. However, there is a paucity of studies on anti-atherosclerosis. Therefore, the anti-atherosclerosis potential of luteolin remains to be elucidated.
Method: ApoE-/- mice were fed with a high-fat diet to induce atherosclerosis in an animal model, where they were treated with oral administration of luteolin for 12 weeks. Primary mouse peritoneal macrophages challenged with oxidized low-density lipoprotein (oxLDL) were used for in vitro mechanistic study. The effectiveness of luteolin in the ApoE-/- mouse model of atherosclerosis was estimated in the aortic sinus and enface, and the underlying mechanisms were explored by molecular modeling study and siRNA-induced gene silencing.
Results: Our results showed that luteolin remarkably attenuated atherosclerosis in high-fat diet-induced ApoE-/- mouse via alleviating inflammation. We further found that luteolin decreased oxLDL-induced inflammation by inhibiting signal transducer and activator of transcription 3 (STAT3) in vitro, respectively. Further molecular modeling analysis indicated that luteolin interacted with STAT3 primarily through hydrogen bond interaction.
Conclusion: Luteolin could be a promising candidate molecule for atherosclerosis, and STAT3 may be a potential therapeutic target that could prevent the development of atherosclerosis.
Keywords: atherosclerosis, luteolin, inflammation, transducer and activator of transcription 3
Introduction
Coronary heart disease, caused by coronary atherosclerosis, is the leading cause of mortality and morbidity worldwide.1,2 Atherosclerosis is considered as a systemic, lipid-driven, large and medium-sized arterial inflammatory disease that leads to the formation of multiple focal plaques.3–5 The development and progression of atherosclerotic plaque involves inflammatory cell recruitment, foam cell formation, apoptosis and necrosis, smooth muscle cell (SMC) proliferation and matrix synthesis, reactive oxygen species, and arterial remodeling.6,7 Among these changes, inflammation plays a leading role in the pathogenesis of atherosclerosis.8 During the inflammatory stage of atherosclerosis, low-density lipoprotein (LDL) that entered the arterial wall was oxidized by excessive ROS and scavenged by macrophages, forming lipid drops, which are characterized as foam cells.9 Oxidized low-density lipoprotein (oxLDL) is one of the most important pathogenic factors leading to atherosclerosis. Mounting evidence has shown that oxLDL also induces cells to release inflammatory factors, which promote the development and progress of atherosclerosis.10,11 However, the mechanism by which oxLDL induces inflammation and lesion progression has not been fully determined, and therapeutic drugs designed for the treatment of atherosclerosis are lacking.
Luteolin is a plant flavonoid extracted from natural herbs, fruits and vegetables, and has been reported to exert potent anti-inflammatory and anti-cancer effects.12–15 The anti-inflammatory and antioxidant activities of luteolin have been well documented.16 Recent studies have indicated that high levels of flavonols in the diet, especially luteolin, are closely related with a decrease in the serum inflammatory cytokine IL-6.16 In addition, H2O2-induced oxidative injury in ischemic cerebrovascular disease can be reversed by luteolin.17 Luteolin has shown much potential in the fight against cancer by several different mechanisms such as inhibition impacts of angiogenesis, inflammation and metastasis.12,13,18 However, the effects of luteolin on the cardiovascular disease have hardly been reported, let alone the effect on atherosclerosis.
In the present study, we clarified the therapeutic effects and molecular mechanism of luteolin on the development of atherosclerosis in ApoE-/- mice fed with high-fat diet (HFD). Our results demonstrated that luteolin could attenuate the development and progression of atherosclerosis in HFD-induced ApoE-/- mice, thereby alleviating inflammatory response and decreasing accumulation of macrophages and lipid droplet. Furthermore, the beneficial effects of luteolin are closely associated with its ability to inhibit phosphorylation of STAT3. Our study suggested that luteolin might be another clinically significant indicator for treating atherosclerosis.
Materials And Methods
Reagents And Cell Culture
Luteolin and oxLDL were obtained from Topscience Co., Ltd. (Shanghai, People’s Republic of China) and Yiyuan Biotechnology Co., Ltd., respectively (Guangzhou, People’s Republic of China). The luteolin was dissolved in dimethyl sulfoxide (DMSO) and 1% sodium carboxyl methylcellulose (CMC-Na) for in vitro experiments and in vivo experiments. Moma-2 (MCA519) was purchased from Bio-Rad (Hercules, CA, USA). p-STAT3 (#9145) and STAT3 (#9139) were purchased from Cell Signaling (Danvers, MA, USA). β-actin (sc-47778) was obtained from Santa Cruz Technology (Delaware Avenue, CA, USA).
Preparation Of Mouse Peritoneal Macrophages
Primary mouse peritoneal macrophages (MPMs) were obtained from C57BL/6 mice and cultured as previously described.19 C57BL/6 mice were intraperitoneally injected with 6% thioglycollate solution (0.3 g beef extract, 1 g tryptone, 0.5 g sodium chloride dissolved in 100 mL ddH2O, and filtrated through 0.22-μm filter membrane, 3 mL per mouse) and kept in a pathogen-free condition for 3 days before the MPMs were isolated. Total MPMs were harvested by washing the peritoneal cavity with PBS containing 30 mM of EDTA (8 mL per mouse), centrifuged, and suspended in RPMI-1640 medium (Gibco/BRL life Technologies, Eggenstein, Germany) with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 U/mL penicillin, and 100 mg/mL streptomycin. Nonadherent cells were removed by washing with medium 3 hrs after seeding. The experiment was conducted after the cells were firmly adhered to the culture plates.
siRNA-Induced Gene Silencing
Specific siRNA sequences were used to achieve gene silencing in cells. STAT3 siRNA was obtained from Gene Pharma Co., Ltd. (Shanghai, People’s Republic of China). Specific siRNA sequences (sense sequence: 5′-AAAUGAAGGUGGUGGAGAAUU-3′; antisense sequence: 5′ UUCUCCACCACCUUCAUUUUU-3′) were used for mouse STAT3. Transfection of siRNA into MPMs was carried out using LipofectAMINE™ 3000 (Invitrogen, Carlsbad, CA), and the transfection procedure was implemented according to the manufacturer’s instructions.
