Back to Journals » OncoTargets and Therapy » Volume 12
Low-Grade Chondrosarcoma In The Sellar Area: Case Report And Literature Review
Authors Zhang Z, Pang LJ, Wang N, Li Z, Cao YW, Hu WH, Liang WH, Jiang JF, Zou H, Qi Y
Received 7 July 2019
Accepted for publication 29 October 2019
Published 10 December 2019 Volume 2019:12 Pages 10763—10770
DOI https://doi.org/10.2147/OTT.S221898
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Zhen Zhang,* Li-Juan Pang,* Ning Wang, Zhong Li, Yu-Wen Cao, Wen-Hao Hu, Wei-Hua Liang, Jin-Fang Jiang, Hong Zou, Yan Qi
Department of Pathology, Shihezi University School of Medicine & The First Affiliated Hospital to Shihezi University School of Medicine, Shihezi, Xinjiang 832002, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yan Qi; Hong Zou
Department of Pathology, Shihezi University School of Medicine & The First Affiliated Hospital to Shihezi University School of Medicine, North 2 Road, Shihezi, Xinjiang 832002, People’s Republic of China
Tel +86 15009932652;
+86 18999737799
Email [email protected]; [email protected]
Abstract: Low-grade chondrosarcoma (LGC) is a very rare intracranial tumor, particularly in the sellar area. Herein, we describe an unusual case of LGC occurring in the sellar area. A 52-year-old man presented with diminution of vision for more than 3 months, but did not exhibit headaches reported in previous cases. MRI showed that the maximum size of the tumor was 7 cm on the left side of the saddle. We characterized the specific pathological characteristics. Histologically, the tumor had polypoid areas and a lobulated growth pattern under low-power examination. At high magnification, the tumor consisted of small cells with hyperchromatic nuclei in the cartilage matrix, with an alternating loose hypocellular zone and rich myxoid area. In our case, LGC needed to be distinguished from chordoma. Immunohistochemically, the tumor cells showed diffuse positivity for S-100 and vimentin, IDH1 was weakly cytoplasm positive. The Ki-67 labeling index was less than 5%. Additionally, AE1/3, EMA, and CK19 were negative, which could be used to exclude chordoma. This case report expands the literature on LGC and will help clinicians and pathologists better understand this entity.
Keywords: low-grade chondrosarcoma, saddle area, differential diagnosis
Introduction
Chondrosarcomas are the second most frequent primary malignant bone tumor, characterized by hyaline cartilaginous neoplastic tissue.1 They account for approximately 25% of all primary bone tumors.2 Common sites for chondrosarcomas include the pelvis, shoulder, and long bones.3 Chondrosarcomas are resistant to radiotherapy and chemotherapy, so the main treatment remains surgery.2 Chondrosarcomas occurring in intracranial regions are rare, representing only 0.15–0.16% of all intracranial tumors.4 In an analysis of 560 patients with cranial chondrosarcomas, Bloch et al5 found that 32% of cases involved the clivus and 27% were at the temporo-occipital junction, and there were no definitive data on the incidence of chondrosarcoma in the sellar region. Only 9 cases of intercranial