Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 13
Loss of HNF1α Function Contributes to Hepatocyte Proliferation and Abnormal Cholesterol Metabolism via Downregulating miR-122: A Novel Mechanism of MODY3
Authors Hu M, Huang X, Han X, Ji L
Received 1 November 2019
Accepted for publication 20 January 2020
Published 2 March 2020 Volume 2020:13 Pages 627—639
DOI https://doi.org/10.2147/DMSO.S236915
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ming-Hui Zou
Mengdie Hu, Xiuting Huang, Xueyao Han, Linong Ji
Department of Endocrinology and Metabolism, Peking University People’s Hospital, Peking University Diabetes Center, Beijing 100044, People’s Republic of China
Correspondence: Linong Ji; Xueyao Han
Department of Endocrinology and Metabolism, Peking University People’s Hospital, Peking University Diabetes Center, Beijing 100044, People’s Republic of China
Tel +86 10-8832 5578
Fax +86 10-8832 4371
Email [email protected]; [email protected]
Purpose: Mutations in hepatocyte nuclear factor 1α (HNF1α) are the cause of maturity-onset diabetes of the young type 3 (MODY3) and involved in the development of hepatocellular adenoma and abnormal lipid metabolism. Previously, we have found that the serum microRNA (miR)-122 levels in MODY3 patients were lower than those in type 2 diabetes mellitus and healthy controls. This study aimed to investigate the mechanism of decreased miR-122 levels in patients with MODY3 and whether low levels of miR-122 mediate tumorigenesis and abnormal lipid metabolism associated with HNF1α deficiency in human hepatocytes.
Methods: The expression of miR-122 was examined by real-time PCR. Dual-luciferase reporter assay was performed to confirm the transcriptional regulation of miR-122 by HNF1α. HepG2 cells were transfected with siRNA or miRNA mimic to downregulate or upregulate the expression of HNF1α or miR-122, respectively. CCK-8 and colony formation assay were used to determine cell proliferation. Lipid accumulation was examined by Oil Red O staining and intracellular triglyceride and cholesterol quantification assays.
Results: HNF1α regulated the expression of miR-122 by directly binding to its promoter. Knockdown of HNF1α in HepG2 cells reduced the expression of miR-122, increased proliferation and promoted intracellular cholesterol accumulation. Overexpression of miR-122 partially rescued the phenotypes associated with HNF1α deficiency in human hepatocytes. Mechanistically, HNF1α modulated cholesterol homeostasis via miR-122-dependent activation of sterol regulatory element-binding protein-2 (SREBP-2) and regulation of proprotein convertase subtilisin/kexin type 9 (PCSK9). Moreover, circulating miR-122 levels were associated with serum cholesterol levels.
Conclusion: Loss of HNF1α function led to hepatocyte proliferation and abnormal cholesterol metabolism by downregulating miR-122. Our findings revealed a novel mechanism that low levels of miR-122 mediate tumorigenesis and abnormal lipid metabolism associated with MODY3. MiR-122 may be a potential therapeutic target for the treatment of MODY3.
Keywords: MODY3, HNF1α, miR-122, cholesterol metabolism, hepatocellular adenoma
Introduction
Maturity-onset diabetes of the young (MODY) is an autosomal dominant form of diabetes.1 Heterozygous mutations in hepatocyte nuclear factor 1α (HNF1α) cause MODY subtype 3 (MODY3), which is one of the most common types.1–3
HNF1α is a homeobox transcription factor expressed in pancreatic islets, liver, kidney and intestine.4 HNF1α deficiency in pancreatic β-cell leads to impaired insulin secretion in response to glucose, resulting in hyperglycaemia.5,6 As HNF1α is a liver-enriched transcription factor (LETF), mutations in HNF1α also lead to abnormalities in liver function.7 Several studies have reported that primary liver neoplasms, especially hepatocellular adenoma (HCA) and liver adenomatosis, are clustered in MODY3 families.8–10 In addition, the lipid profile of patients with MODY3 is distinct from that of type 2 diabetes mellitus (T2DM), which may be attributed to the special lipid metabolism in hepatocytes associated with loss-of-function mutations in HNF1α.11,12 However, the mechanism that HNF1α variants increase the risk of liver neoplasm and abnormal lipid metabolism has not been well known.
Recently, we have found that the serum microRNA-122 (miR-122) levels in MODY3 patients were lower than those in T2DM patients and healthy controls.13 MiR-122 is an abundant liver-specific microRNA (miRNA) that regulates hepatocyte differentiation and proliferation, lipid metabolism and hepatitis C virus replication.14–17 In humans, liver miR-122 expression was found to be suppressed in hepatocellular carcinoma (HCC) and non-alcoholic fatty liver disease (NAFLD).18–22 Mice with deletion of miR-122 display progressive steatohepatitis, liver fibrosis and HCC.23,24 Several studies have demonstrated that HNF1α was involved in the transcriptional regulation of miR-122.15,25 Thus, it is possible that HNF1α regulates the tumorigenic potential and lipid metabolism of hepatocytes by targeting miR-122.
Therefore, we hypothesized that low levels of miR-122 mediate tumorigenesis and abnormal lipid metabolism associated with MODY3. In this study, we examined the effect of HNF1α on the proliferation and lipid metabolism of HepG2 cells and investigated the molecular mechanisms involved.
Materials and Methods
Cell Culture and Transfection
Human hepatic carcinoma cell line HepG2 was obtained from American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were cultured in minimum essential medium (MEM; Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 1% non-essential amino acids solution (NEAA; Gibco), 1% GlutaMAX supplement (Gibco), and 1% penicillin-streptomycin (Gibco) at 37°C in a humidified atmosphere containing 5% CO2.
HepG2 cells were plated in 12-well plates at 2×105 cells per well and grown to 50–60% confluence. Cells were treated with small interference RNA (siRNA; GenePharma, Shanghai, China) targeting HNF1α (siHNF1α) or nonspecific siRNA (siNC) at a concentration of 50 nM in combination with 50 nM of miR-122-5p mimic or negative control oligos (RiboBio, Guangzhou, China) using lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. To avoid the off-target effects, another siRNA sequence was used to confirm the effect of HNF1α. To upregulate or downregulate the expression of miR-122, cells were transfected with 50 nM of miRNA mimic or 100 nM of miRNA inhibitor (RiboBio). Cells were collected after 48 h for further experiments. SiRNA sequences are listed in the Supplementary Table.
Plasmid Construction
To construct HNF1α expression plasmids, the coding sequence for HNF1α was amplified by polymerase chain reaction (PCR) and then cloned into the XhoI/KpnI sites of GV141 vector (Genechem, Shanghai, China). The DNA fragments of miR-122 promoter region containing the putative HNF1α-binding site and mutated binding site were chemically synthesized and inserted into the GV238 luciferase reporter vector (Genechem, Shanghai, China) between the KpnI and XhoI site. Full-length cDNA of HNF1α and mutation of HNF1αArg131Trp (HNF1αR131W) were chemically synthesized and sub-cloned into the 3flag-pcDNA3.1 vector (Hanbio, Shanghai, China). The 3flag-pcDNA3.1 vector was transfected by lipofectamine 3000 according to the manufacturer’s instruction. All constructs were confirmed by DNA sequencing. The primer sequences for PCR amplification of plasmid construction are listed in the Supplementary Table.
Luciferase Reporter Assay
Human embryonic kidney (HEK) 293T cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were transfected with luciferase reporter vectors in combination with HNF1α plasmid or control plasmid using X-tremegene HP transfection reagent (Roche, Basel, Switzerland) according to the manufacturer’s instruction. Cells were collected 48 h after transfection and assayed using the dual-luciferase reporter assay system (Promega, Madison, WI, USA).