Real-Time Quantitative PCR
Total RNA was isolated from cells and artery tissues using TRIZOL (Thermo Fisher, Carlsbad, CA) following the manufacturer’s instructions. Reverse transcription was performed using a two-step M-MLV Platinum RT-qPCR Kit (Invitrogen, Carlsbad, CA) and quantitative PCR was performed using SYBR Green qPCR SuperMix-UDG kit (Thermo Fisher) in Eppendorf Mastercycler ep realplex detection system (Eppendorf, Hamburg, Germany). Primer sequences purchased from Thermo Fisher (Shanghai, People’s Republic of China) are summarized in Table 1. The mRNA levels of target genes were normalized using β-actin.
 |
Table 1 Primers Used For Real-Time qPCR Assay |
Western Immunoblot Analysis
10% SDS-PAGE was used to isolate solutes from cells or arterial tissue and electro-transfer them to nitrocellulose membrane. Each membrane was pre-incubated in Tris-buffered saline (pH 7.6, containing 0.05% Tween 20 and 5% non-fat milk) at room temperature for 1.5 hrs. Then, the membranes were subjected to specific antibodies for incubation. Immune response spectra were observed by enhanced chemiluminescence (Bio-Rad, Hercules, CA) after incubation with secondary antibodies bound by horseradish peroxidase. Image J analysis program (1.38e) was utilized to perform quantitative analysis and normalization on them as a comparison to their respective control groups.
Enzyme-Linked Immunosorbent Assay
After MPMs were treated, the culture medium and protein lysate were collected separately. Then, the mouse tumor necrosis factor alpha (TNF-α) and mouse IL-6 kits (eBioScience, San Diego, CA) were applied for ELISA to determine TNF-α and IL-6 levels in the medium. The total amount of inflammatory factors in the medium was normalized to the total amount of protein in the living cells.
Prediction Of The Binding Mode Between STAT3 SH2 Domain And Luteolin
The SH2 domain of STAT3 was obtained from the Protein Data Bank (PDB) (PDB entry: 1BG1) according to previously reported studies.20–22 The crystal structure of STAT3 SH2 domain was refined by Chimera software.23 The binding mode was predicted by the AutoDock (version 4.2) software with a grid box of 22.5 Å×22.5 Å×22.5 Å, which covered the binding pocket of STAT3.24 Lamarckian genetic algorithm was used for conformational sampling with trials of 100 dockings.
The initial structure of STAT3 SH2 domain bound with luteolin was provided by molecular docking, which was further used for the molecular dynamics (MD) simulations. The restrained electrostatic potential method was performed to calculate the partial atomic charges for luteolin based on HF/6-13G* basis set. The ff14SB force field and general Amber force field were assigned to STAT3 SH2 domain and luteolin, respectively.25,26 The complex was then placed into a water tank with appropriate counter ions. Then, minimization, heating and equilibration of the simulated system were carried out. The minimization step involved 6000 steepest descent minimization cycles, followed by 6000 conjugate gradient minimization cycles. Afterwards, the simulated system was heated up from 0 to 300 K in 300 ps, and unconstrained equilibration was performed under the periodic boundary condition for 500 ps. Lastly, the system was subjected to 100 ns production MD simulation in the isothermal isobaric (NPT) ensemble. During the MD simulation, Langevin dynamics and Berendsen barostat were employed to maintain the temperature and pressure, respectively.27,28 The coordinates were recorded every 5 ps for the subsequent numerical analyses.29 In accordance with previous research procedures, binding energy decomposition was calculated using molecular mechanics/generalized Born surface area method based on 200 snapshots from last 40 ns MD simulation with GB model of 2 (igb=2).30,31
Animal Experiments
Atherosclerosis Model
Male ApoE-/- mice (18–20 g, 8 weeks) were purchased from HFK Bioscience Co., Ltd (Beijing, People’s Republic of China). The mice were placed at constant room temperature with a 12:12 hrs light–dark cycle and fed with a standard rodent diet. The mice were domesticated in the laboratory at least 3 days before the start of the study. Protocols for mouse experiments were approved by the institutional review boards of Zhejiang University School of Medicine, Hangzhou, People’s Republic of China. All animal experiments used for this study were performed according to NIH standards as set forth in the Guide for the Care and Use of Laboratory Animals (DHHS pub. NIH 85–23 Rev. 1985).
ApoE-/- mice were randomly divided into three weight-matched groups. Fourteen mice were fed with normal animal diet containing 70 kcal.% carbohydrate, 20 kcal.% protein and 10 kcal.% fat (MediScience Diets Co., Ltd, Yangzhou, People’s Republic of China, Cat. #MD12031) served as a normal control group (ND), while the remaining 28 mice were fed with a high-fat diet of 0.2% cholesterol containing 20 kcal.% protein, 40 kcal.% fat and 20 kcal.% carbohydrate (HFD, MediScience Diets Co., Ltd, Yangzhou, People’s Republic of China, Cat. #MD12033) for 12 weeks. Starting from the first week, mice fed with high fat were divided into two groups: HFD (n=14) and luteolin-treated HFD (HFD+Lut, n=14). Luteolin was administered orally at 10 mg/kg/2days for 12 weeks. 1% CMC-Na solution was used in the HFD group and the normal control group. Bodyweight was recorded on a weekly basis. Mice were euthanized under ether anesthesia and blood was also collected. Arterial tissues were rapidly frozen in liquid nitrogen for gene and protein expression analysis or embedded in 4% paraformaldehyde for microscopic analysis.
Measure The Level Of Serum Lipid And Biochemical Indicators
Serum lipid compositions were measured by the appropriate kit, including low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), total cholesterol (TCH) and triglycerides (TG) (Nanjing Jiancheng, Jiangsu, People’s Republic of China).
Histological Analysis Of Atherosclerotic Lesions
To analyze aortic sinus plaque lesions, the heart and the proximal aorta were removed and embedded in optimum cutting temperature compound. Frozen sections of 8 μm thickness were collected continuously from the central ventricle to the aortic arch. Oil Red O staining and hematoxylin were used to stain sections or analyze accumulation of lipid droplets. Frozen sections were fixed in ice cold 10% formalin for 10 mins and washed with distilled water 3 times. They were then stained in pre-warmed Oil Red O solution for 10 mins in an oven at 60°C, followed by staining in hematoxylin for 30 s. The slides were rinsed thoroughly in running water for 3 mins.
To perform en face analyses of lesions in the entire aorta, the whole aorta was dissected out, opened longitudinally from the heart to the iliac arteries, and stained with Oil Red O according to the procedures described above.
Immunohistochemistry
Frozen sections were used for Immunohistochemistry. To block endogenous peroxidase activity, slides were incubated with 3% H2O2 for 10 mins. The slides were first sealed with 5% (BSA for 30 mins, and then incubated overnight with p-STAT3 antibody (1:200) at 4°C. The second antibody was combined with horseradish peroxidase and DAB were used for detection.
Immunofluorescence
Frozen sections were used for immunofluorescence. The slide was first sealed with 5% BSA for 30 mins, and then incubated overnight with Moma-2 antibody (1:100) at 4°C. The sections were incubated with second antibody (1:500) combined with appropriate Alexa Fluor 488 at room temperature for 1 hr, and mounted with DAPI (P36935, Life Technologies).
Statistical Analysis
Data were presented as means±SEM. The Student’s t-test or ANOVA multiple comparisons were used to obtain the statistical significance of differences between groups in GraphPad Pro GraphPad, San Diego, CA). The difference was significant at the 0.05 level (p<0.05).
