Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Long Noncoding RNA SNHG1 Knockdown Ameliorates Apoptosis, Oxidative Stress and Inflammation in Models of Parkinson’s Disease by Inhibiting the miR-125b-5p/MAPK1 Axis
Authors Xiao X, Tan Z, Jia M, Zhou X, Wu K, Ding Y, Li W
Received 14 October 2020
Accepted for publication 11 February 2021
Published 22 April 2021 Volume 2021:17 Pages 1153—1163
DOI https://doi.org/10.2147/NDT.S286778
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Taro Kishi
Xiao Xiao,1 Zhiwen Tan,2 Min Jia,3 Xiaoli Zhou,1 Kemei Wu,3 Yanbing Ding,1 Wenjing Li3
1Department of Encephalopathy, Hubei Provincial Hospital of Traditional Chinese Medicine (Affiliated Hospital of Hubei University of Traditional Chinese Medicine, Hubei Institute of Traditional Chinese Medicine), Wuhan City, Hubei Province, People’s Republic of China; 2Department of Encephalopathy, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Enshi City, Hubei Province, People’s Republic of China; 3Department of Neurology, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Enshi City, Hubei Province, People’s Republic of China
Correspondence: Wenjing Li
Department of Neurology, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, No. 158 Wuyang Avenue, Enshi, 445000, Hubei Province, People’s Republic of China
Tel +86 13597789520
Fax +86 718 8263395
Email [email protected]
Background: Parkinson’s disease (PD) is a prevalent neurodegenerative disease. Long noncoding RNA small molecule RNA host gene 1 (SNHG1) has been reported to play critical roles in Parkinson’s disease (PD) progression. The study aimed to further elucidate the mechanism of SNHG1 in PD pathogenesis.
Methods: The levels of SNHG1, miR-125b-5p and mitogen-activated protein kinase 1 (MAPK1) were determined by quantitative real-time polymerase chain reaction (qRT-PCR) or Western blot. Cell viability and apoptosis were evaluated by Cell Counting Kit-8 (CCK-8) assay and flow cytometry, respectively. The activity of Caspase-3 or Caspase-9 was measured using a Caspase-3 or Caspase-9 Assay Kit. The levels of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-1β, lactic dehydrogenase (LDH) activity, reactive oxygen species (ROS) generation and superoxide dismutase (SOD) activity were gauged by enzyme-linked immunosorbent assay (ELISA). Dual-luciferase reporter assay was performed to identify the relationship between miR-125b-5p and SNHG1 or MAPK1. The MPTP-induced PD mouse was used as an in vivo model of PD and MPP+-treated SK-N-SH and MN9D cells were used as in vitro models of PD.
Results: SNHG1 and MAPK1 were significantly up-regulated while miR-125b-5p was down-regulated in the MPTP-induced PD mouse model and MPP+-induced PD cell models. SNHG1 silence or miR-125b-5p overexpression protected against MPP+-evoked apoptosis, oxidative stress and inflammation in SK-N-SH and MN9D cells. Moreover, SNHG1 acted as a molecular sponge of miR-125b-5p, and the protective impact of SNHG1 silence on MPP+-evoked cell damage was reversed by miR-125b-5p inhibition. Furthermore, MAPK1 was a functional target of miR-125b-5p and its overexpression attenuated the effects of miR-125b-5p restoration in MPP+-triggered cell injury. In addition, the behavioral changes in MPTP-induced PD mouse in vivo model were relieved by SNHG1 silence.
Conclusion: SNHG1 knockdown exerted neuroprotective effects in MPP+-evoked cytotoxicity through regulating the miR-125b-5p/MAPK1 axis both in human and mouse PD cell models, highlighting a possible target for PD therapy.
Keywords: Parkinson’s disease, SNHG1, miR-125b-5p, MAPK1, cytotoxicity
Introduction
Parkinson’s disease (PD) is the second most familiar neurodegenerative disease after Alzheimer’s disease (AD).1 PD prevalence increases with age and is regarded as a multifactorial central nervous system (CNS) disorder characterized by rest tremor, rigidity and bradykinesia.2 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 1-methyl-4-phenylpyridinium (MPP+, active metabolite of MPTP), which have neurotoxic effects, are well recognized to produce PD animal or cell models, respectively.3,4 Though massive works have been done to elucidate the pathological mechanisms of PD, the cure rate of PD is still very low. Thus, it is urgent to explore effective remedial methods for PD.
Long non-coding RNAs (lncRNAs) are a cluster of novel RNA molecules containing more than 200 nucleotides (nt), but lacking protein-coding abilities and have vital function in various human diseases.5 LncRNAs participate in the pathogenesis and progression of PD and serve as putative diagnostic biomarkers and therapeutic targets for early PD diagnosis.6,7 For instance, lncRNA HOTAIR accelerated MPTP-induced PD progression via upregulating leucine-rich repeat kinase 2 (LRRK2) expression.8 Nuclear enriched abundant transcript 1 (NEAT1) silence blocked MPP+-triggered cell damage via miR-212-5p in SK-N-SH cells.9 LncRNA small molecule RNA host gene 1 (SNHG1) is believed to be involved in PD pathogenesis and may target miR-153-3p to exacerbate MPP-induced SH-SY5Y cellular toxicity.10 Nevertheless, the role of SNHG1 in the etiology of PD still requires to be expounded furtherly.