chondrosarcomas in the sellar area have been reported in the literature (Table 1).6–14 Common sellar lesions include pituitary adenomas, craniopharyngiomas, and Rathke’s cleft cysts.15 The majority of neoplasms in the sellar and parasellar regions originate from the pituitary, and only 10% are non-pituitary lesions.7 Freda et al reported that among 911 cases of sellar lesions, only 83 were non-pituitary lesions, 11% of which were chondrogenic tumors and chordomas.16
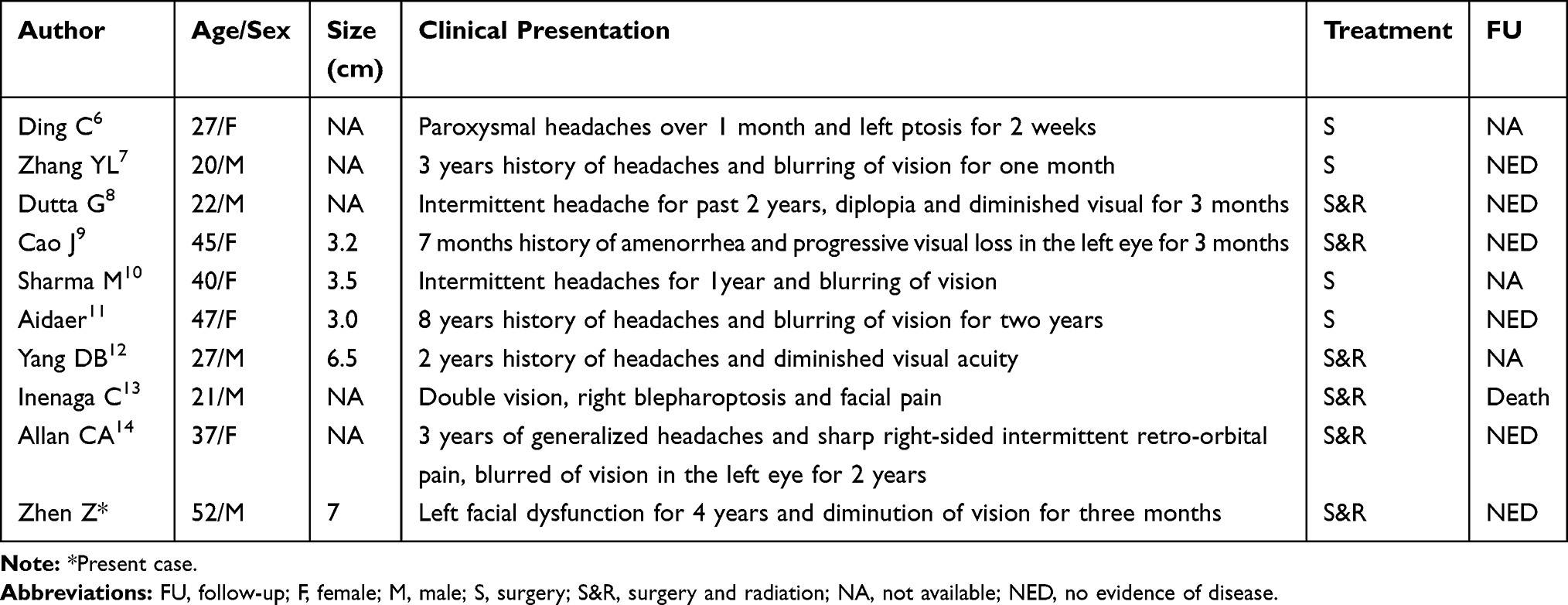 |
Table 1 Review Of Reported 9 Cases Of Chondrosarcoma In Sellar Area |
In this study, a case of LGC in the sellar area, which has rarely been reported in the literature, is presented. The clinical symptoms of intracranial chondrosarcoma depend on tumor size, location, and growth rate. The most common clinical manifestation is headache, but this patient only had reduced vision. We describe the clinicopathological and immunohistochemical features of this case of LGC and present a literature review. Our data provide important insights into the differential diagnosis of LGC in the sellar area.
Case Presentation
A 52-year-old man presented with left facial dysfunction and went to a local hospital in 2016. Brain magnetic resonance imaging (MRI) at the local hospital revealed a mass in the saddle area. The tumor was 6.3 cm × 5.8 cm × 4.5 cm. The patient was referred to a tertiary hospital and was diagnosed with a hypophysoma. He received pharmacotherapy and his facial numbness went into mild remission. In 2017, his MRI showed that the tumor grew from 6.3 cm to 7 cm. He continued treatment with medicine. The patient felt that his eyesight diminished further, and in April of 2018, he was admitted to hospital. The patient had no history of trauma, no family history of any hereditary illness, and his neurological examinations were normal.
Preoperative computer tomography (CT) and MRI showed a huge tumor in the sellar area. A CT scan revealed a hyperintense mass with an indistinct boundary located in the saddle area, sphenoid sinus area, and left temporal lobe; the tumor demonstrated expansive growth (Figure 1A and B). MRI showed a 7 cm × 6 cm × 5 cm giant irregular mass in the left side saddle, sphenoid sinus, and medial temporal lobe. The tumor presented as non-uniform hyperintense signal on T1-weighted images and uniform hyperdense signal on T2-weighted images (Figure 1C and D). Based on preoperative imaging results, the provisional diagnosis was a malignant tumor of the sellar region. A neurosurgeon removed the tumor by transcranial endoscopy. During tumor removal, the surgeon found that the tumor tissue was soft and had a fish-like appearance, with a rich blood supply. The tumor was completely removed piece-by-piece under an endoscope. Postoperative CT imaging on the first day after surgery showed that the tumor was resected (Figure 1D–F). The patient accepted radiation therapy at the local hospital 1 month later (total dose, 50 Gy; 2 Gy once a day for 4 weeks). At a follow-up investigation 1 year after tumor resection, the patient’s condition was generally good.