Results
Luteolin Administration Did Not Affect Hyperlipidemia Profile In HFD-Fed ApoE-/- Mice
Increased serum lipid levels were observed in ApoE-/- mice with HFD during the animal experiment interval, while the 12-week treatment with luteolin did not change the levels of serum LDL-C, HDL-C, TCH or TG in HFD-fed ApoE-/- mice, indicating that luteolin could not affect hyperlipidemia profile (Figure 1).
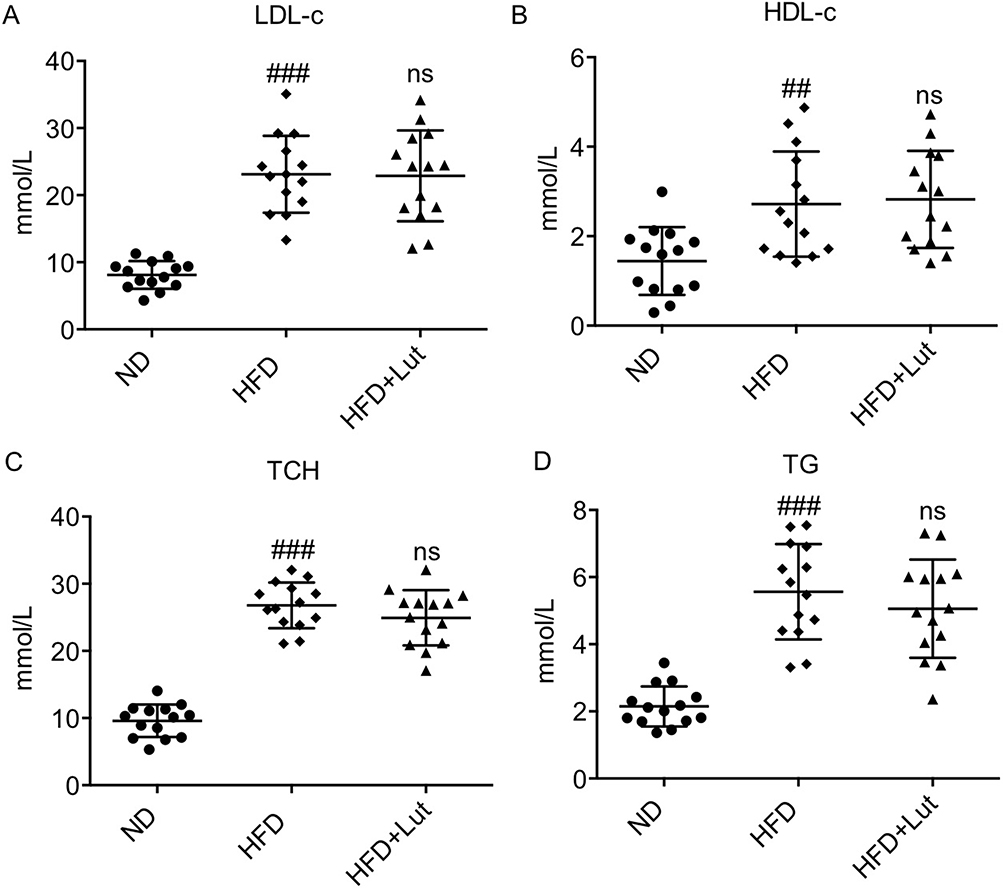 |
Figure 1 Luteolin administration did not affect hyperlipidemia profile in HFD-fed ApoE-/- mice. Notes: Luteolin failed to alter HFD-induced increased serum levels of LDL-C (A), HDL-C (B), TCH (C) and TG (D) (n=14 in each group; ##P<0.01, ###P<0.001, vs ND; ns, no significance, vs HFD). |
Luteolin Administration Prevented The Plaque Development Of Atherosclerosis In HFD-Fed ApoE-/- Mice
We examined the effects of luteolin on the formation of lesion in atherosclerotic aortas of ApoE-/- mice with HFD. The atherosclerotic lesion area in aortic arch was markedly raised. Meanwhile, luteolin administration reduced the atherosclerotic lesion area (Figure 2A). The atherosclerotic lesion area in HFD-fed ApoE-/- mice, as determined by Oil Red O staining of the enface, was significantly increased, while luteolin administration reduced the atherosclerotic lesion area of ApoE-/- mice by about 50% (Figure 2B). Additional histological evaluation using Oil Red O staining suggested that the aortic root plaque areas of luteolin-treated mice were significantly smaller than those in untreated ApoE-/- mice (Figure 2C). These results suggest that luteolin administration prevented the plaque development of atherosclerosis in HFD-fed ApoE-/- mice.
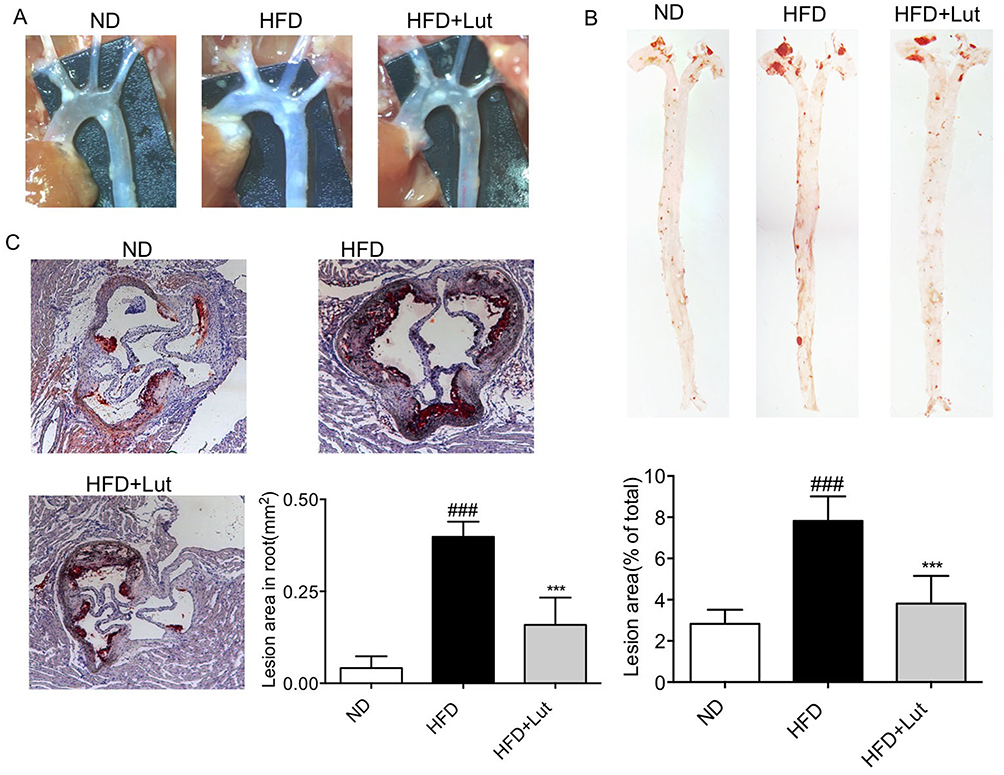 |
Figure 2 Luteolin administration prevented atherosclerotic plaque development in HFD-fed ApoE-/- mice. Notes: Luteolin attenuates atherosclerosis plaque formation in the artery (A, B). (A) The atherosclerotic lesion area in aortic arch is reduced in the administration of luteolin. (B) Oil Red O staining showed that atherosclerosis plaque is reduced in the administration of luteolin and quantification of atherosclerotic plaque lesion area. (C) Oil Red O staining in aortic root showed that lipid accumulation is reduced by administration of luteolin quantification of atherosclerotic plaque lesion area (n=7 in each group; ###P<0.001, vs ND; ***P<0.001, vs HFD;). |
Luteolin Administration Inhibited Inflammation In Atherosclerotic Aortas
Subsequent analysis was performed to clarify whether inflammation is involved in alleviating atherosclerotic plaque formation by luteolin. As shown in Figure 3A–D, the levels of inflammatory factors and adhesion molecules including ICAM-1, VCAM-1, IL-6 and TNF-α were markedly enhanced in atherosclerotic aortas of HFD-fed mice. Compared with HFD-fed mice, these changes were reversed in luteolin-treated mice (Figure 3A–D). Likewise, macrophage infiltration in the atherosclerotic root in untreated mice was markedly higher than that in luteolin-treated mice, as determined by immunostaining for Moma-2 (Figure 3E–G). This indicates that luteolin reduced inflammation in the development of atherosclerosis.