MicroRNAs (miRNAs) are diminutive non-coding RNAs having approximately 23 nt that serve vital gene-regulatory roles by coupling with 3ʹuntranslated regions (3ʹUTR) of mRNAs.11 Evidence is emerging that miRNA dysregulation is implicated in the evolution of neurodegenerative lesion covering PD12,13 and could serve as biomarkers in PD targeted therapy.14 Study has reported that the unbalance of miR-125b-5p level was allied to SH-SY5Y cell viability, autophagy and apoptosis in the MPTP-induced PD model.15 However, the evidence about the action of miR-125b-5p in PD is far from enough.
In the present study, we used MPTP-induced PD mouse model in vivo and MPP+-induced SK-N-SH and MN9D cell models in vitro of PD to explore the possible roles and regulatory mechanisms of SNHG1 in the development of PD.
Materials and Methods
MPTP-Induced PD Mouse Model
This study was conducted with the authorization of the Animal Ethics Committee of Hubei Provincial Hospital of Traditional Chinese Medicine (Affiliated Hospital of Hubei University of Traditional Chinese Medicine, Hubei Institute of Traditional Chinese Medicine). All animal experimental procedures were implemented following the Guidelines of Management and Use for Laboratory Animals of National Institutes of Health (NIH). 6–8 week old C57BL/6J mice (male, n=45) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). The mice in the MPTP group or control group (n=5 for each group) were intraperitoneally injected with 25 mg/kg MPTP (Sigma, St Louis, MO, USA) or isotonic saline solution. After MPTP injection for different times (0, 1, 3, 5 or 7 days), the mice were sacrificed and their ventral midbrains were harvested for subsequent analysis.
To probe the function of SNHG1 in MPTP-stimulated PD mice, mouse midbrain dopaminergic MN9D cells were transduced with SNHG1 lentiviral short hairpin RNA (sh-SNHG1) or empty vector (sh-NC) for 48 h. Then, the transfected cells (1 × 106) were suspended with saline solution and then injected subcutaneously into the left flank of mice. One week after lentivirus inoculation, 25 mg/kg MPTP (Sigma, n=5 for each group) or saline solution (Con, n=5) were intraperitoneally injected into mice for 7 days to establish reliable PD animal models.
Cell Culture and MPP+ Treatment
Human neuroblastoma cell line SK-N-SH and mice dopaminergic neuronal cell line MN9D from Procell (Wuhan, China) were maintained in DMEM medium (Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Gibco) and 1% antibiotics (Gibco) in a moist incubator at 37°C under 5% CO2.
For PD in vitro cell model, SK-N-SH and MN9D cells were stimulated with 1 mM MPP+ (Sigma) at 37°C for 24 h, respectively.
Cell Transfection
The small interfering RNA (siRNA) against human or mouse SNHG1 (hsa-si-SNHG1 and mmu-si-SNHG1), siRNA negative control (hsa-si-NC and mmu-si-NC), lentiviral vector short hairpin RNA against SNHG1 (sh-SNHG1) or control (sh-NC), miR-125b-5p mimic and inhibitor (hsa-miR-125b-5p, mmu-miR-125b-5p, hsa-in-miR-125b-5p and mmu-in-miR-125b-5p) and respective controls (hsa-miR-NC, mmu-miR-NC, hsa-in-miR-NC and mmu-in-miR-NC), mitogen-activated protein kinase 1 (MAPK1) overexpression vector (hsa-MAPK1 and mmu-MAPK1), and pcDNA3.1 empty vector (hsa-pcDNA and mmu-pcDNA) were acquired from Genepharma (Shanghai, China). Above oligonucleotides or vectors were transduced into cells via Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA extraction was conducted with Trizol (Invitrogen). The cDNA was synthesized using specific RT-PCR kit (Takara, Dalian, China). Subsequently, qRT-PCR was managed by the SYBR Green PCR Master Mix (Takara) in PCR amplifier. Relative expression was measured by 2−ΔΔCt method and normalized to GAPDH or U6. The primers synthesized from Sangon Biotech (Shanghai, China) are listed in Table 1.
 |
Table 1 Primers Sequences Used for Quantitative Real-Time PCR (qRT-PCR) |
Cell Counting Kit-8 (CCK-8) Assay
CCK-8 assay was proceeded to detect cell viability. In brief, cells (100 μL) were seeded into 96-well plates and cultivated in a 5% CO2 incubator for 48 h at 37°C. Then, CCK-8 reagent (10 μL, Sangon Biotech) was added to each well and incubated for another 3 h after treatment or/and transfection at 37°C. Finally, the absorbance at 450 nm was determined by a microplate reader (Bio-Rad, Hercules, CA, USA).
Apoptosis Assay
The detection of apoptosis was performed via an Annexin V-FITC/Propidium Iodide (PI) Apoptosis Detection kit (Beyotime, Shanghai, China) following manufacturer’s directions. In brief, SK-N-SH and MN9D cells (5 × 104) with different treatment or/and transfection at 37°C were collected and resuspended in binding buffer. Then, cells were double stained with 5 μL Annexin V-FITC and 5 μL propidium iodide (PI) for 20 min in the dark. Finally, the apoptosis rate of cells was measured using a flow cytometer.
Caspase-3 and Caspase-9 Activity Assay
The activities of Caspase-3 and Caspase-9 were assessed using a Caspase-3 or Caspase-9 Assay Kit (Abcam, Cambridge, UK, USA) referring to the specification. In brief, extracted protein samples from harvested cells were detected via the BCA Protein Assay Kit (Pierce, Appleton, WI, USA). Then, the protein was incubated with 2× reaction buffer containing a specific substrate for 1 h at 37°C in the dark. The absorbance at 405 nm was measured via a microplate reader as fold change to reflect the relative activity.
