Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Lung inflammation caused by inhaled toxicants: a review
Authors Wong J, Magun B, Wood L
Received 7 February 2016
Accepted for publication 27 April 2016
Published 23 June 2016 Volume 2016:11(1) Pages 1391—1401
DOI https://doi.org/10.2147/COPD.S106009
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
John Wong, Bruce E Magun, Lisa J Wood
School of Nursing, MGH Institute of Health Professions, Boston, MA, USA
Abstract: Exposure of the lungs to airborne toxicants from different sources in the environment may lead to acute and chronic pulmonary or even systemic inflammation. Cigarette smoke is the leading cause of chronic obstructive pulmonary disease, although wood smoke in urban areas of underdeveloped countries is now recognized as a leading cause of respiratory disease. Mycotoxins from fungal spores pose an occupational risk for respiratory illness and also present a health hazard to those living in damp buildings. Microscopic airborne particulates of asbestos and silica (from building materials) and those of heavy metals (from paint) are additional sources of indoor air pollution that contributes to respiratory illness and is known to cause respiratory illness in experimental animals. Ricin in aerosolized form is a potential bioweapon that is extremely toxic yet relatively easy to produce. Although the aforementioned agents belong to different classes of toxic chemicals, their pathogenicity is similar. They induce the recruitment and activation of macrophages, activation of mitogen-activated protein kinases, inhibition of protein synthesis, and production of interleukin-1 beta. Targeting either macrophages (using nanoparticles) or the production of interleukin-1 beta (using inhibitors against protein kinases, NOD-like receptor protein-3, or P2X7) may potentially be employed to treat these types of lung inflammation without affecting the natural immune response to bacterial infections.
Keywords: cigarette, mycotoxin, trichothecene, ricin, inflammasome, macrophage, inhibitors
Introduction
Inflammation is a complex biological process that occurs in response to harmful stimuli and whose function is to eliminate the cause of cell injury and initiate the repair process. Lung inflammation occurs in response to bacterial and viral pathogens and environmental pollutants. The sources of indoor pollution include cigarette smoke, mycotoxins, and airborne particulates of asbestos, silica, and heavy metals. Sustained inflammation of the lung, as occurs in response to cigarette smoke, may lead to chronic obstructive pulmonary disease (COPD), which is the third leading cause of death globally and whose prevalence is still rising.1,2 Current therapies for COPD focus on long-acting bronchodilators and do not sufficiently target pulmonary inflammation that underlies the pathogenesis of the disease.3 There exists a critical need to understand the mechanisms that lead to lung inflammation and develop novel strategies to treat COPD. In addition to cigarette smoke, other inhaled toxicants are known to produce lung inflammation. Recent epidemiologic evidence has recognized the importance of air pollution from traffic worldwide and domestic fires that burn biomass fuels in underdeveloped countries.4 In cases of exposure to sublethal amounts of inhaled toxicants, such as mycotoxins and ricin, inflammation is usually resolved when the cause of the cell injury has been eliminated. Although these toxicants belong to the different classes of chemicals, they nevertheless may activate similar biochemical pathways. Elucidating these pathways may serve to identify potential therapeutic targets susceptible to anti-inflammatory treatments.
Several types of cells are involved in lung inflammation, including the epithelial cells that line the airways and alveoli and the immune cells in the blood. Airway epithelial cells are important in the host defense system by acting as a physical barrier and secreting mucus that traps inhaled particles.5 These cells also secrete antimicrobial peptides and proteases that neutralize the danger,6–8 cytokines and chemokines that serve as inflammatory mediators,9–12 and growth factors that promote tissue repair and fibrosis.13 During the acute phase of inflammation, neutrophils rapidly migrate to the lung as first responders, producing reactive oxygen species and secreting serine proteases, matrix metalloproteinases, and other enzymes during degranulation. These products not only degrade invading dangers but also contribute to alveolar destruction.14,15 Resident and recruited macrophages engulf invading particles and secrete inflammatory mediators and various enzymes.16–18 The number of T lymphocytes also increases and may contribute to the pathophysiology of lung inflammation.19,20 The decreased effector function and increased regulatory function of these lymphocytes may account for the reduced host immunity to bacterial infections in COPD patients.21
Produced by epithelial and inflammatory cells, cytokines and chemokines play a central role in the inflammatory process. In particular, tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β) act as initiator cytokines by inducing the increased production of themselves and the synthesis of other cytokines, chemokines, and adhesion molecules, thereby attracting and activating immune cells at the site of inflammation.22–24 TNF-α is initially synthesized as a membrane-bound precursor and proteolytically released from cell surfaces.25 Soluble TNF-α then binds to the TNF receptor and activates the mitogen-activated protein kinase (MAPK) cascade and the nuclear factor-kappa B (NF-κB) pathway after the ligand-bound receptor forms a protein complex with TNF receptor 1-associated death domain protein and TNF receptor-associated factor-2.26,27 MAPKs are phosphorylated and activated by MAPK kinases, which in turn are activated by MAPK kinase kinases.28–30 MAPKs directly phosphorylate and activate transcription factors or they phosphorylate other kinases, which in turn activate transcription factors that lead to the expression of response genes; MAPKs also phosphorylate other substrates that are involved in many biological processes, including inflammation.28,31
Like TNF-α, IL-1β is initially synthesized as pro-IL-1β, an inactive precursor. Pro-IL-1β is then cleaved inside the cell by a protein complex called the inflammasome, which is composed of apoptosis-associated speck-like protein containing caspase recruitment domain, caspase-1, and a member of the nucleotide-binding oligomerization domain (NOD)-like receptor family.32–34 Different NOD-like receptor members respond to different signals. One of these members, NOD-like receptor protein-3 (NLRP3), is recruited in response to tissue damage, metabolic stress, and infection.35,36 Once pro-IL-1β is processed, the mature IL-1β product is secreted and binds to the IL-1 receptor. The ligand-bound receptor forms a complex with myeloid differentiation primary response 88, IL-1 receptor-associated kinase, and TNF receptor-associated factor-6, thereby activating the MAPK cascade and the NF-κB pathway.37–39 Different mechanisms have been proposed for the activation of the inflammasome, including potassium efflux and the generation of reactive oxygen species, but both hypotheses have been challenged.40,41 Other researchers have demonstrated the importance of autophagy and the P2X7 receptor in mediating the processing of IL-1β by the inflammasome.42–44
There is currently no cure for COPD or effective treatment for severe lung inflammation caused by toxicants, such as fungal toxins and ricin. This review article summarizes current research on lung inflammation following exposure to cigarette smoke, mycotoxins, and ricin. The goal of comparing these studies is to determine whether common pathways exist and to identify potential targets for the future development of therapeutics. Indeed, although these toxicants belong to different classes of chemicals that exhibit a variety of pathological effects, some of the biochemical pathways they activate are identical, including the IL-1β pathway, which is increasingly recognized for its importance in lung inflammation.45,46 Elucidation of these mechanisms is facilitated by reviewing the research that has been performed on these different toxicants, and such understanding may facilitate the development of therapeutics that would be useful in treating acute and chronic lung inflammation. Effective strategies that block inflammation may ultimately lead to successful treatment of COPD.