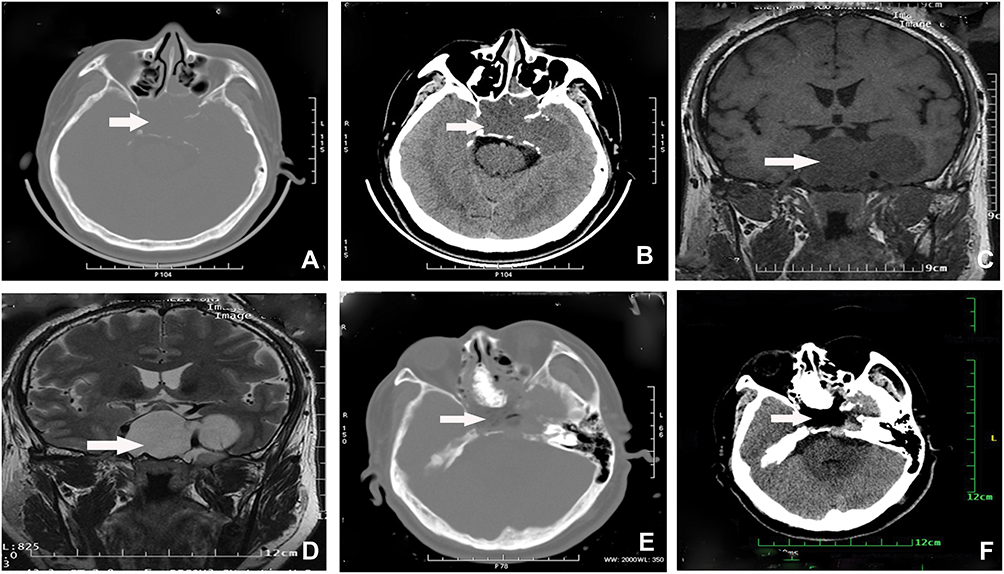 |
Figure 1 (A–D) Preoperative CT and MRI examination showed a large tumor in the sellar area. (A) CT bone window showed Bone thinning in the left region of occipital base, slope, medial lateral plate of left pterygial process, sphenoid bone and left superior ethmoid sinus wall. (B) CT plain scan showed a hypointense mass with an indistinct boundary located in the saddle area, sphenoid sinus area, and left temporal lobe. (C) MRI showed a giant irregular mass in the left saddle area with non-uniform iso-/hypointense on T1-weighted imaging (D) and uniform hyperdense on T2-weighted imaging. (E, F) Postoperative CT imaging examination showed that the tumor was resected. |
Materials And Methods
The resected specimens were fixed in 10% neutral-buffered formalin and routinely processed. Paraffin-embedded blocks were sectioned at a thickness of 5 μm and stained with hematoxylin and eosin. Immunohistochemical investigations were performed using paraffin-embedded tissue samples. The antibodies, clones, working dilutions, and their commercial sources are listed in Table 2.
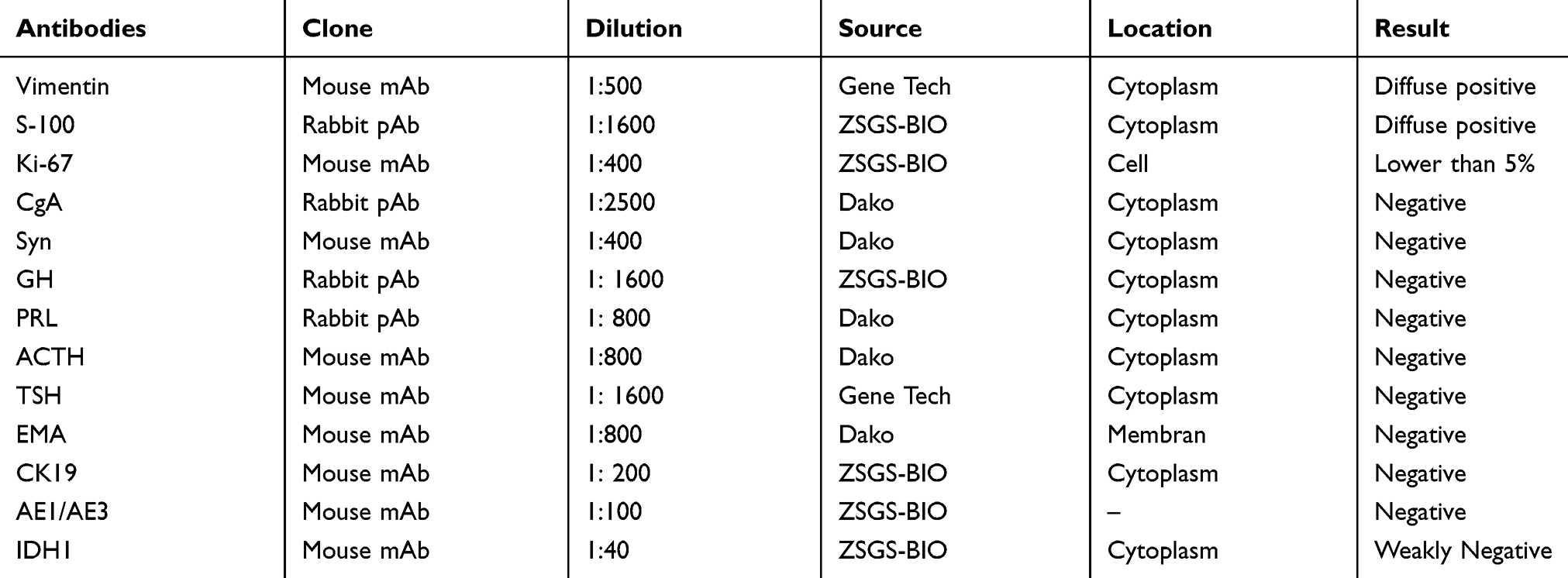 |
Table 2 Antibodies Used In This Case |
Results
Pathological Findings
Grossly, the tumor size was 3.5 cm × 5.1 cm × 2 cm. The main portion of the tumor was semi-translucent and gelatinous. Some areas were polypoid. On sectioning, the tumor was soft, tan-red-whitish, and resembled the flesh of a fish.
