Back to Journals » International Journal of Nanomedicine » Volume 14
Low density lipoprotein mimic nanoparticles composed of amphipathic hybrid peptides and lipids for tumor-targeted delivery of paclitaxel
Authors Qian J, Xu N, Zhou X, Shi K, Du Q, Yin X, Zhao Z
Received 9 May 2019
Accepted for publication 1 August 2019
Published 11 September 2019 Volume 2019:14 Pages 7431—7446
DOI https://doi.org/10.2147/IJN.S215080
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Junyi Qian,1,2 Ningze Xu,1,2 Xu Zhou,1,2 Kaihong Shi,1,2 Qian Du,1,2 Xiaoxing Yin,1,2 Ziming Zhao1,2
1Department of Pharmacy, Xuzhou Medical University, Xuzhou 221004, People’s Republic of China; 2Jiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, Xuzhou Medical University, Xuzhou 221004, People’s Republic of China
Correspondence: Ziming Zhao; Xiaoxing Yin
Department of Pharmacy, Xuzhou Medical University, 209 Tongshan Road, Xuzhou 221004, People’s Republic of China
Tel +86 516 8326 2139
Fax +86 516 8326 2257
Email [email protected]; [email protected]
Background: Low density lipoprotein (LDL) has been regarded as a promising antitumor drug vehicle. However some problems, such as rare source, difficulty of large-scale production, and potential safety concerns, hinder its clinical application.
Purpose: The objective of this study is to develop a biomimetic LDL nanocarrier by replacing the native apolipoprotein B-100 (apoB-100) with an artificial amphipathic peptide and demonstrate its antitumor efficacy.
Methods: The amphipathic hybrid peptide (termed as FPL) consisting of a lipid binding motif of apoB-100 (LBMapoB)-polyethylene glycol (PEG)-folic acid (FA) was synthesized and characterized by 1H NMR and circular dichroism. FPL decorated lipoprotein-mimic nanoparticles (termed as FPLM NPs) were prepared by a modified solvent emulsification method. Paclitaxel (PTX) was incorporated into NPs and its content was quantified by HPLC analysis. The morphology of NPs was observed by transmission electron microscopy (TEM), and the particle size and zeta potential of NPs were determined by dynamic light scattering (DLS). The colloidal stability of FPLM NPs was evaluated in PBS containing bovine serum albumin (BSA). In vitro release of PTX loaded FPLM NPs was evaluated using the dialysis method. Cellular uptake and cytotoxity assayswere evaluated on human cervical cancer cells (HeLa) and lung cancer cells (A549). Tumor inhibition in vivo was investigated in M109 tumor-bearing mice via tail vein injection of Taxol formulation and PTX loaded NPs.
Results: The composition of FPLM NPs, including cholesteryl oleate, glyceryl trioleate, cholesterol, 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), and FPL peptides, was optimized to be 5:1:1:3:10 (w/w). FPLM NPs had a spherical shape with a mean diameter of 83 nm and a negative charge (-12 mV). FPLM NPs with optimum formulation had good colloidal stability in BSA solution.The release of PTX from FPLM NPs was slow and sustained. The uptake of FPLM NPs was higher in folate receptor (FR) overexpressing tumor cells (HeLa cells) than in FR deficient tumor cells (A549 cells). The intracellular distribution indicated that FPLM NPs had the lysosome escape capacity. The internalization mechanism of FPLM NPs was involved with clathrin- and caveolae-mediated endocytosis and FR played a positive role in the internalization of FPLM NPs. The CCK-8 assay demonstrated that FPLM NPs exhibited notably better anti-tumor effect than Taxol formulation in vitro. Moreover, PTX loaded FPLM NPs produced very marked anti-tumor efficiency in M109 tumor-bearing mice in vivo.
Conclusion: FPLM NPs is a promising nanocarrier which can improve the therapeutic effect and reduce the side effects of antitumor drugs.
Keywords: LDL mimic nanoparticles, amphipathic peptide, folic acid, paclitaxel, tumor targeting
Introduction
Drug-delivery systems (DDSs) based on endogenous lipid nanostructures, such as lipoproteins, have gained increasing attention because of their perfect biocompatibility, uniform particle size, unique structure, and long circulation time.1,2 Lipoproteins are carriers of hydrophobic lipids in the blood, in which a hydrophobic core containing triglycerides (TGs) and cholesteryl esters is sealed by a shell composed of phospholipids, cholesterol, and apolipoproteins. Based on the density and particle size, lipoproteins can be divided into chylomicrons, very low-density lipoproteins (VLDLs), low-density lipoproteins (LDLs), and high-density lipoproteins (HDLs).3 It is generally known that the rapid proliferation of tumor cells causes excessive requirement of cholesterol.4 Cholesterol is mainly transported by LDL in human blood circulation; therefore, the LDL receptors (LDLRs) have been shown to be overexpressed in various malignant tumors.5,6 As a result, LDL has been regarded as a promising antitumor drug vehicle.
One major hurdle in developing LDL as a clinical drug carrier lies in the fact that it is acquired from human blood, resulting in a limited supply and potential safety concerns. Moreover, the complex isolation procedure and low extraction efficiency of LDL cause the high cost and the difficulty of large-scale production, which have limited the clinical application of native LDL as a drug-delivery carrier.7 Therefore, efforts have been devoted to develop recombinant LDL in vitro to replace the native LDL. In recent years, a lot of researches have been reported to successfully synthesize recombinant LDL from natural or synthetic lipids and isolated or recombinant apolipoprotein B-100 (apoB-100), which mimic the physiological functions of their native counterparts in animal models as well as humans.8–10 The sole protein component of human plasma LDL is apoB-100, which is a single polypeptide chain of 4563 amino acids and is one of the largest monomeric protein.11 The native apoB-100 protein is difficult to isolate due to its large size and propensity to aggregate. Therefore, the problems relating to the large-scale production, purity, and cost of recombinant full-length apoB-100 are still hard to overcome.12
To solve the problems that hampered the application of LDL in clinic, novel designs of biomimetic LDL nanocarriers were put forward. In biomimetic LDL, the whole apoB-100 was replaced by mimetic peptides, and the core-shell structure of LDL remained. The distinct advantage of mimetic peptides over the full-length apoB-100 was that they are relatively easy and cheap to prepare. A recent study has shown the feasibility of creating a synthetic LDL (sLDL) particle by combining a lipid microemulsion with amphipathic peptides consisting of the apoB receptor domain and lipid anchors. The sLDL could be uptaken by U937 lymphoma cells via LDLR and mimicked the effects of native LDL in the proliferation of U937 cells.13 Nikanjam et al developed paclitaxel prodrug-loaded nano-LDLs by combining a synthetic peptide consisting of a lipid-binding motif (18-amino acid alpha helix) and the LDLR-binding domain of apoB-100 with a lipid emulsion containing phosphatidyl choline, triolein, and paclitaxel oleate. The synthetic nano-LDLs could be taken up via LDLR and serve as a drug-delivery vehicle for targeting glioblastoma multiforme.14
In this study, an amphipathic hybrid peptide was synthesized by grafting a lipid-binding motif of apoB-100 (LBMapoB) onto one end of PEG and introducing folic acid (FA) as a tumor-targeting moiety to the other end of PEG, which was designated as FA-PEG-LBMapoB (FPL). These amphipathic FPL peptides were utilized to construct mimetic LDL nanoparticles (FPLM NPs) instead of apoB-100, in which the LDLR-binding domain of apoB-100 was replaced by FA. To mimic the structure of LDL, cholesteryl oleate and glyceryl trioleate were used to form the lipid core of FPLM NP, and its shell layer was composed of L-α-dioleoyl phosphatidylethanolamine (DOPE) and cholesterol. The inner segments (LBMapoB) of FPLs could insert into the phospholipid layer as anchors, and the outer segments (FA-PEG) exposed to the surface of FPLM NPs. Paclitaxel (PTX) was chosen as a model anticancer drug and encapsulated into the lipid core of FPLM NPs. The FPLM NPs were characterized by Zetasizer and transmission electron microscopy (TEM). The colloidal stability and drug release behavior of FPLM NPs were examined in vitro. The uptake effect of FPLM NPs in carcinoma cells (HeLa and A549) and the cellular uptake mechanisms were investigated. In addition, the cytotoxicity of FPLM NPs was evaluated. The antitumor efficacy of the prepared FPLM NPs was evaluated in terms of tumor volume regression and tumor inhibition rate (TIR) using M109 lung tumor-bearing mice.
