Back to Journals » Drug Design, Development and Therapy » Volume 14
Loxoprofen Sodium Alleviates Oxidative Stress and Apoptosis Induced by Angiotensin II in Human Umbilical Vein Endothelial Cells (HUVECs)
Authors Ji C, Yu Y, Zhang M, Yu W, Dong S ![]()
Received 4 June 2020
Accepted for publication 28 October 2020
Published 18 November 2020 Volume 2020:14 Pages 5087—5096
DOI https://doi.org/10.2147/DDDT.S266175
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Chuanzhao Ji,1 Yang Yu,2 Min Zhang,2 Wenyan Yu,2 Shuo Dong1
1Department of Cardiovascular Medicine Ward 2, Zibo Central Hospital, Zibo, Shandong 255020, People’s Republic of China; 2General Medicine Department of Zibo Central Hospital, Zibo, Shandong 255020, People’s Republic of China
Correspondence: Shuo Dong
Department of Cardiovascular Medicine Ward 2, Zibo Central Hospital, No. 54, Gongqingtuan West Road, Zhangdian District, Zibo, Shandong 255020, People’s Republic of China
Tel/Fax +86-0533-3179175
Email [email protected]
Background and Purpose: Endothelium exerts an important role in releasing vasoactive substances, maintaining the blood flow, regulating the growth of vessels, moderating the process of coagulation, and the balance of fibrinolytic system, the dysfunction of which is reported to result in arterial stiffness. The present study aimed to investigate the effects of loxoprofen sodium against HUVECs injury induced by angiotensin II.
Methods: The injury model on HUVECs was established through incubation with angiotensin II. The expression levels of AT2R, NOX-4, Bax, Bcl-2, and caspase-3 were evaluated using qRT-PCR and Western Blot. DCFH-DA assay was used to detect the production of ROS and ELISA assay was used to evaluate the level of reduced glutathione. Mitochondrial membrane potential (MMP) was measured using dihydrorhodamine 123 assay. MTT and LDH assays were utilized to determine the proliferation ability of HUVECs. The apoptosis rate of HUVECs was evaluated using flow cytometry.
Results: Loxoprofen sodium suppressed endothelial AT2R elevation by angiotensin II. Loxoprofen ameliorated Angiotensin II–induced production of ROS, reduced GSH, and NOX-2 and NOX-4 expression. Furthermore, Loxoprofen mitigated Angiotensin II, reduced mitochondrial membrane potential and improved cell viability, and suppressed LDH release by angiotensin II. Importantly, loxoprofen showed a beneficial role in protecting endothelial apoptosis by mitigating apoptotic machinery including the balanced expression of Bax, Bcl-2, and caspase-3 cleavage.
Conclusion: Loxoprofen sodium might alleviate the high ROS levels and apoptosis induced by angiotensin II in HUVECs.
Keywords: angiotensin II, apoptosis, endothelial cells, ROS
Introduction
Endothelium forms the interphase between the blood vessels and surrounding tissues. The endothelium is extensive in almost all the tissues and plays important roles in whole-body homeostasis by releasing vasoactive substances, maintaining the blood flow, regulating the growth of vessels, moderating the process of coagulation, and balancing the fibrinolytic system. Vascular endothelial cells not only function as the barrier between the inside and outside of a blood vessel but also play essential roles in synthesizing, releasing, transforming, activating, or inactivating vasoactive hormones.1,2 The vasoactive factors released by the vascular endothelial cells, such as nitric oxide, prostaglandin, and endothelin, exert important roles in regulating the vasomotor function of blood vessels, maintaining the homeostasis of vascular walls and the steady flow of blood.3–5 Therefore, the integrity and functional normality of vascular endothelial cells are of great significance in protecting us from cardiovascular diseases, such as arterial stiffness.6–8 Angiotensin II, which is a peptide transformed from angiotensin I, is reported to be involved in the process and development of arterial stiffness by stimulating the vessels to release excessive free radicals, which contribute to the damage inflammation, and dysfunction of vascular endothelial cells.9 Angiotensin II acts by binding two receptors, the angiotensin II type 1 receptor (AT1R) and the angiotensin II type 2 receptor (AT2R). AT1R and AT2R are widely used as the targets of drug development. Most studies show that AT2R often mediates effects opposing those of the AT1R.10
Oxidative stress (OS) induced by angiotensin is responsible for the damage to vascular endothelial cells. OS is a negative regulation reaction produced by radicals, which is regarded as an important factor that results in cell senescence and apoptosis.11 Mitochondrial dysfunction is caused by the extensive OS as the mitochondrial transmembrane potential and ATP levels decrease, which finally results in the apoptosis of cells.12 To protect vascular endothelial cells from the damage of harmful stimulators and decrease the risk of arterial stiffness, alleviating the apoptosis caused by extensive OS might be an effective therapeutic method.