Lactate Dehydrogenase (LDH) Release, Reactive Oxygen Species Activity (ROS) and Superoxide Dismutase (SOD) Activity Assay
The levels of LDH release, ROS and SOD were determined by the LDH Assay Kit (Cytotoxicity) (Abcam), ROS Assay Kit (Beyotime) and SOD Assay Ki (Beyotime), respectively, referring to the directions of manufacturers. The absorbance of samples at 450 nm, 490 nm and 530 nm was tested to reflect SOD, LDH and ROS activity via a microplate reader, respectively.
Enzyme-Linked Immunosorbent Assay (ELISA)
The production levels of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β) and IL-6 in the supernatants of SK-N-SH and MN9D cells were gauged using corresponding human or mouse ELISA kits (Beyotime) as recommended by manufacturers.
Dual-Luciferase Reporter Assay
The human or mouse wide type (WT) and mutated (MUT) sequences of SNHG1 or MAPK1 3ʹUTR with supposed miR-125b-5p targeted sites were inserted into pmirGLO luciferase vectors (LMAI Bio, Shanghai, China) to form plasmids (hsa/mmu-SNHG1 WT, hsa/mmu-SNHG1 MUT, hsa/mmu-MAPK1 3ʹUTR WT or hsa/mmu-MAPK1 3ʹUTR MUT). The constructed luciferase reporter plasmids were co-transfected with miR-125b-5p or miR-NC into cells. 48 h post-transfection at 37°C, cells were collected and the luciferase activity was measured by the Dual-Lucy Assay Kit (Solarbio, Shanghai, China) and data were analyzed based on ratio of Firefly/Renilla activity.
Western Blot Assay
Total protein was extracted via RIPA lysis buffer (Sangon Biotech). The same amount of protein was subjected to 10% SDS-PAGE gel and then transferred onto PVDF membranes (Solarbio). After sealing of 5% skim milk, the membranes were joined with the primary antibodies against MAPK1 (1:2000, CSB-PA013448LA01HU, Cusabio Biotech, Wuhan, China) and β-actin (1:2000, ab8227, Abcam) overnight at 4°C. Thereafter, the membranes were probed with a secondary antibody (1:10,000, ab205718, Abcam). The protein bands were visualized via ECL reagent (Beyotime) and the protein was quantified by ImageJ software.
Statistical Analysis
All data are presented as the mean ± standard deviation (SD) from at least 3 independent experiments. The differences were analyzed by Student’s t-test or one-way analysis of variance (ANOVA) via SPSS software. P less than 0.05 was deemed as statistically significant.
Results
Silencing of SNHG1 Mitigated MPP+-Evoked Neuronal Injury in SK-N-SH and MN9D Cells
In order to probe the function of SNHG1 in PD model, we first explored the influence of MPTP and MPP+ on SNHG1 expression in ventral midbrain of mouse and SK-N-SH and MN9D cells. qRT-PCR data revealed that SNHG1 expression was dramatically elevated by MPTP treatment in a time-dependent manner in ventral midbrains of mouse (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, Figure 1A). Meanwhile, SNHG1 level was significantly increased in SK-N-SH and MN9D cells stimulated with MPP+ (**P < 0.01, ***P < 0.001, Figure 1B and C). SK-N-SH and MN9D cells were exposed to 1 mM MPP+ for 24 h as the treatment conditions for subsequent experiments.
 |
Figure 1 SNHG1 was highly expressed in PD models and participated in PD biological processes. (A) The relative SNHG1 level in MPTP-induced PD mouse model (n=5 mice/group) was examined by qRT-PCR assay. (B and C) MPP+-induced SK-N-SH cells and MN9D cells were tested by qRT-PCR assay. (D) The level of SNHG1 in MPP+-triggered SK-N-SH or MN9D cells transfected with hsa-si-NC, hsa-si-SNHG1, mmu-si-NC or mmu-si-SNHG1 was assessed. (E–N) SK-N-SH and MN9D cells were introduced with or without SNHG1 siRNAs or corresponding si-NCs and then exposed to 1 mM of MPP+ for 24 h. (E and F) Cell viability was analyzed by CCK8 assay. (G and H) Cell apoptosis rate was tested by flow cytometry. (I and J) The activity of Caspase-3 and Caspase-9 was detected by corresponding kits. (K–N) ELISA assay was utilized for LDH, ROS, SOD, TNF-α, IL-1β, and IL-6. Data were expressed as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus corresponding controls analyzed by ANOVA (Figure 1A) or Student’s t-test (Figure 1B-1N). |
Next, a series of loss-of-function assays were performed to explore SNHG1 action in hsa-si-SNHG1-transfected SK-N-SH cells and mmu-si-SNHG1-transfected MN9D cells. As illustrated in Figure 1D (**P < 0.01, ***P < 0.001), SNHG1 was successfully transfected into SK-N-SH and MN9D cells. Then, the impact of MPP+ on cell damage of SK-N-SH and MN9D cells was inspected. As expected, the results revealed that MPP+ stimulation resulted in a conspicuous inhibition in cell viability (*P < 0.05, **P < 0.01, ***P < 0.001, Figure 1E and F), an overt promotion in cell apoptosis (****P < 0.0001, Figure 1G and H) together with increasing levels of pro-apoptotic proteins Caspase-3 and Caspase-9 (*P < 0.05, **P < 0.01, Figure 1I and J). Furthermore, the relative activities of LDH and ROS were raised while the relative activity of SOD was reduced in MPP+-disposed SK-N-SH and MN9D cells (*P < 0.05, **P < 0.01, Figure 1K and L). Moreover, after MPP+ treatment, the levels of TNF-α, IL-1β and IL-6 were remarkably elevated in SK-N-SH and MN9D cell culture medium (*P < 0.05, **P < 0.01, Figure 1M and N). These findings together demonstrated that MPP+ stimulation triggered cytotoxicity in SK-N-SH and MN9D cells. Interestingly, the above effects of MPP+ stimulation on cell viability, apoptosis, oxidative stress and inflammation were all partly reversed by hsa-si-SNHG1 or mmu-si-SNHG1 in corresponding SK-N-SH or MN9D cells (Figure 1E-N). Together, these data manifested that knockdown of SNHG1 allayed MPP+-triggered neurotoxicity in SK-N-SH and MN9D cells. Thus, SNHG1 was required for the neurotoxic effects of MPP+ in these cell lines.