Microscopically, the tumor was located within polyp-like tissues, clearly demarcated from normal tissues in the cartilage matrix (Figure 2A and B). The surface of the polyp was covered with columnar epithelial cells. At medium magnification, loose tissue was detected in mucinous areas (Figure 2C). The chondroid area with calcification demonstrated that tumor cells were interspersed in trabecular bone, suggesting an invasive manner (Figure 2D). The tumor cells were characterized by round nuclei and transparent cytoplasm, with mild cellular atypia (Figure 2E). There was noticeably greater cellularity in the tumor edge areas and the tumor displayed lobulation (Figure 2F). At high magnification, cells in the rich cellular area had small hyperchromatic nuclei and no mitosis (Figure 2G). The chondroid matrix region was juxtaposed with the mucinous area (Figure 2H). Nearly all of the tumor cells featured single round nucleoli, some areas showed cytoplasm vacuoles or vesicular nuclei, and binucleate or multinucleated giant cells were mildly atypia (Figure 2I). The pathological results support the diagnosis of LGC.
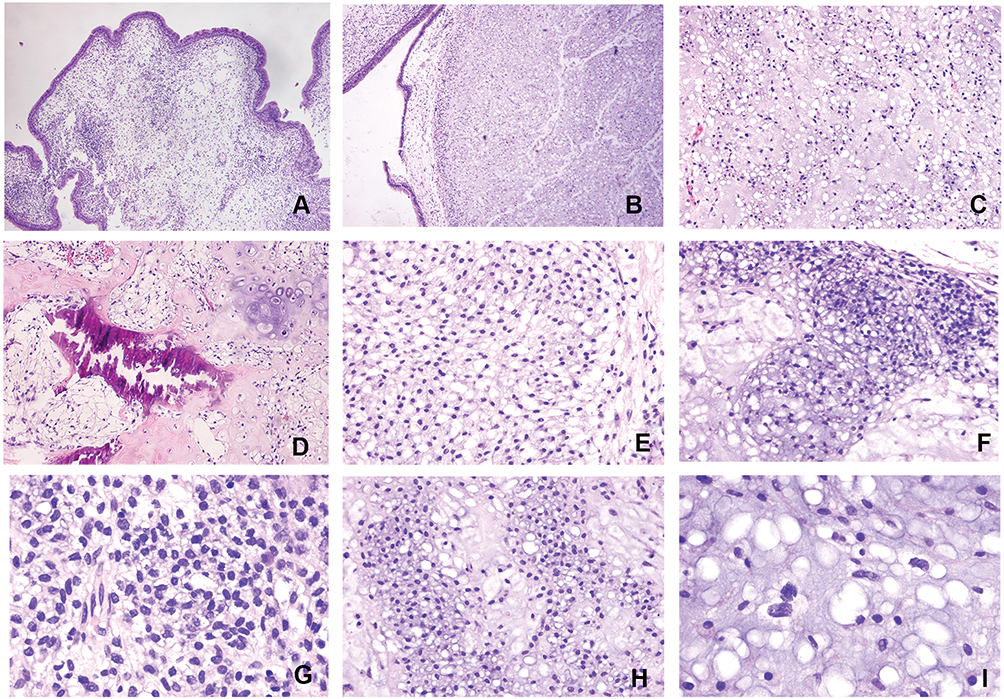 |
Figure 2 Microphotographs showed the histopathological features of the tumor. (A) Part of the tissue showed a polyp-like appearance with some tumor tissue (H&E; ×40). (B) The tumor was clearly demarcated from normal tissues in the cartilage matrix (H&E; ×40). (C) Mucinous areas and had loose tissue (H&E; ×100). (D) Chondroid region and calcification area, tumor cells interspersed in trabecular bone (H&E; ×100). (E) Tumor cell nuclei are round and most of the cells contain transparent cytoplasm, with mild cellular atypia (H&E; ×200). (F) Tumor cells were relatively abundant in the marginal areas of the tumor, the tumor displayed lobulation (H&E; ×200). (G) Tumor cells with small hyperchromatic nuclei and no mitosis in regions rich in cells (H&E; ×400). (H) The chondroid matrix region was juxtaposed with the mucinous area (H&E; ×200). (I) Focal cells with binucleate or multinucleated giant cells were mildly atypia, a few cells had cytoplasm vacuoles or vesicular nuclei (H&E; ×400). |
In this case, the myxoid matrix-rich region displayed lobulation (Figure 3A). Tumor cells formed a web or strands floating in abundant myxoid matrix (Figure 3B). Above the myxoid matrix, cells with cytoplasm vacuoles were detected (Figure 3C). Some areas showed cells that were plasma-rich with eosinophilic cytoplasm and nuclei exhibiting mild variability (Figure 3D). These findings were consistent with chordoma; accordingly, we should distinguish current case from chordoma.