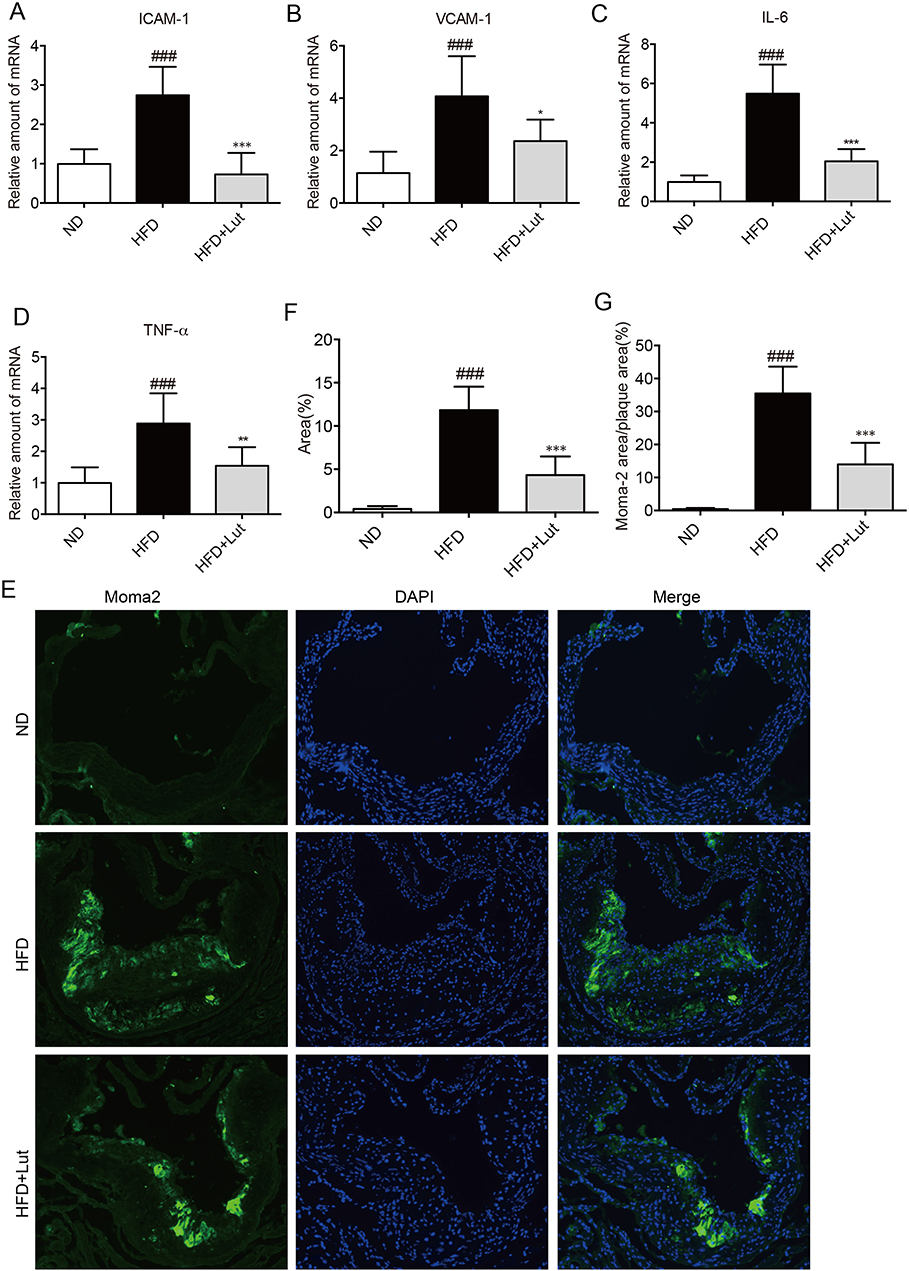 |
Figure 3 Luteolin administration inhibited inflammation in atherosclerotic aortas. Notes: Artery tissues from each group were individually processed for RNA extraction and RT-qPCR analysis. The mRNA levels of ICAM-1, VCAM-1, IL-6 and TNF-α were normalized by β-actin (A–D). (E–G) Immunofluorescence staining with anti-Moma-2 in the artery tissues showed that macrophage infiltration was reduced by luteolin. Representative images (E) and analysis (F–G) of immunofluorescence staining with anti-Moma-2. (n=7 in each group; ###P<0.001, vs ND group; *P<0.05, **P<0.01, ***P<0.001, vs HFD group). |
Increased The Phosphorylation Of STAT3 In Aortas Of HFD-Fed ApoE-/- Mice
Inflammation plays an important role in the development and progression of atherosclerosis. It has been reported that STAT3, which mediates type I interferon signaling, is associated with inflammation. There is also evidence that inhibition of JAK2/STAT3 signal pathway might help prevent LPS-induced inflammation.32,33 Besides, inhibitors of the JAK2/STAT3 pathway have been shown to significantly ameliorate HG/AngII-induced inflammation.34,35 Cupric-ion-oxidized LDL (CuLDL), endothelial cell-oxidized LDL (ELDL) and oxLDL also induced the phosphorylation of JAK2, and then activated STAT3.36 The growing body of evidence indicates that luteolin has a potent ability to inhibit activation of the STAT3 pathway, suggesting that the pharmacological activity of luteolin may be related to its inhibitory effect on STAT3.37–39
Furthermore, we tested the STAT3 activation in atherosclerotic aortas. The phosphorylation of STAT3 was significantly increased in HFD-fed ApoE-/- mice compared to that in ND-fed ApoE-/- mice, as evidenced by immunohistochemical staining (Figure 4A–C). Figure 4D–E also shows that HFD induced phosphorylation of STAT3. Administration with luteolin significantly blocked HFD-increased STAT3 activation in atherosclerotic aortas of ApoE-/- mice, but the protein expression of total STAT3 was not changed.
