Back to Journals » Infection and Drug Resistance » Volume 19
Long-Term Antibiotic-Driven Gut Microbiota Disruption Promotes Toxigenic Clostridioides difficile Proliferation: A Four-Year Retrospective Study of a Single ICU Patient
Authors Jiang X, Wu L, Duan S, Bian J, Lv T, Zheng L, Zhao Y, Shen P, He J, Chen Y ![]()
Received 23 September 2025
Accepted for publication 5 January 2026
Published 13 January 2026 Volume 2026:19 562973
DOI https://doi.org/10.2147/IDR.S562973
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hemant Joshi
Xinrong Jiang,1 Lechi Wu,1 Simiao Duan,1 Junyu Bian,1 Tao Lv,1 Lisi Zheng,1 Yuhong Zhao,2 Ping Shen,1 Jianqin He,1 Yunbo Chen1
1State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China; 2Department of Blood Transfusion, The First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China
Correspondence: Jianqin He; Yunbo Chen, Email [email protected]; [email protected]
Objective: This four-year longitudinal study of a single critically ill patient leverages deep temporal profiling to unravel the dynamic interplay between antibiotic pressure, gut microbiota, and Clostridioides difficile (C. difficile) colonization, providing temporal insights unattainable through cross-sectional designs.
Methods: We performed a retrospective analysis of one critically ill patient (2015– 2019). Sixty-four fecal samples were subjected to toxigenic C. difficile culture and metagenomic sequencing. To isolate short-term effects, we implemented a 7-day retrospective window, categorizing each sample based on antibiotic exposure in the preceding week: no antibiotics, monotherapy, or polypharmacy.
Results: Antibiotic exposure significantly reduced microbial diversity and promoted dysbiosis. Crucially, we identified a transitional C. difficile colonization state (Tcd±) that potentially determines progression to toxigenic (Tcd+) or non-toxigenic (Tcd-) outcomes. Analysis using the 7-day window revealed that intensive antibiotic pressure was strongly associated with successional progression towards toxigenic dominance. Conversely, brief antibiotic-free intervals were linked to partial restoration of microbial network complexity and a competitive landscape favoring non-toxigenic strains.
Conclusion: This deep temporal profiling of a single case provides novel, hypothesis-generating insights. The identification of a transitional colonization state and the association between short-term antibiotic pressure and colonization outcomes define critical dynamics for future validation. These findings highlight the potential of longitudinal data to inform precise antibiotic stewardship strategies in high-risk, critically ill populations.
Keywords: Clostridioides difficile, antibiotics use, gut microbiota, metagenomic, critically ill patient
Introduction
Antibiotics are highly effective for treating infectious diseases, but their use often disrupts gut microbiota homeostasis. This disruption, characterized by decreased diversity, reduced richness, and altered species abundance, predisposes patients to antibiotic-associated diarrhea (AAD).1–5
Clostridioides difficile (C. difficile) is a leading cause of healthcare-associated AAD.6–8 The pathogenesis of C. difficile infection (CDI) is mediated through a multi-step process involving spore colonization in the intestine, germination into vegetative cells, and subsequent production of virulence factors, including toxin A (Tcd A), toxin B (Tcd B), and binary toxin (CDT).9 While it is well-established that antibiotic exposure reduced microbial diversity and creates an ecological niche favorable for C. difficile colonization,10,11 a critical gap remains in understanding the longitudinal dynamics of how the gut microbiota and C. difficile interact under continuous and prolonged antibiotic pressure. Nevertheless, previous studies have primarily relied on cross-sectional comparisons between CDI patients and healthy controls, which are inherently limited by inter-individual variations in age, diet, environment, and comorbidities.11–13 Furthermore, the optimal duration of antibiotic interruption to confer a protective effect remains unknown. Short periods (eg around 7 days) are of particular interest as they may be more readily implemented in clinical practice than prolonged cessation, and could provide a critical window for the recovery of key commensal bacteria.
Longitudinal, deep-sampling studies of single subjects offer a powerful, albeit underutilized, approach to address this gap. By intensively profiling one individual over time, such designs control for inter-personal variation and can reveal intra-host microbial and pathogen dynamics in response to perturbations, generating time-series data for hypothesis generation.14 This case presented a rare, albeit clinically complex, opportunity to apply this principle. The patient’s prolonged four-year Intensive Care Unit (ICU) stay under continuous monitoring, coupled with recurrent infections necessitating complex antibiotic regimens, created a persistent dysbiosis pressure ideal for observing microbial succession. Furthermore, the clinical team’s high index of suspicion for CDI led to the collection of 64 fecal samples over the study period, providing a unique longitudinal dataset. We acknowledge the inherent challenges of a retrospective design, including uneven sampling intervals due to the patient’s altered bowel habits (post-operative watery stools and an ileostomy), which made symptom-triggered sampling difficult. Nevertheless, the overall density and temporal span of the data offer a unique window into long-term dynamics that are virtually impossible to capture in a prospective cohort study. This unique clinical scenario allowed us to pursue several specific objectives. We aimed to characterize the longitudinal impact of varying antibiotic exposure on gut microbiota structure, and to investigate how these microbial community states were linked to the emergence of toxigenic versus non-toxigenic C. difficile. We further sought to identify whether transitional states in the microbiota could be discerned that might precede critical shifts in C. difficile colonization. Finally, by implementing a defined temporal window for analysis, we aimed to dissect the short-term effects of antibiotic pressure from its cumulative impact. Our central hypothesis was that intensive antibiotic pressure would drive the gut microbiota through distinct compositional states, which would in turn be deterministically associated with different C. difficile colonization outcomes.
Methods
Patient’s Character and Collection of Clinical Data
We conducted a case study on an 84-year-old male patient diagnosed with colon cancer who underwent right hemicolectomy followed by fistulization. The patient was hospitalized in the intensive care unit of our institution from February 2015 to April 2019. A total of 64 fecal samples were collected throughout the hospitalization period. (Figure 1) Fecal samples were collected in sterile containers and stored at −80°C until further analysis.