Lung inflammation by cigarette smoke
Cigarette smoking is the major risk factor for COPD and has been estimated to account for more than 50% of cases of COPD worldwide.47 Interestingly, there is no consensus on the mechanisms by which cigarette smoke causes COPD. One reason for this difficulty is the presence of additional environmental factors that may contribute to the development of lung inflammation. These factors include occupational and environmental exposures to dusts and fumes,48 infections in early life,49 genetic predisposition,50–52 and asthma.53,54 Another factor is the frequent contamination of tobacco by toxins from other sources and the presence of microbes that activate toll-like receptors.55,56 Moreover, cigarette smoke contains several thousand distinct compounds,57 further complicating an understanding of their individual contribution to lung disease. In the gas phase of smoke, these chemicals include acetaldehyde, methane, hydrogen cyanide, nitric acid, acetone, acrolein, ammonia, methanol, hydrogen sulfide, hydrocarbons, gas phase nitrosamines, and carbonyl compounds. In the particulate phase, they include carboxylic acids, phenols, humectants, nicotine, terpenoids, paraffin waxes, tobacco-specific nitrosamines, polycyclic aromatic hydrocarbons, catechols, metals, and other inorganic substances. Many of these chemicals are irritants, suspected carcinogens, and agents that promote inflammation.58
Despite these challenges, and in view of the millions of tobacco-related deaths and the accompanying billions of dollars in estimated health care cost each year, extensive research has been conducted to study the biochemical and health effects of cigarette smoking. Exposure to cigarette smoke in vitro induces the release of IL-1β from human airway epithelial cells59 and chemokines from both epithelial cells and neutrophils.59,60 However, there are conflicting data on whether macrophages produce a similar inflammatory response in vivo.61 Components in cigarette smoke also block protein synthesis in macrophages.62–64
COPD is thought to be associated with an innate immune response by macrophages, neutrophils, and epithelial cells and an adaptive immune response by lymphocytes. Because lung inflammation persists after smoking cessation, autoimmunity has been proposed as a mechanism that drives disease progression. Th17 cells are a subset of CD4+ T lymphocytes associated with autoimmune conditions, and these cells increase in numbers in COPD patients. Interestingly, levels of regulatory T-cells, which normally control the proliferation of Th17 cells, are also elevated, suggesting that an imbalance of Th17 and regulatory T subsets may be important.65 However, the presence of autoantibodies remains controversial.66,67
In rodents, cigarette smoke causes activation of MAPKs in the lungs,68 increased numbers of neutrophils, lymphocytes, and macrophages,20,69 and apoptosis of airway epithelial cells.70 Pulmonary inflammation by cigarette smoke is dependent on IL-1 receptor/myeloid differentiation primary response 88 signaling,71 and the release of IL-1β induced by cigarette smoke into the bronchoalveolar lavage fluid is mediated by the P2X7 receptor and the NLRP3-inflammasome.59,72,73 Blocking the NLRP3-inflammasome by knocking out apoptosis-associated speck-like protein containing caspase recruitment domain, caspase 1, or NLRP3 also reduces neutrophilia, providing evidence that the inflammasome is involved in mediating pulmonary inflammation.72 Similarly, knocking out the mitochondrial antiviral signaling molecule, which may play a role in the activation of the inflammasome by some agents by regulating autophagy and the mitochondrial production of reactive oxygen species,74 leads to reduced levels of IL-1β and neutrophilia following exposure to cigarette smoke.75
Consistent with data from animal models, smokers have a fourfold increase in the number of macrophages and other leukocytes into the bronchoalveolar lavage fluid; this increase is positively correlated with smoking history.76 The levels of IL-1β and many biomarkers, such as chemokines, are elevated in the serum of smokers and are believed to play a key role in the development of the chronic inflammation associated with COPD.77 These mediators are mainly produced by macrophages,16,18 which also show an impaired ability to clear apoptotic epithelial cells.70 In contrast, even though cigarette smoke induces the expression of IL-1β by bronchial epithelial cells in vitro,59 IL-1β and components of the inflammasome are not detected in the bronchial biopsies of COPD patients,78 suggesting either that the inflammasome may not play a major role in the central airway of certain COPD patients or their levels may fall below detection levels. IL-33, a member of the IL-1 cytokine family, has also been recently found to be associated with COPD.79,80 Unlike IL-1β, however, IL-33 is processed by neutrophil-derived proteases81,82 rather than the inflammasome.83
The inflammatory response even persists in those who have quit smoking for years,84 probably as a result of autoimmunity or continued microbial infection.55,85,86 Effective anti-inflammatory treatment for COPD is currently lacking, in part because macrophages become resistant to the anti-inflammatory effects of corticosteroids as a result of dysregulated NF-κB activity.87 Intensive research is currently being undertaken to develop potent protease inhibitors in an attempt to improve symptoms.88,89
Lung inflammation by mycotoxins
Fungal spores are ubiquitous in the environment. Containing allergens and mycotoxins, these spores are especially hazardous to those living inside damp buildings or to farmers, malt workers, and wood workers whose occupations include handling of moldy materials.90 Different fungi produce mycotoxins as secondary metabolites, which include various trichothecenes that are synthesized by several species of Fusarium, Myrothecium, Trichoderma, Trichothecium, Cephalosporium, Verticimonosporium, and Stachybotrys.91 Readily absorbed through the skin, gut, and airways, trichothecenes are chemically stable and are neither degraded by elevated heat nor hydrolyzed in the stomach.92 One such trichothecene, the T2 toxin, has been used in aerosolized form in biological warfare because of its toxicity, heat stability, and chemical stability.93
Trichothecenes cause immunosuppression in lymphocytes94 and stimulate the production of IL-1β by macrophages in an NLRP3-inflammasome-dependent manner, mediated by the P2X7 receptor.95,96 In addition, these toxins inhibit protein synthesis by targeting the ribosome, impair mitochondrial function, activate MAPKs, and induce apoptosis in mammalian cells.92,97–99 They also stimulate the expression of genes that are upregulated in response to other ribosome-damaging agents, including many inflammatory cytokines.100–105
Deoxynivalenol, a trichothecene that commonly contaminates cereal grains, inhibits TNF-α signaling,106 activates MAPKs through a unique MAPK kinase kinase called zipper sterile-alpha-motif kinase (ZAK) (Wong, unpublished data, 2011), and induces cytotoxicity and inflammation synergistically with particulates107 and lipopolysaccharide108 to induce cytotoxicity and inflammation. Because similar studies have not been conducted on other trichothecenes, it remains unknown whether these properties are common to other members of this family of compounds.