SNHG1 Was a Molecular Sponge of miR-125b-5p
On the basis of the prediction database LncBase Predicted v.2, wild type SNHG1 was verified to have a putative binding sequence for hsa-miR-125b-5p and mmu-miR-125b-5p (Figure 2A and B). The overexpression efficiency of hsa-miR-125b-5p in MPP+-evoked SK-N-SH cells and mmu-miR-125b-5p in MPP+-evoked MN9D cells was testified by qRT-PCR (***P < 0.001, Figure 2C). To confirm this interaction between SNHG1 and miR-125b-5p, the dual-luciferase reporter assay was implemented, and the results suggested that hsa-miR-125b-5p overexpression resulted in lower luciferase activity (62% reduction) in SK-N-SH cells transfected with wild-type reporter construct (hsa-SNHG1 WT) compared with hsa-miR-NC group (**P < 0.01), while did not affect the luciferase activity of mutant SNHG1 reporter (hsa-SNHG1 MUT) (P > 0.05) (Figure 2D). Similarly, the co-transfection of mmu-miR-125b-5p and mmu-SNHG1 WT in MN9D cells demonstrated the same result (*P < 0.05, 47% reduction of luciferase activity, Figure 2E). The expression of miR-125b-5p in ventral midbrains of mouse was observably reduced after MPTP treatment relative to NC group (**P < 0.01, Figure 2F). Simultaneously, the level of miR-125b-5p was distinctly reduced after MPP+ treatment (**P < 0.01, Figure 2G and H). And miR-125b-5p level was prominently elevated by SNHG1 silence relative to that of si-NC group in MPP+-evoked SK-N-SH and MN9D cells (***P < 0.001, ****P < 0.0001, Figure 2I and J), which implied the targeted sites were functional. Collectively, all above data evidenced that SNHG1 sponged miR-125b-5p in SK-N-SH and MN9D cells.
 |
Figure 2 SNHG1 participated in PD biological processes via miR-125b-5p. (A and B) The target sequence of hsa-SNHG1 (A) or mmu-SNHG1 (B) with miR-125b-5p and the corresponding mutated sites. (C) The efficiency of miR-125b-5p overexpression in MPP+-evoked SK-N-SH and MN9D cells was verified. (D and E) The relative luciferase activity in SK-N-SH and MN9D cells co-transfected with hsa/mmu-SNHG1 WT or MUT luciferase reporter plasmids and miR-125b-5p or miR-NC was detected. (F–H) The relative miR-125b-5p level in MPTP-induced PD model (F), MPP+-induced SK-N-SH (G) and MN9D cells (H) was detected. (I and J) The relative miR-125b-5p level in MPP+-induced SK-N-SH and MN9D cells transfected with SNHG1 siRNAs or corresponding si-NCs was examined. Data were expressed as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus corresponding controls analyzed by Student’s t-test (Figure 2C-2J). |
The Protective Impact of SNHG1 Knockdown on MPP+-Evoked SK-N-SH and MN9D Cell Damage Was Alleviated by miR-125b-5p Inhibition
To verify whether the protective effect of SNHG1 silence on MPP+-triggered cytotoxicity was mediated by miR-125b-5p, SK-N-SH and MN9D cells were transduced with hsa-si-SNHG1, hsa-si-SNHG1+ hsa-in-miR-125b-5p, mmu-si-SNHG1, mmu-si-SNHG1+ mmu-in-miR-125b-5p or corresponding controls, and then treated with 1 mM MPP+. Compared with control groups, hsa-si-SNHG1- or mmu-si-SNHG1-mediated miR-125b-5p increase was overturned by hsa-in-miR-125b-5p or mmu-in-miR-125b-5p introduction in SK-N-SH and MN9D cells under MPP+ stimulation (**P < 0.01, ***P < 0.001, Figure 3A and B). CCK-8 assay testified that SNHG1 silence-induced accelerated effect on cell viability in MPP+-treated SK-N-SH and MN9D cells was attenuated by knockdown of miR-125b-5p (*P < 0.05, **P < 0.01, Figure 3C and D). Meanwhile, SNHG1 depletion caused a striking suppression in cell apoptosis and decrease in Caspase-3 and Caspase-9 activity, and these impacts were abrogated by miR-125b-5p inhibition (*P < 0.05, **P < 0.01, ***P < 0.001, Figure 3E-H). Furthermore, the reduction of LDH and ROS release and the increase in SOD level induced by SNHG1 silence were depressed by deficiency of miR-125b-5p in SK-N-SH and MN9D cells under MPP+ stimulation (*P < 0.05, **P < 0.01, Figure 3I and J). Similarly, the inhibiting action of hsa-si-SNHG1 and mmu-si-SNHG1 on the levels of TNF-α, IL-1β and IL-6 in MPP+-exposed SK-N-SH or MN9D cells was corrected by lessened expression of miR-125b-5p (*P < 0.05, **P < 0.01, Figure 3K and L). These findings indicated that knockdown of SNHG1 attenuated MPP+-induced neurotoxicity via reducing the levels of miR-125b-5p.