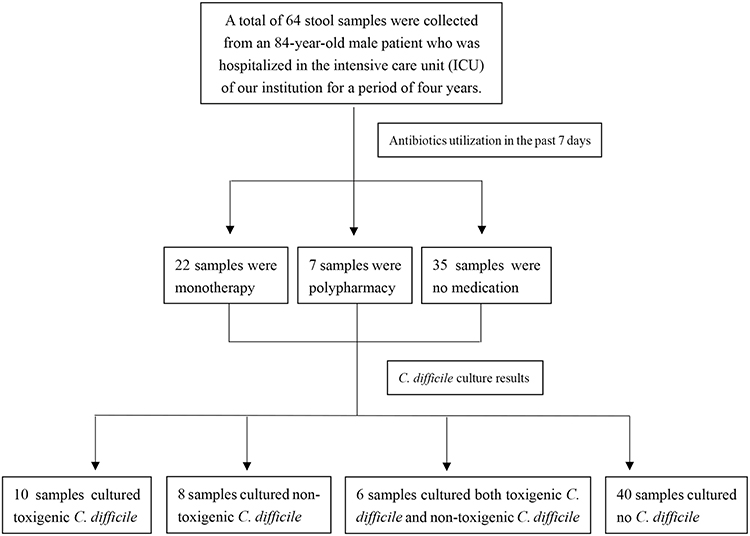 |
Figure 1 Patient and sample flow in the study. This flowchart illustrates a medical study. A total of 64 stool samples were collected from an 84-year-old male patient hospitalized in the intensive care unit (ICU) of the institution for four years. The samples were categorized based on antibiotic utilization in the past 7 days: 22 samples were from monotherapy, 7 from polypharmacy, and 35 from no medication. Subsequently, C. difficile culture was performed on these samples. The results showed that 10 samples cultured toxigenic C. difficile, 8 samples cultured non-toxigenic C. difficile, 6 samples cultured both toxigenic and non-toxigenic C. difficile, and 40 samples cultured no C. difficile. |
Clinical data were retrieved from the electronic medical record system, including demographic information, underlying comorbidities, length of hospital stay, and invasive procedures (such as tracheal intubation, central venous catheterization, urinary catheterization, and gastric tube placement). Comprehensive antibiotic usage, including oral (P.O.) and intravenous (i.v.) administration, was recorded. (Figure 2a–2d)
 |
Figure 2 The antibiotic utilizations and C. difficile culture results from 2015 to 2019. The timeline is divided into four consecutive periods (a–d). Horizontal bars indicate durations of antibiotic administration (iv, intravenous; po, oral; inh, inhalation). Fecal samples collected for metagenomic sequencing are marked by dots, color-coded to reflect concurrent C. difficile culture status: yellow (negative), green (non-toxigenic), red (toxigenic), and half-red/half-green (mixed toxigenic and non-toxigenic colonization). Dates are formatted as YYYY-MM-DD. (a) February 2015–December 2015. Antibiotics: Amikacin (inh, periwinkle), Aztreonam (iv, gray), Biapenem (iv, tan), Cefepime (iv, salmon), Cefoperazone (iv, yellowgreen), Ceftazidime (iv, steelblue), Cefoxitin (iv, thistle), Daptomycin (iv, light pink), Imipenem (iv, light sage), Levofloxacin (iv, slateblue), Linezolid (iv, medium purple), Metronidazole (po, Peru), Piperacillin (iv, skyblue), Tigecycline (iv, indigo), Vancomycin (po, red). (b) January 2016–December 2016. Antibiotics: Ceftazidime (iv, steelblue), Daptomycin (iv, light pink), Imipenem (iv, light sage), Linezolid (iv, medium purple), Piperacillin (iv, skyblue), Vancomycin (iv, darkgreen; po, red). (c) January 2017–December 2017. Antibiotics: Caspofungin (iv, purple), Cefepime (iv, salmon), Ceftazidime (iv, steelblue), Imipenem (iv, light sage), Linezolid (po, saddlebrown), Vancomycin (iv, darkgreen). (d)January 2018–April 2019. Antibiotics: Amikacin (iv, periwinkle), Aztreonam (iv, gray), Biapenem (iv, tan), Cefepime (iv, salmon), Cefoperazone (iv, yellowgreen), Ceftazidime (iv, steelblue), Levofloxacin (iv, slateblue; po, olive), Linezolid (iv, medium purple; po, saddlebrown), Meropenem (iv, sienna), Micafungin (iv, deep pink), Vancomycin (po, red). |
Grouping Criteria
Given the prolonged administration of multiple antibiotics, we aimed to investigate the effects of antibiotic monotherapy versus polypharmacy on the patient’s microbiota and C. difficile colonization dynamics. We systematically documented and categorized all antibiotic usage during the hospitalization period, with particular attention to antimicrobial exposure with the preceding seven days. Based on this temporal classification, we stratified all collected samples into three groups: monotherapy group, polypharmacy group and no medication within the past 7 days group.
To further elucidate the gut microbiota effected by antibiotics on varied C. difficile colonization states, we classified the 64 fecal samples into four categories based on culture results: (1) only toxigenic C. difficile (Tcd+) (n=10); (2) only non-toxigenic C. difficile (Tcd-) (n=8); (3) both toxigenic and non-toxigenic C. difficile (Tcd±) (n=6) and (4) C. difficile-negative (No C. difficile) (n=40).
C. Difficile Detected
Fecal samples were mixed with 95% ethanol and cultured under anaerobic incubation (80% N2, 10% H2, 10% CO2) for 48 h at 37°C on cycloserine-cefoxitin-fructose agar (CCFA; Oxoid Ltd., Basingstoke, United Kingdom) supplemented with 7% sheep blood. C. difficile isolates were confirmed using standard microbiological methods, including colony morphology, odor. Isolated suspicious colonies were identified as C. difficile using Brooke Matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF) (Bruker Daltonik GmbH, Bremen, Germany). Toxin genes was performed by PCR targeting the toxin genes tcdA, tcdB, and binary toxin genes (cdtA and cdtB), as previously described.15
Metagenomics Sequencing and Analysis
DNA Extraction, Sequencing, and Quality Control
Total genomic DNA was extracted from fecal samples using the Qiagen DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. DNA quality and quantity were assessed using a NanoDrop spectrophotometer (Thermo Fisher Scientific, USA) and Qubit fluorometer (Invitrogen, USA).
Libraries were prepared using an Illumina DNA Prep kit, and sequencing was performed on an Illumina NovaSeq 6000 platform, generating an average of 10 Gb of data per sample with paired-end 150 bp reads.
Raw sequencing data underwent a rigorous quality control (QC) pipeline. Fastp was used to remove low-quality bases, adapter sequences, and ambiguous reads. The specific filtering criteria were as follows: reads were discarded if either read contained adapter contamination, more than 10% uncertain nucleotides, or more than 50% low-quality nucleotides (Phred quality score < 5). Subsequently, reads aligning to the human reference genome (GRCh38) were identified and removed using Bowtie2 with parameters: --end-to-end --sensitive -I 200 -X 400 to eliminate host-derived contamination.
Metagenome Assembly, Gene Catalog Construction, and Annotation
High-quality, non-host reads were assembled for each sample using MEGAHIT with the parameter --presets meta-large. Scaffolds were broken at N junctions to obtain contiguous fragments (scaffigs) without Ns. Protein-coding genes were predicted on scaffigs (≥ 500 bp) using MetaGeneMark with default parameters. Predicted genes shorter than 100 nucleotides were filtered out. A non-redundant gene catalog was constructed from all predicted genes using CD-HIT with parameters: -c 0.95 -G 0 -aS 0.9 -g 1 -d 0. This 95% identity threshold was chosen to cluster highly similar sequences while retaining potential functional variants, a standard practice to reduce redundancy.
For taxonomic and functional annotation, the non-redundant gene sequences were aligned against the National Center for Biotechnology Information (NCBI) Non-redundant (NR) database using DIAMOND16 in blastp mode with an e-value cutoff of 1×10−5. Taxonomic profiling was performed using the lowest common ancestor (LCA) algorithm implemented in MEGAN. Functional annotations were conducted by aligning sequences against the Kyoto Encyclopedia of Genes and Genomes (KEGG) and the Virulence Factor Database (VFDB) using DIAMOND with the same e-value cutoff. The best alignment hit was selected for each sequence for functional analysis.
Gene Abundance and Statistical Analysis
Clean reads from each sample were aligned to the final gene catalog using Bowtie2 with parameters --end-to-end --sensitive -I 200 -X 400 to calculate gene abundance. Genes with ≤ 2 reads in a sample were filtered out. The abundance of a gene was calculated as: (r * L) / (Σ (r_i * L_i)), where r is the number of reads aligned to the gene and L is the gene length.
Microbial alpha and beta diversity analyses were performed based on taxonomic abundance profiles. Principal coordinate analysis (PCoA) was conducted using the R “vegan” package based on Bray-Curtis’s dissimilarity. Differential abundance analysis was performed using MetaGenomeSeq and LEfSe (LDA score threshold=4). Correlation networks between microbial taxa and KEGG pathways were constructed using Spearman’s rank correlation. Only robust correlations with |r| > 0.5 and a p-value < 0.05 were retained. Network visualization and analysis were performed using the R “igraph” package.