Loxoprofen sodium is the first non-steroidal anti-inflammatory drug (NSAID), which belongs to the propionic acid group which includes commonly used drugs ibuprofen and naproxen.13,14 The main therapeutic mechanism of loxoprofen sodium is its inhibition on the synthesis of prostaglandin, and its analgesic effect is approximately 10–20 fold of several other NSAIDs such as indomethacin, ketoprofen, or naproxen.15 Recognized as a promising analgesic, anti-inflammatory and antipyretic effects have been observed on the clinical trials on loxoprofen sodium.16
So far, loxoprofen has been shown to be a very effective anti-inflammatory agent without side effects in the gastrointestinal tract.17 A clinical case report indicates combination usage of loxoprofen, and ACE antagonists can produce serious side effects in aged patients.18 However, its effect on vascular cells remains unclear. In the present study, we investigated the anti-apoptotic effects of loxoprofen sodium on vascular endothelial cells and explored its potential therapeutic function in treating arterial stiffness. By investigating the possible mechanism, the present study aimed to provide a fundamental theoretical basis for the clinical application of loxoprofen sodium against arterial stiffness.
Materials and Methods
Cell Culture and Treatments
Human umbilical vein endothelial cells (HUVECs) were purchased from American Type Culture Collection (ATCC, Rockville, MD USA). DMEM medium with 10% fetal bovine serum was used to culture the cells at 37°C with 5% CO2.
Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
The TRIzol reagent (Thermo Fisher Scientific, Waltham, USA) was used to isolate total RNA. Subsequently, 2 μg of total RNA with a Primescript material kit (Thermo Fisher Scientific, Waltham, USA) was used to synthesize cDNA. PCR amplification was performed with TransStart Tip Green qPCR SuperMix kit (Genscript, Nanjing, China). The 2−ΔΔCt method was used to compare the relative expressions of genes.
Western Blotting Assay
HUVECs were collected and lysed in RIPA buffer (Thermo Fisher Scientific, Waltham, USA) to generate crude extracts, and the proteins in the extracts were separated using 15% SDS-PAGE. Subsequently, the separated proteins were transferred to PVDF membranes (Thermo Fisher Scientific, Waltham, USA) using semi-dry transfer. The membranes were blocked by incubation with a 5–10% BSA solution for 1–2 hours. Then, the membranes were incubated with primary antibodies against AT2R, NOX-4, Bax, Bcl-2, caspase-3, and β-actin (Abcam, Cambridge, MA) at 25°C for 2 hours. After washing, the blot was incubated with a horseradish peroxidase-conjugated secondary antibody (1:3000, Abcam, Cambridge, MA) at 25°C for 1–2 hours. Finally, the blots were incubated with ECL reagents (Amersham Pharmacia Biotech, Inc, USA) and visualized with an Amersham Imager 600 (GE, United Kingdom).
2.7-Dichlorodi-Hydrofluorescein Diacetate (DCFH-DA) Staining
The ROS levels were evaluated using a fluorescent staining method with the ROS probe DCFH-DA (Thermo Fisher Scientific, Waltham, USA). HUVECs were collected and washed with PBS buffer and DCFH-DA was transfected into the cells by incubating with the HUVECs for 40 minutes at 37°C in the dark. The fluorescent images representing the ROS levels were observed and captured using an inverted microscope. The mean fluorescent intensity of DCFH-DA was calculated with Image-Pro Plus (version 5.0, Media Cybernetics, USA) to indicate the intracellular ROS levels.
Measurement of Reduced Glutathione
The concentration of reduced glutathione released by HUVECs was evaluated using an ELISA assay. Briefly, 100 μL different dilutions of the standard sample were added to the reaction plate and incubated at 37°C for 30 minutes. Secondly, to each well, 100 μL of test samples was added to the plate after washes and incubated at 37°C for 2 hours. Thereafter, the plates were washed before the addition of 100 μL horseradish peroxidase-labeled secondary antibody and incubated at 37°C for 30 minutes. Afterward, 50 μL developer A and 50 μL developer B were added. The plates were kept in the dark for 15 minutes. Lastly, to each well, 50 μL stop solution was added into the plate to terminate the reaction.19 The optical density of the plate was read at 450 nm using an ELISA reader (EXL808; BioTek Instruments), and the concentration of the test sample was calculated.
Dihydrorhodamine 123 Staining
1 g/L dihydrorhodamine 123 in DMSO was diluted by DMEM medium containing 10% fetal bovine serum to 10 mg/L and was added into the HUVECs cultural medium. The cells were washed with cold PBS for 5 minutes following incubation for 30 minutes at 37°C in the dark and loaded into the flow cytometry to detect the mitochondrial membrane potential.
ATP Production
The cellular ATP level was measured using a commercial ATP determination kit (Thermo Fisher Scientific, USA). Briefly, HUVECs were lysed to obtain the soluble portion. 50 μL of the samples were used to react with the equal volume of the D-luciferin-luciferase reagent. The intensity of chemiluminescence from the reaction was read using a microplate luminometer at 560 nM. The ATP concentrations of the samples were extrapolated from a standard curve, and the results were presented as the fold change relative to the control sample.