 |
Figure 3 SNHG1 knockdown alleviated MPP+-evoked neuronal injury by regulating miR-125b-5p. (A–L) SK-N-SH and MN9D cells were transduced with hsa-si-SNHG1, hsa-si-SNHG1+hsa-in-miR-125b-5p, mmu-si-SNHG1, mmu-si-SNHG1+ mmu-in-miR-125b-5p or homologous controls (hsa-si-NC, hsa-si-SNHG1+hsa-in-miR-NC, mmu-si-NC, or mmu-si-SNHG1+ mmu-in-NC) before MPP+ stimulation (1 mM MPP+, 24 h). The level of miR-125b-5p (A and B), cell viability (C and D), cell apoptosis (E and F), Caspase-3 and Caspase-9 activity (G and H), LDH, ROS or SOD activity (I and J) and inflammatory factor levels (K and L) were examined via corresponding methods. Data were expressed as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding controls analyzed by Student’s t-test. |
MAPK1 Was Targeted and Suppressed by miR-125b-5p
To further explore the action of miR-125b-5p in MPP+-evoked cytotoxicity, DIANA TOOLS microT-CDS (v5.0) software was utilized to identify its molecular targets. As illustrated in Figure 4A and B, the 3ʹUTR sequence of MAPK1 contained putative miR-125b-5p complementary sites in SK-N-SH and MN9D cells. Transient transfection of hsa-miR-125b-5p or mmu-miR-125b-5p observably lessened the luciferase activity of MAPK1 3ʹUTR luciferase reporter (hsa-MAPK1 3ʹUTR WT and mmu-MAPK1 3ʹUTR WT) (**P < 0.01) but barely affected the luciferase activity in the MAPK1 mutant reporter (hsa-MAPK1 3ʹUTR MUT and mmu-MAPK1 3ʹUTR MUT) (P > 0.05) in corresponding SK-N-SH and MN9D cells (Figure 4C and D). The protein level of MAPK1 in ventral midbrains of mouse after MPTP treatment and in MPP+-stimulated SK-N-SH or MN9D cells was visibly increased (***P < 0.001, Figure 4E-G). Moreover, MAPK1 level was distinctly reduced by miR-125b-5p overexpression in SK-N-SH and MN9D cells (**P < 0.01, ***P < 0.001, Figure 4H). In addition, the level of MAPK1 was significantly decreased by hsa-si-SNHG1 or mmu-si-SNHG1 introduction, which was respectively inverted by hsa-in-miR-125b-5p or mmu-in-miR-125b-5p introduction in MPP+-induced SK-N-SH or MN9D cells (**P < 0.01, ***P < 0.001, Figure 4I and J).
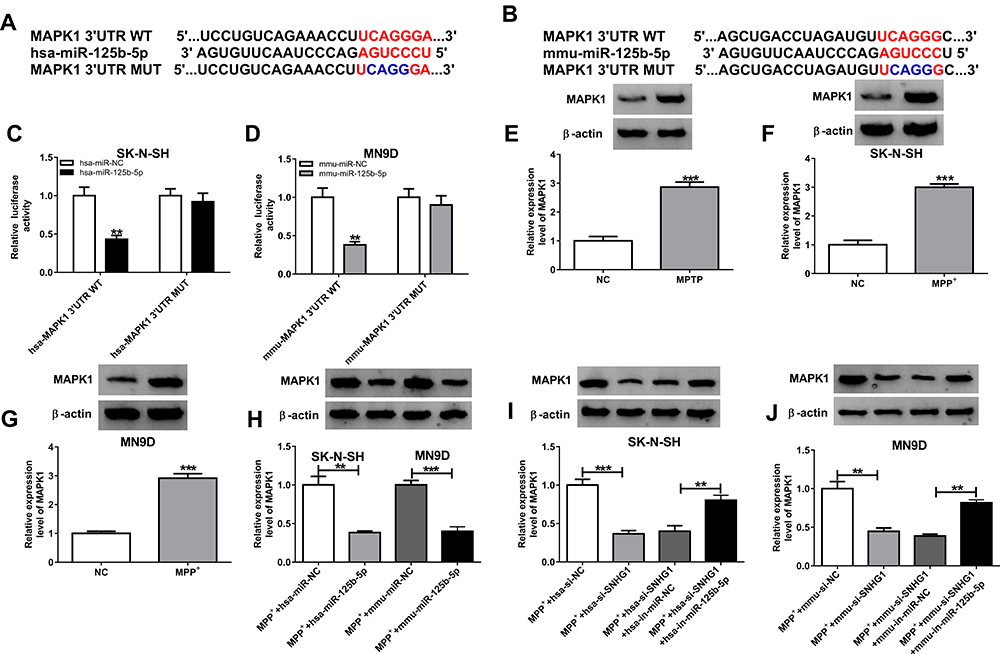 |
Figure 4 MAPK1 was a direct target of miR-125b-5p. (A and B) The predicted binding sequence between MAPK1 and miR-125b-5p was displayed. (C and D) The targeting relationship between MAPK1 and miR-125b-5p was confirmed via dual-luciferase reporter assay. (E–G) The protein level of MAPK1 in MPTP-induced PD model (E), MPP+-induced SK-N-SH cells (F), MPP+-induced MN9D cells (G) was tested by Western blot. (H) The expression of MAPK1 was measured in SK-N-SH and MN9D cells transfected with miR-NC or miR-125b-5p. (I and J) The level of MAPK1 was tested in MPP+-stimulated SK-N-SH and MN9D cells transfected with hsa/mmu-si-SNHG1, hsa/mmu-si-SNHG1 + hsa/mmu-in-miR-125b-5p or corresponding controls (hsa/mmu-si-NC or hsa/mmu-si-SNHG1 + hsa/mmu-in-miR-NC). Data were expressed as mean ± SD from at least three independent experiments. **P < 0.01, ***P < 0.001 versus corresponding controls analyzed by Student’s t-test (Figure 4C-4J). |
Overexpression of miR-125b-5p Mitigated MPP+-Triggered Neuronal Injury by Targeting MAPK1
To explore whether MAPK1 participated in the regulation by miR-125b-5p of MPP+-stimulated cytotoxicity, the effect of miR-125b-5p and/or MAPK1 overexpression was examined in SK-N-SH and MN9D cells. As displayed in Figure 5A and B (*P < 0.05, **P < 0.01), the down-regulation of MAPK1 level caused by miR-125b-5p overexpression was abated by the introduction of MAPK1 overexpression plasmid in MPP+-triggered SK-N-SH and MN9D cells. Moreover, miR-125b-5p overexpression-mediated viability enhancement (*P < 0.05, **P < 0.01, Figure 5C and D), apoptosis repression (*P < 0.05, **P < 0.01, ***P < 0.001, Figure 5E-H) and inflammation reduction (*P < 0.05, **P < 0.01, Figure 5K and L) were distinctly reversed by the up-regulation of MAPK1 expression. Concurrently, MAPK1 overexpression also abrogated the decrease of LDH and ROS and the enhancement of SOD induced by miR-125b-5p introduction (*P < 0.05, **P < 0.01, Figure 5I and J). These data together evidenced that miR-125b-5p ameliorated MPP+-triggered neurotoxicity via sponging MAPK1.