Materials and methods
Materials
Cys-LBMapoB peptide (CQELQRYLSLVGQVYSTLVTYISDWWTL, supplied at >95% purity) was purchased from Sangon Co. (Shanghai, China). Glyceryl trioleate, cholesterol, cholesteryl oleate, colchicine, and chlorpromazine hydrochloride (CPZ) were bought from Tokyo Chemical Industry (TCI; Tokyo, Japan). DOPE, MAL-PEG-NHS (MW: 1K, 2K, 5K Da), and mPEG-MAL (MW: 2K Da) were purchased from Ruixi Co. (Shanxi, China). PTX was obtained from Haoxuan Biotech (Shanxi, China). Fluorescein isocyanate (FITC), FA, BSA, and nystatin were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Lyso-Tracker Red and BCA protein assay kit were obtained from Thermo Fisher Scientific (Waltham, MA, USA). DMEM, RPMI-1640 medium, FBS, penicillin/streptomycin, and trypsin were obtained from Gibco Co. (Grand Island, NE, USA).
Cell lines and animals
The cell lines, including A549 human lung carcinoma cells and HeLa human cervical carcinoma cells, and Chinese hamster lung (CHL) cells were obtained from the cell bank of Chinese Academy of Sciences (Shanghai, China). The Madison 109 (M109) murine lung carcinoma cells were from American type Culture Collection (ATCC, Manassas, VA, USA). HeLa cells, M109 cells, and CHL cells were cultured in DMEM medium supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin. A549 cells were cultured in RPMI-1640 medium supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin. All cells were maintained with 5% CO2 at 37°C under fully humidified atmosphere. Male BALB/c mice (6–8 weeks) were supplied by the laboratory animal center of Xuzhou Medical University (Jiangsu, China). All animal experiments were approved by Xuzhou Medical University and performed according to the guiding principles for the care and use of experiment animals of Xuzhou Medical University.
The subcutaneous tumor models were established by subcutaneously inoculating 100 μL of M109 cell suspension with the density of 1×107cells/mL into the right forelimb armpits of BALB/c mice (20±2 g). Then, the tumor-bearing mice were kept under standard conditions with free access to food and water.
Synthesis and characterization of FPL amphipathic peptides
MAL-PEG2K-NHS (20 mg) and FA (5 mg) were dissolved in 10 mL of PBS buffer (pH 8.0, 0.1 M) and stirred at room temperature for 12 hrs. The pH of the solution was adjusted to 6.5, then Cys-LBMapoB peptides (16 mg) were added and the reaction was stirred under a nitrogen atmosphere for another 6 hrs. The mixture was separated by a protein chromatography (Shanghai Huxi Analysis Instrument) with a Sephadex G25 column. The portion of FPL peptides was dialyzed (MWCO: 3000 Da) against deionized(DI) water thrice with an interval of 6 hrs. After lyophilization, FPL2K peptides were collected as a yellow powder. FPL1K and FPL5K were synthesized in the same manner by replacing MAL-PEG2K-NHS with MAL-PEG1K-NHS and MAL-PEG5K-NHS separately. As a control without FA group, mPEG-LBMapoB (PL) peptides were synthesized with mPEG-MAL instead of MAL-PEG-NHS.
1H NMR measurement of FPL was performed on an AVANCE DMX 500 spectrometer (Bruker, Germany). The solvent was DMSO-d6 and the internal reference was tetramethylsilane(TMS).
CD spectra were measured at 190–250 nm of wavelength on a J-815 spectropolarimeter (Jasco, Japan). LBMapoB and FPL samples were dissolved in PBS (0.05 M, pH 7.4) at a concentration of 1.0 mg/mL. The helicity of the polypeptide was calculated by the following equation:15

Preparation and characterization of FPLM NPs
FPLM NPs were prepared by a modified solvent emulsification method.16 The lipid formulation was composed of cholesteryl oleate (10 mg), glyceryl trioleate (2 mg), cholesterol (2 mg), and DOPE (6 mg). These lipid mixtures with or without PTX (3 mg) were dissolved in 2 mL of chloroform and methanol mixture (2:1, v/v). The aqueous solution of FPL (10 mL, 2 mg/mL) was added to the vial under high-speed stirring. The mixture was ultrasonicated using a SCIENTZ-IID sonifier (SCIENTZ, China) with parameters of 200 w, duty cycle =50, working time =3 s, interval =3 s. Then, the suspension was transferred to a rotary evaporator, and the organic solvents were removed at 50°C. After filtration through a millipore filter (0.22 μm), the blank or PTX-loaded FPLM NPs suspension was obtained. LBMapoB decorated lipoprotein-mimic nanoparticles (LM NPs) and mPEG-LBMapoB decorated lipoprotein-mimic nanoparticles (PLM NPs) suspensions were prepared by replacing FPL with LBMapoB and mPEG-LBMapoB (PL) separately.
The particle size and zeta potential of FPLM NPs were measured with a Nicomp 380/ZLS particle size/zeta potential analyzer (Particle Sizing System, USA). The samples were dissolved in PBS (0.1 M, pH 7.4) at a concentration of 1.0 mg/mL.
The morphology of FPLM NPs was studied by a Tecnai Spirit G2 TWIN transmission electron microscope (FEI, USA). The nanosuspension (1.0 mg/mL) was dropped onto a copper grid and evaporated naturally at room temperature before observation.
To optimize the formulation of FPLM NPs, the colloidal stabilities of FPLM NPs under physiological condition were evaluated. BSA solution (45 mg/mL) in pH 7.4 PBS was chosen to mimic the blood environment. Various NPs with different compositions were suspended at a concentration of 1 mg/mL in BSA solution at 37°C, 100 rpm. At prearranged time intervals, the particle sizes of these NPs were measured with the Nicomp 380/ZLS particle size/zeta potential analyzer.
PTX-loading capacity and release behavior
PTX content incorporated in FPLM NPs was quantified by HPLC analysis. Freeze-dried PTX-loaded NPs were sonicated in acetonitrile for 3 mins and filtered through a 0.22 μm pore-size membrane filter (Millipore). The concentration of PTX in the filtrate was determined by HPLC (LC-20AT, Shimadzu, Japan) using a reverse-phase column (Zorbax SB-C18, 5 μm pore size, 4.6×250 mm, Agilent, USA). The mobile phase was acetonitrile/water (45/55, v/v), the flow rate was 1 mL/min, and the detection wavelength was 227 nm.
The PTX-loading content (LC) was calculated:
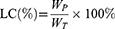
where WP is the amount of PTX loaded in the NPs and WT is the total amount of NPs and the loaded PTX.
In vitro drug release behavior was evaluated using the dialysis method. 1 mL of PTX-loaded FPLM NPs (PTX concentration was 300 μg/mL) was transferred into a dialysis bag (MWCO: 3000 Da), then immersed in a beaker with 50 mL of pH 7.4 PBS containing 1 M sodium salicylate, and shook gently (100 rpm) at 37°C. As a comparison, 1 mL of PTX solution in Cremophor EL and ethanol (1:1, v/v) with the same concentration of 300 μg/mL was conducted in the same way. At predefined time intervals, 1 mL of release medium was collected and fresh medium of equal volume was added. The samples were diluted with 1 mL of acetonitrile and filtrated through a 0.22 μm pore-size membrane filter (Millipore). The amount of PTX in the filtrates was measured with HPLC as described above.