MTT Assay
Cells were seeded into plates (Corning, Corelle City, NY, USA) at 37°C for 24 hours. Then, 10 μL of 5 mg/mL MTT solution (Thermo Fisher Scientific, Waltham, USA) was mixed into the medium. Four hours later, the formazan was added to about 200 μL of dimethyl sulfoxide (DMSO, Genview, Beijing, China). The OD value at 490 nm was recorded using a microplate reader (BMG LABTECH). The value (ODcontrol-ODtreatment)/OD control was used to represent the suppressive rate.
LDH Release Assay
The injury on HUVECs was determined using the LDH-Cytotoxicity Colorimetric Assay Kit II (#K313-500, BioVision, USA). Briefly, HUVECs were seeded in a 24-well plate and a 10 μL medium solution per well was added into the cells. Subsequently, the cells were mixed with 100 μL LDH Reaction Mix to incubate for 30 minutes at room temperature. A microplate reader (EXL808; BioTek Instruments) was used to measure the absorbance at 450 nm.
Flow Cytometer Assay
HUVECs were collected into 1.5 mL tubes. 10 μL fluorescently labeled Annexin V reagent and 5 μL PI reagent was added to each tube. The tubes were incubated for 10 minutes under room temperature. Approximately, 200 μL cells were added into the flow tube containing 2 mL PBS and tested using the flow cytometry (Thermo Fisher Scientific, Waltham, USA). Three independent assays were performed.
Statistical Analysis
Mean ± standard deviation (SD) was displayed to show the data. Prism (version 6, GraphPad, USA) was used to analyze the data. Students’ t-test and one-way analysis of variance (ANOVA) were utilized for the contrast among different groups. P<0.05 was regarded as a statistically significant difference between the groups.
Results
Loxoprofen Suppresses the Expression of Endothelial AT2R
To establish the injured HUVECs model in-vitro, HUVECs were incubated with 10 µM angiotensin II. Two concentrations (10 and 20 μg/mL) of loxoprofen sodium, the chemical structure of which is shown in Figure 1A, were added to the culture medium to explore its effects on the expression of AT2R. To test the cytotoxicity of loxoprofen, we performed an MTT experiment with different doses of loxoprofen treatment for 24 hours. As shown in Figure 1B, the range of 0.1 to 20 μg/mL loxoprofen had no obvious influence on the viability of HUVECs, while 100 and 200 μg/mL loxoprofen reduced their viability to 88- and 82%, respectively (Figure 1B). Therefore, 10 and 20 μg/mL loxoprofen were used in all the following experiments. As shown in Figure 2A and B, the stimulation of angiotensin II induced about 2.8- and 2.4-fold AT2R mRNA and protein level, respectively. However, these inductions were significantly inhibited by the addition of loxoprofen, with higher doses (20 μg/mL) reducing AT2R mRNA and proteins to 1.7- and 1.6-fold, respectively.
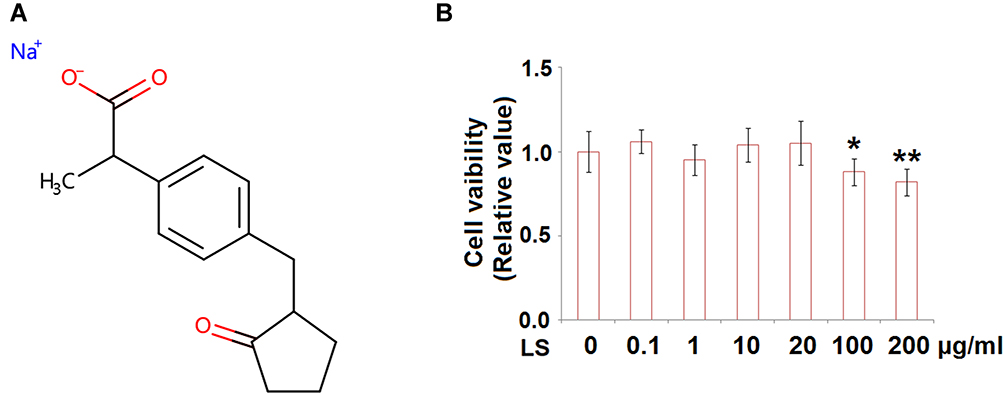 |
Figure 1 (A) Molecular structure of Loxoprofen sodium. (B) Cytotoxicity of Loxoprofen. Cells were treated with Loxoprofen sodium (0, 0.1, 1, 10, 20, 100, 200 μg/mL) for 24 hours. Cell viability was measured using MTT assay (N=3, *, **, P<0.05, 0.01 vs vehicle group). |
 |
Figure 2 Loxoprofen sodium reduced the expression of Ang-II receptor type 2 (AT2R) in human umbilical vein endothelial cells (HUVECs). Cells were treated with 10 µM angiotensin II in the presence or absence of Loxoprofen sodium (10, 20 μg/mL) for 24 h. (A) mRNA of AT2R (N=5); (B) Protein of AT2R (****, P<0.0001 vs vehicle group; #, ##, P<0.05, 0.01 vs Ang-II treatment group, N=3). |
Loxoprofen Suppresses Endothelial ROS Elevation by Angiotensin II
Following the stimulation with angiotensin II, the levels of ROS in HUVECs were greatly elevated, the result is shown in Figure 3A. Angiotensin II increased ROS production 3.5-fold, but 10 and 20 μg/mL loxoprofen ameliorated its induction to only 2.4- and 1.6-fold, respectively. As shown in Figure 3B, the concentration of reduced glutathione (GSH) in angiotensin II-treated HUVECs was reduced to 53%, of that of control. However, 10 and 20 μg/mL loxoprofen doses responsively recovered 78% and 93% of reduced GSH, respectively. Results in Figure 4A and B show the mRNA and protein expression levels of ROS related protein, NOX-2, and NOX-4, in treated HUVECs. Consistently, angiotensin II treatment increased both the expressions of NOX-2 and NOX-4, which were reduced by 10 and 20 μg/mL loxoprofen.