Data and Code Availability
The raw sequencing data have been deposited in the NCBI SRA under BioProject accession number PRJNA1237222. The custom scripts used for statistical analysis and figure generation are available from the corresponding author upon reasonable request.
Statistical Analysis
Continuous variables were presented as median and interquartile range (IQR), while categorical variables were presented as frequencies and percentages. The normality of the data distribution was assessed using the Shapiro–Wilk test, and the homogeneity of variances was assessed using Levene’s test. The Wilcoxon rank-sum test was used to compare continuous variables between two independent groups, while the Kruskal–Wallis H-test was used to compare continuous variables between multiple independent groups. Post hoc tests (Bonferroni correction) were performed to adjust for multiple comparisons. The Chi-square test or Fisher’s exact test was used to compare the categorical variables. Spearman’s rank test was used for the correlation analysis. P-value ≤ 0.05 was considered statistically significant. Statistical analysis was performed with SPSS 27.0 (SPSS, Chicago, IL, USA) and R v4.1.2, following the standard procedures for data input, test selection, and result output.
Results
Inpatient Clinical Course and Therapeutic Strategy
We present the case of an 84-year-old gentleman with an extended hospitalization course, initially admitted for subsequent therapeutic interventions following surgical resection of colon cancer. The patient underwent radical colon resection on September 30, 2014, and performed an exploratory laparotomy with abdominal irrigation and terminal ileostomy on October 6, 2014. Following the secondary surgical intervention, the patient was subsequently transferred to the ICU encompassed comprehensive therapeutic regimens.
During the ICU hospitalization, the patient developed multiple infections, including: pulmonary infections caused by Acinetobacter baumannii, Pseudomonas aeruginosa, and Serratia marcescens; bloodstream infections attributed to Klebsiella pneumoniae, Staphylococcus aureus, and Enterococcus faecalis; as well as intestinal infections. The antimicrobial treatment regimen encompassed various classes of agents, including β-lactams, glycopeptides, nitroimidazoles, oxadixyl, peptide antibiotics, echinocandins, and several other categories.
The diagnosis of CDI in clinical practice is primarily based on the presence of characteristic clinical manifestations like diarrhea, abdominal pain and fever combined with elevated inflammatory markers.17–19 However, in this particular case, the presence of concurrence systemic infections may compromise the diagnostic specificity of fever and inflammatory markers for CDI. Consequently, gastrointestinal symptoms assumed particular diagnostic significance, necessitating long-term clinical monitored. Moreover, the patient’s terminal ileostomy resulted in altered stool patterns that differed substantially from those observed in patients with intact colon function. This postoperative change, as outlined in the Infectious Diseases Society of America (IDSA) and European Society of Clinical Microbiology and Infectious Diseases (ESCMID) clinical practice guidelines, may potentially mimic certain symptoms.20,21 The patient demonstrated multiple established risk factors for CDI, including advanced age and prolonged antibiotic exposure, particularly to β-lactams with a predominance of third-generation cephalosporins.22 In light of these considerations, clinicians maintained a high index of suspicion, implementing a protocol of empirical stool testing for C. difficile, which ultimately yielded 64 analyzable stool samples.
In this clinical scenario, despite the diagnostic uncertainty regarding CDI, clinicians initiated empirical vancomycin therapy during specific periods in the early 2015, early 2016 and late 2017. Notably, our longitudinal analysis revealed persistent positive for toxigenic C. difficile following these therapeutic interventions. This observation suggests empirical antimicrobial treatment may not consistent achieve optimal clinical outcomes, as evidenced by the sustained risk of toxigenic C. difficile pathogenicity despite therapeutic measures.
Diversity Metrics and Differential Abundance