 |
Figure 5 MAPK1 overexpression reversed the effects of miR-125b-5p on MPP+-triggered neuronal injury. (A–L) SK-N-SH cells were transduced with hsa-miR-NC, hsa-miR-125b-5p, hsa-miR-125b-5p + hsa-pcDNA, hsa-miR-125b-5p + hsa-MAPK1 and MN9D cells were transduced with mmu-miR-NC, mmu-miR-125b-5p, mmu-miR-125b-5p + mmu-pcDNA or mmu-miR-125b-5p + mmu-MAPK1 and then treated with 1 mM MPP+. MAPK1 level (A and B), cell viability (C and D), cell apoptosis (E and F) and Caspase-3 or Caspase-9 activity (G and H) were examined via corresponding methods. LDH, ROS or SOD activity (I and J) and inflammatory factor levels (K and L) were tested by corresponding kits. Data were expressed as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 versus corresponding controls analyzed by Student’s t-test. |
SNHG1 Knockdown Mitigated Behavioral Deficits in MPTP‐Induced PD Mice
To study the in vivo effects of SNHG1, we established SNHG1 knockdown mice by lentivirus transduction, and then the mice were treated with MPTP. As shown in Figure 6A and B (*P < 0.05, **P < 0.01), the numbers of counters (5 min) and latency to fall (sec) were decreased in MPTP treated mice by using spontaneous motor activity test and rotarod test, implying the behavioral deficits of MPTP-treated mice, while the above decreased effects were reversed by SNHG1 knockdown. Furthermore, the levels of SNHG1 and MAPK1 were increased while miR-125b-5p level was declined in MPTP-induced PD mice, while these alterations were all overturned by silence of SNHG1 (**P < 0.01, ***P < 0.001, ****P < 0.0001, Figure 6C-E). In conclusion, SNHG1 knockdown attenuated MPTP-induced neuronal damage in PD in vivo model mice.
 |
Figure 6 SNHG1 silence attenuated behavioral deficits in MPTP‐induced PD model mice. The mice transduced with sh-SNHG1 or not were treated with MPTP. (A) The numbers of counters (5 min) of mice was evaluated by spontaneous motor test. (B) The time of latency to fall was evaluated by rotational behavior. (C–E) The levels of SNHG1, miR-125b-5p and MAPK1 were examined. Data were expressed as mean ± SD from at least three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 versus corresponding controls analyzed by Student’s t-test. |
Discussion
PD is a progressive chronic age-related neurological disease which impairs the action capability of human being.2 Until now, PD cannot be completely cured and the pathogenesis of PD is still unclear. Therefore, it is indispensable to find available therapeutic regimen to restrain PD development and thus to improve life quality of PD sufferers. This study elucidated the action mode of lncRNA SNHG1/miR-125b-5p/MAPK1 axis in PD etiopathology.
LncRNAs are pivotal regulators in diverse malignant diseases.16 SNHG1 has been demonstrated to accelerate tumorigenesis and development and could act as a useful tumor biomarker for cancer diagnosis, prognosis and treatment in multifold cancers.17 Also, SNHG1 was reported to be a vital regulatory molecule in the brain actions and pathophysiology of CNS disorders, including PD. For example, SNHG1 promoted glioma progression by sponging miR-194 to up-regulate PHLDA1.18 Silence of SNHG1 exerted its neuronal protective effects by repressing KRENEN1 via miR-137 in the AD in vitro cell model.19 Particularly, SNHG1 was deemed to participate in PD progression. For instance, SNHG1 aggravated MPP-induced cellular toxicity in SH-SY5Y cells via modulating miR-153-3p.10 The raised SNHG1 level was found in MPP+-evoked cell model and brain tissues of PD sufferers, and SNHG1 facilitated PD evolvement by miR-7.20 Here, we proposed a prominent up-regulation of SNHG1 in MPTP-stimulated animal model and MPP+-treated SK-N-SH and MN9D cell models of PD. We also exposed that SNHG1 silence exerted a protective effect on MPP+-triggered apoptosis, oxidative stress and inflammation in SK-N-SH and MN9D cells.