 |
Figure 3 Loxoprofen sodium prevented Ang-II-induced oxidative stress in human umbilical vein endothelial cells (HUVECs). (A) Production of ROS (N=3); (B) the levels of reduced GSH (***, ****, P<0.001, 0.0001 vs vehicle group; #, ##, P<0.05, 0. 01 vs Ang-II treatment group, N=3). |
 |
Figure 4 Loxoprofen sodium prevented Ang-II-induced expression of NOX-4 in human umbilical vein endothelial cells (HUVECs). (A) mRNA of NOX-2 and NOX-4 (N=5); (B) protein of NOX-2 and NOX-4 (****, P<0.0001 vs vehicle group; #, ##, P<0.05, 0.01 vs Ang-II treatment group, N=3). |
Loxoprofen Ameliorates Mitochondrial Function and Protects Endothelial Viability
To explore the effects of loxoprofen sodium on the mitochondrial function and cell viability of injured HUVECs, dihydrorhodamine 123 assay, and MTT assay were performed. As shown in Figure 5A, the mitochondrial membrane potential in HUVECs was reduced to 46% percent of the control by the stimulation of angiotensin II but was elevated to 73% and 89% by incubating with 10 or 20 μg/mL loxoprofen, respectively. Meanwhile, the results in Figure 5B show that exposure to angiotensin II resulted in the ATP production being reduced to 68% of the control group, however, treatment with loxoprofen sodium significantly recovered the ATP production to 83- and 95% of the control group, respectively. Figure 6A shows that cell viability was reduced from 100% to 62% by the introduction of angiotensin II and promoted to 81% and 93% by incubating with 10 and 20 μg/mL loxoprofen sodium, respectively. The release of LDH was used to represent the cell death rate, the result is shown in Figure 6B. The release of LDH was promoted from 6.9% to 39.8% following the stimulation of angiotensin II, but reduced to 22.3% and 15.8% by the introduction of 10 and 20 μg/mL loxoprofen sodium, respectively.
 |
Figure 5 (A) Loxoprofen sodium prevented Ang-II-induced reduction of mitochondrial membrane potential (Δψm) in human umbilical vein endothelial cells (HUVECs). Mitochondrial membrane potential (Δψm) was measured by dihydrorhodamine 123 (N=3). (B) Loxoprofen recovered angiotensin II reduced ATP production. Cells were treated with 10 µM angiotensin II in the presence or absence of loxoprofen sodium (10, 20 μg/mL) for 24 hours (****, P<0.0001 vs vehicle group; #, ##, P<0.05, 0.01 vs Ang-II treatment group, N=3). |
 |
Figure 6 Loxoprofen sodium prevented Ang-II-induced reduction of cell viability and release of lactate dehydrogenase (LDH) in HUVECs. Cells were treated with 10 µM angiotensin II in the presence or absence of Loxoprofen sodium (10, 20 μg/mL) for 24 h. (A) Cell viability (N=3); (B) release of LDH (****, P<0.0001 vs vehicle group; #, ##, ###, P<0.05, 0.01, 0.001 vs Ang-II treatment group, N=3). |
Loxoprofen Protects Angiotensin II-Induced Endothelial Apoptosis
Flow cytometry was used to detect the apoptosis rate of HUVECs, and the results are shown in Figure 7A and B. The cell apoptosis rate was promoted from 7.9% to 32.1% after the stimulation with angiotensin II and was suppressed to 20.9% and 10.5% following the introduction of 10 and 20 μg/mL loxoprofen sodium, respectively. To verify the effects of loxoprofen sodium on apoptosis, the expression levels of apoptotic proteins were evaluated using Western Blot and qRT-PCR experiments. As shown in Figure 8A and B, Bax and cleaved caspase-3 were significantly up-regulated in HUVECs by the administration of angiotensin II, the expressions of which were greatly suppressed by the introduction of 10 and 20 μg/mL loxoprofen sodium. The expression of Bcl-2 was inhibited by angiotensin II, and elevated by 10 and 20 μg/mL loxoprofen sodium.