Initially, we assessed the effects of antibiotic administration on the taxonomic composition and ecological structure of the patient’s gut microbiota. This comprehensive analysis was conducted using α diversity metric, β diversity measures, and species abundance profiling. Our findings revealed a significant reduction in all measured diversity indices (ACE, Chao1, Shannon, Simpson, and observed species) following antibiotic exposure compared to samples without recent antibiotic treatment (within 7 days; Kruskal–Wallis H-test with Bonferroni correction, p < 0.001), consistent withsubstantial antibiotic-induced dysbiosis. (Table 1) Notably, samples from polypharmacy regimens were observed to have lower indices, suggesting a potential trend towards a more pronounced impact of polypharmacy on gut microbial communities, although this difference did not reach statistical significance. To further characterize temporal variations in microbial community structure, we performed principal co-ordinates analysis (PCoA) based on β-diversity metrics. (Figure 3a) Interestingly, the analysis using the unweighted UniFrac distance algorithm revealed relative temporal stability in the patient’s gut microbiome composition, with no significant clustering patterns observed across different antibiotic treatment conditions. This suggests that while antibiotic administration significantly reduced microbial diversity, the overall community structure maintained a degree of stability throughout the treatment period.
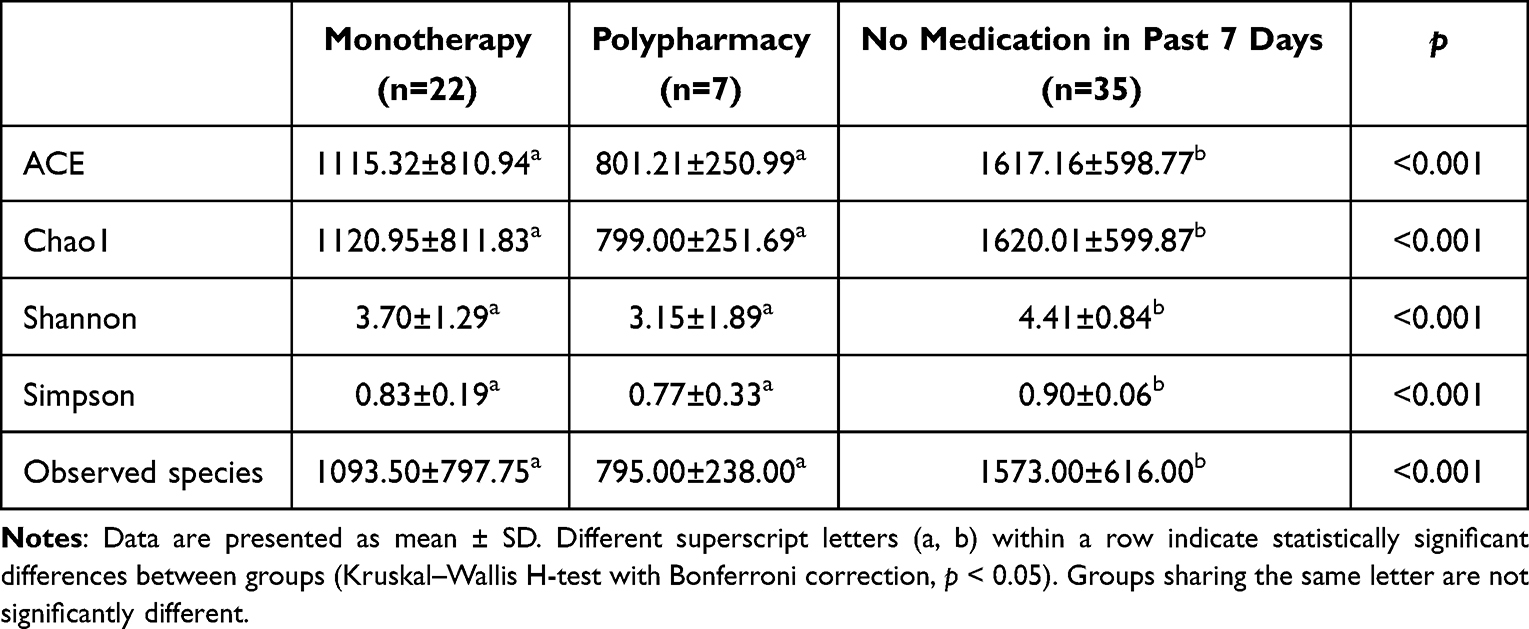 |
Table 1 Comparison of Gut Microbiota Alpha-Diversity Indices Across Antimicrobial Treatment Conditions |
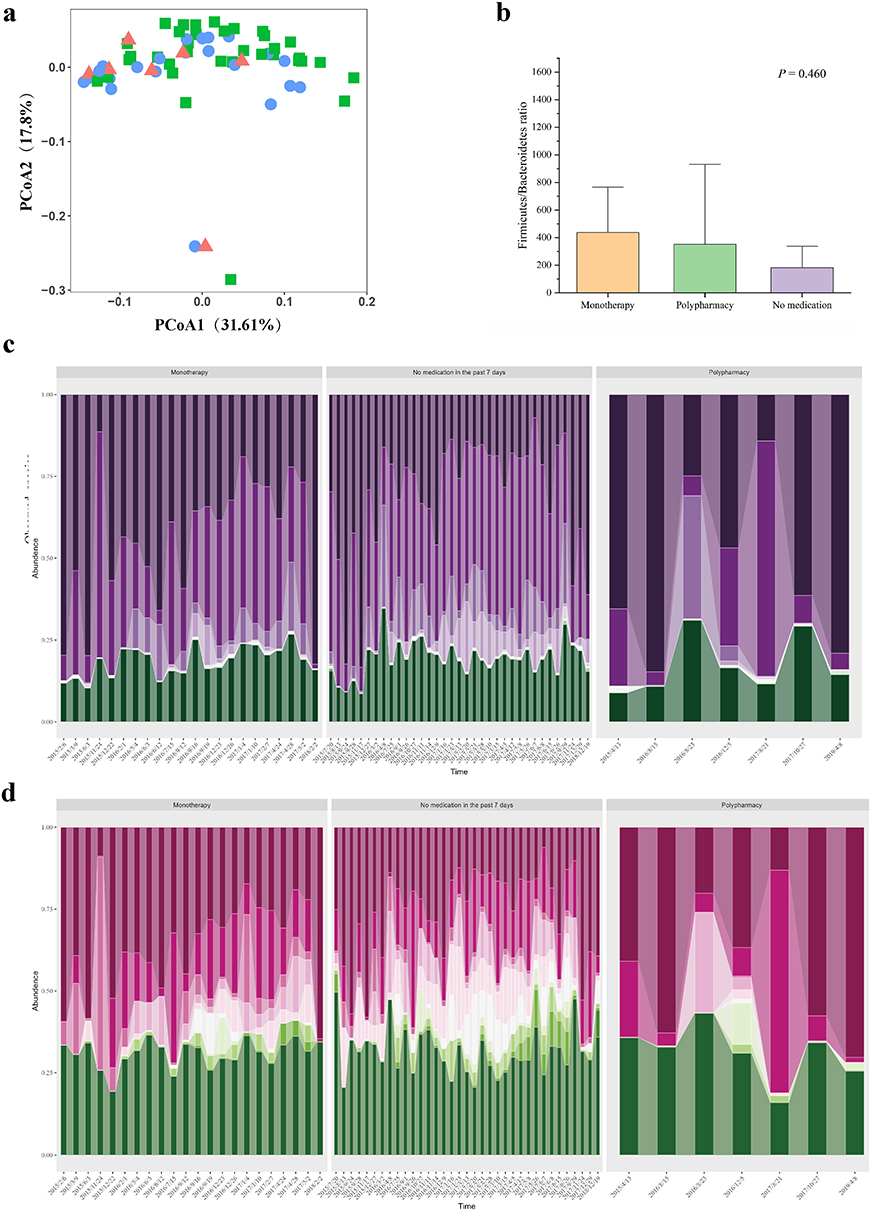 |
Figure 3 Gut microbiota changes associated with antibiotic treatments. (a) Principal coordinates analysis (PCoA) based on unweighted UniFrac distances, colored by recent antibiotic exposure history. Samples are categorized as monotherapy (blue circles), polypharmacy (red triangles), or no medication within the past 7 days (green squares). (b) Firmicutes/Bacteroidetes ratio across treatment groups. The monotherapy group shows the highest median ratio, followed by polypharmacy, while the no-medication group exhibits the lowest ratio. No statistically significant differences were observed between groups. (c and d) Temporal dynamics of microbial composition at the (c) phylum and (d) family levels. Stacked area charts depict the relative abundances of the top 10 most abundant taxa; remaining taxa are grouped as “Others”. Samples are stratified by antibiotic exposure as in (a). Taxa are arranged in descending order of mean relative abundance from bottom to top. Phylum order (c): Pseudomonadota, Bacteroidota, Bacillota, Actinomycetota, Thermoplasmatota, Spirochaetota, Cyanobacteria, Chloroflexi, Fusobacteriota, Planctomycetota. Family order (d): Enterobacteriaceae, Moraxellaceae, Staphylococcaceae, Lachnospiraceae, Ruminococcaceae, Erysipelotrichaceae, Veillonellaceae, Comamonadaceae, Coriobacteriaceae, Bifidobacteriaceae. These profiles illustrate structural shifts in the gut microbiota in response to varying degrees of antibiotic pressure, highlighting changes in the dominance and succession of major taxonomic groups. |
To characterize temporal dynamics of gut microbiota composition, we conducted a comprehensive analysis of microbial relative abundance. Our investigation initially focused on the Firmicutes (Bacillota)/Bacteroidetes ratio, a well-established indicator of gut microbiota balance. (Figure 3b) The analysis revealed a markedly elevated ratio, indicative of significant dysregulation of Bacillota proportions. In contrast, the no-medication group demonstrated the lowest ratio, approximating values observed in healthy individuals, although these differences did not reach statistical significance.23 Building upon these findings of microbial imbalance, we performed detailed taxonomic profiling at both phylum and family levels. Quantitative analysis revealed significant alterations in specific microbial taxa following antibiotic administration (p < 0.05). (Table 2) At phylum level, we observed a substantial decrease in the relative abundance of the Bacillota and Actinomycetota, while Pseudomonadota demonstrated a significant increase (p < 0.05). (Figure 3c) Furthermore, our analysis identified pronounced shifts in key microbial families: dysbiosis-associated taxa, including Enterobacteriaceae, showed significant proliferation, whereas protective symbionts such as Clostridiaceae, Bifidobacteriaceae, Veillonellaceae, and Lachnospiraceae exhibited depletion (p < 0.05).22,24 (Figure 3d) These findings collectively revealed that antibiotic exposure is associated with substantial maladaptation in gut microecological composition, characterized by depletion of beneficial taxa and expansion of potential pathogenic organisms. This pattern of dysbiosis aligns with established literature documenting antibiotic-induced microbiota alterations in healthy individuals.25,26
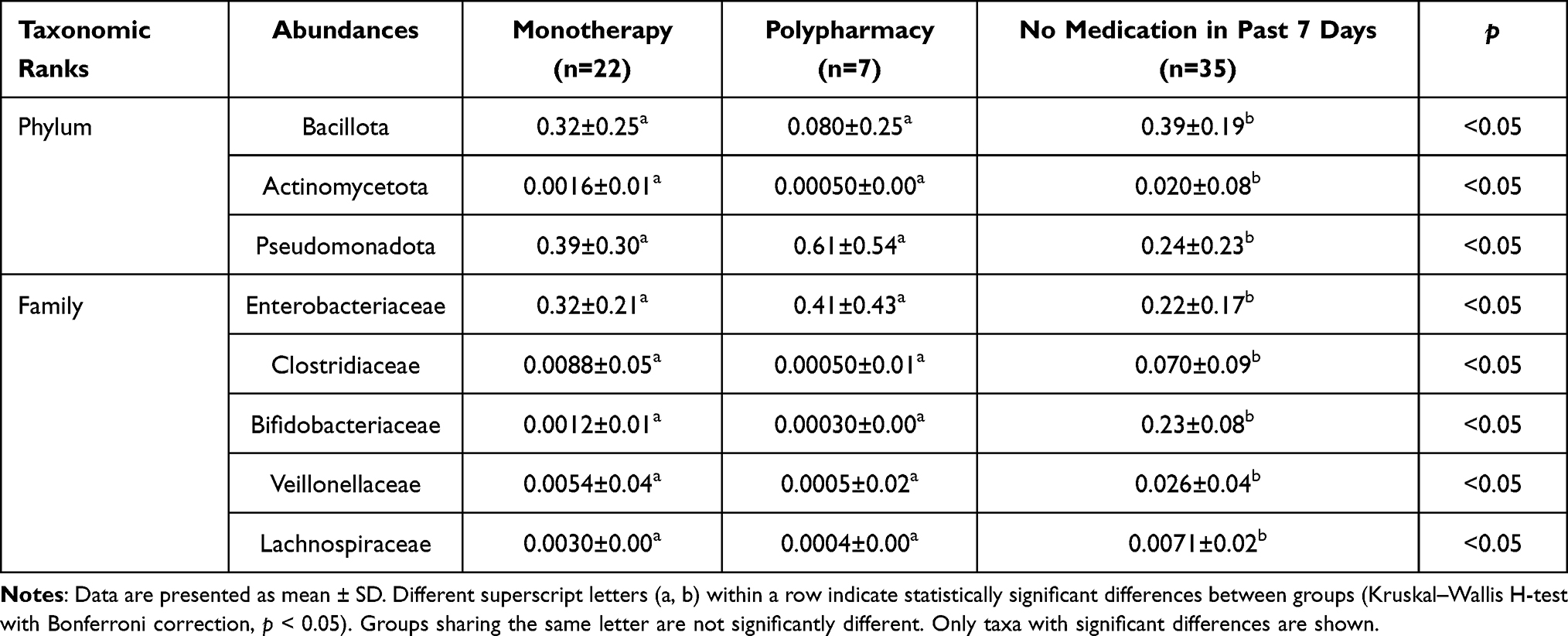 |
Table 2 Differential Relative Abundance of Selected Microbial Taxa at Phylum and Family Levels Across Antimicrobial Treatment Conditions |