Cellular uptake
To study the cellular uptake of NPs in vitro, PLM and FPLM NPs were labeled by FITC. Briefly, PLM and FPLM NPs (20 mg) were separately dissolved in 0.1 M carbonate buffer (pH 9.0) and FITC (1.0 mg) was added. The reaction was stirred in dark at room temperature for 12 hrs. Then, the mixture was dialyzed (MWCO: 3000 Da) against DI water five times within 24 hrs. After lyophilization in dark, FITC-labeled PLM (FITC-PLM) NPs and FITC-labeled FPLM (FITC-FPLM) NPs were obtained. To demonstrate the tumor targeting of FPLM NPs, HeLa cells and A549 cells were seeded at 1×105 cells/well onto 24-well plates and incubated for 12 hrs before use. FITC-PLM NP and FITC-FPLM NP nanosuspensions were added (the final concentration in the medium was 500 μg/mL) and co-incubated with cells for 4 hrs. After washing by cold PBS thrice, the cells were lysed with RIPA lysis buffer. The cell lysates were centrifuged at 3000 rpm for 10 mins and the fluorescence intensities of FITC in supernatants were determined (λex =492 nm, λem =530 nm) by a CMax Plus microplate reader (Molecular Devices, USA). To study the effect of incubation time and NP concentration on cellular uptake, HeLa cells were incubated with FITC-FPLM NPs at varying concentrations (50~1500 μg/mL) for different time (0.5~4 hrs). After that, the cells were washed with cold PBS thrice and lysed with RIPA lysis buffer. The fluorescence measurements of FITC were conducted according to the above-mentioned steps. Fluorescence intensity was normalized with respect to total protein content. The protein content was determined using bicinchoninic acid (BCA) protein assay kit according to the method specified by the manufacturer.
To clarify the intracellular distribution of FPLM NPs, HeLa cells were seeded in 35 mm glass dishes (Nest, China) at a density of 5×103 cells/well and incubated in DMEM containing 5% FBS. After 12-hr incubation, the medium in dishes was replaced by serum-free DMEM medium and FITC-FPLM NPs were co-incubated with cells for different time. The lysosomes and nucleus of cells were separately stained with Lyso-Tracker Red and Hoechest 33,342 following the manufacturer’s protocol. The cells were washed 3 times with cold PBS and fixed with 4% paraformaldehyde solution. The confocal laser scanning microscope(CLSM) observation was performed using an LSM 510 confocal laser scanning microscope (Carl Zeiss, Germany).
To understand of the mechanism of the cellular internalization of FPLM NPs, various endocytosis inhibitors (including 10 mM NaN3, 10 μg/mL CPZ, 2.5 μg/mL genistein, 25 μg/mL colchicine, and 10 μg/mL nystatin) were introduced. In addition to these inhibitors, free folate (final concentration of 1 mM) was added to reveal whether the uptake was related to folate receptors (FRs). HeLa cells were seeded at 5×103 cells/well onto 96-well plates and incubated for 12 hrs before use. The above endocytosis inhibitors and free FA were added and co-incubated with HeLa cells. After 1 hr, the medium in wells was replaced by serum-free DMEM medium. Then, FITC-FPLM NPs were added (final concentration of 200 μg/mL) and co-incubated with cells for another 2 hrs. After that, the cells were washed by cold PBS thrice and lysed with RIPA lysis buffer. The fluorescence intensities of FITC and the protein contents of cells were determined as described above.
Cytotoxicity assay in vitro
In vitro antiproliferation activities of PTX-loaded NPs against HeLa cells and A549 cells were estimated by WST-8 assay. The cells were seeded at 5×103 cells/well onto 96-well plates and incubated for 12 hrs before use. The cells were co-incubated with PTX-loaded PLM NPs, PTX-loaded FPLM NPs, and Taxol formulations, and the cytotoxicity was determined as a function of PTX concentration at 24-hr post-incubation. In addition, blank NPs were tested in HeLa, A549, and CHL cells to evaluate the toxicities of the nanocarriers. The cell viability was determined using Cell Counting Kit-8 (CCK-8, Sigma-Aldrich) according to the method specified by the manufacturer.
In vivo antitumor efficacy of PTX-loaded FPLM NPs
To evaluate the antitumor efficacy of PTX-loaded NPs in vivo, M109 tumor-bearing mice were established as above and randomly grouped (n=5). When the tumor volume increased to approximately 100 mm3, the mice were intravenously injected with saline, blank FPLM NPs, Taxol formulation, PTX-loaded PLM NPs, and PTX-loaded FPLM NPs, separately. All of the mice were injected with PTX formulations every 2 days for a total of three injections with a PTX dosage of 5 mg/kg each time. Tumor volumes were calculated as V = (L × W2)/2 (L, the longest dimension; W, the shortest dimension). All mice were sacrificed on day 12 after the first treatment, and all of the tumor xenografts were dissected and weighed. For further examination, tumors were fixed with 4% paraformaldehyde and embedded in paraffin followed by being sectioned. Finally, the slices were subjected to H&E staining for determination of tumor cell necrosis.
Statistical analysis
All data were given as mean ± SD. Statistical significance was tested by two-tailed Student’s t-test or one-way ANOVA. Statistical significance was noted as follows: *p<0.05; **p<0.01; ***p<0.001.
Results and discussion
Synthesis and characterization of FPL amphipathic peptides
ApoB-100 contains a number of physiologically important domains, for example, LDL receptor-binding regions and N-glycosylation sites. In addition, apoB-100 contains putative lipid-binding regions which might have a role in stabilizing LDL structure.17,18 However, apoB-100 protein is difficult to prepare by either separation method or recombination method due to its large molecular size and propensity to aggregate. In this study, an amphipathic hybrid peptide, which was derived from apoB-100 but had a much simpler structure, was designed to take the place of apoB-100 in LDL. The hydrophobic segment of the FPL peptide was a lipid-binding motif of apoB-100 (LBMapoB) reported by Gordon et al.19 LBMapoB is largely composed of hydrophobic amino acids, such as Val, Leu, and Ile, plus some polar amino acids, such as Ser and Thr. It forms an α-helix secondary structure in aqueous buffer, which is similar to the class A amphipathic helical peptide except for the larger hydrophobic face and the uncharged hydrophilic face. Thus, LBMapoB was predicted to insert deeper into a phospholipid bilayer than Class A amphipathic helices.18 PEG was chosen as the hydrophilic segment of FPL, for the reason that PEG could not only improve the dispersion stability of FPLM NPs, but also help to prevent the absorption of serum proteins and prolong the circulation time of FPLM NPs.20
Although LDLR has been proven to be a useful target point for delivery drugs to some tumors, it has a limitation for antitumor therapy because there are still many tumors that do not overexpress the LDLR, whereas some normal tissues do. Moreover, a number of other receptors have proven to be more tumor specific than LDLR, including EGFR, FR, and αvβ3 integrins.21 Therefore, FA was introduced into the FPL peptide as a tumor-targeting moiety instead of native LDLR.
The molecular structure of FPL amphipathic peptide was verified by 1H NMR. As shown in Figure 1, the characteristic peak of PEG (-CH2-CH2-O-) appeared at 3.5 ppm (a in Figure 1), whereas that of FA (the pteridine group) was mainly found at 8.4 ppm (b in Figure 1). The peaks near 7.0 (c in Figure 1) were contributed to the benzene rings of tyrosines in LBMapoB peptide.
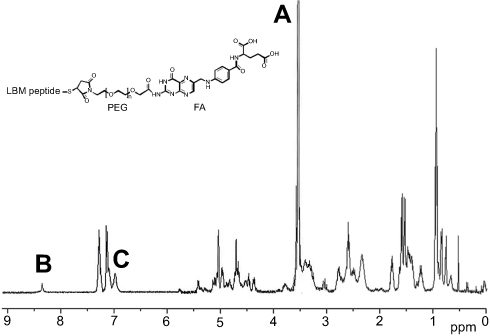 |
Figure 1 1H NMR spectrum of FPL hybrid peptide (A) the characteristic peak of PEG; (B) the characteristic peak of FA; (C) the characteristic peak of LBMapoB.Abbreviations: FPL, FA-PEG-LBMapoB; FA, folic acid; LBMapoB, lipid-binding motif of apoB-100. |
As mentioned earlier, the amphipathic α-helical structure was crucial for the lipid-binding capacity of LBMapoB; therefore, it was necessary to clarify whether the grafting of FA-PEG disturbed the α-helix secondary structure. The CD spectra of LBMapoB (Figure 2) exhibited a typical α-helix pattern and the calculated helicity was 58.9%. In the CD spectra of FPL, the double negative peaks of α-helix at 208 nm and 220 nm also appeared, but the peak intensities had a little decrease. The calculated helicity of FPL was 48.7%, which meant that the α-helix of LBMapoB was basically unchanged after PEG conjugation.
 |
Figure 2 CD spectra of LBMapoB and FPL peptides in trifluoroethanol. Abbreviations: CD, circular dichroism; FPL, FA-PEG-LBMapoB; LBMapoB, lipid-binding motif of apoB-100. |
Preparation and characterization of FPLM NPs
The lipid composition of FPLM NPs, including cholesteryl oleate, glyceryl trioleate, cholesterol, and DOPE, was optimized to be 5:1:1:3 (w/w), based on the reference of native LDL and stability.7 When the lipid composition was fixed, FPLM NPs with different weight ratio of FPL2K peptides were prepared and compared. As shown in Figure 3A, the particle size of FPLM NPs decreased from ca. 130 nm to ca. 80 nm as the FPL ratio increased from 20% to 50%. When the FPL ratio exceeded 50%, the change in particle size was negligible. The weight ratio of apoB-100 in native LDL is about 20%, but FPLM NPs prepared with FPL at the same ratio was much larger than native LDL. This might be due to that apoB-100 had stronger hydrophobicity and lipid-binding capacity, which let the lipid core be more condensed. In order to mimic the native LDL, the weight ratio of FPL peptides needed to be much higher than that of apoB-100. Because PEG had an important effect on the in vitro and in vivo stability of NPs, the comparison of FPLM NPs prepared from three FPL peptides (FPL1K, FPL2K, and FPL5K) with the same formulation ratio was conducted. It was apparent that FPLM NPs consisting of longer PEG had a larger particle size (Figure 3B).