 |
Figure 7 Loxoprofen sodium prevented Ang-II-induced apoptosis in HUVECs. Cells were treated with 10 µM angiotensin II in the presence or absence of Loxoprofen sodium (10, 20 μg/mL) for 24 h. (A) Cell apoptosis was measured by the flow cytometer assay (N=3); (B) quantification of apoptosis (****, P<0.0001 vs vehicle group; ##, ###, P<0.01, 0.001 vs Ang-II treatment group, N=3). |
 |
Figure 8 Loxoprofen sodium prevented Ang-II- induced reduction of Bcl-2 and an increase in expression of Bax and cleavage of caspase-3. Cells were treated with 10 µM angiotensin II in the presence or absence of loxoprofen sodium (10, 20 μg/mL) for 24 h. (A) Expression of Bax and Bcl-2 (N=3); (B) cleavage of caspase-3; (****, P<0.0001 vs vehicle group; ##, ###, P<0.01, 0.001 vs Ang-II treatment group, N=3). |
Discussion
Vascular endothelial cells (VEC) are not only the mechanical barrier between the circulating blood and vascular smooth muscle cells but also one of the most important endocrine organs in our body, by which only a small fraction of materials is permitted to enter the vascular walls.20,21 Two types of bioactive factors are released by VECs: the anti-arteriosclerosis factors, such as nitric oxide, prostaglandin, and tissue plasminogen activators, and pro-arteriosclerosis factors, such as endothelin-1, angiotensin II, and prostaglandin and tissue plasminogen inhibitors. A steady balance is kept between these factors under normal physiological conditions to maintain the steady blood flow. However, cardiovascular diseases, such as arterial stiffness, will develop when the function of endothelium is impaired.22,23 Angiotensin II is reported to be one of the main factors that contribute to the process and development of arterial stiffness. The infiltration of inflammatory cells into the injured VECs could be induced by angiotensin II, which will be produced by inflammatory cells in turn. A malignant cycle is formed to impact the function of VECs.24 In addition, the synthesis of NF-κB can be promoted by angiotensin II, which is a transcriptional factor that up-regulates the expression of adherence factors, inflammatory factors, epoxidase-2, and angiotensinogen.25,26 It is reported that Angiotensin II–induced mitochondrial dysfunction reduces endothelial NO bioavailability and promotes ROS generation,27 therefore Angiotensin II–impaired mitochondrial function could be one of the mechanisms of endothelial dysfunction. Besides, the renin-angiotensin system (RAS) can be activated by angiotensin II to inhibit the function of progenitor cells, which has been reported to be beneficial to the recovery of VEC function and alleviates the symptoms of arterial stiffness.28 In the present study, angiotensin II was used to establish the in-vitro HUVEC injury model, which was verified to be characterized as an up-regulated AT2R, elevated ROS levels, impaired mitochondrial membrane potential, reduced ATP production, reduced cell viability and a higher apoptosis rate. The angiotensin II–injured HUVECs were used to evaluate the protective effects of loxoprofen sodium in this model. Our data shows loxoprofen promoted cell viability and inhibited LDH release, indicating that loxoprofen plays a protective role against angiotensin II–induced HUVEC injury. A representative schematic of the molecular mechanisms is shown in Figure 9. As illustrated in Figure 9, loxoprofen mitigates AT2R receptor expression on the endothelial surface which is known to be a protective receptor of Angiotensin II. Loxoprofen appears to regulate intracellular signaling by modulating Bax/Bcl-2 ratio on the mitochondrial compartment and balanced expression of NOX-2 and NOX-4. As a result, loxoprofen works as a beneficial antioxidant agent to protect Angiotensin II triggered endothelial apoptosis.
 |
Figure 9 A representative schematic of the molecular mechanisms. |
To further explore the potential therapeutic effects of loxoprofen sodium against arterial stiffness, the in-vivo animal models will be well designed to evaluate the systemic preclinical pharmacodynamic function of loxoprofen sodium in our future work. As the most important effector molecule in the renin angiotensin-aldosterone-system, angiotensin II is reported to induce excessive oxidative stress reactions in HUVECs.29 Rajagopalan30 reported that the diastolic reactivity of artery vessels against acetylcholine decreased greatly following the administration of angiotensin II into normal rats, which was found to be related to the production of superoxide anions.