Quantitative Real-Time PCR
Total RNA was extracted from HepG2 cells using TRIzol reagent (Invitrogen) and MiniBEST universal RNA extraction kit (Takara, Shiga, Japan), treated with dsDNase to eliminate genomic DNA. For mRNA detection, total RNA was reverse-transcripted by maxima H minus first-strand cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instruction. Bulge-Loop miRNA primer and starter kit (RiboBio) was used to detect the expression of has-miR-122-5p. Real-time PCR was performed on the ViiA 7 real-time PCR system using powerup SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA, USA). Relative mRNA expression levels were calculated using the 2−ΔΔCT method (with β-actin or 18s rRNA used as the reference gene). Relative miRNA expression levels were normalized to U6 small nuclear RNA. The primer sequences for real-time PCR assay are listed in the Supplementary Table.
Cell Proliferation and Colony Formation Assay
Cell proliferation was determined using the cell counting kit-8 (CCK-8; Dojindo, Kumamoto, Japan). Briefly, HepG2 cells were plated in 96-well plates at 5×103 per well and transfected with siHNF1α with or without miR-122 mimic as mentioned above. Before incubation (0 h) and after 24, 48, 72, 96 h of incubation, CCK-8 solution was added at a 1:10 dilution to each well and incubated in the dark at 37°C for 1 h. The optical density (OD) value of each well was then measured at a wavelength of 450 nm using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). Cell proliferation was calculated by the following equation: relative proliferation = time t (ODtest − ODblank)/time 0 h (ODtest − ODblank).
For colony formation assay, HepG2 cells were isolated by trypsin and plated in 6-well plates at a density of 2×103 per well and incubated for 10 days at 37°C in a humidified atmosphere containing 5% CO2. The plates with colonies were washed twice with phosphate buffer saline and fixed in 4% phosphate-buffered paraformaldehyde for 30 min. Colonies were stained with 0.1% crystal violet stain solution for 30 min and countered.
Oil Red O Staining
HepG2 cells plated on the cover slides were treated with or without 200 μM free fatty acid (FFA) mixture (oleate [O-7501, Sigma-Aldrich, St. Louis, MO, USA] and palmitate [P-9767, Sigma-Aldrich] at the ratio of 2:1) for 24 h. Lipids were stained with Oil Red O stain kit (Solarbio, Beijing, China) according to the manufacturer’s instruction. Briefly, the cover slides were washed with 60% isopropanol and stained with Oil Red O. Slides were counterstained with Mayer’s hematoxylin. Oil Red O staining images were taken using a light microscope (Olympus Optical, Tokyo, Japan).
Triglyceride and Cholesterol Measurement
Intracellular triglyceride and cholesterol contents were assayed using tissue triglyceride and cholesterol assay kit (Applygen Technologies, Beijing, China). Protein concentration was determined using a BCA protein assay kit (Thermo Fisher Scientific). All experiments were performed according to the manufacturer’s protocol. The contents of triglyceride and cholesterol were normalized to total protein concentration from each well.
Western Blot
Cells were lysed in RIPA lysis buffer containing protease inhibitors (Thermo Fisher Scientific). Cell protein lysates were centrifuged at 14,000 rpm for 20 min at 4°C and the supernatants were collected. Protein concentrations were determined using a BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein from samples were resolved on 4–20% sodium dodecyl sulfate–polyacrylamide gel (SDS-PAGE; Applygen Technologies) and transferred onto 0.45 μm polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking for nonspecific binding with 5% non-fat dry milk for 1 h at room temperature, the membranes were incubated with antibody HNF1α (1:500, ab96777, Abcam, Cambridge, MA, USA), SCAP (1:2000, ab125186, Abcam), SREBP-2 (1:500, ab30682, Abcam), PCSK9 (1:1000, ab181142, Abcam), or β-actin (1:1000, #4970, Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C and followed by an incubation with secondary antibody (1:3000, #7074, Cell Signaling Technology) for 1 h at room temperature. Protein was visualized with enhanced chemiluminescence (ECL; Thermo Fisher Scientific) and membranes were imaged with GE Healthcare imaging system (Waukesha, WI, USA). Band intensities were quantified using Image J software.
Statistics Analysis
Data were presented as the mean ± standard deviation (SD) from at least three independent experiments. Comparisons between two groups were performed with Student’s t-test. Multiple group comparisons were analyzed with a one-way analysis of variance (ANOVA). Associations between circulating miR-122 and lipid profiles were analyzed using Spearman correlation coefficient and multiple linear regression. P < 0.05 was considered statistically significant. All statistical analyses were performed with the SPSS 24.0 statistical software package (Chicago, IL, USA).
Results
HNF1α Regulates miR-122 Expression by Directly Binding to Its Promoter
Our previous study has found that the serum miR-122 levels in MODY3 patients were lower than those in type 2 diabetes mellitus and healthy controls (Figure 1A).13 As miR-122 is a liver-specific miRNA, we investigated that whether loss of HNF1α function downregulated the expression of miR-122 in hepatocytes. Two siRNAs were separately transfected into HepG2 cells to suppress HNF1α expression. As shown in Figure 1B, cells transfected with siHNF1α significantly downregulated the protein levels of HNF1α compared with the negative control. MiRNA real-time PCR showed that transfected with two siHNF1α both resulted in a decrease in the expression of miR-122 in HepG2 cells (Figure 1C). Mutation in HNF1α (R131W, the most common mutation in MODY3 patients in our previous study) also reduced the expression of miR-122 in hepatocytes (Figure 1D). However, transfection of miR-122 mimic into HepG2 cells did not significantly alter the protein levels of HNF1α (Figure 1E).
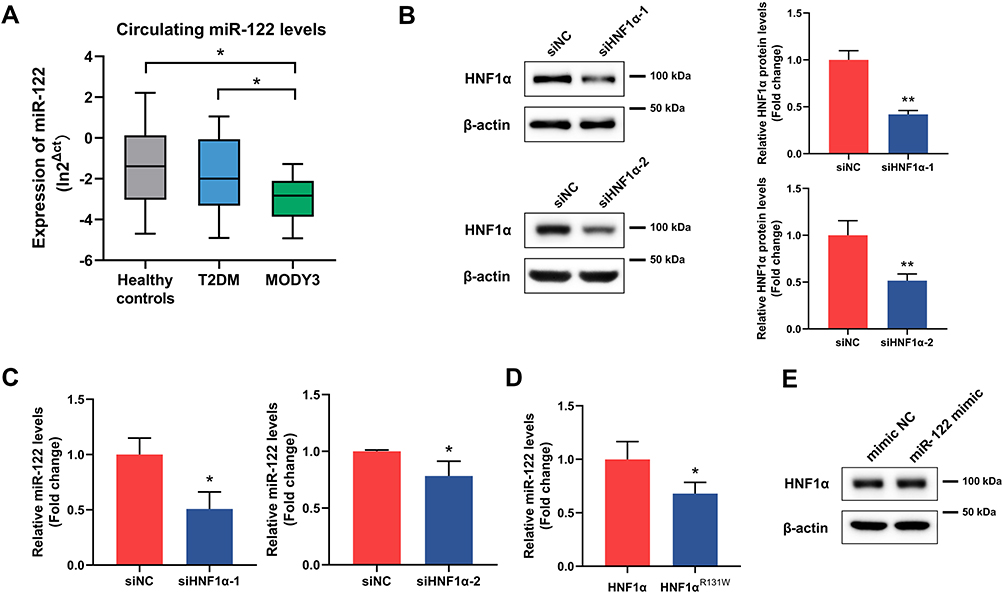 |
Figure 1 Loss of HNF1α function downregulated the expression of miR-122. (A) The expression of serum miR-122 in healthy control, T2DM and MODY3. (B) Protein levels of HNF1α in HepG2 cells transfected with two siHNF1α sequences (siHNF1α-1 and siHNF1α-2) or siNC for 48 h. (C) RNA levels of miR-122 in HepG2 cells transfected with siHNF1α or siNC for 48 h. (D) RNA levels of miR-122 in HepG2 cells transfected with HNF1α plasmid or HNF1αR131W plasmid for 48 h. (E) Protein levels of HNF1α in HepG2 cells transfected with miR-122 mimic or negative control oligos (mimic NC) for 48 h. Data represent mean ± SD of three independent experiments; *P < 0.05 and **P < 0.01 as indicated. Abbreviations: miR-122, microRNA-122; T2DM, type 2 diabetes mellitus; MODY3, maturity-onset diabetes of the young type 3; HNF1α, hepatocyte nuclear factor 1α; siHNF1α, specific siRNA targeting HNF1α; siNC, nonspecific siRNA. |
To further investigate the mechanism of decreased miR-122 levels in patients with MODY3, we examined the transcription regulation effect of HNF1α on miR-122. UCSC and JASPAR were used to analyze the 2-kb region upstream of the transcription start site of human miR-122. Bioinformatics analysis revealed that the miR-122 promoter region contains three HNF1α-binding motifs (Figure 2A). To confirm whether miR-122 is under the transcriptional control of HNF1α, a luciferase reporter vector containing the miR-122 promoter with wild-type or mutated binding sites (Figure 2B) was cotransfected with an HNF1α plasmid or control plasmid into HEK293T cells. As shown in Figure 2C, HNF1α overexpression significantly increased the luciferase activity of the miR-122 promoter reporter. Mutation of the putative binding sites resulted in decreased luciferase activity, suggesting that HNF1α was involved in the transcriptional regulation of miR-122. Taken together, these results suggested that HNF1α regulated the expression of miR-122 by directly binding to its promoter. MiR-122 may act as an effector of HNF1α in physiological processes in the liver.