Following intranasal delivery in animals, mycotoxins are not only localized in the lung but are also distributed to the liver, kidney, and spleen.105 These toxins elicit recruitment of alveolar macrophages and neutrophils, pulmonary hemorrhage, cytokine production, and damage to multiple organs.109,110 In fact, it has been reported that toxicity following inhalation of a toxic dose of mycotoxin leads to systemic effects exclusive of lung injury,111 but the systemic effects of a sublethal dose of mycotoxins were not addressed by these authors. Even when mycotoxins are ingested, they can cause chronic inflammation of the lungs.112,113 Mycotoxins may also trigger COPD in farm animals.114 Unfortunately, no effective treatment is currently available for exposure to mycotoxin.91
Lung inflammation by ricin
Found in the beans of the castor plant Ricinus communis, ricin is a ribosome-inactivating protein that is relatively easy to purify using simple procedures. Although ricin aerosols are not naturally occurring, the inhalation of ricin is the subject of many studies because of its high toxicity and potential to be exploited as an agent of bioterrorism. Ricin is listed as a biological select agent by the Centers of Disease Control and a category B priority pathogen for the study of the biodefense strategic plan of the US National Institutes of Health. In addition, ricin is being engineered as a component of immunotoxins to target and destroy cancer cells.115,116
Similar to lung inflammation caused by cigarette smoke and mycotoxins, effective treatment for ricin intoxication is lacking. Ricin is poorly absorbed through intact skin but can readily enter the body by ingestion, injection, or inhalation. In the case of ricin poisoning caused by inhalation, symptoms include fever, dyspnea, tightness in the chest, cough, and nausea.117,118 Ricin intoxication induces an early massive migration of inflammatory cells (especially neutrophils) to the lungs and causes apoptosis and necrosis of airway epithelial cells.119 In addition, and unlike cigarette smoke and mycotoxins, ricin causes apoptosis of alveolar macrophages.119 Severe poisoning following inhalation of ricin causes interstitial pneumonia, alveolar edema, and respiratory failure, leading to death within days.120 Exposure to a sublethal dose of ricin results in fibrosis and hemorrhage restricted to the lung tissue.121
The tissue distribution of ricin following pulmonary delivery in animal studies can be measured by several methods. Using enzyme-linked immunosorbent assay, ricin is localized to the lungs.122 More sensitive methods, such as protein radiolabeling123,124 and detection of ricin-specific damage in the ribosomal RNA,125 show that inhaled ricin is also distributed to the kidney, heart, spleen, and blood. The spread of ricin to extrapulmonary tissues, likely the result of destruction of the barrier function of epithelial cells, may contribute to its systemic effects and lethality.
The lethality of ricin is caused by its ability to kill cells rapidly at low concentrations and induce extensive inflammation. Because ricin inhibits protein synthesis by damaging ribosomes, it causes cells to undergo apoptosis.126 Similar to cigarette smoke and mycotoxins, ricin activates the NF-κB and MAPK pathways and increases the expression of inflammatory genes in airway epithelial cells121 and macrophages.127 Like deoxynivalenol and several other ribosome-damaging agents, including anisomycin, Shiga toxin, and ultraviolet radiation, ricin activates the MAPK cascade through ZAK.128,129
In animal studies, ricin causes alveolar macrophages to undergo apoptosis119 and induces the expression of genes involved with the immune response, inflammation (including cytokine signaling), and wound healing.125,130,131 Depletion of macrophages from mice prior to administration of pulmonary ricin reduces the expression of pulmonary IL-1β and subsequent inflammatory responses, demonstrating a central role for both macrophages and IL-1β in the inflammatory process.132 Similar results were obtained following administration of ricin into lungs of IL-1β-deficient mice.132
A causal relationship may exist between the apoptosis of macrophages and the inflammatory response when cells are exposed to ricin. Exposure of murine macrophages in vitro to zVAD, a chemical inhibitor of apoptosis, blocks the expression of inflammatory genes in macrophages,127 suggesting that caspase activity is required for ricin-mediated gene expression. Because ricin and other inhibitors of protein translation are capable of activating the NLRP3-mediated inflammasome,41,133 the ability of zVAD to block the production of IL-1β may result from inhibition of caspase-1.
When inhaled, chemicals that are not biologically derived can also lead to lung inflammation. Volatile organic compounds that can be produced from household items, office supplies, and craft materials (such as formaldehyde, benzene, and perchloroethylene) affect the lung by various mechanisms. One of these, toluene diisocyanate, is capable of activating the inflammasome in a mouse model.134 Asbestos, crystalline silica, alloy particles, and carbon nanotubes can also activate MAPKs135–139 and the inflammasome.140–144 Macrophages may play an important role in the inflammatory response to the inhalation of these particulates.145,146
Summary
Despite extensive research that has been conducted to study lung inflammation induced by toxicants, effective treatment is lacking. Although cigarette smoke, mycotoxins, and ricin represent different classes of agents, they nevertheless induce similar gene expression profiles, produce a similar list of biomarkers, damage the airway epithelium, and involve macrophages in their pathogenesis. Recent advances in the targeting of macrophages using nanoparticle-based delivery of small interfering RNA147 or simvastatin have been reported,148 but the therapeutic value of these strategies has not been tested on lung inflammatory diseases.
The inhaled toxicants described in this review all activate the MAPK cascade, inhibit protein synthesis, and utilize the NLRP3-inflammasome to process IL-1β (Figure 1). Because MAPK and IL-1β are known to play important roles in toxicant-induced lung inflammation, inhibitors of MAPKs and the inflammasome may be effective in blocking the harmful effects of these agents. In recent years, several MAPK inhibitors have been developed to treat many human inflammatory diseases. These agents produce fewer side effects, such as severe infection, compared with therapeutics that directly inhibit cytokines, such as IL-1β.149 However, many of these inhibitors are either still too toxic or ineffective in clinical settings,149,150 probably as a result of complex positive and negative feedback from different members of the MAPK cascade and the presence of broad effects on downstream targets. Similarly, although hundreds of potential inhibitors against NF-κB have been identified, their toxicities are well known.151,152 As a result, MAPK kinase kinases are an attractive therapeutic target because specific members of this family are activated by selective stimuli.153 As discussed earlier, ricin acts exclusively through ZAK, a MAPK kinase kinase. Whether cigarette smoke and mycotoxins other than deoxynivalenol have specificity for activation of ZAK is unknown. Kinase profiling has identified small-molecule kinase inhibitors, such as nilotinib and sorafenib, which have strong affinity for ZAK.154–156 Sorafenib has been shown to inhibit ZAK activity in vitro.157 These agents have been successfully employed to block the inflammatory effects of ricin.133 Another novel compound, INNO-406, is a ZAK inhibitor158 that may prove effective against ZAK-mediated toxicants. Identifying the MAPK kinase kinases that signal lung inflammation in response to cigarette smoke and mycotoxins may facilitate the development of effective therapeutics. For example, researchers have identified transforming growth factor beta-activated kinase-1, another MAPK kinase kinase, which is involved in the cigarette smoke-induced inflammatory response of airway smooth muscle cells in vitro. Further research into the potential role of transforming growth factor beta-activated kinase-1 would be warranted.159 Similarly, several P2X7 antagonists are currently being explored for the treatment of various inflammatory diseases.160 The possible role of P2X7 in ricin intoxication has not yet been reported.