Endothelial cells express different types of NOX protein including NOX-1, NOX-2, NOX-4, and NOX-5. Among them, NOX-2 and NOX-4 are the dominant forms in endothelial cells.31 The expression levels of NOX-2 and NOX-4 could be up-regulated in the endothelial progenitor cells by angiotensin II to produce excessive reactive oxygen but reversed by telmisartan, an antagonist of angiotensin.32 Tsuneki33 verified that the ROS were induced and NOX-2 was up-regulated in the HUVECs incubated with 100 nM angiotensin II and reversed by the introduction of Coenzyme Q10. In the present study, by stimulating HUVECs with 10 µM angiotensin II, the production of ROS was significantly promoted and the release of GSH was significantly reduced, which was consistent with previous reports. NOX-4, an oxidase of NADPH, was up-regulated by angiotensin II. These data indicated that the ROS levels in HUVECs were greatly elevated by angiotensin II, which was a factor that induced HUVEC injury. Following the introduction of loxoprofen sodium, the production of ROS was suppressed, the releasing of GSH was elevated and the expression level of NOX-4 was inhibited. These data indicate that the promoted ROS levels by angiotensin II were significantly reversed by loxoprofen sodium, which implied that the HUVECs, injured by angiotensin II might be repaired by loxoprofen sodium.
Loxoprofen is a potent and non-selective inhibitor of cyclooxygenases including COX-1 and COX-2, two rate-limiting enzymes for the production of prostaglandin (PG) and thromboxane (TX). Loxoprofen has been shown to suppress both cyclooxygenases COX-1 and COX-2 in both in vitro and in vivo experiments.34–36 In endothelial cells, COX-1-derived vasoconstrictor PGs contribute to endothelial dysfunction, but COX-2 derived contractile factors play complicated roles. It has been reported that COX-2 plays a beneficial role in hypertensive patients, but its inhibition in coronary artery disease improves vascular function and flow-mediated dilatation.37 The beneficial role of Loxoprofen on Angiotensin II–induced endothelial injury could be mediated by the inhibition of endothelial COX-1 and COX-2 to mitigate the imbalance of inflammation and vasotone. According to our data, the mechanistic role of loxoprofen could rely on its suppression of endothelial AT2R receptor expression. Secondly, it is reported that Angiotensin II–induced mitochondrial dysfunction reduces endothelial NO bioavailability and promotes ROS generation,27 therefore, Angiotensin II impaired mitochondrial function could be one of the mechanisms of endothelial dysfunction. A previous study showed that COX-1/COX-2 inhibition raises cellular levels of arachidonic acid, which can lead to the inhibition of mitochondrial oxidative phosphorylation (OXPHOS) and increase in generation of mitochondrial-derived reactive oxygen species.38 In our study, loxoprofen treatment indeed ameliorated impaired mitochondrial membrane potential and reduced ROS production, suggesting loxoprofen could protect mitochondrial function to promote cell survival. In future work, the effects of loxoprofen sodium on ROS levels and flow-mediated vasodilation in animal models should be investigated to validate its pharmacological effect on endothelial dysfunction.
AT2R is one of the functional receptors of angiotensin II, which has been proven to mediate the progress of apoptosis. The function of mitrisin-activated protein kinase phosphatase (MAKP-1) can be promoted following the activation of AT2R, which induces the dephosphorylation of anti-apoptotic proteins, such as Bcl-2, and the activation of pro-apoptotic proteins, such as caspase-3. Therefore, in this way, the apoptosis of HUVECs occurs.39 In addition, the level of ceramide can be promoted by the activation of AT2R to induce the apoptosis of the neurogenic PC12 cells.40 Neuron growth factor (NGF) is reported to activate Bcl-2 through activating the ERK signal pathway, which is inhibited by the angiotensin II/AT2R signal pathway.41 In the present study, the apoptosis rate of HUVECs was significantly promoted by angiotensin II, which also induced the up-regulation of AT2R. Simultaneously, Bcl-2 was down-regulated whereas Bax, caspase-3 were up-regulated, which was consistent with previous reports. Following the introduction of loxoprofen sodium, the apoptotic state of HUVECs was significantly alleviated. These data indicated that the apoptosis induced by angiotensin II/AT2R signal pathway was greatly reversed by loxoprofen sodium. Besides the receptors, the availability of circulatory angiotensin II is also critical to vascular function. ACE antagonists have been used to modulate the vascular function.42 ACE converts the inactive angiotensin I to the active angiotensin II or inactivate kinins, while ACE antagonists bind to ACE and decrease circulating Angiotensin II by inhibiting ACE activity. Several ACE antagonists, such as zofenoprilat (ZOF), enalaprilat, lisinopril, and captopril, have been employed to suppress the level angiotensin II in endothelial cells.43,44 One of the limitations of this study is that we could not use ACE antagonists as a positive group to block Angiotensin II pathway in our loxoprofen treatment experiment. Furthermore, the findings of loxoprofen in cultured endothelial cells should be tested in animal models. A future experiment in vivo is required to validate the regulatory effect of loxoprofen in the vascular system. Collectively, our data shows solid evidence that the COX inhibitor drug loxoprofen sodium possesses a protective effect to repair endothelial injury caused by angiotensin II.