We hypothesized that antibiotic exposure was strongly associated with the colonization and growth of toxigenic C. difficile culturing. To investigate the relationship between antibiotic use and toxigenic C. difficile dynamics, we analyzed microbiota variation across different C. difficile culture outcome. Our analysis revealed that 83.3% (20/24) of C. difficile-positive samples were associated with antibiotic exposure within the preceding 30 days, while 54.2% (13/24) had recent antibiotic exposure within 15 days. Notably, all toxigenic C. difficile-positive samples (10/10) demonstrated antibiotic exposure within30 days, with 70.0% (7/10) showing recent exposure within 15 days.
To further elucidate the impact of microbial diversity alteration on toxigenic C. difficile (Tcd+), non-toxigenic C. difficile (Tcd-), and co-colonization with both toxigenic and non-toxigenic C. difficile (Tcd±), we conducted comparative analyses of microbial diversity across these three and C. difficile-negative sample groups. (Figure 4a) We findings demonstrated that samples with the lowest diversity consistently yielded toxigenic C. difficile growth. Quantitative analysis revealed significantly lower diversity in Tcd+ samples compared to Tcd- samples (p < 0.05). In addition, samples collected one month prior to toxigenic C. difficile detection (pre-Tcd+) had significantly reduced diversity compared to those collected one month prior to non-toxigenic C. difficile detection (pre-Tcd) (p < 0.05), suggesting that antibiotic-induced loss of diversity associated with toxigenic C. difficile colonization. At the taxonomic level, we observed distinct microbial composition patterns preceding different C. difficile outcome. The pre-Tcd-period was characterized by significantly lower Pseudomonadota and higher levels of Bacillota, Bacteroidota and Actinomycetota compared to the pre-Tcd+ period (all p < 0.05), reflecting a more favorable microbial composition. Interestingly, the Tcd± group demonstrated intermediate characteristics in both diversity and relative abundance profiles, positioned between the Tcd- and Tcd+ groups.
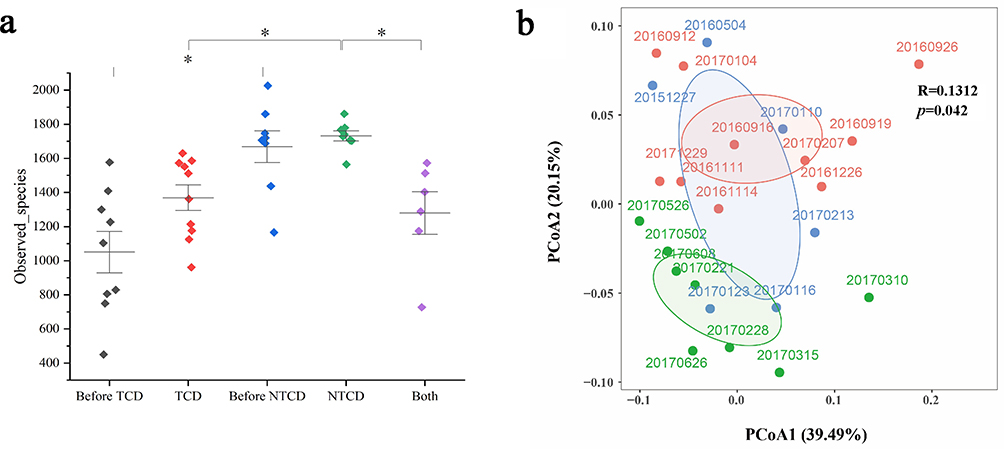 |
Figure 4 Alterations in gut microbiota composition stratified by C. difficile culture status. (a) Comparisons of alpha diversity (observed species) among groups defined by subsequent C. difficile culture results. The experimental groups include: pre-toxigenic C. difficile (samples prior to toxigenic strain detection), toxigenic C. difficile (Tcd+), pre-non-toxigenic C. difficile (samples prior to non-toxigenic strain detection), non-toxigenic C. difficile (Tcd-), and co-colonization by both toxigenic and non-toxigenic strains (Tcd±). Data are presented as distinct colored diamonds for each condition with vertical error bars. Statistically significant inter-group differences (p < 0.05) are marked by asterisks above connecting lines. (b) Beta diversity analysis visualized via principal coordinates analysis (PCoA). Samples are categorized and colored by C. difficile culture outcome: Tcd+ (red), Tcd- (green), and Tcd± (blue). Individual sampling dates are annotated for each point. Confidence ellipses encapsulate the distribution of each sample group. The statistical summary indicates a significant, albeit weak, relationship between microbial community structure and C. difficile status (R = 0.1312, p = 0.042). |
The Impact of Antibiotic Use on the Temporal Dynamics of the Intestinal Microbiota
To characterize the longitudinal dynamics of gut microbiota over four-year observation period, we performed PCoA analysis to examine microbial community variations associated with different C. difficile culture outcomes. Our analyses of C. difficile-positive samples revealed distinct clustering patterns between the Tcd+ and Tcd- group. However, the samples from Tcd± group exhibited a degree of overlap with the samples from Tcd+ and Tcd- group. Interestingly, samples from the mixed colonized group Tcd± demonstrated partial overlap with both Tcd+ and Tcd- clusters in PCoA space. (Figure 4b) Notably, temporal analysis of Tcd± samples revealed a progressive transition in microbial community structure. Specifically, samples collected on 20151227 and 20160504, which preceded of toxigenic strains (20160912 to 20170104), exhibited closer proximity to the Tcd+ group in the principal coordinate space. Subsequent samples (20170110, 20,170,116, 20170123, and 20170213) demonstrated a gradual transition from Tcd+-like to Tcd--like microbial profiles, as evidenced by their shifting positions in the PCoA plot. These findings suggest the possibility that antibiotic-induced alterations in gut microbiota composition may facilitate distinct C. difficile colonization patterns. The observed transitional phase in microbial community structure suggests that the trajectory of C. difficile colonization outcomes may be influenced by the timing and duration of antibiotic exposure, with potential implications for clinical management.