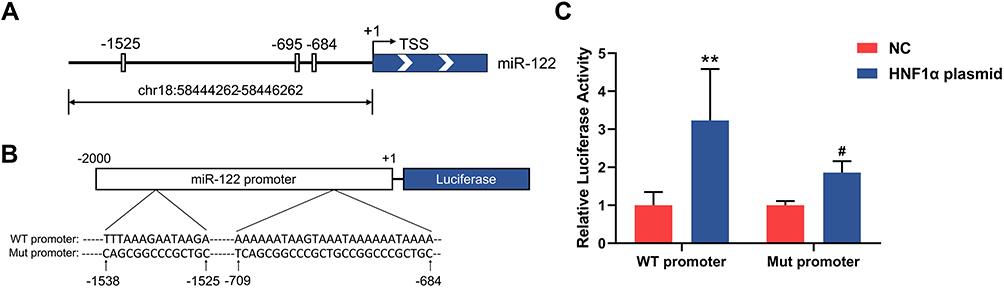 |
Figure 2 HNF1α regulates the expression of miR-122 by directly binding to its promoter. (A) Schematic representation of putative HNF1α-binding motifs in the miR-122 promoter from −2kb to the TSS (indicated as +1). The grey boxes depict putative HNF1α-binding sites, resided in the proximal promoter at −684nt, −695nt and −1525nt 5ʹ to TSS. (B) Schematic model for luciferase reporter vectors containing WT promoter or Mut promoter. (C) Luciferase activities in HEK293T cells cotransfected with luciferase reporter vectors containing WT promoter or Mut promoter and HNF1α plasmid or control plasmid. **P < 0.01 vs control cells; #P < 0.05 vs cells transfected with WT promoter. Abbreviations: miR-122, microRNA-122; HNF1α, hepatocyte nuclear factor 1α; TSS, transcription start site; WT promoter, miR-122 promoter with wild-type binding site; Mut promoter, miR-122 promoter with mutated binding site; NC, control plasmid. |
Inhibition of HNF1α Promotes Hepatocyte Proliferation by Suppressing miR-122
HCA is a rare, benign liver tumor that is derived from proliferation of mature hepatocytes.26 We hypothesized that HNF1α knockdown may promote hepatocyte proliferation by downregulating the expression of miR-122. As shown in Figure 3A, inhibition of HNF1α in HepG2 cells significantly promoted cell proliferation. However, this effect was partially restored by miR-122 overexpression. In addition, HNF1α knockdown led to a significant increase in colony formation, whereas cotransfection with miR-122 mimic abolished this effect on colony formation (Figure 3B). Collectively, these data indicated that inhibition of HNF1α promoted hepatocyte proliferation by suppressing miR-122.
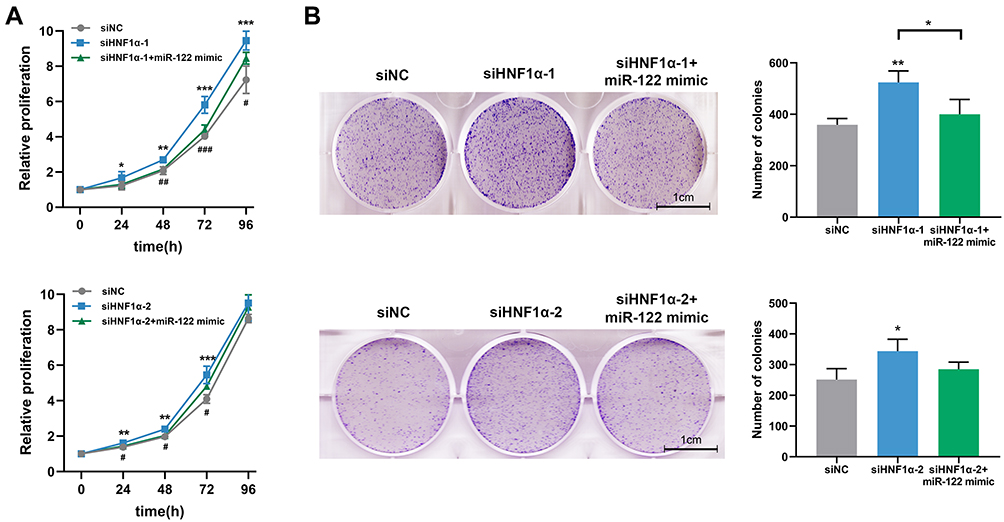 |
Figure 3 Inhibition of HNF1α promotes hepatocyte proliferation by suppressing miR-122. (A) Cell proliferation was evaluated by the CCK-8 assay. Data represent mean ± SD of four independent experiments. (B) Representative graphs and quantification of cell colonies analyzed by the colony formation assay. Data represent mean ± SD of three independent experiments; *P < 0.05, **P < 0.01 and ***P < 0.001, siNC vs siHNF1α or as indicated; #P < 0.05, ##P < 0.01 and ###P < 0.001, siHNF1α vs siHNF1α+miR-122 mimic.Abbreviations: miR-122, microRNA-122; HNF1α, hepatocyte nuclear factor 1α; siHNF1α, specific siRNA targeting HNF1α; siNC, nonspecific siRNA. |
HNF1α Knockdown Elevates Hepatic Cholesterol Levels by Downregulating miR-122
Although it has been reported that HNF1α inactivation promoted lipogenesis in hepatocytes, no research has been conducted to explore whether this effect was dependent on decreased miR-122 expression.27–29 We performed the Oil Red O staining assay and triglyceride and cholesterol quantification assays to examine the changes in lipid metabolism in hepatocytes. Unexpectedly, Oil Red O staining showed that although FFA treatment led to a significant accumulation of lipid droplets, there was little or no difference in lipid deposition in HepG2 cells treated with siHNF1α or miR-122 mimic (Figure 4A). Similar results were observed in the intracellular triglyceride content measurement (Figure 4B). We further examined the content of total and free cholesterol in hepatocytes. The results indicated that HNF1α knockdown increased the intracellular total cholesterol and free cholesterol content of HepG2 cells (Figure 4C and D). However, transfection of miR-122 mimic ameliorated cholesterol accumulation induced by HNF1α deficiency. Our results demonstrated that HNF1α knockdown increased hepatic cholesterol content, but not triglyceride content, by downregulating miR-122.