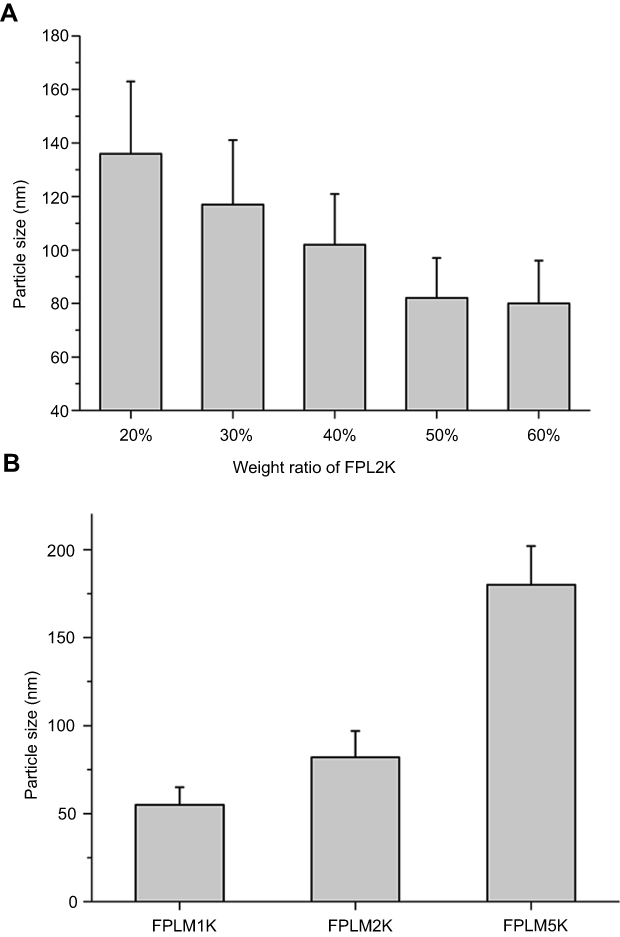 |
Figure 3 Particle sizes of FPLM NPs with various compositions. (A) The effect of FPL weight ratio on the particle size of FPLM NPs and (B) the effect of PEG length on the particle size of FPLM NPs. All data are shown as means ± SD (n=3). Abbreviations: FPLM NPs, FPL decorated lipoprotein-mimic nanoparticles; FPL, FA-PEG-LBMapoB; LBMapoB, lipid-binding motif of apoB-100. |
The colloidal stability of FPLM NPs under physiological condition was monitored via particle size measurement using dynamic light scattering (DLS). BSA was chosen as a model protein because albumin is the most abundant protein in serum and a major component in the protein corona surrounding nanoparticles.22,23 When suspended in BSA solution (Figure 4A), the particle size of FPLM NPs composed of 20%, 30%, and 40% of FPL peptides increased rapidly after 4 hrs, while FPLM NPs with 50% and 60% peptide ratio remained stable for 24 hrs. Considering the particle size, stability, and drug LC comprehensively, 50% peptide ratio was chosen as the optimized formulation. In addition, the stabilities of LM NPs without PEG modification and FPLM NPs with different lengths of PEG were compared. As shown in Figure 4B, LM NPs were found to precipitate at 8 hrs in the presence of BSA, and the size of FPLM1K NPs increased by 200% within 24 hrs. The sizes of FPLM2K NPs and FPLM5K NPs had no remarkable change in 24 hrs, which demonstrated that they had better colloidal stability and resistance to serum protein adsorption. In general, nanoparticles with the size of <200 nm are considered to be able to penetrate through the blood vessels of tumors due to enhanced permeability and retention (EPR) effect.24 Considering the particle size and serum stability, FPLM2K NPs were more suitable for antitumor drug carrier and were applied in the following studies. The optimized compositions of FPLM NPs and PLM NPs are illustrated in Table 1.
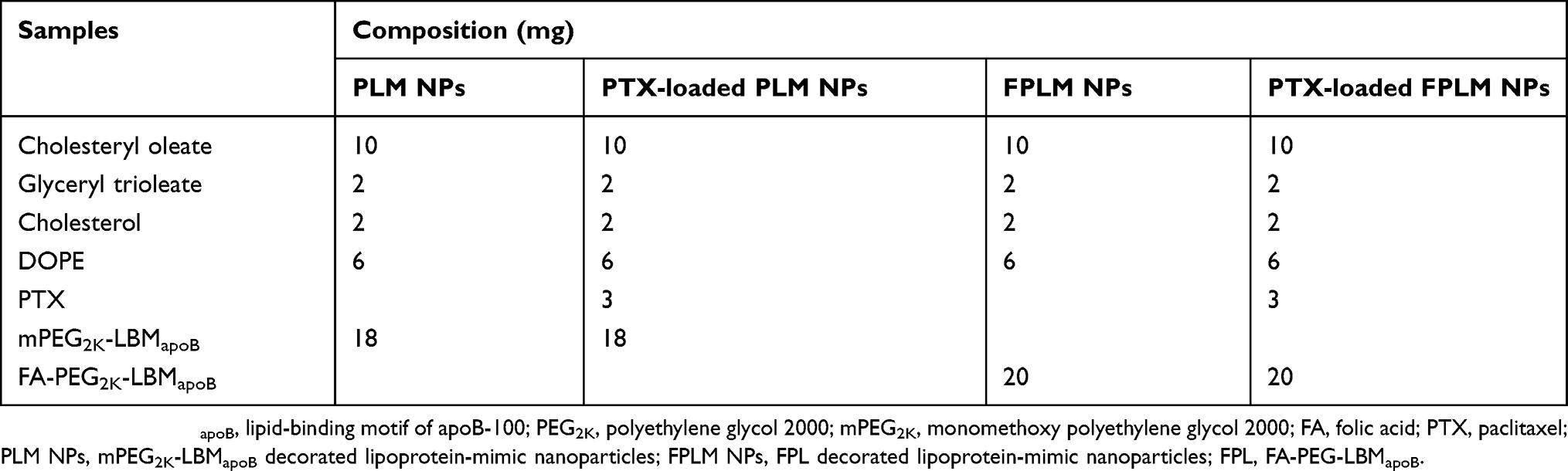 |
Table 1 Formulation compositions of blank PLM NPs, PTX-loaded PLM NPs, blank FPLM NPs, and PTX-loaded FPLM NPs |
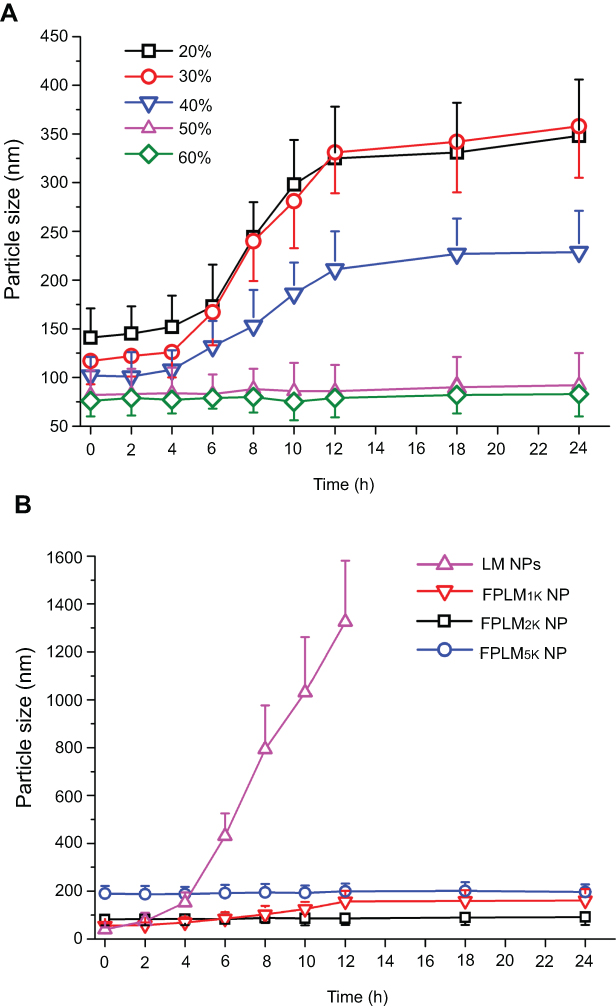 |
Figure 4 Colloidal stabilities of FPLM NPs with various compositions in 4.5% BSA solution. (A) The effect of peptide ratio on the stability of FPLM NPs and (B) the effect of peptide composition on the stability of NPs. All data are shown as means ± SD (n=3).Abbreviations: LBMapoB, lipid-binding motif of apoB-100; LM NPs, LBMapoB decorated lipoprotein-mimic nanoparticles; FPLM NPs, FPL decorated lipoprotein-mimic nanoparticles; FPL,FA-PEG-LBMapoB. |
The particle size and zeta potential of the optimized FPLM NPs in PBS (0.1 M, pH7.4) are shown in Table 2. FPLM NPs had a mean size of 83 nm and a negative charge (ca. −12 mV). PLM NPs without FA groups had a slightly smaller size than FPLM NPs. Compared with negative FPLM NPs, the zeta potential of PLM was near neutral due to the absence of carboxyl groups on FA. After PTX encapsulation, the particle size of NPs got a slight increase and the change of zeta potential was not significant.