Alterations in Intestinal Microbiota Metabolism and Virulence Factors After Antibiotic Utilization
To further elucidate the functional consequences of antibiotic exposure on C. difficile colonization dynamics, we conducted comprehensive analysis of metabolic pathways and virulence factors using KEGG and VFDB databases. (Figure 5) Our pathway analysis revealed significant upregulation of several KEGG pathways in both the antibiotics-treated group and toxigenic C. difficile group (p < 0.05), including carbohydrate metabolism, signal transduction, metabolism of other amino acids, xenobiotics biodegradation and antimicrobial resistance pathways. Conversely, we observed significant downregulation in glycan biosynthesis and metabolism, as well as DNA replication and repair (p < 0.05). These parallel alterations in metabolic pathway activity are consistent with the view that antibiotic exposure on the metabolic landscape favoring toxigenic C. difficile colonization.
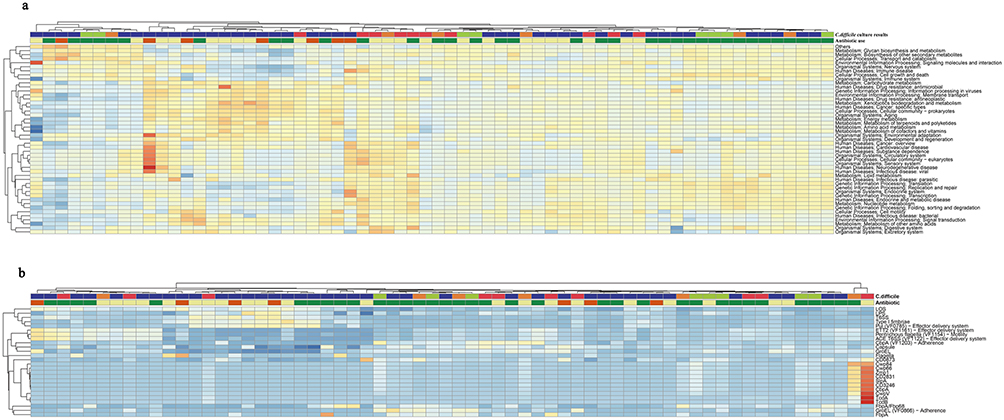 |
Figure 5 The heatmap of the correlations between different antibiotics utilization and C. difficile culture results according to KEGG and VFDB. Hierarchically clustered heatmaps display the relative abundance of microbial functions, with columns representing individual samples and rows representing functional features. The color intensity of each cell reflects the relative abundance, with a gradient from blue (low) to yellow/red (high). Two annotation bars above each heatmap indicate the sample metadata:1 C. difficile culture result (red, toxigenic; green, non-toxigenic; Orange, mixed; purple, negative) and2 recent antibiotic exposure (yellow, monotherapy; dark orange, polypharmacy; dark green, no medication within 7 days). (a) Metabolic and cellular pathways. The heatmap shows the relative abundance of KEGG orthologs grouped into level 2 categories, including Metabolism (eg, Glycan biosynthesis, Energy metabolism), Environmental Information Processing (eg, Membrane transport, Signal transduction), Cellular Processes, Human Diseases, and Organismal Systems. (b) Virulence and survival factors. The heatmap shows the relative abundance of virulence-associated genes and systems from the Virulence Factor Database (VFDB), including effector delivery systems (eg, T6SS, T4SS), adhesins (eg, Fbp88, FbpA), motility factors, and toxin genes. |
Furthermore, our virulence factor analysis revealed distinct expression patterns across different colonization states. Besides, we found that virulence factors varied from each group. Among those top ten expressed virulence factors, LOS, T6SS and type I fimbriae were decreased from January 10, 2017, to June 26, 2017, when antibiotic use was lowest. Notably, among C. difficile-specific virulence factors in this period, we identified upregulation of groEL, fbpA, cwpV, cwp 66, cwp 84, zmp 1, cbpA, slpA, CD3246, and CD2831 (p < 0.05), while tcdA and tcdB genes showed non-significant downward trends. Contrary to conventional understanding, the non-toxigenic C. difficile colonization phase was characterized by heightened expression of virulence factors associated with host attachment, colonization, nutrient acquisition, immune evasion and biofilm formation. This unexpected finding suggests that non-toxigenic strains may employ alternative survival strategies to maintain ecological competitiveness within the gut microbiota, potentially as a mechanism for persistence in a competitive environment.
Comparison of the Correlation Networks Between Microbiota and Metabolic Pathway After the Utilization of Antibiotics
To investigate the relationship between microbial composition and metabolic functionality, we constructed correlation network analyzing the association between relative abundance and metabolic pathways. Our analysis revealed distinct network complexity across different antibiotic exposure conditions. Both the monotherapy (Figure 6a) and polypharmacy (30 nodes, 32 edges) (Figure 6b) groups demonstrated a less complex network topology compared to the no-medication group (prior 7-day antibiotic-free period) (36 nodes, 38 edges) (Figure 6c). Network centrality analysis identified Pseudomonadota and Bacillota as hub taxa across all conditions, characterized by extensive connectivity and high betweenness centrality, indicating their pivotal roles in maintaining gut microbiome stability and functionality. Notably, in the no-medication group, Pseudomonadota and Bacillota exhibited stronger negative correlations with associated KEGG pathways compared to antibiotic-exposed groups, which may indicate more robust regulatory interactions in the absence of antimicrobial pressure. Furthermore, the antibiotic-naive state was characterized by a more balanced microbial community structure, with diverse taxa participating in multiple functional networks encompassing core metabolic processes, genetic information processing, and environmental adaptation pathways. This intricate network architecture suggests enhanced functional redundancy and ecosystem resilience in the absence of antibiotic perturbation.