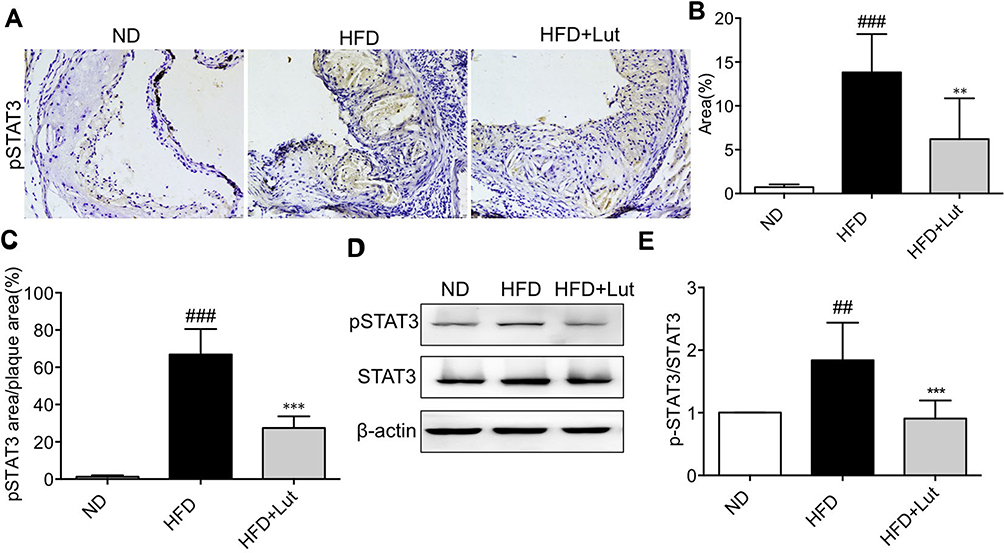 |
Figure 4 STAT3 phosphorylation in aortas of HFD-fed ApoE-/- mice was increased. Notes: (A–C) Representative microscopic images and analysis of p-STAT3 staining in artery tissues showed that HFD increased p-STAT3 immunoreactivity in ApoE-/- mouse arteries. Administration of luteolin significantly reduced p-STAT3. (D, E) Western blot analysis of p-STAT3 in artery tissues showed that administration of luteolin significantly reduced p-STAT3 level without changing STAT3 levels. (n=7 in each group; ##P<0.01, ###P<0.001, vs ND group; **P<0.01, ***P<0.001, HFD group). |
Luteolin Suppressed Inflammation In oxLDL-Stimulated Macrophages
Given that luteolin can alleviate atherosclerosis by decreasing inflammation in mice and that macrophages play an important role in inflammatory atherosclerosis, we investigated the anti-inflammatory effects of luteolin in oxLDL-stimulated primary macrophages. Firstly, oxLDL incubation induced STAT3 phosphorylation in macrophages (Figure 5A). Western blot analysis showed that luteolin dose-dependently inhibited the phosphorylation of STAT3 in oxLDL-induced macrophages (Figure 5A). Then, the oxLDL increased expression of pro-inflammatory genes, IL-6 and TNF-α, in both mRNA and protein levels, while pretreatment with luteolin dose-dependently decreased their overexpressions (Figure 5B–D). Real-time qPCR assay showed that luteolin also reduced the oxLDL-induced mRNA expression of adhesion molecules VCAM-1 and ICAM-1 in macrophages (Figure 5B). These results suggest that luteolin suppressed oxLDL-induced inflammation and may inhibit the phosphorylation of STAT3. Existing studies have demonstrated that the SH2 domain of STAT3 is the most druggable binding site because the STAT3 signal pathway is activated upon the phosphorylation of Tyr-705, which is located in the STAT3 SH2 domain.21,22,40 In this study, molecular modeling was employed to predict the possible interaction between STAT3 SH2 domain and luteolin. As shown in Figure 5E, the root-mean square deviations (RMSDs) of all the protein backbone atoms (Cα) of STAT3 SH2 domain, and the heavy atoms of luteolin were plotted to monitor the dynamic stability of the complex. The RMSDs of the Cα of the STAT3 SH2 domain and the heavy atoms of luteolin have a small fluctuation after 40 and 35 ns MD simulation, respectively. This observation suggests that a series of dynamic stable complexes were obtained. After that, these complexes from MD simulation were used for the binding free energy decomposition into each residue to highlight the importance of each residue in protein-ligand recognition. As shown in Figure 5F, the most contributed residues were Glu-594, Ser-636, Glu-638, Arg-609 and Ile-634. Structural analysis suggests that the primary interactions between STAT3 SH2 and luteolin were hydrogen bond with Glu-594, Ser-636, Arg-609 (Figure 5G and H). Overall, our results indicate that SH2 domain of STAT3 might be a binding site of luteolin and that luteolin could be a promising lead compound for the discovery of novel STAT3 inhibitors.