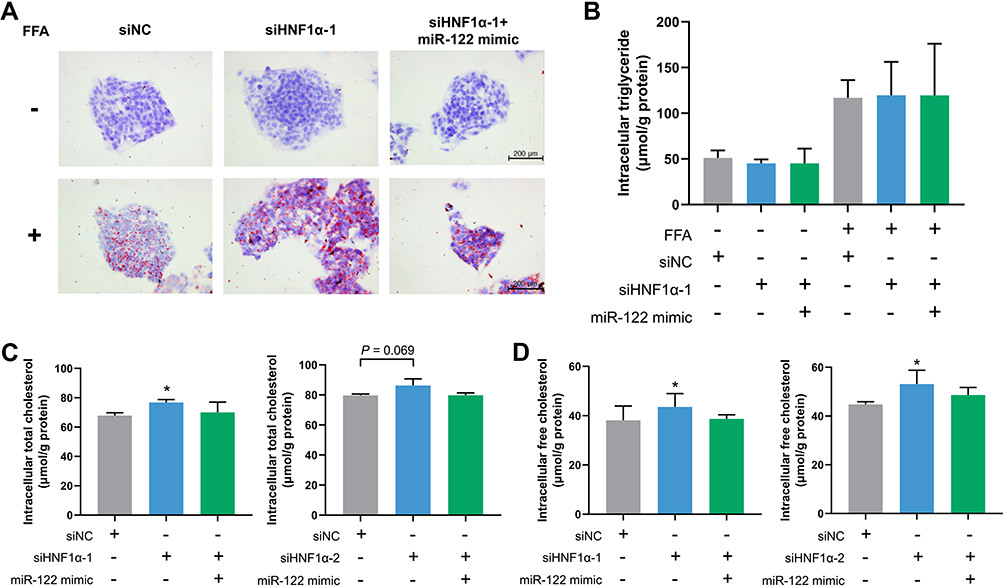 |
Figure 4 HNF1α knockdown elevates hepatic cholesterol levels by downregulating miR-122. (A) Oil Red O staining in HepG2 cells transfected with siHNF1α or siNC and miR-122 mimic or negative control oligos for 48 h in the presence or absence of FFA. (B–D) Intracellular triglyceride content (B), total cholesterol (C) and free cholesterol (D) content in HepG2 cells transfected with siHNF1α or siNC and miR-122 mimic or negative control oligos for 48 h. Data represent mean ± SD of three independent experiments; *P < 0.05 vs control cells. Abbreviations: FFA, free fatty acid; miR-122, microRNA-122; HNF1α, hepatocyte nuclear factor 1α; siHNF1α, specific siRNA targeting HNF1α; siNC, nonspecific siRNA. |
HNF1α Modulates Cholesterol Homeostasis via miR-122-Mediated SREBP-2 Activation and PCSK9 Regulation
To further explore the molecular mechanism underlying the effect of HNF1α on hepatic cholesterol metabolism, we investigated the expression of genes involved in cholesterol metabolism. Sterol regulatory element-binding protein-2 (SREBP-2) is a transcription regulator that activates genes involved in cholesterol synthesis and uptake.30 Its function is modulated by SREBP cleavage-activating protein (SCAP), an escort protein necessary for SREBP-2 transport and activation.30 As shown in Figure 5A, HNF1α knockdown increased the expression of SCAP and the mature nuclear form of SREBP-2; however, these affects were rescued by overexpression of miR-122. We next investigated the expression of proprotein convertase subtilisin/kexin type 9 (PCSK9), a secreted protein that degrades hepatic low-density lipoprotein receptor (LDLR), thereby reducing cholesterol uptake and increasing plasma low-density lipoprotein cholesterol (LDL-c) levels.31,32 It has been reported that the expression of PCSK9 is regulated by HNF1α and SREBP-2.33,34 As shown in Figure 5A, the protein levels of PCSK9 were significantly decreased after HNF1α knockdown. However, addition of miR-122 partially restored the expression of PCSK9.
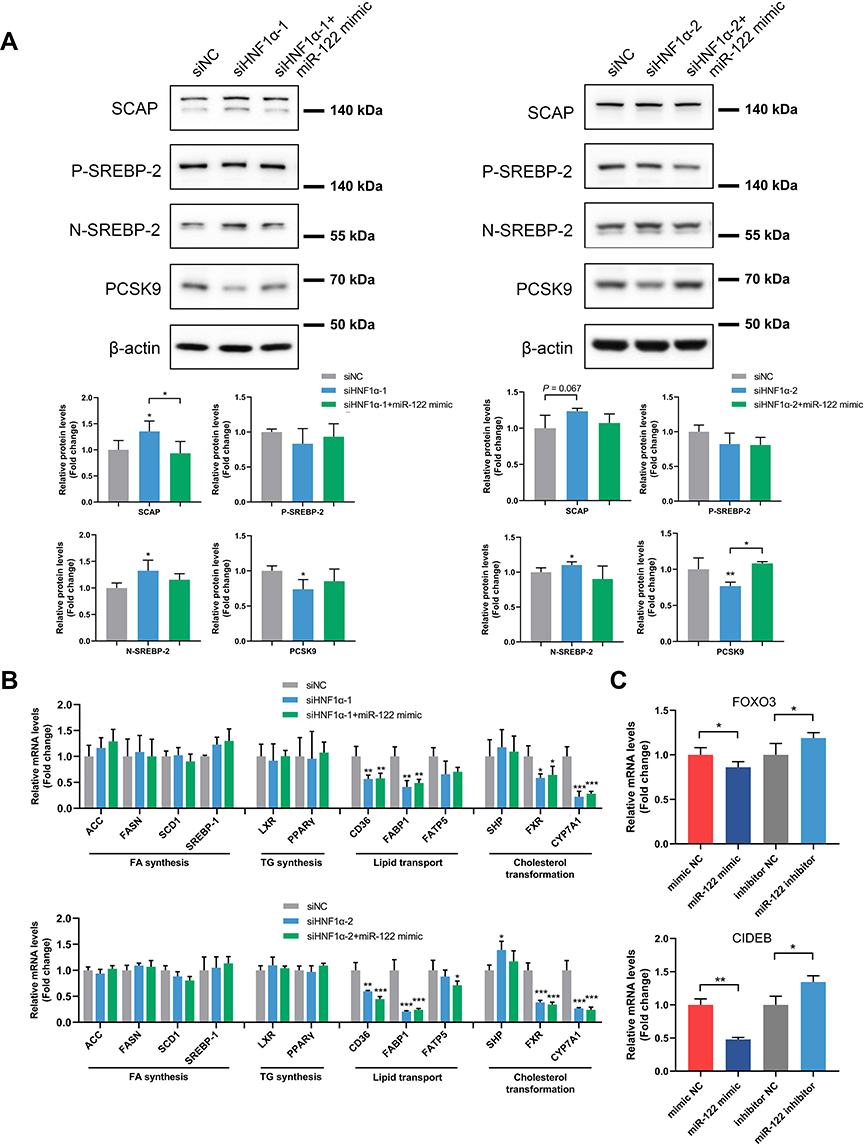 |
Figure 5 HNF1α modulates cholesterol homeostasis via miR-122-mediated SREBP-2 activation and PCSK9 regulation. (A) Western blot analysis of SCAP/SREBP-2 signaling and PCSK9 in HepG2 cells transfected with siHNF1α or siNC and miR-122 mimic or negative control oligos for 48 h. (B) Expression levels of genes related to FA synthesis (ACC, FASN, SCD1 and SREBP-1), TG synthesis (LXR and PPARγ), lipid transport (CD36, FABP1 and FATP5) and cholesterol transformation (SHP, FXR and CYP7A1) in HepG2 cells transfected with siHNF1α or siNC and miR-122 mimic or negative control oligos for 48 h. (C) Expression of FOXO3 or CIDEB after overexpression or inhibition of miR-122. Data represent mean ± SD of three independent experiments; *P < 0.05, **P < 0.01 and ***P < 0.001 vs control cells or as indicated. Abbreviations: miR-122, microRNA-122; HNF1α, hepatocyte nuclear factor 1α; siHNF1α, specific siRNA targeting HNF1α; siNC, nonspecific siRNA; SREBP, sterol regulatory element-binding protein; SCAP, SREBP cleavage-activating protein; P-SREBP-2, precursor SREBP-2; N-SREBP-2, nuclear SREBP-2; PCSK9, proprotein convertase subtilisin/kexin type 9; FA, fatty acid; TG, triglyceride; ACC, acetyl-CoA carboxylase; FASN, fatty acid synthase; SCD1, stearoyl-coenzyme A desaturase 1; LXR, liver X receptor; PPARγ, peroxisome proliferator-activated receptor γ; FABP1, fatty acid-binding protein 1; FATP5, fatty acid transport protein 5; SHP, small heterodimer partner; FXR, farnesoid X receptor; CYP7A1, cholesterol 7-alpha hydroxylase; FOXO3, forkhead box O3; CIDEB, cell death-inducing DFFA-like effector B. |