MicroRNAs are believed to play important roles via serving as the targets of lncRNAs thereby to regulate target genes expression and implicate in PD pathogenesis.11,21,22 miR-135b exerts a protective role in MPP-intoxicated PD cell model by inhibition of apoptosis and neuroinflammation by sponging FoxO123 or GSK3.24 Meanwhile, BDNF-AS knockdown could increase SH-SY5Y cell viability, block autophagy and apoptosis in MPTP-induced PD models via miR-125b-5p.15 This study uncovered that the elevated level of miR-125b-5p alleviated MPP+-triggered cytotoxicity in PD cell models. Furthermore, we first authenticated that miR-125b-5p was targeted by SNHG1 and SNHG1 knockdown relieved MPP+-evoked neuronal damage by increasing miR-125b-5p expression.
Studies have verified the character of MAPK1 in various cancers, brain functions and CNS disorders. For example, MAPK1 was increased and acted as a tumor inhibitor through serving as the target of miRNAs in gastric cancer,25 hepatocellular carcinoma26 and lung adenocarcinoma.27 Meanwhile, miR-129-1 acted as tumor depressor and impeded cell cycle progress of glioblastoma (GBM) cells via MAPK1 and IGF2BP3.28 Besides, MAPK1 level was elevated in intermediate and late AD stages and the alteration of MAPK1 expression was related to the pathogenesis of AD.29 Knockdown of MAPK1 mitigated MPP+-treated SH-SY5Y cell injury via lncRNA AL049437/miR-205-5p/MAPK1 pathway.30 We first substantiated that MAPK1 was functionally targeted by miR-125b-5p in MPP+-triggered PD cells, and miR-125b-5p upregulation relieved MPP+-evoked cytotoxicity via suppressing MAPK1. Meanwhile, we demonstrated the function of SNHG1 as a sponge of miR-125b-5p to control MAPK1 expression and SNHG1 knockdown could efficaciously improve behavioral changes in MPTP-induced PD animal model. Even though our research illuminated the regulatory mechanism of SNHG1 in PD progression, there were still some disadvantages. For example, this study lacked the data support of clinical tissues from PD patients. Meanwhile, we noted that miR-125b-5p inhibition could only partially relieve the effects of si-SNHG1 on MAPK1 expression, suggesting that there might exist other factors that were regulated by SNHG1/miR-125b-5p axis. This progress was worthy of further studying, and we will explore this in our future studies.
In a word, we identified that SNHG1 silence protected against MPP+-evoked injury in MN9D and SK-N-SH cells by targeting miR-125b-5p/MAPK1 axis. These data provided a novel regulatory mechanism about SNHG1 in PD pathogenesis and proposed feasible targets for PD remedy.
Ethics Approval and Consent to Participate
Our study was approved by the Ethics Committee of Hubei Provincial Hospital of Traditional Chinese Medicine (Affiliated Hospital of Hubei University of Traditional Chinese Medicine, Hubei Institute of Traditional Chinese Medicine) and was conducted in accordance with the Guidelines of Management and Use for Laboratory Animals of National Institutes of Health (NIH).
Funding
There is no funding to report.
Disclosure
The authors declare that they have no financial conflicts of interest and conflicts of interest in this work.
References
1. Elbaz A, Carcaillon L, Kab S, et al. Epidemiology of Parkinson’s disease. Rev Neurol (Paris). 2016;172:14–26. doi:10.1016/j.neurol.2015.09.012
2. Tysnes OB, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm (Vienna). 2017;124:901–905. doi:10.1007/s00702-017-1686-y
3. Haque ME, Thomas KJ, D’souza C, et al. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A. 2008;105(5):1716–1721. doi:10.1073/pnas.0705363105
4. Cui X, Li M, He Z, et al. MiR -302b-5p enhances the neuroprotective effect of IGF -1 in methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease by regulating inducible nitric-oxide synthase. Cell Biochem Funct. 2020;38(8):1025–1035. doi:10.1002/cbf.3534
5. Maass PG, Luft FC, Bahring S. Long non-coding RNA in health and disease. J Mol Med (Berl). 2014;92(4):337–346. doi:10.1007/s00109-014-1131-8
6. Lv Q, Wang Z, Zhong Z, et al. Role of Long Noncoding RNAs in Parkinson’s disease: putative biomarkers and therapeutic targets. Parkinsons Dis. 2020;2020:5374307. doi:10.1155/2020/5374307
7. Chen L, Guo X, Li Z, et al. Relationship between long non-coding RNAs and Alzheimer’s disease: a systematic review. Pathol Res Pract. 2019;215:12–20. doi:10.1016/j.prp.2018.11.012