Acknowledgments
This study is supported by the “Shandong Scientific Support Project (No. 1632050033)”.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mack JJ, Iruela-Arispe ML. NOTCH regulation of the endothelial cell phenotype. Curr Opin Hematol. 2018;25(3):212. doi:10.1097/MOH.0000000000000425
2. Suburo AM, D’Amore PA. Development of the endothelium. Handb Exp Pharmacol. 2006;71.
3. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: pathogenesis and clinical management. BMJ. 2018;360:j5492. doi:10.1136/bmj.j5492
4. Zhang J, Zhao WS, Wang X, Xu L, Yang XC. Palmitic acid increases endothelin-1 expression in vascular endothelial cells through the induction of endoplasmic reticulum stress and protein kinase C signaling. Cardiology. 2018;140(3):133. doi:10.1159/000490093
5. Nava E, Llorens S. The local regulation of vascular function: from an inside-outside to an outside-inside model. Front Physiol. 2019;10:729. doi:10.3389/fphys.2019.00729
6. Del Campo L, Sanchez-Lopez A, Salaices M, et al. Vascular smooth muscle cell-specific progerin expression in a mouse model of Hutchinson-Gilford progeria syndrome promotes arterial stiffness: therapeutic effect of dietary nitrite. Aging Cell. 2019;18:e12936. doi:10.1111/acel.12936
7. Xue C, Zhang T, Xie X, et al. Substrate stiffness regulates arterial-venous differentiation of endothelial progenitor cells via the Ras/Mek pathway. Biochim Biophys Acta Mol Cell Res. 2017;1864(10):1799. doi:10.1016/j.bbamcr.2017.07.006
8. Mozos I, Luca CT. Crosstalk between oxidative and nitrosative stress and arterial stiffness. Curr Vasc Pharmacol. 2017;15(5):446. doi:10.2174/1570161115666170201115428
9. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292(1):C82. doi:10.1152/ajpcell.00287.2006
10. Kaschina E, Namsolleck P, Unger T. AT2 receptors in cardiovascular and renal diseases. Pharmacol Res. 2017;125(Pt A):39–47. doi:10.1016/j.phrs.2017.07.008
11. Luzzatto L, Nannelli C, Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Hematol Oncol Clin North Am. 2016;30(2):373. doi:10.1016/j.hoc.2015.11.006
12. Kang Z, Qiao N, Liu G, Chen H, Tang Z, Li Y. Copper-induced apoptosis and autophagy through oxidative stress-mediated mitochondrial dysfunction in male germ cells. Toxicol in Vitro. 2019;61:104639. doi:10.1016/j.tiv.2019.104639
13. Hamaguchi M, Seno T, Yamamoto A, et al. Loxoprofen Sodium, a non-selective NSAID, reduces atherosclerosis in mice by reducing inflammation. J Clin Biochem Nutr. 2010;47(2):138.
14. Zhao D, Chen Z, Hu S, et al. Efficacy and safety of loxoprofen hydrogel transdermalpatch versus loxoprofen tablet in Chinese patients with myalgia: a double-blind, double-dummy, parallel-group, randomized, controlled, non-inferiority trial. Clin Drug Investig. 2019;39(4):369. doi:10.1007/s40261-019-00756-x
15. Sekiguchi M, Shirasaka M, Konno S, Kikuchi S. Analgesic effect of percutaneously absorbed non-steroidal anti-inflammatory drugs: an experimental study in a rat acute inflammation model. BMC Musculoskelet Disord. 2008;9(1):15. doi:10.1186/1471-2474-9-15
16. Azuma A, Kudoh S, Nakashima M, Nagatake T. Antipyretic and analgesic effects of zaltoprofen for the treatment of acute upper respiratory tract infection: verification of a noninferiority hypothesis using loxoprofen sodium. Pharmacology. 2011;87(3–4):204. doi:10.1159/000324532
17. Yamakawa N, Suemasu S, Kimoto A, et al. Low direct cytotoxicity of loxoprofen on gastric mucosal cells. Biol Pharm Bull. 2010;33(3):398–403. doi:10.1248/bpb.33.398
18. Kurata C, Uehara A, Sugi T, Yamazaki K. Syncope caused by nonsteroidal anti-inflammatory drugs and angiotensin-converting enzyme inhibitors. Jpn Circ J. 1999;63(12):1002–1003. doi:10.1253/jcj.63.1002
19. Zhang H, Yang N, Wang T, Dai B, Shang Y. Vitamin D reduces inflammatory response in asthmatic mice through HMGB1/TLR4/NFkappaB signaling pathway. Mol Med Rep. 2018;17:2915.
20. Sturtzel C. Endothelial cells. Adv Exp Med Biol. 2017;1003:71.
21. Jiang F. Autophagy in vascular endothelial cells. Clin Exp Pharmacol Physiol. 2016;43(11):1021. doi:10.1111/1440-1681.12649
22. Gryglewski RJ, Chlopicki S, Uracz W, Marcinkiewicz E. Significance of endothelial prostacyclin and nitric oxide in peripheral and pulmonary circulation. Med Sci Monit. 2001;7:1.