We also assessed the expression levels of genes related to fatty acid synthesis, triglyceride synthesis, lipid transport and cholesterol transformation. We found that the expression of genes related to lipid transport, including CD36 and FABP1, and cholesterol transformation, such as FXR and CYP7A1, were decreased after HNF1α knockdown (Figure 5B). However, the addition of miR-122 did not alter the mRNA levels of these genes, suggesting that HNF1α regulates these genes in a miR-122-independent manner. Taken together, our results indicated that HNF1α modulated cholesterol homeostasis via miR-122-mediated SREBP-2 activation and PCSK9 regulation.
In order to investigate the mechanism by which miR-122 regulates SREBP-2 and PCSK9. We used miRNA mimic or inhibitor to upregulate or downregulate the expression of miR-122. We found that forkhead box O3 (FOXO3) and cell death-inducing DFFA-like effector B (CIDEB) were regulated by miR-122 (Figure 5C). It has been reported that FOXO3 inhibited PCSK9 promoter activity or suppressed PCSK9 transcription by recruiting histone deacetylase sirtuin 6.35,36 The bioinformatics analysis and luciferase reporter assays also showed that miR-122 directly targeted FoxO3.37 Therefore, miR-122 regulated the expression of PCSK9 partially via directly regulation of FOXO3. CIDEB has emerged as an important regulator of cholesterol metabolism.38 Loss of CIDEB reduced the activation of SREBP-2.39 We observed that overexpression of miR122 significantly downregulated the expression of CIDEB whereas inhibition of miR-122 upregulated CIDEB. Therefore, miR-122 regulated the nuclear form of SREBP-2 might partially through CIDEB.
Circulating miR-122 Levels Were Positively Associated with the Serum Cholesterol Levels
Given that PCSK9 expression is regulated by miR-122 in hepatocytes, we evaluated the associations between circulating miR-122 and cholesterol levels in 107 volunteers (data from our previous study). Linear regression analysis showed that the levels of miR-122 were positively correlated with the LDL-c levels (Figure 6A; Spearman correlation coefficient = 0.201, P = 0.046; standardized β coefficient = 0.221, P = 0.041 after adjusting for age, gender, body mass index and glycated hemoglobin). The levels of miR-122 were also positively associated with the total cholesterol levels (Figure 6B; Spearman correlation coefficient = 0.177, P = 0.077; standardized β coefficient = 0.222, P = 0.028, after adjusting for age, gender, body mass index and glycated hemoglobin). No association was found between circulating miR-122 and serum triglyceride or high-density lipoprotein cholesterol (HDL-c) levels (Table 1).
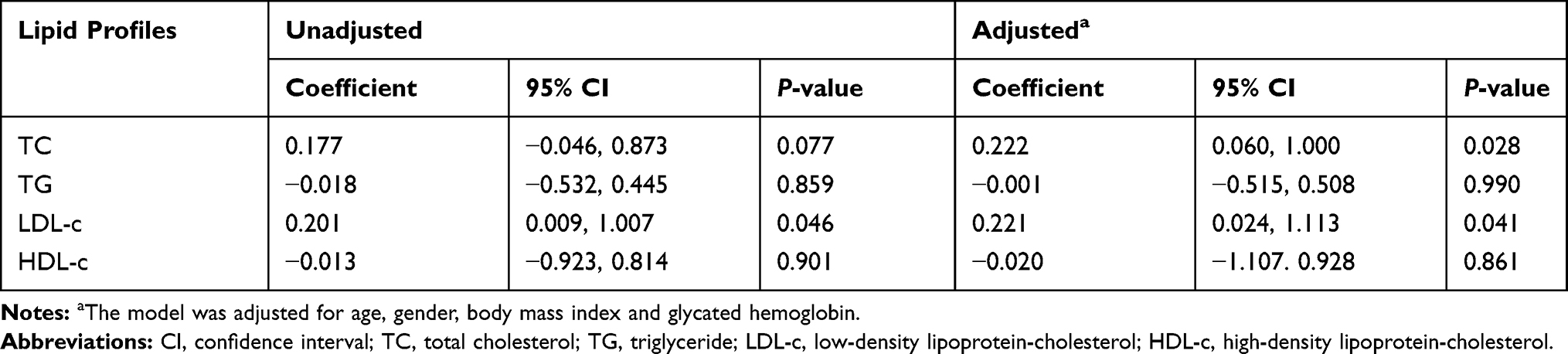 |
Table 1 Associations Between Circulating miR-122 and Serum Lipid Profiles |
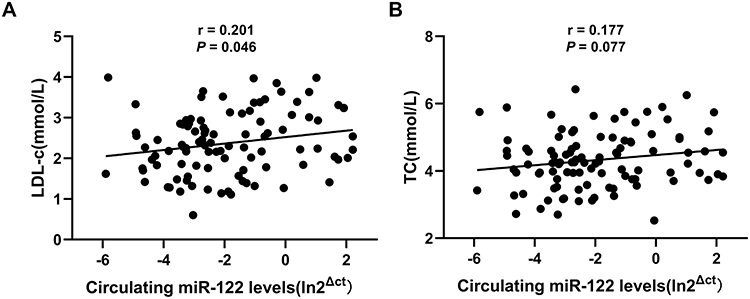 |
Figure 6 Circulating miR-122 levels were positively associated with the serum cholesterol levels. The correlation between circulating miR-122 and LDL-c (A) or TC (B) was analyzed by Spearman correlation coefficient (n=107). Abbreviations: miR-122, microRNA-122; LDL-c, low-density lipoprotein-cholesterol; TC, total cholesterol. |
Discussion
In the current study, for the first time, we explain the mechanism of decreased miR-122 levels in patients with MODY3. Low levels of miR-122 in MODY3 patients were attributed to the downregulated expression of miR-122 in hepatocytes by loss of function in HNF1α. We also demonstrated that HNF1α knockdown led to hepatocyte proliferation and abnormal cholesterol metabolism by downregulating miR-122, and overexpression of miR-122 could rescue the phenotypes associated with HNF1α deficiency in human hepatocytes. Our findings suggest that low levels of miR-122 in hepatocytes mediate tumorigenesis and abnormal lipid metabolism associated with MODY3 (Figure 7).