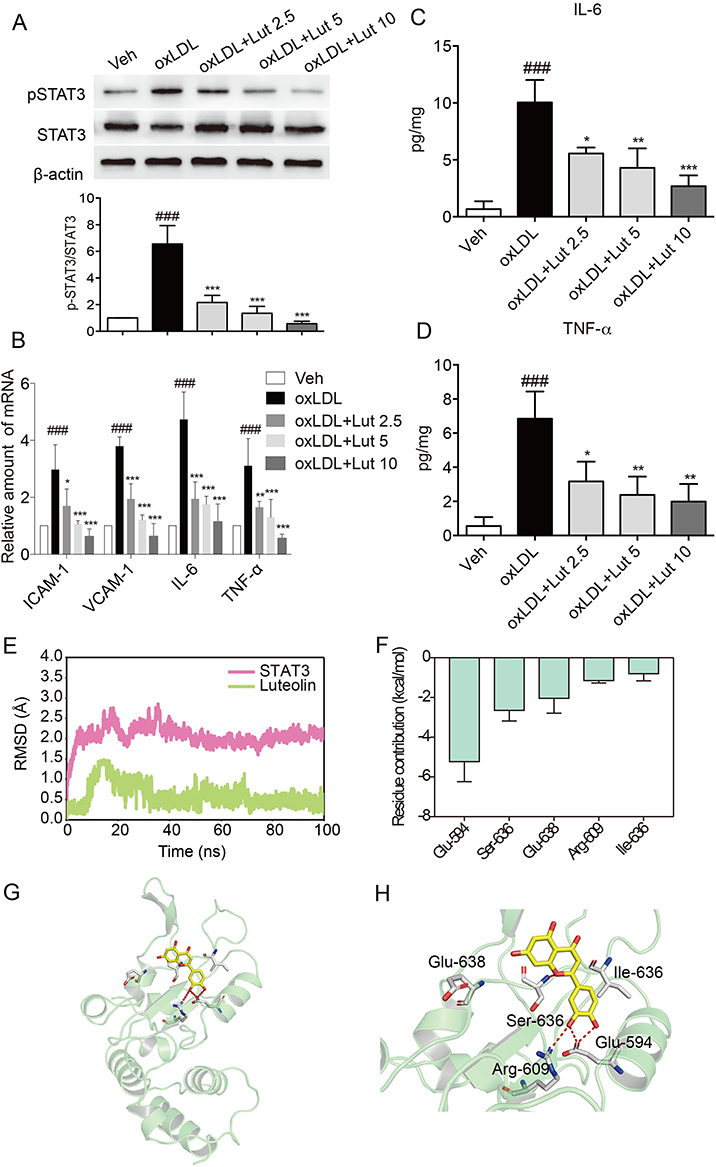 |
Figure 5 Luteolin suppressed inflammation in oxLDL-stimulated macrophages. Notes: (A) Luteolin suppressed oxLDL-induced activation of STAT3. Primary macrophages were pretreated with luteolin (Lut, 2.5, 5 or 10 μM), or vehicle (DMSO, 1 μL) for 1 hr and then stimulated with oxLDL (50 µg/mL) for 15 mins. Total proteins were extracted to detect the levels of p-STAT3 and STAT3. (B) Luteolin inhibited oxLDL-induced increase in mRNA levels of inflammatory molecules. Macrophages were pretreated with Lut (2.5, 5 or 10 μM) or vehicle (DMSO, 1 μL) for 1 hr and then stimulated with oxLDL (50 µg/mL) for 6 hrs. Figure showing mRNA levels of ICAM-1, VCAM-1, IL-6 and TNF-α. (C, D) Luteolin inhibited oxLDL-induced release of cytokines. Macrophages were pretreated with Lut (2.5, 5 or 10 μM) or vehicle (DMSO, 1 μL) for 1 hr and then stimulated with oxLDL (50 µg/mL) for 24 hrs. The levels of IL-6 (C) and TNF-α (D) in the culture medium were detected by ELISA. (E) RMSD curves for the 100 ns molecular dynamics simulation. (F) Top 5 contributing residues of STAT3 SH2 domain to luteolin binding. (G) Overview of contributed residues of STAT3 SH2 domain to luteolin binding. (H) Detailed view of STAT3 SH2 domain to Luteolin binding. (n=3 in each group; ###P<0.001, vs vehicle group; *P<0.05, **P<0.01, ***P< 0.001, vs oxLDL group). |
Luteolin Suppressed oxLDL-Induced Inflammation Via Inhibiting Activation Of STAT3
To confirm that the anti-inflammatory effect of luteolin is STAT3-dependent in oxLDL-challenged macrophage, we knocked down the expression of STAT3 prior to oxLDL exposure. Compared with the control group, transfection specific siRNA into macrophages decreased protein abundance by more than 70% (Figure 6A). As shown in Figure 6B–D, oxLDL failed to increase the mRNA expression of adhesion molecules and pro-inflammatory factors (including ICAM-1, VCAM-1, IL-6 and TNF-α) in STAT3-knockdown primary macrophages. In addition, luteolin was not able to reduce oxLDL-induced secretion of IL-6 and TNF-α (Figure 6C and D). These results suggest that luteolin suppressed oxLDL-induced inflammation via inhibiting STAT3 activation.
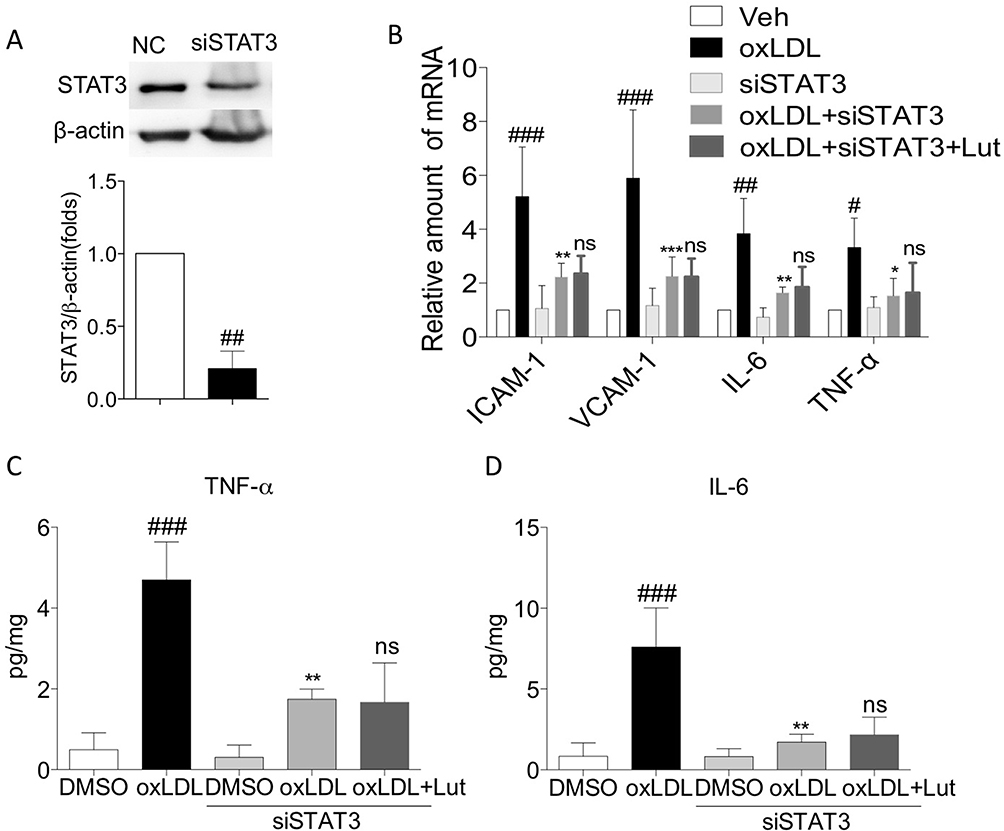 |
Figure 6 Luteolin suppressed oxLDL-induced inflammation via inhibiting activation of STAT3. Notes: Primary macrophages treated with siRNA for STAT3. (A) Western blot analysis for silencing STAT3. (B) Pretreated with luteolin (10µM) for 1 hr, then stimulated with oxLDL (50 µg/mL) for 6 hrs, mRNA levels of ICAM-1, VCAM-1, IL-6 and TNF-α were detected by RT-qPCR analysis. (C, D) Pretreated with luteolin (10µM) for 1 hr, then stimulated with oxLDL (50 µg/mL) for 24 hrs, culture medium was used to detect the levels of IL-6 (C) and TNF-α (D) by ELISA (n=3 in each group; #P< 0.05, ##P<0.01, ###P<0.001, vs vehicle group; *P<0.05, **P<0.01, ***P<0.001, vs oxLDL group; ns, no significance, vs oxLDL+siSTAT3 group). |
Discussion
As a chronic inflammatory disease, atherosclerosis involves a variety of cell types at different stages of plaque formation, including lymphocytes, endothelial cells, SMCs and monocytes/macrophages.41 Previous studies have shown that monocyte-derived macrophages and other immune mediators play an essential role in atherosclerosis.3,42 The proliferation of fibro-fatty plaque and SMCs that causes narrowed artery constitutes the fundamental lesion of atherosclerosis.43 However, the mechanism underlying the progression of lesion has not yet been fully defined, and the therapeutic drugs for atherosclerosis are insufficient. Thus, it is imperative that novel and effective agents be identified to facilitate the treatment of atherosclerosis.