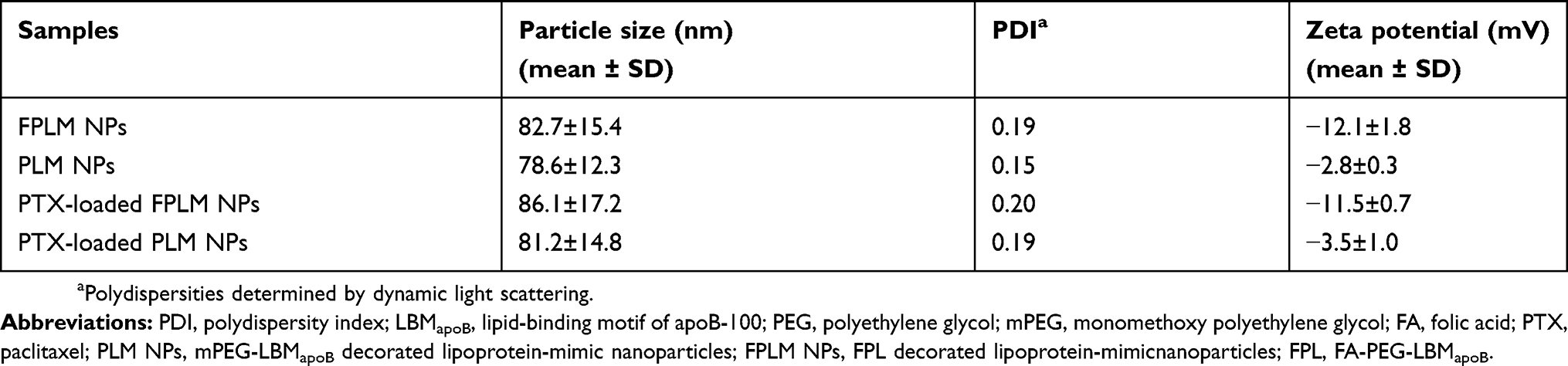 |
Table 2 The particle sizes and zeta potentials of FPLM NPs in PBS (0.1 M, pH7.4) |
As shown in Figure 5, the morphology of FPLM NPs exhibited a regular spherical shape. The darker and compact core consisted of multiple lipids, and the flexible PEG-FA chains formed the lighter and cloud-like shell. The particle size of FPLM NPs estimated by TEM was basically consistent with the result of DLS.
 |
Figure 5 TEM image of FPLM NPs.Abbreviations: TEM, transmission electron microscope; LBMapoB, lipid-binding motif of apoB-100; FPLM NPs, FPL decorated lipoprotein-mimicnanoparticles; FPL, FA-PEG-LBMapoB. |
PTX-loading capacity and release behavior
Two drug-loading methods, thin-film dispersion-ultrasonic method and solvent emulsification method, were compared. The PTX LC of the latter method (6.8%) was higher than the thin-film dispersion-ultrasonic method (5.3%); therefore, PTX-loaded FPLM NPs were prepared by the solvent emulsification method.
The poor solubility of PTX in water (about 0.3 μg/mL) makes it difficult to satisfy the sink condition and detection precision at the same time. Therefore, some additives, such as tween-80 and sodium salicylate, were added in the release medium to improve the water solubility of PTX.25,26 Considering that tween-80 might disturb the stability of FPLM NPs, sodium salicylate solution (1 M) in pH 7.4 PBS was utilized as the drug release medium in this study. The maximum concentration of PTX released from FMLP NPs was 6 μg/mL, and the solubility of PTX in this medium was around 28 μg/mL, which produced a good sink condition for release.
As shown in Figure 6, the average cumulative release percent of PTX dissolved in Cremophor EL and ethanol (1:1, v/v) at 2 hrs and 6 hrs was 63.1% and 92.2%, which meant that the release of PTX in this formulation was very fast and basically completed within 6 hrs. Unlike Taxol formulation, the in vitro release profile of PTX-loaded FPLM NPs exhibited a biphasic and sustained release pattern. A rapid release during the initial period (approximately 10% within initial 0.5 hrs) could be contributed to the drugs adsorbed onto the outer PEG layers of NPs, which were easy to spread into the release medium. The following stage was a slow and continuous release process, in which the average cumulative release percent at 12 hrs, 24 hrs, and 48 hrs was 36.3%, 41.8%, and 52.3%, separately. The release behavior of PTX-loaded FPLM NPs suggested that the leakage of PTX from the NPs in blood circulation was minor and it could provide a long-term drug therapy once entered the tumors.
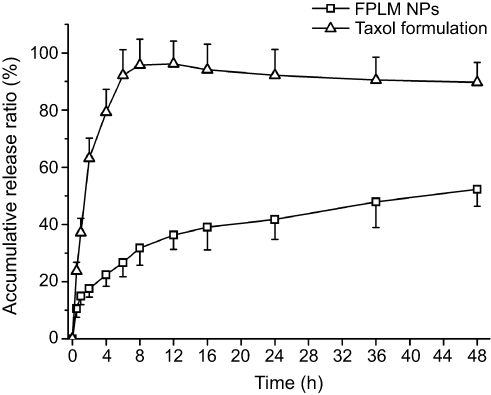 |
Figure 6 In vitro release profiles of PTX from FPLM NPs and Taxol formulation in pH 7.4 PBS containing 1 M sodium salicylate at 37°C. All data are shown as means ± SD (n=3).Abbreviations: LBMapoB, lipid-binding motif of apoB-100; FPLM NPs, FPL decorated lipoprotein-mimicnanoparticles; FPL, FA-PEG-LBMapoB. |
Cellular uptake
To clarify the targeting effect of FA groups attached to the surface of the LNPs, two different types of tumor cells were used (HeLa cells and A549 cells), which were FR-positive (FR+) and FR-negative (FR-) cell lines, respectively.27,28 FITC was conjugated to PLM NPs and FPLM NPs as the fluorescent marker to detect the cellular uptake kinetics and the intracellular distribution of NPs. Figure 7A exhibits the uptake of PLM NPs and FPLM NPs by HeLa cells and A549 cells separately. The results clearly indicated that the intracellular fluorescent intensity of FPLM NPs in HeLa cells was remarkably higher than that in A549 cells (p=0.005). In addition, the uptake of FPLM NPs by HeLa cells was around 5 times as much as that of PLM NPs, while there was no significant difference between the uptake of FPLM NPs and PLM NPs by A549 cells. It confirmed the effectiveness on improving cellular uptake and the selectivity of FA modification in FR expressing tumor cells.