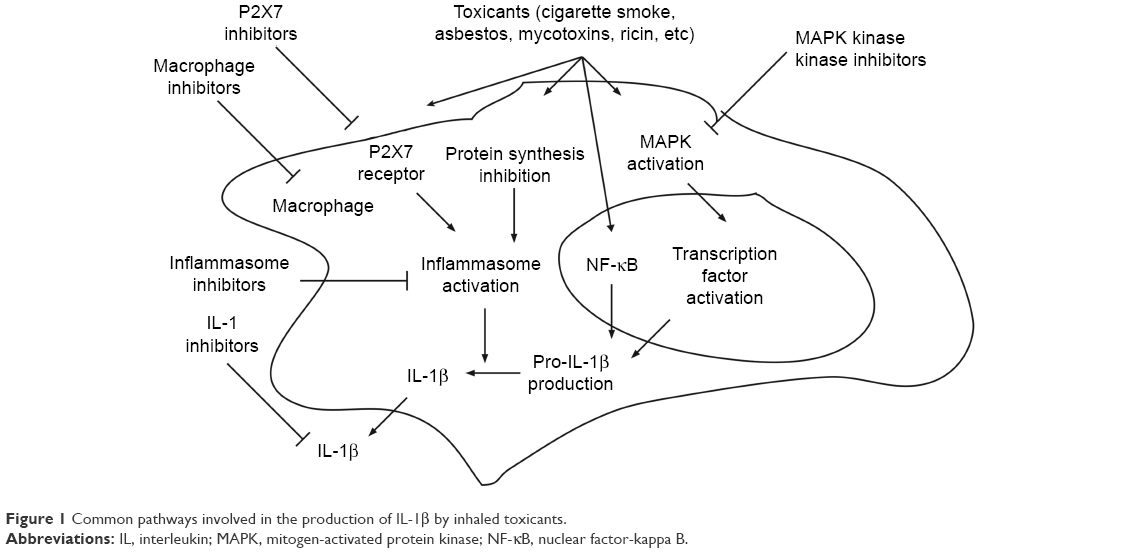 | Figure 1 Common pathways involved in the production of IL-1β by inhaled toxicants. |
Inhalation of toxicants leads to the production of multiple cytokines and other mediators, which in turn produce multiple downstream inflammatory effects. Potential therapeutics is likely to have higher success when directed at upstream, rather than downstream, targets. Like IL-1β, TNF-α is also widely recognized as an initiator cytokine, and both IL-1β and TNF-α are produced after the inhalation of many toxicants (Table 1) and seem to be important in cigarette smoke-induced emphysema and small airway remodeling in mice.161 However, anti-TNF-α therapy is ineffective in reducing symptoms of COPD in patients,162 and TNF-α does not seem to play an important role in ricin intoxication.132 Because several translation inhibitors, including deoxynivalenol, inhibit TNF-α signaling,106 further research is warranted to investigate whether other ribosome-targeting toxicants share the same mechanism that could explain the lack of involvement of TNF-α.
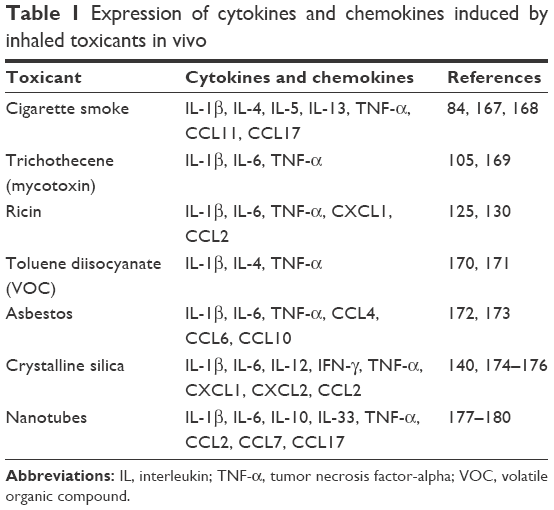 | Table 1 Expression of cytokines and chemokines induced by inhaled toxicants in vivo |
While it is still unknown whether cigarette smoke and other mycotoxins act through ZAK, it is clear that, like ricin, they stimulate the processing of IL-1β using NLRP3. By selective targeting of NLRP3, the production of IL-1β via other members of the inflammasome family may remain normal, thereby reducing the chance of immunosuppression. Several NLRP3 inhibitors, including parthenolide,163 glyburide,164 5-chloro-2-methoxy-N-[2-(4-sulfamoylphenyl)ethyl]benzamide,165 and isoliquiritigenin,166 are currently under investigation. The selective targeting of toxicant-mediated production of IL-1β by MAPK kinase kinase inhibitors and inhibitors against specific NOD-like receptor members may thus lead to the development of novel therapeutic strategies that may be employed for treatment of lung inflammatory disease.
In conclusion, although acute and chronic lung inflammation is known to contribute to the serious effects of cigarette smoke, mycotoxins, ricin, and other inhaled toxicants, effective anti-inflammatory treatments are lacking. By looking beyond cigarette smoke and reviewing the current understanding of how different toxicants induce the inflammatory response, this paper has identified several promising targets to treat COPD and lung inflammation. In particular, ZAK, P2X7, and NLRP3 are unique targets that foster the production of IL-1β by specific stimuli that inhibits protein translation. Selective targeting may interrupt respiratory inflammation while simultaneously permitting a normal immune response to respiratory tract infections that frequently accompany COPD, thereby reducing the risk of severe pneumonia.
Acknowledgments
This work was supported by grants AI105933.5 (to BEM) and 1R01NR013171-01A1 (to LJW) from the US National Institutes of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
Tashkin DP, Murray RP. Smoking cessation in chronic obstructive pulmonary disease. Respir Med. 2009;103(7):963–974. | ||
Parikh R, Shah TG, Tandon R. COPD exacerbation care bundle improves standard of care, length of stay, and readmission rates. Int J Chron Obstruct Pulmon Dis. 2016;11:577–583. | ||
Brusselle G, Bracke K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2014;11(Suppl 5):S322–S328. | ||
Laumbach RJ, Kipen HM. Respiratory health effects of air pollution: update on biomass smoke and traffic pollution. J Allergy Clin Immunol. 2012;129(1):3–11; quiz 12–13. | ||
Adler KB, Li Y. Airway epithelium and mucus: intracellular signaling pathways for gene expression and secretion. Am J Respir Cell Mol Biol. 2001;25(4):397–400. | ||
McCray PB Jr, Bentley L. Human airway epithelia express a beta-defensin. Am J Respir Cell Mol Biol. 1997;16(3):343–349. | ||
Kota S, Sabbah A, Chang TH, et al. Role of human beta-defensin-2 during tumor necrosis factor-alpha/NF-kappaB-mediated innate antiviral response against human respiratory syncytial virus. J Biol Chem. 2008;283(33):22417–22429. | ||
Aarbiou J, Rabe KF, Hiemstra PS. Role of defensins in inflammatory lung disease. Ann Med. 2002;34(2):96–101. | ||
Hellermann GR, Nagy SB, Kong X, Lockey RF, Mohapatra SS. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir Res. 2002;3:22. | ||
Takizawa H, Tanaka M, Takami K, et al. Increased expression of inflammatory mediators in small-airway epithelium from tobacco smokers. Am J Physiol Lung Cell Mol Physiol. 2000;278(5):L906–L913. | ||
Mills PR, Davies RJ, Devalia JL. Airway epithelial cells, cytokines, and pollutants. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S38–S43. | ||
Herfs M, Hubert P, Poirrier AL, et al. Proinflammatory cytokines induce bronchial hyperplasia and squamous metaplasia in smokers: implications for chronic obstructive pulmonary disease therapy. Am J Respir Cell Mol Biol. 2012;47(1):67–79. | ||
Takizawa H, Tanaka M, Takami K, et al. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med. 2001;163(6):1476–1483. | ||
Meijer M, Rijkers GT, van Overveld FJ. Neutrophils and emerging targets for treatment in chronic obstructive pulmonary disease. Expert Rev Clin Immunol. 2013;9(11):1055–1068. | ||
Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;48(5):531–539. | ||
Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD. 2004;1(1):59–70. | ||
Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(5 Pt 2):S29–S32. | ||
Pappas K, Papaioannou AI, Kostikas K, Tzanakis N. The role of macrophages in obstructive airways disease: chronic obstructive pulmonary disease and asthma. Cytokine. 2013;64(3):613–625. | ||
Barnes PJ. Mediators of chronic obstructive pulmonary disease. Pharmacol Rev. 2004;56(4):515–548. | ||
O’Donnell R, Breen D, Wilson S, Djukanovic R. Inflammatory cells in the airways in COPD. Thorax. 2006;61(5):448–454. | ||
Kalathil SG, Lugade AA, Pradhan V, et al. T-regulatory cells and programmed death 1+ T cells contribute to effector T-cell dysfunction in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2014;190(1):40–50. | ||
Mills KH, Dunne A. Immune modulation: IL-1, master mediator or initiator of inflammation. Nat Med. 2009;15(12):1363–1364. | ||
Sedger LM, McDermott MF. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants – past, present and future. Cytokine Growth Factor Rev. 2014;25(4):453–472. | ||