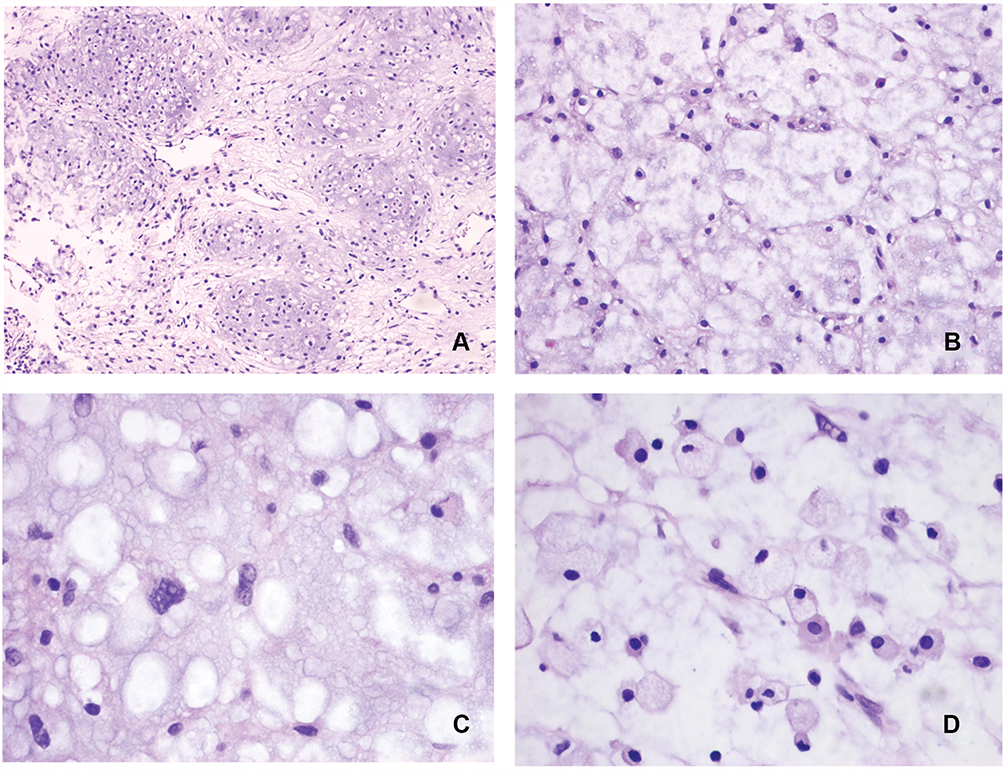 |
Figure 3 (A) The myxoid matrix-rich region displayed lobulation (H&E; ×100). (B) Tumor cells formed a web or strands floating in abundant myxoid matrix (H&E; ×200). (C) Cells with cytoplasm vacuoles in myxoid matrix (H&E; ×400). (D) Tumor cells were plasma-rich with eosinophilic cytoplasm, nuclei exhibited mild variability (H&E; ×400). |
Based on immunostaining, tumor cells were negative for AE1/AE3 (Figure 4A) and epithelial membrane antigen (EMA) (Figure 4B), while epithelial cells were positive for these markers. These results helped to differentiate between chondrosarcoma and chordoma. The tumor cells were positively stained for S-100 (Figure 4C) and exhibited diffuse positive cytoplasmic staining for vimentin (Figure 4D), supporting the diagnosis of chondrosarcoma. Compared with the positive control group, the tissue sample from this patient was negative for Cytokeratin 19 (CK19) (Figure 4E), which would exclude a diagnosis of chordoma, which is typically characterized by strong and diffuse CK19 expression. Only scattered proliferating cells were positive for the proliferative marker Ki-67; the labeling index was less than 5% (Figure 4F). The expression of IDH1 was weakly positive (Figure 4G). The remaining antibodies (Chromogranin A, Synaptophysin, Growth Hormone, Adrenocorticotropin, PRL, and TSH) were all negative (Table 1).