 |
Figure 7 Cellular uptake characteristic of FITC-FPLM NPs. (A) The uptake comparison of FITC-PLM NPs and FITC-FPLM NPs in HeLa cells (FR+) and A549 cells (FR-); (B) Concentration-dependent uptake of FITC-FPLM NPs by HeLa cells. (C) Time-dependent uptake of FITC-FPLM NPs by HeLa cells. All data are shown as means ± SD (n=6).Abbreviations: LBMapoB, lipid-binding motif of apoB-100; PLM NPs, mPEG-LBMapoB decorated lipoprotein-mimic nanoparticles; FPLM NPs, FPL decorated lipoprotein-mimicnanoparticles; FPL, FA-PEG-LBMapoB. |
As shown in Figure 7B and C, the uptake of FPLM NPs was concentration dependent and time dependent. Increasing the concentration of NPs in the medium resulted in an increasing uptake of FPLM NPs in the range of 50~500 μg/mL. When the concentration of NPs exceeded 500 μg/mL, the uptake mediated by FR of HeLa cells was close to saturation. The uptake of FPLM NPs also increased with the incubation time during 4 hrs, and then began to decrease as the incubation time extended. It might due to the reason that the uptake rate of FITC-FPLM NPs was slower than the degradation or quenching rate of FITC in cells after 4 hrs.
The fluorescent images of the internalization process and the intracellular distribution of FITC-FPLM NPs are shown in Figure 8. FPLM NPs mainly adsorbed on the cell membranes and did not yet enter the endosomes at 0.5 hrs. After 1 hr of incubation, most of FPLM NPs occurred in endosomes because a majority of green fluorescence were overlapped with the red fluorescence (appearing yellow). It demonstrated that the internalization of FPLM NPs was mainly via the receptor-mediated endocytosis.29 At 4 hrs, the green fluorescence representing FPLM NPs spread over the whole cytoplasm, which indicated that FPLM NPs had endosome escape ability. The endosome escape ability of FPLM NPs might contribute to the DOPE in the components of NPs. DOPE is a fusogenic lipid that undergoes phase transition from lamellar to inverted hexagonal phase at endosomal pH, thereby causing endosome destabilization and releasing the drug to the cytoplasm.30,31 FPL amphipathic peptides might also play a role in the endosome escape of FPLM NPs for the reason that they have the ability to insert into the endosome membrane and induce the disorder of the endosome membrane.
 |
Figure 8 The intracellular distribution of FITC-FPLM NPs in HeLa cells at different coincubation times (0.5 hrs, 1 hr, and 4 hrs).Abbreviations: FITC, fluorescein isothiocyanate; LBMapoB, lipid-binding motif of apoB-100; FPLM NPs, FPL decorated lipoprotein-mimicnanoparticles; FPL, FA-PEG-LBMapoB. |
To understand the uptake mechanism of FPLM NPs by tumor cells, the effects of adenosine triphosphate (ATP) depletion and other endocytosis inhibitors were evaluated quantitatively (Figure 9). We first examined the effect of ATP depletion by preincubation of the cells with sodium azide (NaN3). ATP depletion reduced the uptake of FPLM NPs by ca. 56%, indicating that the internalization of FPLM NPs by HeLa cells was energy dependent. The influence of temperature on the internalization of FPLM NPs was also studied. The uptake of FPLM NPs at 4°C was decreased by ca. 72%, compared to that incubated at 37°C. These results supported that the uptake of FPLM NPs was a process of active endocytosis. Blocking clathrin-coated pit formation with CPZ remarkably inhibited the uptake of FPLM NPs (approximately 69% decrease), suggesting that the uptake of FPLM NPs by HeLa cells relied on the clathrin-mediated endocytosis. Furthermore, pretreatment of cells with nystatin, a caveolae disrupting agents, also had an inhibitory effect on the uptake of FPLM NPs. In contrast, pretreatment with colchicine, a disrupting agent of microtubule required for macropinocytosis, showed an unapparent effect on the uptake of FPLM NPs. Moreover, the preincubation with excessive free folate (1 mM) also caused a decrease (approximately 50%) of cellular internalization. These results indicated that FPLM NPs utilized not only clathrin-mediated but also caveolae-mediated endocytosis pathways to enter HeLa cells, and FR played an important role in this process. Suen et al revealed that 50 nm and 120 nm folate-decorated polymeric nanoparticles were internalized via both clathrin- and caveolae-mediated endocytosis, while the 250 nm folate-decorated nanoparticles were only internalized via caveolae-mediated pathway in retinal pigment epithelium.32 Dalal et al found that the endocytosis pathway of FA-modified nanoprobes could shift from caveolae- to clathrin-mediated endocytosis as the surface FA density increased. It could be concluded that low FA density offered modular interaction with cell surface FRs that directed the nanoprobes toward caveolae and promoted caveolae-mediated endocytosis. In contrast, high FA density offered strong binding of nanoprobes with cell surface via multiple receptors and induced signaling pathway for clathrin-mediated rapid endocytosis.33 These conclusions could explain why the FPLM NPs prepared in our study, which had a small size (<100 nm) and a high FA density, were internalized via both clathrin- and caveolae-mediated endocytosis.
 |
Figure 9 Investigation of the cellular internalization mechanism of FPLM NPs in HeLa cells by using inhibitors of endocytosis. The internalization ratio was normalized to that of the control (FPLM NPs internalization in the absence of inhibitors). All data are shown as means ± SD (n=6).Abbreviations: CPZ, chlorpromazine hydrochloride; LBMapoB, lipid-binding motif of apoB-100; FPLM NPs, FPL decorated lipoprotein-mimicnanoparticles; FPL, FA-PEG-LBMapoB. |
Cytotoxicity assay in vitro
The degree of tumor cell proliferation was determined by WST-8 assay to evaluate the therapeutic potential of the PTX-loaded NPs. As shown in Figure 10A and B, all tested PTX formulations effectively reduced the viability of tumor cells, and their antiproliferation effect increased with the concentration of PTX. In FR+ HeLa cells, the introduction of FA dramatically increased the antiproliferation activity. FPLM NPs exhibited notably better antitumor effect than PLM NPs and Taxol formulation at almost all concentrations, and PLM NPs had similar antitumor efficiency with Taxol formulation. For example, the treatment with FPLM NPs exhibited ca. 33% of cell viability at a PTX concentration of 0.1 μg/mL, while Taxol formulation showed ca. 60% cell viability at the same concentration (p=0.006). In FR-A549 cells, there was no significant difference in the level of cell viabilities between PLM NPs, FPLM NPs, and Taxol formulation. The cytotoxicity of blank PLM NPs and FPLM NPs was evaluated in both tumor cells and normal cells (Figure 10C and D). The results exhibited that blank PLM NPs and blank FPLM NPs had no antiproliferation activity either to tumor cells or to normal cells.