Striz I, Brabcova E, Kolesar L, Sekerkova A. Cytokine networking of innate immunity cells: a potential target of therapy. Clin Sci (Lond). 2014;126(9):593–612. | ||
Horiuchi K, Kimura T, Miyamoto T, et al. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol. 2007;179(5):2686–2689. | ||
Darnay BG, Aggarwal BB. Early events in TNF signaling: a story of associations and dissociations. J Leukoc Biol. 1997;61(5):559–566. | ||
Liu ZG. Molecular mechanism of TNF signaling and beyond. Cell Res. 2005;15(1):24–27. | ||
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. | ||
Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18(45):6087–6093. | ||
Garrington TP, Johnson GL. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11(2):211–218. | ||
Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75(1):50–83. | ||
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. | ||
Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14(1):10–22. | ||
Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. | ||
Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol. 2010;30(5):628–631. | ||
Leemans JC, Cassel SL, Sutterwala FS. Sensing damage by the NLRP3 inflammasome. Immunol Rev. 2011;243(1):152–162. | ||
Muroi M, Tanamoto K. TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-kappaB. J Leukoc Biol. 2008;83(3):702–707. | ||
O’Neill LA. Signal transduction pathways activated by the IL-1 receptor/toll-like receptor superfamily. Curr Top Microbiol Immunol. 2002;270:47–61. | ||
Bowie A, O’Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal generators for pro-inflammatory interleukins and microbial products. J Leukoc Biol. 2000;67(4):508–514. | ||
Bauernfeind F, Bartok E, Rieger A, Franchi L, Nunez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187(2):613–617. | ||
Vyleta ML, Wong J, Magun BE. Suppression of ribosomal function triggers innate immune signaling through activation of the NLRP3 inflammasome. PLoS One. 2012;7(5):e36044. | ||
Petrovski G, Ayna G, Majai G, et al. Phagocytosis of cells dying through autophagy induces inflammasome activation and IL-1beta release in human macrophages. Autophagy. 2011;7(3):321–330. | ||
Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol. 2007;179(3):1913–1925. | ||
Solle M, Labasi J, Perregaux DG, et al. Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001;276(1):125–132. | ||
Ather JL, Martin RA, Ckless K, Poynter ME. Inflammasome activity in non-microbial lung inflammation. J Environ Immunol Toxicol. 2014;1(3):108–117. | ||
De Nardo D, De Nardo CM, Latz E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol. 2014;184(1):42–54. | ||
Tam A, Sin DD. Pathobiologic mechanisms of chronic obstructive pulmonary disease. Med Clin North Am. 2012;96(4):681–698. | ||
Trupin L, Earnest G, San Pedro M, et al. The occupational burden of chronic obstructive pulmonary disease. Eur Respir J. 2003;22(3):462–469. | ||
Shaheen SO, Barker DJ, Holgate ST. Do lower respiratory tract infections in early childhood cause chronic obstructive pulmonary disease? Am J Respir Crit Care Med. 1995;151(5):1649–1651; discussion 1651–1652. | ||
Lomas DA, Silverman EK. The genetics of chronic obstructive pulmonary disease. Respir Res. 2001;2(1):20–26. | ||
Mayer AS, Newman LS. Genetic and environmental modulation of chronic obstructive pulmonary disease. Respir Physiol. 2001;128(1):3–11. | ||
Fischer BM, Voynow JA, Ghio AJ. COPD: balancing oxidants and antioxidants. Int J Chron Obstruct Pulmon Dis. 2015;10:261–276. | ||
Sparrow D, O’Connor G, Weiss ST. The relation of airways responsiveness and atopy to the development of chronic obstructive lung disease. Epidemiol Rev. 1988;10:29–47. | ||
Ulrik CS, Backer V. Nonreversible airflow obstruction in life-long nonsmokers with moderate to severe asthma. Eur Respir J. 1999;14(4):892–896. | ||
Pauly JL, Smith LA, Rickert MH, Hutson A, Paszkiewicz GM. Review: Is lung inflammation associated with microbes and microbial toxins in cigarette tobacco smoke? Immunol Res. 2010;46(1–3):127–136. | ||
Pauly JL, Paszkiewicz G. Cigarette smoke, bacteria, mold, microbial toxins, and chronic lung inflammation. J Oncol. 2011;2011:819129. | ||
Centers for Disease Control and Prevention (US), National Center for Chronic Disease Prevention and Health Promotion (US), Office on Smoking and Health (US). How tobacco smoke causes disease. 3, chemistry and toxicology of cigarette smoke and biomarkers of exposure and harm. In: Sidransky D, Norman LA, McCarthy A, Taylor PL, editors. The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Atlanta, GA: Centers for Disease Control and Prevention (US); 2010. Available from: http://www.ncbi.nlm.nih.gov/books/NBK53014/. Accessed June 10, 2016. | ||
Fowles J, Dybing E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob Control. 2003;12(4):424–430. | ||
Mortaz E, Henricks PA, Kraneveld AD, Givi ME, Garssen J, Folkerts G. Cigarette smoke induces the release of CXCL-8 from human bronchial epithelial cells via TLRs and induction of the inflammasome. Biochim Biophys Acta. 2011;1812(9):1104–1110. | ||
Mortaz E, Adcock IM, Ito K, Kraneveld AD, Nijkamp FP, Folkerts G. Cigarette smoke induces CXCL8 production by human neutrophils via activation of TLR9 receptor. Eur Respir J. 2010;36(5):1143–1154. | ||
Smith LA, Paszkiewicz GM, Hutson AD, Pauly JL. Inflammatory response of lung macrophages and epithelial cells to tobacco smoke: a literature review of ex vivo investigations. Immunol Res. 2010;46(1–3):94–126. | ||
Holt PG, Keast D. The effect of tobacco smoke on protein synthesis in macrophages. Proc Soc Exp Biol Med. 1973;142(4):1243–1247. | ||
Leffingwell CM, Low RB. Cigarette smoke components and alveolar macrophage protein synthesis. Arch Environ Health. 1979;34(2):97–102. | ||
Yeager H Jr. Alveolar cells: depression effect of cigarette smoke on protein synthesis. Proc Soc Exp Biol Med. 1969;131(1):247–250. | ||
Vargas-Rojas MI, Ramirez-Venegas A, Limon-Camacho L, Ochoa L, Hernandez-Zenteno R, Sansores RH. Increase of Th17 cells in peripheral blood of patients with chronic obstructive pulmonary disease. Respir Med. 2011;105(11):1648–1654. | ||
Greene CM, Low TB, O’Neill SJ, McElvaney NG. Anti-proline-glycine-proline or antielastin autoantibodies are not evident in chronic inflammatory lung disease. Am J Respir Crit Care Med. 2010;181(1):31–35. | ||
Rinaldi M, Lehouck A, Heulens N, et al. Antielastin B-cell and T-cell immunity in patients with chronic obstructive pulmonary disease. Thorax. 2012;67(8):694–700. | ||