23. Suzuki Y, Sano H, Tomczyk M, Brzoska T, Urano T. Activities of wild-type and variant tissue-type plasminogen activators retained on vascular endothelial cells. FEBS Open Bio. 2016;6(5):469. doi:10.1002/2211-5463.12057
24. Xu H, Du S, Fang B, et al. VSMC-specific EP4 deletion exacerbates angiotensin II-induced aortic dissection by increasing vascular inflammation and blood pressure. Proc Natl Acad Sci U S A. 2019;116(17):8457. doi:10.1073/pnas.1902119116
25. Wang C, Luo H, Xu Y, Tao L, Chang C, Shen X. Salvianolic acid B-alleviated angiotensin II induces cardiac fibrosis by suppressing NF-κB pathway in vitro. Med Sci Monit. 2018;24:7654. doi:10.12659/MSM.908936
26. Si W, Xie W, Deng W, et al. Angiotensin II increases angiogenesis by NF-κB–mediated transcriptional activation of angiogenic factor AGGF1. FASEB J. 2018;32(9):5051. doi:10.1096/fj.201701543RR
27. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II–mediated mitochondrial dysfunction. Circ Res. 2008;102(4):488–496. doi:10.1161/CIRCRESAHA.107.162800
28. Durik M, Seva Pessoa B, Roks AJ. The renin–angiotensin system, bone marrow and progenitor cells. Clin Sci (Lond). 2012;123(4):205. doi:10.1042/CS20110660
29. Yang M, Kahn AM. Insulin-stimulated NADH/NAD+ redox state increases NAD(P)H oxidase activity in cultured rat vascular smooth muscle cells. Am J Hypertens. 2006;19(6):587. doi:10.1016/j.amjhyper.2005.11.017
30. Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone.. J Clin Invest. 1996;97(8):1916. doi:10.1172/JCI118623
31. Drummond GR, Sobey CG. Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol Metab. 2014;25(9):452–463. doi:10.1016/j.tem.2014.06.012
32. Li H, Liu Q, Wang N, Xu J. Correlation of different NADPH oxidase homologues with late endothelial progenitor cell senescence induced by angiotensin II: effect of telmisartan. Intern Med. 2011;50(16):1631. doi:10.2169/internalmedicine.50.5250
33. Tsuneki H, Tokai E, Suzuki T, et al. Protective effects of coenzyme Q10 against angiotensin II-induced oxidative stress in human umbilical vein endothelial cells. Eur J Pharmacol. 2013;701(1–3):218. doi:10.1016/j.ejphar.2012.12.027
34. Noguchi M, Kimoto A, Gierse JK, et al. Enzymologic and pharmacologic profile of loxoprofen sodium and its metabolites. Biological & Pharmaceutical Bulletin. 2005;28(11):2075–2079. doi:10.1248/bpb.28.2075
35. Hamaguchi M, Seno T, Yamamoto A, et al. Loxoprofen sodium, a non-selective NSAID, reduces atherosclerosis in mice by reducing inflammation. J Clin Biochem Nutr. 2010;47(2):138–147. doi:10.3164/jcbn.10-33
36. Riendeau D, Salem M, Styhler A, Ouellet M, Mancini JA, Li CS. Li CS: evaluation of loxoprofen and its alcohol metabolites for potency and selectivity of inhibition of cyclooxygenase-2. Bioorg Med Chem Lett. 2004;14(5):1201–1203. doi:10.1016/j.bmcl.2003.12.047
37. Félétou M, Huang Y, Vanhoutte PM. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol. 2011;164(3):894–912.
38. Fosslien E. Cardiovascular complications of non-steroidal anti-inflammatory drugs. Ann Clin Lab Sci. 2005;35(4):347–385.
39. Calo LA, Schiavo S, Davis PA, et al. Angiotensin II signaling via type 2 receptors in a human model of vascular hyporeactivity: implications for hypertension. J Hypertens. 2010;28:111.
40. Tian M, Lin X, Wu L, Lu J, Zhang Y, Shi J. Angiotensin II triggers autophagy and apoptosis in PC12 cell line: an in vitro Alzheimer’s disease model. Brain Res. 2019;1718:46.
41. Anand U, Yiangou Y, Sinisi M, et al. Mechanisms underlying clinical efficacy of Angiotensin II type 2 receptor (AT2R) antagonist EMA401 in neuropathic pain: clinical tissue and in vitro studies. Mol Pain. 2015;11:38.
42. Rajagopalan S, Harrison DG. Reversing endothelial dysfunction with ACE inhibitors. A new trend. Circulation. 1996;94(3):240–243.
43. Desideri G, Grassi D, Croce G, et al. Different effects of angiotensin-converting enzyme inhibitors on endothelin-1 and nitric oxide balance in human vascular endothelial cells: evidence of an oxidant-sensitive pathway. Mediators Inflamm. 2008;2008:305087.
44. Evangelista S, Antioxidant MS. cardioprotective properties of the sulphydryl angiotensin-converting enzyme inhibitor zofenopril. J Int Med Res. 2005;33(1):42–54.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.