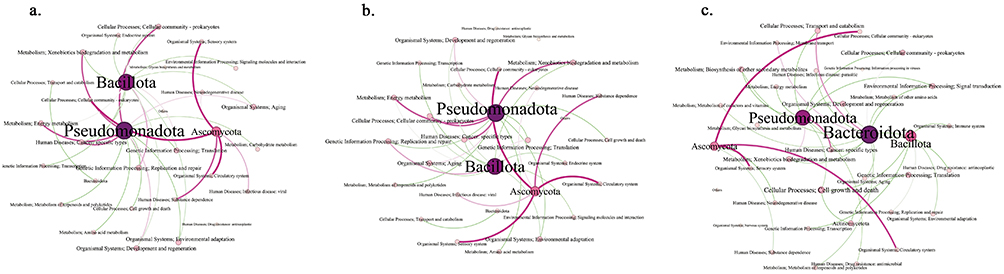 |
Figure 6 The correlation networks pertaining to differential microbiota and KEGG metabolic pathways in different antibiotics situations. (a–c) monotherapy group, polypharmacy group, no medication group. Correlation networks depict significant associations (Spearman’s |r| > 0.5, p < 0.05) for the (a) Monotherapy, (b) Polypharmacy, and (c) No medication within the past 7 days groups. In each network: Nodes represent microbial taxa (large, purple) or KEGG pathways (small, colored by category). Edges represent significant correlations: red for positive and green for negative associations. The thickness of each edge is proportional to the absolute value of the correlation coefficient (|r|), indicating the strength of the relationship. Node size is proportional to the number of connections (degree centrality). KEGG pathways are color-coded by functional category: Metabolism (blue), Genetic Information Processing (Orange), Environmental Information Processing (green), Cellular Processes (red), Organismal Systems (purple), Human Diseases (brown). (a) Monotherapy: Key taxa include Pseudomonadota, Bacillota, Bacteroidota, and Ascomycota. Representative pathways involve Xenobiotics biodegradation, Energy metabolism, and Cellular community-prokaryotes. (b) Polypharmacy: Dominant taxa include Pseudomonadota, Bacillota, and Ascomycota. Notable pathways include Energy metabolism, Transcription, and Cell growth and death. (c) No medication: Key taxa include Pseudomonadota, Bacteroidota, Bacillota, Ascomycota, and Actinomycetota. Prominent pathways include Biosynthesis of secondary metabolites, Glycan biosynthesis, and Immune system. The comparative analysis demonstrates that antibiotic pressure, particularly polypharmacy, restructures microbial-functional interactions and reduces network complexity compared to the untreated state. |
Discussion
This longitudinal case study provides a unique perspective on the dynamic interplay between antibiotic pressure and the gut ecosystem in a critically ill patient. Our data indicate that antibiotic exposure, particularly polypharmacy, was associated with marked ecological shifts, including reduced microbial diversity and altered taxonomic composition, which coincided with an environment conducive to toxigenic C. difficile colonization. Interestingly, these changes occurred against a backdrop of relative overall community stability at the individual level.
It is crucial to frame the interpretation of these findings within the inherent limitations of this work. The single-subject design, while yielding rich longitudinal data, limits the generalizability of our conclusions and precludes causal inferences. The retrospective nature led to uneven temporal sampling, potentially introducing observational bias, and the absence of samples during the 2018–2019 period represents a significant data gap. Furthermore, the patient’s complex clinical course—encompassing disease progression, nutritional status, and concurrent medications—presents potential confounding factors that could have contributed to the observed microbiota changes. These constraints underscore the strictly hypothesis-generating nature of our study.
Within this context, the observed stability in beta diversity provides a critical backdrop against which more specific, pathology-relevant dynamics unfolded. A central finding of this study was the identification of a distinct transitional colonization state (Tcd±). The temporal dynamics and intermediate microbial features of this state suggest it may represent a critical juncture that determines progression towards toxigenic or non-toxigenic dominance. Furthermore, our analysis, which used 7-day intervals to define exposure, revealed that periods of reduced antibiotic pressure were associated with partial microbial network recovery and a competitive landscape that appeared to favor non-toxigenic strains. While these observations require validation in larger cohorts, they generate the hypothesis that strategic antibiotic cessation, even of relatively short duration, might help steer the microbiota away from states that favor toxigenic C. difficile.27 This aligns with the principle of antibiotic stewardship but frames our specific finding as a testable concept rather than a clinical recommendation. Consistent with previous findings, we observed characteristic microbial shifts following antibiotic administration and preceding toxigenic C. difficile detection, marked by depletion of protective taxa (Clostridiaceae, Bifidobacteriaceae, Veillonellaceae, and Lachnospiraceae) and proliferation of potentially pathogens Gammaproteobacteria. Our findings suggest that polypharmacy may exert more pronounced effects on gut microbiota than monotherapy, though this difference did not reach statistically significant due to limited sample size. It is well established that β-lactam antibiotics, particularly third-generation cephalosporins, constitute major risk factors for CDI,22 and were extensively used in this patient. While previous studies have demonstrated that short-term antibiotic courses can induce profound, long-lasting (6–24 months) microbiota alteration in healthy adults.28
Our findings align with current IDSA and ESCMID, which recommend cautious consideration of empirical CDI antibiotic prophylaxis.20,21 However, in this case, a standard prophylactic regimen of oral vancomycin (125mg four times daily for more than 10 days) failed to prevent toxigenic C. difficile colonization. This observation highlights the challenges in patients with multiple risk factors for non-infectious diarrhea, including antibiotic use, proton pump inhibitors or NSAIDS administration, enteral nutrition, short bowel syndrome, special bowel surgeries, hypoalbuminemia, and malnutrition.8,29–31 The clinical differentiation between CDI and other causes of diarrhea in such complex cases remains problematic, potentially compromising the efficacy of both prophylaxis or treatment. These findings underscore the need for more refined clinical guidelines that can better distinguish CDI from other diarrheal etiologies in high-risk populations. The development of such guidelines, incorporating specific diagnostic algorithms and treatment initiation criteria, could significantly improve clinical management. However, this will require further large-scale, prospective studies to establish evidence-based recommendations tailored to complex clinical scenarios. Intriguingly, our analysis revealed enhanced expression of virulence factors associated with adhesion and colonization during the phase of Tcd-, suggesting that non-toxigenic strains may employ alternative strategies, potentially enhancing their ability to compete for ecological niches and persist within the gut, even in the absence of toxin production. These finding merits further investigation as it could reveal novel mechanisms of microbial competition relevant to CDI.
Collectively, this detailed longitudinal profile highlights the potential of deep temporal data to reveal complex dynamics, such as the transitional state (Tcd±) and the competitive strategies of non-toxigenic strains, which are typically obscured in cohort studies. Our work does not offer definitive clinical strategies but rather proposes specific, testable hypotheses about microbial succession and critical intervention windows. Future prospective studies in high-risk populations are warranted to validate these dynamics and explore their potential for informing more personalized, microbiota-aware patient management.
Conclusion
This four-year longitudinal study of a single ICU patient underscores the power of deep temporal profiling to unravel complex host-microbe dynamics inaccessible to cross-sectional designs. Our analysis revealed that within a backdrop of relative community-level stability, intensive antibiotic pressure was associated with marked ecological shifts that created a niche permissive for C. difficile colonization. A central finding was the identification of a critical transitional colonization state (Tcd±), which may represent a determinative juncture in the progression towards toxigenic or non-toxigenic dominance. Furthermore, our data suggest that even brief antibiotic-free intervals may facilitate partial microbial network recovery and shift competitive dynamics in favor of non-toxigenic strains.
These observations are inherently hypothesis-generating. This study does not offer definitive clinical recommendations but provides a novel perspective on CDI pathogenesis by highlighting the transitional state and the potential impact of short-term antibiotic pressure as specific, testable concepts. Future prospective studies in high-risk populations are warranted to validate these dynamics and to explore their utility in informing precision antibiotic stewardship strategies.
Data Sharing Statement
The data that support the findings of this study are openly available in National Center for Biotechnology Information at https://dataview.ncbi.nlm.nih.gov/object/PRJNA1237222?reviewer=m9utl596dmab3c7mvao1kqsmr2.