Marumo S, Hoshino Y, Kiyokawa H, et al. P38 mitogen-activated protein kinase determines the susceptibility to cigarette smoke-induced emphysema in mice. BMC Pulm Med. 2014;14:79. | ||
van der Vaart H, Postma DS, Timens W, ten Hacken NH. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax. 2004;59(8):713–721. | ||
Mukaro VR, Hodge S. Airway clearance of apoptotic cells in COPD. Curr Drug Targets. 2011;12(4):460–468. | ||
Doz E, Noulin N, Boichot E, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. 2008;180(2):1169–1178. | ||
Eltom S, Belvisi MG, Stevenson CS, et al. Role of the inflammasome-caspase1/11-IL-1/18 axis in cigarette smoke driven airway inflammation: An insight into the pathogenesis of COPD. PLoS One. 2014;9(11):e112829. | ||
Eltom S, Stevenson CS, Rastrick J, et al. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLoS One. 2011;6(9):e24097. | ||
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153(2):348–361. | ||
Kang MJ, Yoon CM, Kim BH, et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J Clin Invest. 2015;125(6):2458–2462. | ||
Karimi R, Tornling G, Grunewald J, Eklund A, Skold CM. Cell recovery in bronchoalveolar lavage fluid in smokers is dependent on cumulative smoking history. PLoS One. 2012;7(3):e34232. | ||
Barnes PJ. The cytokine network in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2009;41(6):631–638. | ||
Di Stefano A, Caramori G, Barczyk A, et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. 2014;69(6):516–524. | ||
Shang J, Zhao J, Wu X, Xu Y, Xie J, Zhao J. Interleukin-33 promotes inflammatory cytokine production in chronic airway inflammation. Biochem Cell Biol. 2015;93(4):359–366. | ||
Xia J, Zhao J, Shang J, et al. Increased IL-33 expression in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2015;308(7):L619–L627. | ||
Lefrancais E, Roga S, Gautier V, et al. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc Natl Acad Sci U S A. 2012;109(5):1673–1678. | ||
Bae S, Kang T, Hong J, et al. Contradictory functions (activation/termination) of neutrophil proteinase 3 enzyme (PR3) in interleukin-33 biological activity. J Biol Chem. 2012;287(11):8205–8213. | ||
Luthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31(1):84–98. | ||
Shiels MS, Katki HA, Freedman ND, et al. Cigarette smoking and variations in systemic immune and inflammation markers. J Natl Cancer Inst. 2014;106(11):dju 294. Print 2014. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | ||
Taraseviciene-Stewart L, Douglas IS, Nana-Sinkam PS, et al. Is alveolar destruction and emphysema in chronic obstructive pulmonary disease an immune disease? Proc Am Thorac Soc. 2006;3(8):687–690. | ||
Barnes PJ. Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol. 2009;71:451–464. | ||
von Nussbaum F, Li VM. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Bioorg Med Chem Lett. 2015;25(20):4370–4381. | ||
Liang G, Bowen JP. Development of trypsin-like serine protease inhibitors as therapeutic agents: Opportunities, challenges, and their unique structure-based rationales. Curr Top Med Chem. 2016;16(13):1506–1529. | ||
Eduard W. Fungal spores: a critical review of the toxicological and epidemiological evidence as a basis for occupational exposure limit setting. Crit Rev Toxicol. 2009;39(10):799–864. | ||
Bennett JW, Klich M. Mycotoxins. Clin Microbiol Rev. 2003;16(3):497–516. | ||
Rocha O, Ansari K, Doohan FM. Effects of trichothecene mycotoxins on eukaryotic cells: a review. Food Addit Contam. 2005;22(4):369–378. | ||
Paterson RR. Fungi and fungal toxins as weapons. Mycol Res. 2006;110(Pt 9):1003–1010. | ||
Berek L, Petri IB, Mesterhazy A, Teren J, Molnar J. Effects of mycotoxins on human immune functions in vitro. Toxicol In Vitro. 2001;15(1):25–30. | ||
Kankkunen P, Rintahaka J, Aalto A, et al. Trichothecene mycotoxins activate inflammatory response in human macrophages. J Immunol. 2009;182(10):6418–6425. | ||
Kankkunen P, Valimaki E, Rintahaka J, et al. Trichothecene mycotoxins activate NLRP3 inflammasome through a P2X7 receptor and Src tyrosine kinase dependent pathway. Hum Immunol. 2014;75(2):134–140. | ||
Konigs M, Lenczyk M, Schwerdt G, Holzinger H, Gekle M, Humpf HU. Cytotoxicity, metabolism and cellular uptake of the mycotoxin deoxynivalenol in human proximal tubule cells and lung fibroblasts in primary culture. Toxicology. 2007;240(1–2):48–59. | ||
Islam Z, Gray JS, Pestka JJ. p38 Mitogen-activated protein kinase mediates IL-8 induction by the ribotoxin deoxynivalenol in human monocytes. Toxicol Appl Pharmacol. 2006;213(3):235–244. | ||
Ciacci-Zanella JR, Jones C. Fumonisin B1, a mycotoxin contaminant of cereal grains, and inducer of apoptosis via the tumour necrosis factor pathway and caspase activation. Food Chem Toxicol. 1999;37(7):703–712. | ||
Dong W, Azcona-Olivera JI, Brooks KH, Linz JE, Pestka JJ. Elevated gene expression and production of interleukins 2, 4, 5, and 6 during exposure to vomitoxin (deoxynivalenol) and cycloheximide in the EL-4 thymoma. Toxicol Appl Pharmacol. 1994;127(2):282–290. | ||
Meky FA, Hardie LJ, Evans SW, Wild CP. Deoxynivalenol-induced immunomodulation of human lymphocyte proliferation and cytokine production. Food Chem Toxicol. 2001;39(8):827–836. | ||
Ouyang YL, Azcona-Olivera JI, Pestka JJ. Effects of trichothecene structure on cytokine secretion and gene expression in murine CD4+ T-cells. Toxicology. 1995;104(1–3):187–202. | ||
Wang H, Yadav JS. Global gene expression changes underlying Stachybotrys chartarum toxin-induced apoptosis in murine alveolar macrophages: evidence of multiple signal transduction pathways. Apoptosis. 2007;12(3):535–548. | ||
Schmeits PC, Katika MR, Peijnenburg AA, van Loveren H, Hendriksen PJ. DON shares a similar mode of action as the ribotoxic stress inducer anisomycin while TBTO shares ER stress patterns with the ER stress inducer thapsigargin based on comparative gene expression profiling in Jurkat T cells. Toxicol Lett. 2014;224(3):395–406. | ||
Amuzie CJ, Harkema JR, Pestka JJ. Tissue distribution and proinflammatory cytokine induction by the trichothecene deoxynivalenol in the mouse: comparison of nasal vs oral exposure. Toxicology. 2008;248(1):39–44. | ||
Hirano S, Kataoka T. Deoxynivalenol induces ectodomain shedding of TNF receptor 1 and thereby inhibits the TNF-alpha-induced NF-kappaB signaling pathway. Eur J Pharmacol. 2013;701(1–3):144–151. | ||