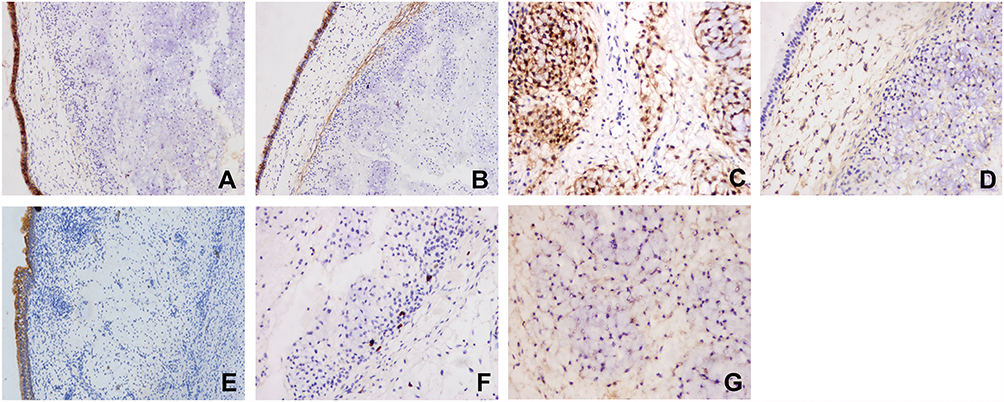 |
Figure 4 Immunohistochemical findings. (A and B) Tumor cells were negative for AE1/3 (A) and epithelial membrane antigen (original magnification ×200) (B). (C) Tumor cells were positively stained by IHC for S-100 (original magnification ×200). (D) Vimentin immunohistochemical staining showed diffuse positivity (original magnification ×200). (E) The IHC of CK19 was negative (original magnification ×100). (F) IHC for Ki-67 showed a labeling index of <5% (original magnification ×200). (G) IHC of IDH1 showed a weakly positive cytoplasm in some area (original magnification ×200). |
Discussion
Chondrosarcoma is a malignant tumor that originates from mesenchymal cells, which differentiate into chondrocytes; it is usually seen in conjunction with the sphenoethmoid bone, spheno-occipital bone, and temporal-occipital bone.7 From a pathological point of view, chondrosarcoma includes four subtypes: conventional chondrosarcoma, mesenchymal chondrosarcoma, clear cell chondrosarcoma, and dedifferentiated chondrosarcoma. Chondrosarcomas are mostly divided into 3 histological grades, grade I (well differentiated), grade II (moderately differentiated), and grade III (poorly differentiated).10 Grade I and II chondrosarcomas have good prognosis, whereas grade III chondrosarcomas are associated with a high recurrence rate and metastasis.9 Intracranial chondrosarcomas are commonly of a low-grade and arise from the skull base,1 but are rarely observed in the saddle region. There are about 9 reports of chondrosarcoma in the sellar region. Patients with sellar chondrosarcoma usually present with a long history of headache, diplopia, or visual impairment; however, our patient had no obvious headaches, making detection difficult. Therefore, the tumor grew to 7 cm when the patient was diagnosed with LGC; it was the largest such tumor reported to date. Yang et al12 reported a case that was initially suspected to be a pituitary tumor but was pathologically diagnosed as a sellar chondrosarcoma after surgery; the case had some resemblance to the present case, which was also initially misdiagnosed as a pituitary tumor. Pituitary tumors account for about 15% of primary intracranial neoplasms;15 they are benign tumors, and pathological results can distinguish between pituitary tumors and chondrosarcomas. Cao et al9 reported the first case of a sellar chondrosarcoma in which the initial symptom was amenorrhea. Ding et al6 reported a sellar chondrosarcoma cause by Ollier disease. Zhang et al7 reported a rare case with headache and blurred vision as the initial presenting symptoms, the difference is that our patient did not have these symptoms. Only Inenaga et al13 reported a patient who died after the tumor was discovered and treated for 3 years. After treatment, symptoms are typically relieved and patients are generally in a good condition. At 1 year of follow-up, there was no evidence of metastasis in this patient.