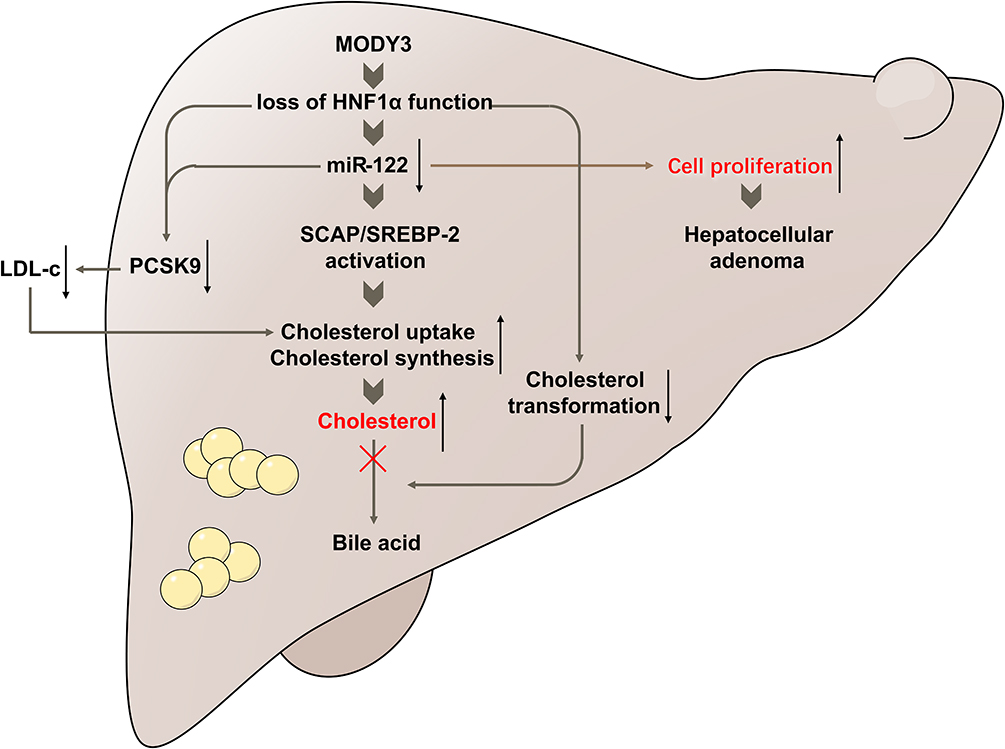 |
Figure 7 Schematic representation of the mechanisms of abnormal cholesterol metabolism and increased risk of liver adenoma associated with MODY3. Loss of HNF1α function downregulated the expression of miR-122, thereby promoted hepatocyte proliferation and increased the risk of liver adenoma. In terms of lipid metabolism, HNF1α knockdown resulted in accumulated hepatic cholesterol via miR-122-mediated activation of the SCAP/SREBP-2 pathway. Suppression of CYP7A1 led to impaired transformation of cholesterol into bile acid and accumulation of cholesterol in human hepatocytes. In addition, HNF1α knockdown repressed the expression of PCSK9 via a direct effect on the promoter or an indirect effect mediated by miR-122, which ultimately resulted in increased cholesterol uptake and decreased plasma LDL-c levels. |
MiR-122 is the most abundant liver-specific miRNA and has been shown to be regulated by LETFs, including HNF1α, HNF3α, HNF3β, HNF4α and HNF6α.15,25,40 Laudadio et al have shown a positive feedback loop between HNF6α and miR-122 that controls hepatocyte differentiation.40 Wei et al have also reported that HNF4α regulated gluconeogenesis and lipid metabolism through regulating miR-122.41 In the present study, we confirmed that HNF1α regulated the expression of miR-122 by directly binding to its promoter. Mir-122 is a target gene of HNF1α. This may partially explain the decrease in serum miR-122 levels in MODY3 patients. Therefore, miR-122 may act as an effector of HNF1α in various physiological processes in the liver.
Several studies have reported that primary hepatocellular neoplasms, especially HCA, are clustered in MODY3 families.8–10 HCA is a rare, benign liver tumor that is derived from the proliferation of mature hepatocytes.26 HNF1α-inactivated HCA is the most common subtype of HCA, phenotypically characterized by marked steatosis without significant cytologic abnormalities.42,43 Given that miR-122 is regarded as a tumor suppressor miRNA that inhibits cell proliferation, it is possible that miR-122 loss is involved in the mechanism of HNF1α-inactivated HCA. Our results confirmed that HNF1α knockdown promoted hepatocyte proliferation by downregulating miR-122, indicating that suppressed miR-122 expression may account for the increased risk of liver adenoma associated with MODY3. Although transformation of HCA to HCC is thought to be a rare phenomenon in HNF1α-inactivated HCA, HCC has been reported in some cases of MODY3, suggesting that inactivation of HNF1α may also play a role in hepatocarcinogenesis.10,43,44 Whether miR-122 mediates malignant transformation of hepatocytes requires further investigation.
Analysis of histopathological and clinical data showed that liver adenoma in MODY3 families was mostly characterized by steatosis.45 Nakamura et al have reported a case of a Japanese girl with MODY3 in whom liver dysfunction and steatosis occurred in early childhood.46 In addition, the lipid profiles of MODY3 patients are distinct from those of T2DM patients and nondiabetic controls, possibly owing to the abnormal lipid metabolism in hepatocytes associated with loss-of-function mutations in HNF1α.11,12 Several studies have also investigated the effect of HNF1α and miR-122 on lipid metabolism in vivo. HNF1α-null mice and HNF1α-mutant mice displayed a phenotype of hepatic steatosis and disorders in lipid metabolism.27,29,47 Although temporary inhibition of miR-122 by antisense oligonucleotides or antagomirs has been shown to reduce serum cholesterol, mice with deletion of miR-122 by liver-specific knockout or germline knockout displayed a progressive steatohepatitis phenotype.16,23,24,48 Herein, we found that HNF1α knockdown increased hepatic cholesterol accumulation, which was restored by miR-122 overexpression. Our findings suggest that HNF1α regulated hepatic cholesterol metabolism via miR-122. However, we did not observe any significant change in lipid droplets accumulation or intracellular triglyceride content in HepG2 cells after inhibition of HNF1α. This may be because we transiently transfected cells with siRNA or miRNA mimic to knockdown HNF1α or overexpress miR-122, respectively. Considering the optimal time frame for small RNA effect, we treated cells with siRNA or miRNA mimic for 48 h and then found alterations in the expression of genes involved in the lipid metabolism. In fact, alterations in gene expression often occurred prior to phenotypic changes. A previous study has observed a marked lipid overload in HepG2 cells after transfection with siHNF1α for 7 days.49 Therefore, further studies are needed to verify the relationships among HNF1α, miR-122 and lipid metabolism in stable cell lines or a genetic mouse model.
Intracellular cholesterol homeostasis is regulated by multiple pathways including cholesterol biosynthesis, uptake and transformation. SREBPs are membrane-bound transcription factors that play a central role in lipid metabolism.50 SREBP-2 mainly regulates the expression of genes involved in cholesterol synthesis and uptake.30 SREBP-2 activation is tightly modulated by the escort protein, SCAP. We found that HNF1α knockdown increased the expression of SCAP and activated SREBP-2; however, these affects were rescued by miR-122 overexpression. Thus, HNF1α modulates intracellular cholesterol homeostasis via the SCAP/SREBP-2 pathway by regulating miR-122.