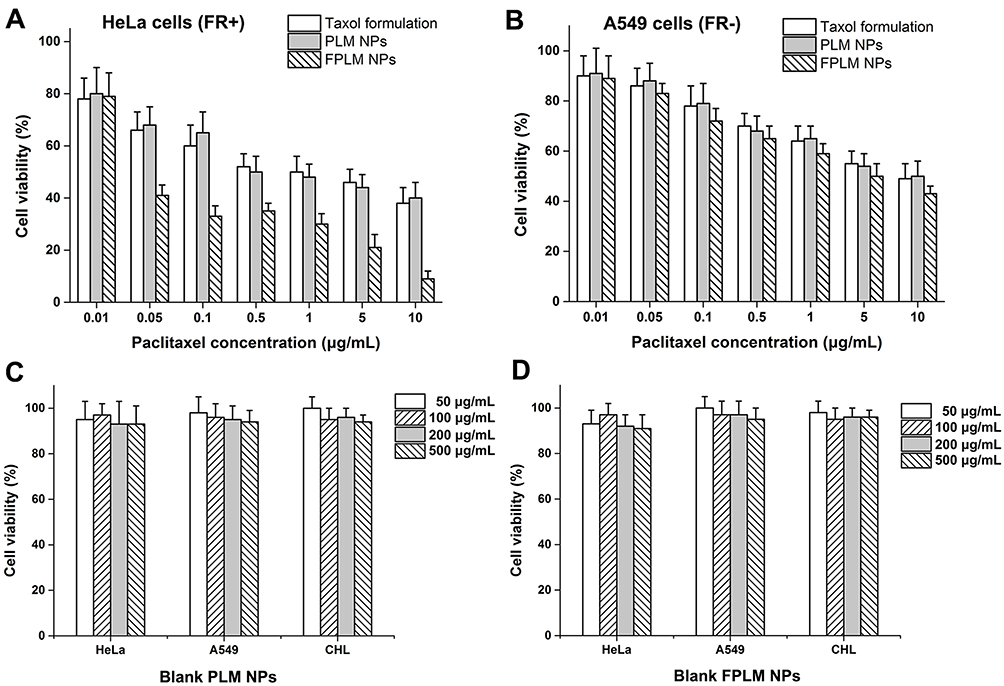 |
Figure 10 In vitro cytotoxic effect of PTX-loaded PLM NPs, PTX-loaded FPLM NPs, and Taxol formulation against (A) HeLa and (B) A549 cells after incubation for 24 hrs at various concentrations of PTX. The viability of HeLa cells, A549 cells, and CHL cells treated with (C) blank PLM NPs and (D) blank FPLM NPs for 24 hrs at different concentrations of NPs. All data are shown as means ± SD (n=6).Abbreviations: LBMapoB, lipid-binding motif of apoB-100; PLM NPs, mPEG-LBMapoB decorated lipoprotein-mimic nanoparticles; FPLM NPs, FPL decorated lipoprotein-mimicnanoparticles; FPL, FA-PEG-LBMapoB. |
Vivo antitumor efficacy of PTX-loaded FPLM NPs
To evaluate antitumor efficacy, the tumor growth curve and TIR of M109 tumor-bearing mice were assessed after three times injection of Taxol and PTX NPs formulations at a dose of 5 mg/kg. Figure 11A shows that the control groups treated with saline and the blank FPLM NPs exhibited a rapid tumor growth rate and had a 16.5-fold increase in the tumor volume within 12 days. The mice treated with PTX-loaded FPLM NPs and Taxol formulation showed a 5.3-fold and 8.2-fold volume increase separately, which indicated that the antitumor effect of FPLM NPs on the suppression of tumor growth was remarkably superior to that of Taxol formulation (p=0.026). The introduction of FA showed an apparent targeting effect in vivo because the suppression of tumor growth by PLM NPs incorporating the same amounts of PTX (8.4-fold volume increase) was far worse than FPLM NPs. The results of the dissected tumor weight and the TIR on day 12 are shown in Figure 11B, and the FPLM NP group achieved the highest TIR (67.7%) in all experimental groups, which were consistent with the tumor growth curves. The body weights of mice in most groups were in normal range except Taxol formulation, suggesting that Taxol formulation caused obvious side effects (Figure 11C).
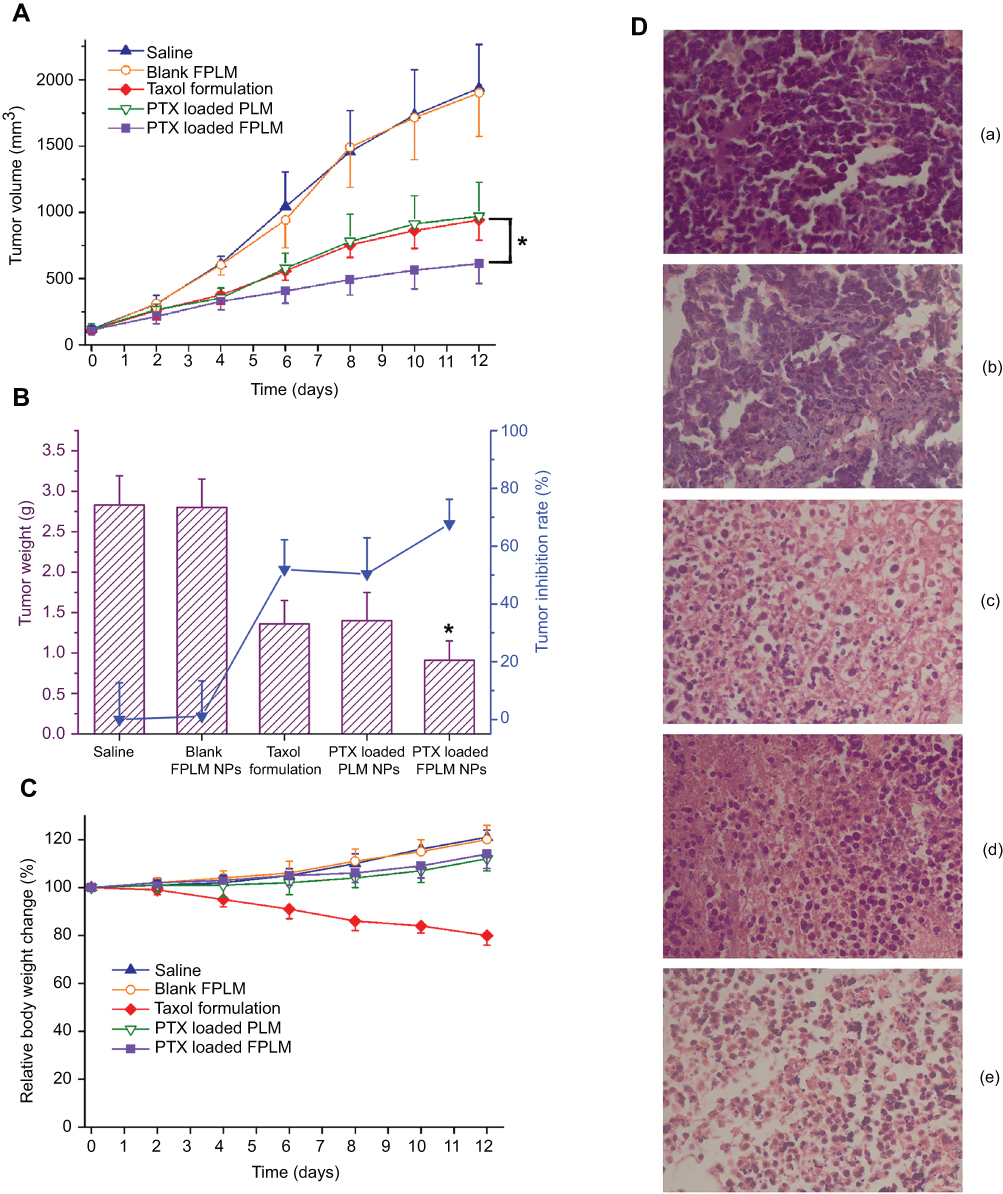 |
Figure 11 In vivo therapeutic efficacy of the different PTX formulations at a dose of 5 mg/kg in M109 lung tumor-bearing mice. (A) Changes of tumor volumes in mice during the experimental period; (B) The dissected tumor weights and the tumor inhibition rates on day 12; (C) Relative body weight changes of tumor-bearing mice during the treatment. Error bars represent SDs (n=5). (D) Images of tumor sections separated from mice stained by H&E on day 12 (a: saline; b: blank FPLM; c: Taxol formulation; d: PTX-loaded PLM; e: PTX-loaded FPLM). Notes: *p<0.05, compared with Taxol formulation group.Abbreviations: LBMapoB, lipid-binding motif of apoB-100; FPLM NPs, FPL decorated lipoprotein-mimicnanoparticles; FPL, FA-PEG-LBMapoB. |
To identify the cell death induced by PTX-loaded NPs and Taxol formulations, H&E staining was used to detect the histological changes of tumor after 12 days of treatment. The H&E staining images (Figure 11D) demonstrated that FPLM NPs effectively delivered PTX into tumor tissues and induced a massive necrosis of tumor cells. The tumors of mice treated with Taxol formulation and PLM NPs also showed signs of necrocytosis, but the necrosis proportions of both Taxol and PLM NPs were not as high as FPLM NPs.