Owing to the pathological features of the myxoid matrix, small round nuclei, and cytoplasm vacuoles, LGC can easily be misdiagnosed as chordoma17 and extraskeletal myxoid chondrosarcoma (EMC),18 but the biological behaviors, treatment, and prognosis of these diseases differ, emphasizing the importance of accurate identification. Chordomas and chondrosarcomas are two major malignant bone neoplasms occurring at the skull base;17 chordomas display distinct lobulation, the tumor cells and matrix are separated by fibrous bands, tumor cells form a web or strands, and the fibrous septum is a classic feature. Immunohistochemical analyses of epithelial markers have been used to distinguish between chordomas and chondrosarcomas;17 chordomas are ectodermal tumors positive for immunophenotypic markers, such as EMA and CK, and chondrosarcomas are mesodermal tumors negative for epithelial markers, like EMA and CK.19 Therefore the current case can rule out chordoma. EMC is an extremely rare malignant tumor showing malignant chondroblast-like cells in a background of myxoid matrix.20 It is characterized by hypocellular myxoid nodules separated by fibrous septa of variable thicknesses;21 immunohistochemistry is positive for vimentin and synaptophysin and negative for S-100 and EMA. In our patient, the diffuse positive signals of vimentin and S-100 and negative signal of synaptophysin helped exclude EMC. Interestingly, our data showed that expression of IDH1 protein weakly positive cytoplasm in some areas. 2013 WHO mentioned that “only a small percentage of IDH1 (approximately 20%) can be identified using the specific IDH1 antibody” in chondrosarcomas. Matthias et al22 found that chondrosarcomas in skull-base and facial bones, IDH1 mutations rate was respectively 85.7% (24/28), IDH1 may serve as a marker for diagnosis of chondrosarcoma. In current case, IDH1 was weakly positive. Zhang et al7 reported a chondrosarcoma in the sellar area that IDH1 was nagetive. Nakagawa et al23 reported about 50% of conventional and dedifferentiated chondrosarcomas harbor IDH mutations, IDH mutations in chondrosarcoma was considered a potential therapeutic target.
Cao et al9 found that the dura mater, arachnoid, and brain parenchyma are potential origins of intracranial chondrosarcomas other than chondroid tissues or chondroid bone. The treatment of choice for LGC is simple total excision and postoperative adjuvant radiotherapy, which is usually curative, and rare reports have described the occurrence of metastasis after complete excision. The prognostic factors for chondrosarcoma include the size of the surgical excision and whether adjuvant radiotherapy is used after surgery. Bloch et al5 demonstrated that the 5-year mortality rate for patients treated with surgery alone is 26%, and postoperative adjuvant radiation therapy reduced this rate dramatically to 4%. Some recent studies have evaluated potential treatment approaches chondrosarcoma. Chih-Yang Lin et al24 indicated that BDNF enhances the migration of chondrosarcoma, suggesting that BDNF may become a new target for treating chondrosarcoma metastasis. Jia-Xue Zhu et al25 showed that SF2523 can potently inhibit chondrosarcoma cell growth in vitro and in vivo. Such studies provide a framework and potential new target for treatment along with strategies for continued research on the mechanism of chondrosarcoma.
Consent To Publish
Written informed consent was obtained from the patient for publication of this case report and the accompanying images. The images did not contain the patient records and information. This study was approved by the Clinical Research Ethics board of the First Affiliated Hospital, Shihezi University School of Medicine.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (grant no. 81860471), the Outstanding Youth Science and Technology Talent Cultivation Plan of Shihezi University (grant no. 2015ZRKXJQ07), and the International Cooperation Projects of Shihezi University (grant no. GJHZ201710).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Little A, Chung C, Perez-Ordonez B, et al. High-grade intracranial chondrosarcoma presenting with haemorrhage. J Clin Neurosci. 2013;20(10):1457–1460. doi:10.1016/j.jocn.2012.10.036
2. Chen X, Yu LJ, Peng HM, et al. Is intralesional resection suitable for central grade 1 chondrosarcoma: A systematic review and updated meta-analysis. Eur J Surg Oncol. 2017;43(9):1718–1726. doi:10.1016/j.ejso.2017.05.022
3. Lee FY, Mankin M, Fondren G, et al. Chondrosarcoma of bone: an assessment of outcome. J Bone Joint Surg Am. 1999;81(3):326–338. doi:10.2106/00004623-199903000-00004
4. Chen F, Chen B, Wang H, et al. Intracranial nonskull-based chondrosarcoma arising from the sagittal sinus: a case report and review of the literature. World Neurosurg. 2018;120:234–239. doi:10.1016/j.wneu.2018.08.239
5. Bloch OG, Jian BJ, Yang I, et al. A systematic review of intracranial chondrosarcoma and survival. J Clin Neurosci. 2009;16(12):1547–1551. doi:10.1016/j.jocn.2009.05.003
6. Ding C, Chen W, Liu F, et al. Skull base chondrosarcoma caused by ollier disease: a case report and literature review. World Neurosurg. 2019;127:103–108. doi:10.1016/j.wneu.2019.03.037
7. Zhang Y, Huang J, Zhang C, et al. An extended endoscopic endonasal approach for sellar area chondrosarcoma: a case report and literature review. World Neurosurg. 2019;127:469–477.