The effects of HNF1α on cholesterol metabolism have been investigated in genetic models.47 HNF1α-knockout mice have been reported to have a defect in bile acid transport, increased bile acid and liver cholesterol synthesis, and impaired high-density lipoprotein metabolism.47 However, in contrast to our findings, that study showed that the expression of Cyp7a1 was elevated in the livers of HNF1α-knockout mice, and the content of free cholesterol in the liver was lower than that in wild-type mice.47 This discrepancy may be attributed to the species-related differences in cholesterol metabolism between rodents and humans. A previous study has demonstrated that HNF1α bound to and transactivated the human, but not the rat, CYP7A1 promoter.51 In addition, the liver X receptor-mediated feed-forward regulation of Cyp7a1 by cholesterol in mice is absent in humans.52 It is likely that HNF1α knockdown in human hepatocytes represses the expression of human CYP7A1 via a direct effect on its promoter, leading to impaired transformation of cholesterol into bile acid and accumulation of cholesterol in human hepatocytes.
Patients with MODY3 have been reported to have lower LDL-c levels than T2DM patients and healthy controls.11,13 PCSK9 is a major regulator of cholesterol homeostasis that interacts with hepatic LDLR and mediates endosomal and lysosomal degradation, resulting in increased plasma LDL-c levels.31,32 In the liver, PCSK9 is regulated by HNF1α and SREBP-2.33,34 We confirmed that HNF1α regulated PCSK9 in our study. In addition, it has been demonstrated that inhibition of miR-122 was associated with lower serum cholesterol in our work and other studies.16,53,54 Our study first found that miR-122 regulated PCSK9 in hepatocytes, which may explain the decreased serum cholesterol level after inhibition of miR-122. Therefore, HNF1α regulated the expression of PCSK9 not only by directly affecting its promoter activity but also by an indirect effect via miR-122. However, it is noteworthy that LDL-c is a well-established risk factor for cardiovascular disease (CVD).55 Inhibition of PCSK9 is associated with reduction in plasma LDL-c levels and decreased risk of CVD.56 Therefore, further studies are required to reveal the associations among MODY3, NAFLD, CVD and PCSK9.
We are the first to demonstrate that low levels of miR-122 may mediate tumorigenesis and abnormal lipid metabolism associated with MODY3; however, we used HNF1α-knockdown cell model to confirm our hypothesis. This is a limitation in our study because patients with MODY3 have heterozygous mutations in the HNF1α gene. Although the majority of the mutations in the HNF1α gene identified in individuals with MODY3 result in loss of function of HNF1α protein, the clinical manifestation is highly variable in MODY3, which may not be explained only by loss of function of HNF1α.57 Another limitation is that we investigated this molecular mechanism in vitro and it should be further confirmed in vivo. Data of liver function and lipid profiles in animal studies will help us further understand the nature of MODY3. Further studies are warranted to elucidate the underlying mechanism.
Conclusion
In summary, our study revealed that loss of HNF1α function in hepatocytes led to increased hepatocyte proliferation and abnormal hepatic cholesterol metabolism by downregulating the expression of miR-122. Low levels of miR-122 partially explain the abnormal lipid metabolism and increased risk of liver neoplasms in patients with MODY3. Thus, miR-122 may be a potential therapeutic target for the treatment of MODY3.
Acknowledgments
This study was funded by National Key Research and Development Program (2016YFC1304901), Beijing Science and Technology Committee Funding (Z141100007414002 and D131100005313008), and the National High-Technology Research and Development Program of China (863 Program 2012AA02A509).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. 2001;345(13):971–980. doi:10.1056/NEJMra002168
2. Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384(6608):455–458. doi:10.1038/384455a0
3. Ellard S, Bellanne-Chantelot C, Hattersley AT. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia. 2008;51(4):546–553. doi:10.1007/s00125-008-0942-y
4. Blumenfeld M, Maury M, Chouard T, Yaniv M, Condamine H. Hepatic nuclear factor 1 (HNF1) shows a wider distribution than products of its known target genes in developing mouse. Development. 1991;113(2):589–599.
5. Pontoglio M, Sreenan S, Roe M, et al. Defective insulin secretion in hepatocyte nuclear factor 1alpha-deficient mice. J Clin Invest. 1998;101(10):2215–2222. doi:10.1172/JCI2548
6. Yamagata K, Nammo T, Moriwaki M, et al. Overexpression of dominant-negative mutant hepatocyte nuclear factor-1 alpha in pancreatic beta-cells causes abnormal islet architecture with decreased expression of E-cadherin, reduced beta-cell proliferation, and diabetes. Diabetes. 2002;51(1):114–123. doi:10.2337/diabetes.51.1.114
7. Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part I: the hepatocyte nuclear factor network and liver-specific gene expression. Pharmacol Rev. 2002;54(1):129–158. doi:10.1124/pr.54.1.129
8. Bacq Y, Jacquemin E, Balabaud C, et al. Familial liver adenomatosis associated with hepatocyte nuclear factor 1alpha inactivation. Gastroenterology. 2003;125(5):1470–1475. doi:10.1016/j.gastro.2003.07.012
9. Reznik Y, Dao T, Coutant R, et al. Hepatocyte nuclear factor-1 alpha gene inactivation: cosegregation between liver adenomatosis and diabetes phenotypes in two maturity-onset diabetes of the young (MODY)3 families. J Clin Endocrinol Metab. 2004;89(3):1476–1480. doi:10.1210/jc.2003-031552
10. Willson JS, Godwin TD, Wiggins GA, Guilford PJ, McCall JL. Primary hepatocellular neoplasms in a MODY3 family with a novel HNF1A germline mutation. J Hepatol. 2013;59(4):904–907. doi:10.1016/j.jhep.2013.05.024
11. McDonald TJ, McEneny J, Pearson ER, et al. Lipoprotein composition in HNF1A-MODY: differentiating between HNF1A-MODY and type 2 diabetes. Clin Chim Acta. 2012;413(9–10):927–932. doi:10.1016/j.cca.2012.02.005
12. Bellanne-Chantelot C, Levy DJ, Carette C, et al. Clinical characteristics and diagnostic criteria of maturity-onset diabetes of the young (MODY) due to molecular anomalies of the HNF1A gene. J Clin Endocrinol Metab. 2011;96(8):E1346–E1351. doi:10.1210/jc.2011-0268
13. Huang X, Gong S, Ma Y, et al. Lower circulating miR-122 level in patients with HNF1A variant-induced diabetes compared with type 2 diabetes. J Diabetes Res. 2018;2018:7842064. doi:10.1155/2018/7842064
14. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12(9):735–739. doi:10.1016/S0960-9822(02)00809-6
15. Xu H, He JH, Xiao ZD, et al. Liver-enriched transcription factors regulate microRNA-122 that targets CUTL1 during liver development. Hepatology. 2010;52(4):1431–1442. doi:10.1002/hep.23818
16. Krutzfeldt J, Rajewsky N, Braich R, et al. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438(7068):685–689. doi:10.1038/nature04303
17. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science (New York, NY). 2005;309(5740):1577–1581. doi:10.1126/science.1113329
18. Bai S, Nasser MW, Wang B, et al. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem. 2009;284(46):32015–32027. doi:10.1074/jbc.M109.016774