Capasso L, Longhin E, Caloni F, Camatini M, Gualtieri M. Synergistic inflammatory effect of PM10 with mycotoxin deoxynivalenol on human lung epithelial cells. Toxicon. 2015;104:65–72. | ||
Zhou HR, Harkema JR, Yan D, Pestka JJ. Amplified proinflammatory cytokine expression and toxicity in mice coexposed to lipopolysaccharide and the trichothecene vomitoxin (deoxynivalenol). J Toxicol Environ Health A. 1999;57(2):115–136. | ||
Lichtenstein JH, Molina RM, Donaghey TC, et al. Pulmonary responses to Stachybotrys chartarum and its toxins: mouse strain affects clearance and macrophage cytotoxicity. Toxicol Sci. 2010;116(1):113–121. | ||
Pang VF, Lambert RJ, Felsburg PJ, Beasley VR, Buck WB, Haschek WM. Experimental T-2 toxicosis in swine following inhalation exposure: effects on pulmonary and systemic immunity, and morphologic changes. Toxicol Pathol. 1987;15(3):308–319. | ||
Creasia DA, Thurman JD, Wannemacher RW Jr, Bunner DL. Acute inhalation toxicity of T-2 mycotoxin in the rat and guinea pig. Fundam Appl Toxicol. 1990;14(1):54–59. | ||
Liu C, Shen H, Yi L, et al. Oral administration of aflatoxin G(1) induces chronic alveolar inflammation associated with lung tumorigenesis. Toxicol Lett. 2015;232(3):547–556. | ||
Guindon-Kezis KA, Mulder JE, Massey TE. In vivo treatment with aflatoxin B1 increases DNA oxidation, base excision repair activity and 8-oxoguanine DNA glycosylase 1 levels in mouse lung. Toxicology. 2014;321:21–26. | ||
Caloni F, Cortinovis C. Toxicological effects of aflatoxins in horses. Vet J. 2011;188(3):270–273. | ||
Parikh SA, Litzow MR. Philadelphia chromosome-negative acute lymphoblastic leukemia: therapies under development. Future Oncol. 2014;10(14):2201–2212. | ||
Barta SK, Zou Y, Schindler J, et al. Synergy of sequential administration of a deglycosylated ricin A chain-containing combined anti-CD19 and anti-CD22 immunotoxin (Combotox) and cytarabine in a murine model of advanced acute lymphoblastic leukemia. Leuk Lymphoma. 2012;53(10):1999–2003. | ||
Audi J, Belson M, Patel M, Schier J, Osterloh J. Ricin poisoning: a comprehensive review. JAMA. 2005;294(18):2342–2351. | ||
Bradberry SM, Dickers KJ, Rice P, Griffiths GD, Vale JA. Ricin poisoning. Toxicol Rev. 2003;22(1):65–70. | ||
Brown RF, White DE. Ultrastructure of rat lung following inhalation of ricin aerosol. Int J Exp Pathol. 1997;78(4):267–276. | ||
Roy CJ, Hale M, Hartings JM, Pitt L, Duniho S. Impact of inhalation exposure modality and particle size on the respiratory deposition of ricin in BALB/c mice. Inhal Toxicol. 2003;15(6):619–638. | ||
Wong J, Korcheva V, Jacoby DB, Magun BE. Proinflammatory responses of human airway cells to ricin involve stress-activated protein kinases and NF-kappaB. Am J Physiol Lung Cell Mol Physiol. 2007;293(6):L1385–L1394. | ||
Cook DL, David J, Griffiths GD. Retrospective identification of ricin in animal tissues following administration by pulmonary and oral routes. Toxicology. 2006;223(1–2):61–70. | ||
Doebler JA, Wiltshire ND, Mayer TW, et al. The distribution of [125I]ricin in mice following aerosol inhalation exposure. Toxicology. 1995;98(1–3):137–149. | ||
Ramsden CS, Drayson MT, Bell EB. The toxicity, distribution and excretion of ricin holotoxin in rats. Toxicology. 1989;55(1–2):161–171. | ||
Wong J, Korcheva V, Jacoby DB, Magun B. Intrapulmonary delivery of ricin at high dosage triggers a systemic inflammatory response and glomerular damage. Am J Pathol. 2007;170(5):1497–1510. | ||
Griffiths GD, Leek MD, Gee DJ. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J Pathol. 1987;151(3):221–229. | ||
Korcheva V, Wong J, Lindauer M, Jacoby DB, Iordanov MS, Magun B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol Immunol. 2007;44(10):2761–2771. | ||
Wang X, Mader MM, Toth JE, et al. Complete inhibition of anisomycin and UV radiation but not cytokine induced JNK and p38 activation by an aryl-substituted dihydropyrrolopyrazole quinoline and mixed lineage kinase 7 small interfering RNA. J Biol Chem. 2005;280(19):19298–19305. | ||
Jandhyala DM, Ahluwalia A, Obrig T, Thorpe CM. ZAK: a MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell Microbiol. 2008;10(7):1468–1477. | ||
David J, Wilkinson LJ, Griffiths GD. Inflammatory gene expression in response to sub-lethal ricin exposure in Balb/c mice. Toxicology. 2009;264(1–2):119–130. | ||
DaSilva L, Cote D, Roy C, et al. Pulmonary gene expression profiling of inhaled ricin. Toxicon. 2003;41(7):813–822. | ||
Lindauer ML, Wong J, Iwakura Y, Magun BE. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signaling. J Immunol. 2009;183(2):1419–1426. | ||
Lindauer M, Wong J, Magun B. Ricin toxin activates the NALP3 inflammasome. Toxins (Basel). 2010;2(6):1500–1514. | ||
Liang J, Zhao H, Yao L, et al. Phosphatidylinositol 3-kinases pathway mediates lung caspase-1 activation and high mobility group box 1 production in a toluene-diisocyanate induced murine asthma model. Toxicol Lett. 2015;236(1):25–33. | ||
Carter AB, Tephly LA, Venkataraman S, et al. High levels of catalase and glutathione peroxidase activity dampen H2O2 signaling in human alveolar macrophages. Am J Respir Cell Mol Biol. 2004;31(1):43–53. | ||
Geist LJ, Powers LS, Monick MM, Hunninghake GW. Asbestos stimulation triggers differential cytokine release from human monocytes and alveolar macrophages. Exp Lung Res. 2000;26(1):41–56. | ||
Li P, Liu T, Kamp DW, et al. The c-Jun N-terminal kinase signaling pathway mediates chrysotile asbestos-induced alveolar epithelial cell apoptosis. Mol Med Rep. 2015;11(5):3626–3634. | ||
Li X, Hu Y, Jin Z, Jiang H, Wen J. Silica-induced TNF-alpha and TGF-beta1 expression in RAW264.7 cells are dependent on Src-ERK/AP-1 pathways. Toxicol Mech Methods. 2009;19(1):51–58. | ||
Jiang Y, Zhang H, Wang Y, et al. Modulation of apoptotic pathways of macrophages by surface-functionalized multi-walled carbon nanotubes. PLoS One. 2013;8(6):e65756. | ||
Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320(5876):674–677. | ||
Cassel SL, Eisenbarth SC, Iyer SS, et al. The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A. 2008;105(26):9035–9040. | ||
Luna-Gomes T, Santana PT, Coutinho-Silva R. Silica-induced inflammasome activation in macrophages: role of ATP and P2X7 receptor. Immunobiology. 2015;220(9):1101–1106. | ||
Caicedo MS, Desai R, McAllister K, Reddy A, Jacobs JJ, Hallab NJ. Soluble and particulate Co-Cr-Mo alloy implant metals activate the inflammasome danger signaling pathway in human macrophages: a novel mechanism for implant debris reactivity. J Orthop Res. 2009;27(7):847–854. | ||