8. Dutta G, Singh D, Singh H, et al. Pituitary fossa chondrosarcoma: an unusual cause of a sellar suprasellar mass masquerading as pituitary adenoma. Surg Neurol Int. 2018;9:76. doi:10.4103/sni.sni_455_17
9. Cao J, Li G, Sun Y, et al. Sellar chondrosarcoma presenting with amenorrhea: a case report. Medicine (Baltimore). 2018;97(27):e11274. doi:10.1097/MD.0000000000011274
10. Sharma M, Madan M, Manjari M, et al. Pituitary chondrosarcoma presenting as a sellar and suprasellar mass with parasellar extension: an unusual presentation. Iran J Pathol. 2016;11(2):161–166.
11. Aidaer AL, Z C, Wang YP, et al. High-grade chondrosarcoma of the sella region: a case report. Chin J Pathol. 2016;45(7):486–487.
12. Yang DB, YW, Gao WD, et al. Sellar chondrosarcoma misdiagnosed as pituitary adenoma: a case report. CMINSJ. 2008;13(1):38.
13. Inenaga C, Morii K, Tamura T, et al. Mesenchymal chondrosarcoma of the sellar region. Acta Neurochir (Wien). 2003;145(7):
14. Allan CA, Kaltsas G, Evanson J, et al. Pituitary chondrosarcoma: an unusual cause of a sellar mass presenting as a pituitary adenoma. J Clin Endocrinol Metab. 2001;86(1):386–391. doi:10.1210/jcem.86.1.7111
15. Pennacchietti V, Garzaro M, Grottoli S, et al. Three-dimensional endoscopic endonasal approach and outcomes in sellar lesions: a single-center experience of 104 cases. World Neurosurg. 2016;89:121–125. doi:10.1016/j.wneu.2016.01.049
16. Freda PU, Wardlaw SL, Post KD. Unusual causes of sellar/parasellar masses in a large transsphenoidal surgical series. J Clin Endocrinol Metab. 1996;81(10):3455–3459. doi:10.1210/jcem.81.10.8855784
17. Kitamura Y, Sasaki H, Yoshida K. Genetic aberrations and molecular biology of skull base chordoma and chondrosarcoma. Brain Tumor Pathol. 2017;34(2):78–90. doi:10.1007/s10014-017-0283-y
18. Zhang L, Wang R, Xu R, et al. Extraskeletal myxoid chondrosarcoma: a comparative study of imaging and pathology. Biomed Res Int. 2018;2018:9684268.
19. Almefty K, Pravdenkova S, Colli BO, et al. Chordoma and chondrosarcoma: similar, but quite different, skull base tumors. Cancer. 2007;110(11):2457–2467. doi:10.1002/(ISSN)1097-0142
20. Qin Y, Zhang HB, Ke CS, et al. Primary extraskeletal myxoid chondrosarcoma in cerebellum: a case report with literature review. Medicine (Baltimore). 2017;96(47):e8684. doi:10.1097/MD.0000000000008684
21. Shao R, Lao IW, Wang L, et al. Clinicopathologic and radiologic features of extraskeletal myxoid chondrosarcoma: a retrospective study of 40 Chinese cases with literature review. Ann Diagn Pathol. 2016;23:14–20. doi:10.1016/j.anndiagpath.2016.04.004
22. Tallegas M, Miquelestorena-Standley E, Labit-Bouvier C, et al. IDH mutation status in a series of 88 head and neck chondrosarcomas: different profile between tumors of the skull base and tumors involving the facial skeleton and the laryngotracheal tract. Hum Pathol. 2019;84:183–191. doi:10.1016/j.humpath.2018.09.015
23. Nakagawa M, Nakatani F, Matsunaga H, et al. Selective inhibition of mutant IDH1 by DS-1001b ameliorates aberrant histone modifications and impairs tumor activity in chondrosarcoma. Oncogene. 2019. doi:10.1038/s41388-019-0929-9
24. Lin CY, Chen HJ, Li TM, et al. beta5 integrin up-regulation in brain-derived neurotrophic factor promotes cell motility in human chondrosarcoma. PLoS One. 2013;8(7):e67990. doi:10.1371/journal.pone.0067990
25. Zhu JX, Xiao JR. SF2523 inhibits human chondrosarcoma cell growth in vitro and in vivo. Biochem Biophys Res Commun. 2019;511(3):559–565. doi:10.1016/j.bbrc.2019.02.080
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.