Conclusion
The present study successfully developed a novel LDL-mimic nanocarrier, FPLM NPs. These NPs could assemble into a core-shell structure, in which cholesteryl oleate and TG acted as the lipid core, and DOPE, cholesterol, and FPL amphipathic peptides acted as the shell. FPL was composed of a lipid-binding motif of apoB-100, PEG, and folate. The hydrophobic LBMapoB segments inserted into the phospholipid layer, and the hydrophilic FA-PEG segments on the surface of NPs had the functions of increasing the stability of NPs in blood and tumor targeting. The uptake of FPLM NPs by HeLa cells (overexpressing FR) significantly increased compared with that of PLM NPS without FA, and it was much higher in FR-positive tumor cells than in FR-negative tumor cells. The intracellular distribution indicated that FPLM NPs had the lysosome escape capacity. FA modification played a positive role in the specific receptor-mediated endocytosis process of the FPLM NPs, and their internalization pathways involved both clathrin- and caveolae-mediated endocytosis. The results of antiproliferative effect in vitro and tumor inhibition experiment in vivo confirmed that FPLM NPs exhibited excellent tumor-targeting efficacy. Based on these conclusions, FPLM NPs is expected to be a biocompatible and efficient antitumor drug nanocarrier.
Acknowledgments
The research was financially supported by The National Natural Science Foundation of China (No. 81773643, No. 81603302, and No. 81102381). This work was also supported by The Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 17KJB350014) and The Natural Science Foundation of Jiangsu Province (No.20150215).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lacko AG, Nair M, Prokai L, Mcconathy WJ. Prospects and challenges of the development of lipoprotein-based formulations for anti-cancer drugs. Exp Opin Drug Deliv. 2007;4(6):665–675. doi:10.1517/17425247.4.6.665
2. Ng KK, Lovell JF, Zheng G. Lipoprotein inspired nanoparticles for cancer theranostics. Acc Chem Res. 2011;44(10):1105–1113. doi:10.1021/ar200017e
3. Jr AM G, Pownall H, Havel RJ. Introduction to the plasma lipoproteins. Methods Enzymol. 1986;128:3–40.
4. Kuzu OF. The role of cholesterol in cancer. Cancer Res. 2016;76(8):1–8. doi:10.1158/0008-5472.CAN-16-0584
5. Corbin IR, Li H, Chen J, et al. Low-density lipoprotein nanoparticles as magnetic resonance imaging contrast agents. Neoplasia. 2006;8(6):488–498. doi:10.1593/neo.05835
6. Firestone RA. Low-density lipoprotein as a vehicle for targeting antitumor compounds to cancer cells. Bioconjugate Chem. 1994;5:105–113. doi:10.1021/bc00026a002
7. Rensen PCN, Vrueh RLAD, Kuiper J, Bijsterbosch MK, Biessen EAL, Berkel TJCV. Recombinant lipoproteins: lipoprotein-like lipid particles for drug targeting. Adv Drug Deliv Rev. 2014;7(2):251–276.
8. Lundberg B. Preparation of drug–low density lipoprotein complexes for delivery of antitumoral drugs via the low density lipoprotein pathway. Cancer Res. 1987;47:4105–4108.
9. Chu ACY, Tsang SY, Lo EHK, Fung KP. Low density lipoprotein as a targeted carrier for doxorubicin in nude mice bearing human hepatoma HepG2 cells. Life Sci. 2001;70:591–601. doi:10.1016/S0024-3205(01)01441-2
10. Kader A, Davis PJ, Kara M, Liu H. Drug targeting using low density lipoprotein (LDL): physicochemical factors affecting drug loading into LDL particles. J Control Release. 1998;55(2–3):231–243.
11. Chen SH, Yang CY, Chen PF, et al. The complete cDNA and amino acid sequence of human apolipoprotein B-100. J Biol Chem. 1986;261(28):12918–12921.
12. Nikanjam M, Blakely EA, Bjornstad KA, Shu X, Budinger TF, Forte TM. Synthetic nano-low density lipoprotein as targeted drug delivery vehicle for glioblastoma multiforme. Int J Pharm. 2007;328(1):86–94. doi:10.1016/j.ijpharm.2006.07.046
13. Baillie G, Owens M, Halbert GW. A synthetic low density lipoprotein particle capable of supporting U937 proliferation in vitro. J Lipid Res. 2002;43(1):69–73.
14. Nikanjam M, Gibbs AR, Hunt CA, Budinger TF, Forte TM. Synthetic nano-LDL with paclitaxel oleate as a targeted drug delivery vehicle for glioblastoma multiforme. J Control Release. 2007;124(3):163–171. doi:10.1016/j.jconrel.2007.09.007
15. Chen YH, Yang JT, Martinez HM. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry. 1972;11:4120–4131. doi:10.1021/bi00772a015
16. Lee JY, Kim JH, Bae KH, et al. Low-density lipoprotein-mimicking nanoparticles for tumor-targeted theranostic applications. Small. 2015;11(2):222–231. doi:10.1002/smll.201303277
17. Knott TJ, Pease RJ, Powell LM, et al. Complete protein sequence and identification of structural domains of human apolipoprotein B. Nature. 1986;323(6090):734–738. doi:10.1038/323734a0
18. Yang CY, Chen SH, Gianturco SH, et al. Sequence, structure, receptor-binding domains and internal repeats of human apolipoprotein B-100. Nature. 1986;323(6090):738–742. doi:10.1038/323738a0
19. Gordon SM, Pourmousa M, Sampson M, et al. Identification of a novel lipid binding motif in apolipoprotein B by the analysis of hydrophobic cluster domains. BBA-Biomembranes. 2017;1859:135–145. doi:10.1016/j.bbamem.2016.10.019
20. Du XJ, Wang JL, Liu WW, et al. Regulating the surface poly(ethylene glycol) density of polymeric nanoparticles and evaluating its role in drug delivery in vivo. Biomaterials. 2015;69:1–11. doi:10.1016/j.biomaterials.2015.07.048
21. Zheng G, Chen J, Li H, Glickson JD. Rerouting lipoprotein nanoparticles to selected alternate receptors for the targeted delivery of cancer diagnostic and therapeutic agents. Proc Natl Acad Sci U S A. 2005;102:17757–17762. doi:10.1073/pnas.0508677102
22. Lazzari S, Moscatelli D, Codari F, Salmona M, Morbidelli M, Diomede L. Colloidal stability of polymeric nanoparticles in biological fluids. J Nanopart Res. 2012;14:920–929. doi:10.1007/s11051-012-0920-7
23. Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci USA. 2008;105:14265–14270. doi:10.1073/pnas.0805135105
24. Maruyama K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv Drug Deliv Rev. 2011;63(3):161–169. doi:10.1016/j.addr.2010.09.003
25. Zhang P, Huang Y, Liu H, et al. A PEG-Fmoc conjugate as a nanocarrier for paclitaxel. Biomaterials. 2014;35(25):7146–7156. doi:10.1016/j.biomaterials.2014.01.026
26. Lee J, Lee SC, Acharya G, Chang CJ, Park K. Hydrotropic solubilization of paclitaxel: analysis of chemical structures for hydrotropic property. Pharm Res. 2003;20(7):1022–1030. doi:10.1023/A:1024458206032
27. Lu Y, Low PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2012;64(supp_S):342–352. doi:10.1016/j.addr.2012.09.020
28. Chen Y, Tezcan O, Li D, et al. Overcoming multidrug resistance using folate receptor-targeted and pH-responsive polymeric nanogels containing covalently entrapped doxorubicin. Nanoscale. 2017;9(29):10404–10419. doi:10.1039/C7NR03592F
29. Jiang LQ, Wang TY, Wang Y,
30. Koltover I, Salditt T, Radler JO, Safinya CR. An inverted hexagonal phase of cationic liposome–DNA complexes related to DNA release and delivery. Science. 1998;281:78–81. doi:10.1126/science.281.5373.78
31. Hafez IM, Cullis PR. Roles of lipid polymorphism in intracellular delivery. Adv Drug Deliv Rev. 2001;47:139–148. doi:10.1016/S0169-409X(01)00103-X
32. Suen WLL, Chau Y. Size-dependent internalisation of folate-decorated nanoparticles via the pathways of clathrin and caveolae-mediated endocytosis in ARPE-19 cells. J Pharm Pharmacol. 2014;66(4):564–573. doi:10.1111/jphp.12134
33. Dalal C, Saha A, Jana NR. Nanoparticle multivalency directed shifting of cellular uptake mechanism. J Phys Chem C. 2016;120(12):6778–6786. doi:10.1021/acs.jpcc.5b11059
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.