19. Tsai WC, Hsu PW, Lai TC, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49(5):1571–1582. doi:10.1002/hep.22806
20. Kutay H, Bai S, Datta J, et al. Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem. 2006;99(3):671–678. doi:10.1002/(ISSN)1097-4644
21. Cheung O, Puri P, Eicken C, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48(6):1810–1820. doi:10.1002/hep.22569
22. Latorre J, Moreno-Navarrete JM, Mercader JM, et al. Decreased lipid metabolism but increased FA biosynthesis are coupled with changes in liver microRNAs in obese subjects with NAFLD. Int J Obes (Lond). 2017;41(4):620–630. doi:10.1038/ijo.2017.21
23. Hsu SH, Wang B, Kota J, et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J Clin Invest. 2012;122(8):2871–2883. doi:10.1172/JCI63539
24. Tsai WC, Hsu SD, Hsu CS, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122(8):2884–2897. doi:10.1172/JCI63455
25. Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28(40):3526–3536. doi:10.1038/onc.2009.211
26. Nault JC, Bioulac-Sage P, Zucman-Rossi J. Hepatocellular benign tumors-from molecular classification to personalized clinical care. Gastroenterology. 2013;144(5):888–902. doi:10.1053/j.gastro.2013.02.032
27. Akiyama TE, Ward JM, Gonzalez FJ. Regulation of the liver fatty acid-binding protein gene by hepatocyte nuclear factor 1alpha (HNF1alpha). Alterations in fatty acid homeostasis in HNF1alpha-deficient mice. J Biol Chem. 2000;275(35):27117–27122. doi:10.1074/jbc.M004388200
28. Rebouissou S, Imbeaud S, Balabaud C, et al. HNF1alpha inactivation promotes lipogenesis in human hepatocellular adenoma independently of SREBP-1 and carbohydrate-response element-binding protein (ChREBP) activation. J Biol Chem. 2007;282(19):14437–14446. doi:10.1074/jbc.M610725200
29. Lee YH, Magnuson MA, Muppala V, Chen SS. Liver-specific reactivation of the inactivated Hnf-1alpha gene: elimination of liver dysfunction to establish a mouse MODY3 model. Mol Cell Biol. 2003;23(3):923–932. doi:10.1128/MCB.23.3.923-932.2003
30. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. doi:10.1172/JCI0215593
31. Benjannet S, Rhainds D, Essalmani R, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem. 2004;279(47):48865–48875. doi:10.1074/jbc.M409699200
32. Maxwell KN, Breslow JL. Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A. 2004;101(18):7100–7105. doi:10.1073/pnas.0402133101
33. Ai D, Chen C, Han S, et al. Regulation of hepatic LDL receptors by mTORC1 and PCSK9 in mice. J Clin Invest. 2012;122(4):1262–1270. doi:10.1172/JCI61919
34. Jeong HJ, Lee HS, Kim KS, Kim YK, Yoon D, Park SW. Sterol-dependent regulation of proprotein convertase subtilisin/kexin type 9 expression by sterol-regulatory element binding protein-2. J Lipid Res. 2008;49(2):399–409. doi:10.1194/jlr.M700443-JLR200
35. Li Z, Liu Q. Hepatitis C virus regulates proprotein convertase subtilisin/kexin type 9 promoter activity. Biochem Biophys Res Commun. 2018;496(4):1229–1235. doi:10.1016/j.bbrc.2018.01.176
36. Tao R, Xiong X, DePinho RA, Deng CX, Dong XC. FoxO3 transcription factor and Sirt6 deacetylase regulate low density lipoprotein (LDL)-cholesterol homeostasis via control of the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene expression. J Biol Chem. 2013;288(41):29252–29259. doi:10.1074/jbc.M113.481473
37. Song G, Zhu L, Ruan Z, Wang R, Shen Y. MicroRNA-122 promotes cardiomyocyte hypertrophy via targeting FoxO3. Biochem Biophys Res Commun. 2019;519(4):682–688. doi:10.1016/j.bbrc.2019.09.035
38. Su L, Zhou L, Chen FJ, et al. Cideb controls sterol-regulated ER export of SREBP/SCAP by promoting cargo loading at ER exit sites. EMBO J. 2019;38(8). doi:10.15252/embj.2018100156.
39. Li JZ, Lei Y, Wang Y, et al. Control of cholesterol biosynthesis, uptake and storage in hepatocytes by Cideb. Biochim Biophys Acta. 2010;1801(5):577–586. doi:10.1016/j.bbalip.2010.01.012
40. Laudadio I, Manfroid I, Achouri Y, et al. A feedback loop between the liver-enriched transcription factor network and miR-122 controls hepatocyte differentiation. Gastroenterology. 2012;142(1):119–129. doi:10.1053/j.gastro.2011.09.001
41. Wei S, Zhang M, Yu Y, et al. HNF-4alpha regulated miR-122 contributes to development of gluconeogenesis and lipid metabolism disorders in Type 2 diabetic mice and in palmitate-treated HepG2 cells. Eur J Pharmacol. 2016;791:254–263. doi:10.1016/j.ejphar.2016.08.038
42. Cho SJ, Ferrell LD, Gill RM. Expression of liver fatty acid binding protein in hepatocellular carcinoma. Hum Pathol. 2016;50:135–139. doi:10.1016/j.humpath.2015.12.002
43. Zucman-Rossi J, Jeannot E, Nhieu JT, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology. 2006;43(3):515–524. doi:10.1002/(ISSN)1527-3350
44. Bluteau O, Jeannot E, Bioulac-Sage P, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32(2):312–315. doi:10.1038/ng1001
45. Haddouche A, Bellanne-Chantelot C, Rod A, et al. Liver adenomatosis in patients with hepatocyte nuclear factor-1 alpha maturity onset diabetes of the young (HNF1A-MODY): clinical, radiological and pathological characteristics in a French series. J Diabetes. 2019.
46. Nakamura A, Ishidu K, Tajima T. Early onset of liver steatosis in a Japanese girl with maturity-onset diabetes of the young type 3 (MODY3). J Clin Res Pediatr Endocrinol. 2012;4(2):104–106. doi:10.4274/Jcrpe
47. Shih DQ, Bussen M, Sehayek E, et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat Genet. 2001;27(4):375–382. doi:10.1038/86871
48. Esau C, Davis S, Murray SF, et al. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3(2):87–98. doi:10.1016/j.cmet.2006.01.005
49. Pelletier L, Rebouissou S, Paris A, et al. Loss of hepatocyte nuclear factor 1alpha function in human hepatocellular adenomas leads to aberrant activation of signaling pathways involved in tumorigenesis. Hepatology. 2010;51(2):557–566. doi:10.1002/hep.23362
50. Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124(1):35–46. doi:10.1016/j.cell.2005.12.022
51. Chen J, Cooper AD, Levy-Wilson B. Hepatocyte nuclear factor 1 binds to and transactivates the human but not the rat CYP7A1 promoter. Biochem Biophys Res Commun. 1999;260(3):829–834. doi:10.1006/bbrc.1999.0980
52. Chiang JY, Kimmel R, Stroup D. Regulation of cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription by the liver orphan receptor (LXRalpha). Gene. 2001;262(1–2):257–265. doi:10.1016/S0378-1119(00)00518-7
53. Moore KJ, Rayner KJ, Suarez Y, Fernandez-Hernando C. The role of microRNAs in cholesterol efflux and hepatic lipid metabolism. Annu Rev Nutr. 2011;31:49–63. doi:10.1146/annurev-nutr-081810-160756
54. Wen J, Friedman JR. miR-122 regulates hepatic lipid metabolism and tumor suppression. J Clin Invest. 2012;122(8):2773–2776. doi:10.1172/JCI63966
55. Silverman MG, Ference BA, Im K, et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. 2016;316(12):1289–1297. doi:10.1001/jama.2016.13985
56. Sabatine MS, Giugliano RP, Keech AC, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–1722. doi:10.1056/NEJMoa1615664
57. Bellanne-Chantelot C, Carette C, Riveline JP, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes. 2008;57(2):503–508. doi:10.2337/db07-0859
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.