Meunier E, Coste A, Olagnier D, et al. Double-walled carbon nanotubes trigger IL-1beta release in human monocytes through Nlrp3 inflammasome activation. Nanomedicine. 2012;8(6):987–995. | ||
Li M, Gunter ME, Fukagawa NK. Differential activation of the inflammasome in THP-1 cells exposed to chrysotile asbestos and Libby “six-mix” amphiboles and subsequent activation of BEAS-2B cells. Cytokine. 2012;60(3):718–730. | ||
Sweeney S, Berhanu D, Misra SK, Thorley AJ, Valsami-Jones E, Tetley TD. Multi-walled carbon nanotube length as a critical determinant of bioreactivity with primary human pulmonary alveolar cells. Carbon N Y. 2014;78:26–37. | ||
De Backer L, Naessens T, De Koker S, et al. Hybrid pulmonary surfactant-coated nanogels mediate efficient in vivo delivery of siRNA to murine alveolar macrophages. J Control Release. 2015;217:53–63. | ||
Tang J, Lobatto ME, Hassing L, et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv. 2015;1(3):e1400223. | ||
Paunovic V, Harnett MM. Mitogen-activated protein kinases as therapeutic targets for rheumatoid arthritis. Drugs. 2013;73(2):101–115. | ||
Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–692. | ||
Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25(51):6887–6899. | ||
Madonna R, De Caterina R. Relevance of new drug discovery to reduce NF-kappaB activation in cardiovascular disease. Vascul Pharmacol. 2012;57(1):41–47. | ||
Craig EA, Stevens MV, Vaillancourt RR, Camenisch TD. MAP3Ks as central regulators of cell fate during development. Dev Dyn. 2008;237(11):3102–3114. | ||
Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–1051. | ||
Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26(1):127–132. | ||
Manley PW, Drueckes P, Fendrich G, et al. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 2010;1804(3):445–453. | ||
Vin H, Ching G, Ojeda SS, et al. Sorafenib suppresses JNK-dependent apoptosis through inhibition of ZAK. Mol Cancer Ther. 2014;13(1): 221–229. | ||
Rix U, Remsing Rix LL, Terker AS, et al. A comprehensive target selectivity survey of the BCR-ABL kinase inhibitor INNO-406 by kinase profiling and chemical proteomics in chronic myeloid leukemia cells. Leukemia. 2010;24(1):44–50. | ||
Pera T, Atmaj C, van der Vegt M, Halayko AJ, Zaagsma J, Meurs H. Role for TAK1 in cigarette smoke-induced proinflammatory signaling and IL-8 release by human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2012;303(3):L272–L278. | ||
Baudelet D, Lipka E, Millet R, Ghinet A. Involvement of the P2X7 purinergic receptor in inflammation: an update of antagonists series since 2009 and their promising therapeutic potential. Curr Med Chem. 2015;22(6):713–729. | ||
Churg A, Zhou S, Wang X, Wang R, Wright JL. The role of interleukin-1beta in murine cigarette smoke-induced emphysema and small airway remodeling. Am J Respir Cell Mol Biol. 2009;40(4):482–490. | ||
Rennard SI, Fogarty C, Kelsen S, et al. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(9):926–934. | ||
Juliana C, Fernandes-Alnemri T, Wu J, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285(13):9792–9802. | ||
Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. | ||
Marchetti C, Chojnacki J, Toldo S, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. 2014;63(4):316–322. | ||
Honda H, Nagai Y, Matsunaga T, et al. Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J Leukoc Biol. 2014;96(6):1087–1100. | ||
Barbieri SS, Zacchi E, Amadio P, et al. Cytokines present in smokers’ serum interact with smoke components to enhance endothelial dysfunction. Cardiovasc Res. 2011;90(3):475–483. | ||
Cozen W, Diaz-Sanchez D, James Gauderman W, et al. Th1 and Th2 cytokines and IgE levels in identical twins with varying levels of cigarette consumption. J Clin Immunol. 2004;24(6):617–622. | ||
Flemming J, Hudson B, Rand TG. Comparison of inflammatory and cytotoxic lung responses in mice after intratracheal exposure to spores of two different Stachybotrys chartarum strains. Toxicol Sci. 2004;78(2):267–275. | ||
Maestrelli P, di Stefano A, Occari P, et al. Cytokines in the airway mucosa of subjects with asthma induced by toluene diisocyanate. Am J Respir Crit Care Med. 1995;151(3 Pt 1):607–612. | ||
Johnson VJ, Yucesoy B, Luster MI. Prevention of IL-1 signaling attenuates airway hyper responsiveness and inflammation in a murine model of toluene diisocyanate-induced asthma. J Allergy Clin Immunol. 2005; 116(4):851–858. | ||
Zhang Y, Lee TC, Guillemin B, Yu MC, Rom WN. Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J Immunol. 1993;150(9):4188–4196. | ||
Kodavanti UP, Andrews D, Schladweiler MC, Gavett SH, Dodd DE, Cyphert JM. Early and delayed effects of naturally occurring asbestos on serum biomarkers of inflammation and metabolism. J Toxicol Environ Health A. 2014;77(17):1024–1039. | ||
Porter DW, Ye J, Ma J, et al. Time course of pulmonary response of rats to inhalation of crystalline silica: NF-kappa B activation, inflammation, cytokine production, and damage. Inhal Toxicol. 2002;14(4):349–367. | ||
Hubbard AK, Timblin CR, Shukla A, Rincon M, Mossman BT. Activation of NF-kappaB-dependent gene expression by silica in lungs of luciferase reporter mice. Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L968–L975. | ||
Johnston CJ, Driscoll KE, Finkelstein JN, et al. Pulmonary chemokine and mutagenic responses in rats after subchronic inhalation of amorphous and crystalline silica. Toxicol Sci. 2000;56(2):405–413. | ||
Rydman EM, Ilves M, Koivisto AJ, et al. Inhalation of rod-like carbon nanotubes causes unconventional allergic airway inflammation. Part Fibre Toxicol. 2014;11:48. | ||
Kido T, Tsunoda M, Kasai T, et al. The increases in relative mRNA expressions of inflammatory cytokines and chemokines in splenic macrophages from rats exposed to multi-walled carbon nanotubes by whole-body inhalation for 13 weeks. Inhal Toxicol. 2014;26(12):750–758. | ||
Kobayashi N, Naya M, Mizuno K, Yamamoto K, Ema M, Nakanishi J. Pulmonary and systemic responses of highly pure and well-dispersed single-wall carbon nanotubes after intratracheal instillation in rats. Inhal Toxicol. 2011;23(13):814–828. | ||
Han SG, Andrews R, Gairola CG. Acute pulmonary response of mice to multi-wall carbon nanotubes. Inhal Toxicol. 2010;22(4):340–347. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.