8. Liu S, Cui B, Dai ZX, et al. Long Non-coding RNA HOTAIR promotes parkinson’s disease induced by MPTP through up-regulating the Expression of LRRK2. Curr Neurovasc Res. 2016;13:115–120. doi:10.2174/1567202613666160316155228
9. Liu R, Li F, Zhao W. Long noncoding RNA NEAT1 knockdown inhibits MPP(+)-induced apoptosis, in fl ammation and cytotoxicity in SK-N-SH cells by regulating miR-212-5p/RAB3IP axis. Neurosci Lett. 2020;731:135060. doi:10.1016/j.neulet.2020.135060
10. Zhao J, Geng L, Chen Y, et al. SNHG1 promotes MPP(+)-induced cytotoxicity by regulating PTEN/AKT/mTOR signaling pathway in SH-SY5Y cells via sponging miR-153-3p. Biol Res. 2020;53:1. doi:10.1186/s40659-019-0267-y
11. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi:10.1016/j.cell.2009.01.002
12. Hebert SS, De Strooper B. Alterations of the microRNA network cause neurodegenerative disease. Trends Neurosci. 2009;32:199–206. doi:10.1016/j.tins.2008.12.003
13. Singh A, Sen D. MicroRNAs in Parkinson’s disease. Exp Brain Res. 2017;235(8):2359–2374. doi:10.1007/s00221-017-4989-1
14. Leggio L, Vivarelli S, L’episcopo F, et al. microRNAs in Parkinson’s disease: from pathogenesis to novel diagnostic and therapeutic approaches. Int J Mol Sci. 2017;18(12):2698. doi:10.3390/ijms18122698
15. Fan Y, Zhao X, Lu K, et al. LncRNA BDNF-AS promotes autophagy and apoptosis in MPTP-induced Parkinson’s disease via ablating microRNA-125b-5p. Brain Res Bull. 2020;157:119–127. doi:10.1016/j.brainresbull.2020.02.003
16. Tang Y, Cheung BB, Atmadibrata B, et al. The regulatory role of long noncoding RNAs in cancer. Cancer Lett. 2017;391:12–19. doi:10.1016/j.canlet.2017.01.010
17. Thin KZ, Tu JC, Raveendran S. Long non-coding SNHG1 in cancer. Clin Chim Acta. 2019;494:38–47. doi:10.1016/j.cca.2019.03.002
18. Liu L, Shi Y, Shi J, et al. The long non-coding RNA SNHG1 promotes glioma progression by competitively binding to miR-194 to regulate PHLDA1 expression. Cell Death Dis. 2019;10:463. doi:10.1038/s41419-019-1698-7
19. Wang H, Lu B, Chen J. Knockdown of lncRNA SNHG1 attenuated Abeta25-35-induced neuronal injury via regulating KREMEN1 by acting as a ceRNA of miR-137 in neuronal cells. Biochem Biophys Res Commun. 2019;518:438–444. doi:10.1016/j.bbrc.2019.08.033
20. Cao B, Wang T, Qu Q, et al. Long Noncoding RNA SNHG1 promotes neuroinflammation in Parkinson’s disease via regulating miR-7/NLRP3 pathway. Neuroscience. 2018;388:118–127. doi:10.1016/j.neuroscience.2018.07.019
21. Oe S, Kimura T, Yamada H. Regulatory non-coding RNAs in nervous system development and disease. Front Biosci (Landmark Ed). 2019;24:1203–1240. doi:10.2741/4776
22. Goh SY, Chao YX, Dheen ST, et al. Role of MicroRNAs in Parkinson’s Disease. Int J Mol Sci. 2019;20. doi:10.3390/ijms20225649
23. Zeng R, Luo DX, Li HP, et al. MicroRNA-135b alleviates MPP(+)-mediated Parkinson’s disease in in vitro model through suppressing FoxO1-induced NLRP3 inflammasome and pyroptosis. J Clin Neurosci. 2019;65:125–133. doi:10.1016/j.jocn.2019.04.004
24. Zhang J, Liu W, Wang Y, et al. miR-135b plays a neuroprotective role by targeting GSK3beta in MPP(+)-Intoxicated SH-SY5Y cells. Dis Markers. 2017;2017:5806146. doi:10.1155/2017/5806146
25. Luo M, Liang C. LncRNA LINC00483 promotes gastric cancer development through regulating MAPK1 expression by sponging miR-490-3p. Biol Res. 2020;53:14. doi:10.1186/s40659-020-00283-6
26. Fu X, Zhang J, He X, et al. Circular RNA MAN2B2 promotes cell proliferation of hepatocellular carcinoma cells via the miRNA-217/MAPK1 axis. J Cancer. 2020;11(11):3318–3326. doi:10.7150/jca.36500
27. Wang M, Liao Q, Zou P. PRKCZ-AS1 promotes the tumorigenesis of lung adenocarcinoma via sponging miR-766-5p to modulate MAPK1. Cancer Biol Ther. 2020;21(4):364–371. doi:10.1080/15384047.2019.1702402
28. Kouhkan F, Mobarra N, Soufi-Zomorrod M, et al. MicroRNA-129-1 acts as tumour suppressor and induces cell cycle arrest of GBM cancer cells through targeting IGF2BP3 and MAPK1. J Med Genet. 2016;53(1):24–33. doi:10.1136/jmedgenet-2015-103225
29. Gerschutz A, Heinsen H, Grunblatt E, et al. Neuron-specific alterations in signal transduction pathways associated with Alzheimer’s disease. J Alzheimers Dis. 2014;40(1):135–142. doi:10.3233/JAD-131280
30. Zhang L, Wang J, Liu Q, et al. Knockdown of long non-coding RNA AL049437 mitigates MPP+-induced neuronal injury in SH-SY5Y cells via the microRNA-205-5p/MAPK1 axis. Neurotoxicology. 2020;78:29–35. doi:10.1016/j.neuro.2020.02.004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.