In this study, we investigated the effects of luteolin on the development and progression of atherosclerosis in ApoE-/- mice with HFD. As aforementioned, with HFD for 12 weeks, we observed that accelerated atherosclerotic lesions were established in ApoE-/- mice, characterized by the accumulation of macrophages and lipid droplet (Figures 2 and 3). Interestingly, administration of luteolin significantly ameliorated the progression of HFD-induced atherosclerosis and decreased the accumulation of macrophages and lipid droplet in the lesion, without the effects on the serum TCH, TG, HDL-C and LDL-C level in ApoE-/- mice (Figure 1–3). Furthermore, these findings suggest that luteolin plays an important protective role in the pathogenesis of atherosclerosis.
Inflammation plays a vital role throughout the development of atherosclerosis.8,44 Many cell types, including T-lymphocytes, macrophages, monocytes, SMC and mast cells, exist in atherosclerotic plaques from the earliest lesions to ruptured plaques, accompanied by the secretion of many inflammatory factors.8 In the present study, we found that administration of luteolin obviously decreased macrophages infiltration and mRNA expression of inflammatory factors (ICAM-1, VCAM-1, TNF-α and IL-6) in HFD-induced ApoE-/- mice (Figure 3). In addition, in oxLDL-stimulated macrophages, similar results were observed (Figure 5). These findings suggest that administration of luteolin attenuates atherosclerosis via inhibition of the inflammatory response. To further explore the molecular mechanism of luteolin in alleviating atherosclerosis, we found that phosphorylation of STAT3 was significantly elevated in HFD-induced ApoE-/- mice and was inhibited by administration of luteolin. Interestingly, the expression of total STAT3 was not changed (Figure 4D). Not only that, the same pattern was also observed in oxLDL-stimulated macrophages (Figure 5A). In addition, the results of molecular modeling indicate that SH2 domain of STAT3 could be a binding site of luteolin. In STAT3-knockdown primary macrophages, inflammatory response was mitigated, but luteolin did not reduce the expression and release of inflammatory factors (Figure 6). These results suggest that luteolin suppressed oxLDL-induced inflammation via inhibiting STAT3 activation.
It is noteworthy that various drug interventions, such as calcium channel blockers, nicotinic acid, and angiotensin-converting enzyme inhibitors and statins, can target ROS and inflammation to eliminate the development of atherosclerosis.41 In the present study, it was shown that luteolin could effectively prevent the development of atherosclerosis. And we found the anti-inflammatory activity of luteolin in vivo and in vitro studies. Also, luteolin showed a slightly stronger STAT3 kinase inhibition, which may be due to the fact that the SH2 domain of STAT3 was targeted to disrupt STAT3-STAT3 dimerization, as indicated by our molecular modeling study. However, we failed to report the effects of luteolin on other molecular modifications (i.e., oxidative stress, extracellular matrix, SMC proliferation and cell foam). Taken together, these results suggest that luteolin might be a potential therapeutic option for subsequent studies in developing preventions of atherosclerosis. Here, we observed that the reduction of luteolin on all these abnormalities seemed to be related to its inhibition of STAT3 activation, implying that the activation of STAT3 promotes the development of atherosclerosis.
Acknowledgments
Financial support was provided by the Natural Science Foundation of Zhejiang Province of China (LYY19H310006), Chinese Medical and Health Research Project of Zhejiang Province (2015ZA025) and Medical and Health Research Project of Zhejiang Province (2013KYB062).
Disclosure
The authors declare that there are no competing or financial interests in this work.
References
1. Heidenreich PA, Trogdon JG, Khavjou OA, et al. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123(8):933–944. doi:10.1161/CIR.0b013e31820a55f5
2. Murray CJ, Lopez AD. Measuring the global burden of disease. N Engl J Med. 2013;369(5):448–457. doi:10.1056/NEJMra1201534
3. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. doi:10.1016/j.cell.2011.04.005
4. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. doi:10.1038/nature10146
5. Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–1844. doi:10.1161/CIRCULATIONAHA.106.676890
6. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114(12):1852–1866. doi:10.1161/CIRCRESAHA.114.302721
7. Park S, Lakatta EG. Role of inflammation in the pathogenesis of arterial stiffness. Yonsei Med J. 2012;53(2):258–261. doi:10.3349/ymj.2012.53.2.258
8. Conti P, Shaik-Dasthagirisaeb Y. Atherosclerosis: a chronic inflammatory disease mediated by mast cells. Cent Eur J Immunol. 2015;40(3):380–386.
9. Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320(14):915–924. doi:10.1056/NEJM198904063201407
10. Moore KJ, Freeman MW. Scavenger receptors in atherosclerosis: beyond lipid uptake. Arterioscler Thromb Vasc Biol. 2006;26(8):1702–1711. doi:10.1161/01.ATV.0000229218.97976.43
11. Shi Y, Cosentino F, Camici GG, et al. Oxidized low-density lipoprotein activates p66Shc via lectin-like oxidized low-density lipoprotein receptor-1, protein kinase C-beta, and c-Jun N-terminal kinase kinase in human endothelial cells. Arterioscler Thromb Vasc Biol. 2011;31(9):2090–2097. doi:10.1161/ATVBAHA.111.229260
12. Imran M, Rauf A, Abu-Izneid T, et al. Luteolin, a flavonoid, as an anticancer agent: a review. Biomed Pharmacother. 2019;112:108612. doi:10.1016/j.biopha.2019.108612
13. Yang PW, Lu ZY, Pan Q, et al. MicroRNA-6809-5p mediates luteolin-induced anticancer effects against hepatoma by targeting flotillin 1. Phytomedicine. 2018;57:18–29. doi:10.1016/j.phymed.2018.10.027
14. Fei J, Liang B, Jiang C, Ni H, Wang L. Luteolin inhibits IL-1beta-induced in fl ammation in rat chondrocytes and attenuates osteoarthritis progression in a rat model. Biomed Pharmacother. 2019;109:1586–1592. doi:10.1016/j.biopha.2018.09.161
15. Hoult JR, Paya M. Pharmacological and biochemical actions of simple coumarins: natural products with therapeutic potential. Gen Pharmacol. 1996;27(4):713–722. doi:10.1016/0306-3623(95)02112-4
16. Fan X, Du K, Li N, et al. Evaluation of anti-nociceptive and anti-inflammatory effect of luteolin in Mice. J Environ Pathol Toxicol Oncol. 2018;37(4):351–364. doi:10.1615/JEnvironPatholToxicolOncol.2018027666
17. Zhang Z, Xu P, Yu H, Shi L. Luteolin protects PC-12 cells from H2O2-induced injury by up-regulation of microRNA-21. Biomed Pharmacother. 2019;112:108698. doi:10.1016/j.biopha.2019.108698
18. Yin H, Wang L, Wu M, Liu Y, Li N, Chen T. Cyanidin-3-O-glucoside chloride acts synergistically with luteolin to inhibit the growth of colon and breast carcinoma cells. Pharmazie. 2019;74(1):54–61. doi:10.1691/ph.2019.8746
19. Wang L, Huang Z, Huang W, et al. Inhibition of epidermal growth factor receptor attenuates atherosclerosis via decreasing inflammation and oxidative stress. Sci Rep. 2017;8:45917. doi:10.1038/srep45917
20. Becker S, Groner B, Muller CW. Three-dimensional structure of the Stat3beta homodimer bound to DNA. Nature. 1998;394(6689):145–151. doi:10.1038/28101
21. Li H, Liu A, Zhao Z, et al. Fragment-based drug design and drug repositioning using multiple ligand simultaneous docking (MLSD): identifying celecoxib and template compounds as novel inhibitors of signal transducer and activator of transcription 3 (STAT3). J Med Chem. 2011;54(15):5592–5596. doi:10.1021/jm101330h
22. Zhang Y, Wang L, Deng Y, et al. Fraxetin suppresses proliferation of non-small-cell lung cancer cells via preventing activation of signal transducer and activator of transcription 3. Tohoku J Exp Med. 2019;248(1):3–12. doi:10.1620/tjem.248.3
23. Pettersen EF, Goddard TD, Huang CC, et al. UCSF chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084
24. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi:10.1002/jcc.21256
25. Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11(8):3696–3713. doi:10.1021/acs.jctc.5b00255
26. Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25(9):1157–1174. doi:10.1002/jcc.20035
27. Loncharich RJ, Brooks BR, Pastor RW. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers. 1992;32(5):523–535. doi:10.1002/bip.360320508
28. Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. doi:10.1063/1.448118
29. Roe DR, Cheatham TE
30. Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, Roitberg AE. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput. 2012;8(9):3314–3321. doi:10.1021/ct300418h
31. Ying Y, Jin J, Ye L, Sun P, Wang H, Wang X. Phloretin prevents diabetic cardiomyopathy by dissociating keap1/Nrf2 complex and inhibiting oxidative stress. Front Endocrinol (Lausanne). 2018;9:774. doi:10.3389/fendo.2018.00420
32. Yang Y, Zhao J, Song X, et al. Amygdalin reduces lipopolysaccharide-induced chronic liver injury in rats by down-regulating PI3K/AKT, JAK2/STAT3 and NF-kappaB signalling pathways. Artif Cells Nanomed Biotechnol. 2019;47(1):2688–2697. doi:10.1080/21691401.2019.1634084
33. Zhang H, Sha J, Feng X, et al. Dexmedetomidine ameliorates LPS induced acute lung injury via GSK-3beta/STAT3-NF-kappaB signaling pathway in rats. Int Immunopharmacol. 2019;74:105717. doi:10.1016/j.intimp.2019.105717
34. Chen J, Zhang W, Xu Q, et al. Ang-(1–7) protects HUVECs from high glucose-induced injury and inflammation via inhibition of the JAK2/STAT3 pathway. Int J Mol Med. 2018;41(5):2865–2878. doi:10.3892/ijmm.2018.3507
35. Qin Z, Bagley J, Sukhova G, et al. Angiotensin II-induced TLR4 mediated abdominal aortic aneurysm in apolipoprotein E knockout mice is dependent on STAT3. J Mol Cell Cardiol. 2015;87:160–170. doi:10.1016/j.yjmcc.2015.08.014
36. Maziere C, Conte MA, Maziere JC. Activation of JAK2 by the oxidative stress generated with oxidized low-density lipoprotein. Free Radic Biol Med. 2001;31(11):1334–1340. doi:10.1016/S0891-5849(01)00649-9
37. Cummins CB, Wang X, Nunez Lopez O, et al. Luteolin-mediated inhibition of hepatic stellate cell activation via suppression of the STAT3 pathway. Int J Mol Sci. 2018;19(6):1567. doi:10.3390/ijms19061567
38. Sonoki H, Tanimae A, Endo S, et al. Kaempherol and luteolin decrease claudin-2 expression mediated by inhibition of STAT3 in lung adenocarcinoma A549 cells. Nutrients. 2017;9(6):597. doi:10.3390/nu9060597
39. Palombo R, Savini I, Avigliano L, et al. Luteolin-7-glucoside inhibits IL-22/STAT3 pathway, reducing proliferation, acanthosis, and inflammation in keratinocytes and in mouse psoriatic model. Cell Death Dis. 2016;7(8):e2344. doi:10.1038/cddis.2016.201
40. Xiao H, Bid HK, Jou D, et al. A novel small molecular STAT3 inhibitor, LY5, inhibits cell viability, cell migration, and angiogenesis in medulloblastoma cells. J Biol Chem. 2015;290(6):3418–3429. doi:10.1074/jbc.M114.616748
41. Hao L, Zhu G, Lu Y, et al. Deficiency of cathepsin K prevents inflammation and bone erosion in rheumatoid arthritis and periodontitis and reveals its shared osteoimmune role. FEBS Lett. 2015;589(12):1331–1339. doi:10.1016/j.febslet.2015.04.008
42. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi:10.1038/ni.2001
43. Kolodgie FD, Virmani R, Burke AP, et al. Pathologic assessment of the vulnerable human coronary plaque. Heart. 2004;90(12):1385–1391. doi:10.1136/hrt.2004.041798
44. Rosenfeld ME. Inflammation and atherosclerosis: direct versus indirect mechanisms. Curr Opin Pharmacol. 2013;13(2):154–160. doi:10.1016/j.coph.2013.01.003
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.