Ethics Approval and Consent to Participate
This retrospective study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China (No.【2020-651】). As per the standard protocol of our ICU at the time of patient admission, a comprehensive General Informed Consent Form for ICU Treatment was signed by the legally authorized representative of the deceased patient. This form explicitly included consent for the use of biological specimens (including feces) collected during routine clinical care for scientific research purposes. Given the historical nature of the case and the full anonymization of all data presented in this manuscript, the requirement for obtaining additional, specific consent for publication was waived by the ethics committee.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by The National Key Research and Development Program of China under Grant 2024YFC2309902 and 2020YFE0204300.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
1. Mekonnen SA, Merenstein D, Fraser CM, Marco ML. Molecular mechanisms of probiotic prevention of antibiotic-associated diarrhea. Curr Opin Biotechnol. 2020;61:226–14. doi:10.1016/j.copbio.2020.01.005
2. Nasiri MJ, Goudarzi M, Hajikhani B, Goudarzi M, Ghazi H, Pouriran R. Clostridioides (Clostridium) difficile infection in hospitalized patients with antibiotic-associated diarrhea: a systematic review and meta-analysis. Anaerobe. 2018;50:32–37. doi:10.1016/j.anaerobe.2018.01.011
3. Slimings C, Riley TV. Antibiotics and hospital-acquired Clostridium difficile infection: update of systematic review and meta-analysis. J Antimicrob Chemother. 2014;69(4):881–891. doi:10.1093/jac/dkt477
4. Tabak YP, Srinivasan A, Yu KC, et al. Hospital-level high-risk antibiotic use in relation to hospital-associated Clostridioides difficile infections: retrospective analysis of 2016–2017 data from US hospitals. Infect Control Hosp Epidemiol. 2019;40(11):1229–1235. doi:10.1017/ice.2019.236
5. Vincent C, Manges AR. Antimicrobial use, human gut microbiota and Clostridium difficile colonization and infection. Antibiotics. 2015;4(3):230–253. doi:10.3390/antibiotics4030230
6. Lessa FC, Mu Y, Bamberg WM, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. 2015;372(9):825–834. doi:10.1056/NEJMoa1408913
7. Kelly CR, Fischer M, Allegretti JR, et al. ACG clinical guidelines: prevention, diagnosis, and treatment of clostridioides difficile infections. Off J Am College Gastroenterol. 2021;116(6):1124–47.
8. Hempel S, Newberry SJ, Maher AR, et al. Probiotics for the prevention and treatment of antibiotic-associated diarrhea: a systematic review and meta-analysis. JAMA. 2012;307(18):1959–1969. doi:10.1001/jama.2012.3507
9. Gonzales-Luna AJ, Carlson TJ, Garey KW. Gut microbiota changes associated with Clostridioides difficile infection and its various treatment strategies. Gut Microbes. 2023;15(1):2223345. doi:10.1080/19490976.2023.2223345
10. Berkell M, Mysara M, Xavier BB, et al. Microbiota-based markers predictive of development of Clostridioides difficile infection. Nat Commun. 2021;12(1):2241. doi:10.1038/s41467-021-22302-0
11. Samarkos M, Mastrogianni E, Kampouropoulou O. The role of gut microbiota in Clostridium difficile infection. Eur J Intern Med. 2018;50:28–32. doi:10.1016/j.ejim.2018.02.006
12. Asnicar F, Berry SE, Valdes AM, et al. Microbiome connections with host metabolism and habitual diet from 1098 deeply phenotyped individuals. Nat Med. 2021;27(2):321–332. doi:10.1038/s41591-020-01183-8
13. Shanahan F, Ghosh TS, O’Toole PW. The healthy microbiome-what is the definition of a healthy gut microbiome? Gastroenterology. 2021;160(2):483–494. doi:10.1053/j.gastro.2020.09.057
14. David LA, Materna AC, Friedman J, et al. Host lifestyle affects human microbiota on daily timescales. Genome Biol. 2014;15(7):R89. doi:10.1186/gb-2014-15-7-r89
15. Chen Y-B, Gu S-L, Wei Z-Q, et al. Molecular epidemiology of Clostridium difficile in a tertiary hospital of China. J Med Microbiol. 2014;63(Pt 4):562–569. doi:10.1099/jmm.0.068668-0
16. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Meth. 2015;12(1):59–60. doi:10.1038/nmeth.3176
17. Guery B, Galperine T, Barbut F. Clostridioides difficile: diagnosis and treatments. BMJ. 2019;366:l4609. doi:10.1136/bmj.l4609
18. Wanahita A, Goldsmith EA, Marino BJ, Musher DM. Clostridium difficile infection in patients with unexplained leukocytosis. Am J Med. 2003;115(7):543–546. doi:10.1016/S0002-9343(03)00420-0
19. Singh M, Vaishnavi C, Kochhar R, Mahmood S. Toxigenic Clostridium difficile isolates from clinically significant diarrhoea in patients from a tertiary care centre. Indian J Med Res. 2017;145(6):840–846. doi:10.4103/ijmr.IJMR_192_16
20. van Prehn J, Reigadas E, Vogelzang EH, et al. European Society of Clinical Microbiology and Infectious Diseases: 2021 update on the treatment guidance document for Clostridioides difficile infection in adults. Clin Microbiol Infect. 2021;27 Suppl 2:S1–s21. doi:10.1016/j.cmi.2021.09.038
21. McDonald LC, Gerding DN, Johnson S, et al. Clinical practice guidelines for clostridium difficile infection in adults and children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clinl Infect Dis. 2018;66(7):e1–e48. doi:10.1093/cid/cix1085
22. Zhang L, Dong D, Jiang C, Li Z, Wang X, Peng Y. Insight into alteration of gut microbiota in Clostridium difficile infection and asymptomatic C. difficile colonization. Anaerobe. 2015;34:1–7. doi:10.1016/j.anaerobe.2015.03.008
23. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi:10.1038/4441022a
24. Schubert AM, Sinani H, Schloss PD. Antibiotic-induced alterations of the murine gut microbiota and subsequent effects on colonization resistance against Clostridium difficile. mBio. 2015;6(4). doi:10.1128/mBio.00974-15
25. de Vos WM, Tilg H, Van Hul M, Cani PD. Gut microbiome and health: mechanistic insights. Gut. 2022;71(5):1020–1032. doi:10.1136/gutjnl-2021-326789
26. Lloyd-Price J, Mahurkar A, Rahnavard G, et al. Strains, functions and dynamics in the expanded human microbiome project. Nature. 2017;550(7674):61–66. doi:10.1038/nature23889
27. Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118(2):229–241. doi:10.1016/j.cell.2004.07.002
28. Chilton CH, Pickering DS, Freeman J. Microbiologic factors affecting Clostridium difficile recurrence. Clin Microbiol Infect. 2018;24(5):476–482. doi:10.1016/j.cmi.2017.11.017
29. Wanden-Berghe C, Patino-Alonso MC, Galindo-Villardón P, Sanz-Valero J. Complications associated with enteral nutrition: CAFANE study. Nutrients. 2019;11(9):2041. doi:10.3390/nu11092041
30. Yde J, Larsen HM, Laurberg S, Krogh K, Moeller HB. Chronic diarrhoea following surgery for colon cancer-frequency, causes and treatment options. Int J Colorectal Dis. 2018;33(6):683–694. doi:10.1007/s00384-018-2993-y
31. Hwang TL, Lue MC, Nee YJ, Jan YY, Chen MF. The incidence of diarrhea in patients with hypoalbuminemia due to acute or chronic malnutrition during enteral feeding. Am J Gastroenterol. 1994;89(3):376–378.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Wheat-Dependent Exercise-Induced Anaphylaxis Patients on a Wheat-Free Diet Exhibit a Gut Microbiota Composition More Similar to Healthy Individuals
Du Z, Li L, Liu J, Wang H, Li J, Xu Y, Cui L, Yin J
Journal of Asthma and Allergy 2026, 19:464532
Published Date: 29 January 2026