Back to Journals » OncoTargets and Therapy » Volume 13
Locally Advanced Pancreatic Ductal Adenocarcinoma: Challenges and Progress
Authors Barcellini A ![]() , Peloso A
, Peloso A ![]() , Pugliese L
, Pugliese L ![]() , Vitolo V
, Vitolo V ![]() , Cobianchi L
, Cobianchi L ![]()
Received 4 August 2020
Accepted for publication 30 November 2020
Published 10 December 2020 Volume 2020:13 Pages 12705—12720
DOI https://doi.org/10.2147/OTT.S220971
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianmin Xu
Amelia Barcellini,1 Andrea Peloso,2 Luigi Pugliese,3 Viviana Vitolo,1 Lorenzo Cobianchi3,4
1National Center of Oncological Hadrontherapy (Fondazione CNAO), Pavia, Italy; 2Divisions of Transplantation and Visceral Surgery, Department of Surgery, University of Geneva, Geneva, Switzerland; 3General Surgery, Foundation IRCCS San Matteo Hospital, Pavia, Italy; 4Department of Clinical, Surgical, Diagnostic and Pediatric Sciences, Foundation IRCCS San Matteo Hospital, University of Pavia, Pavia, Italy
Correspondence: Amelia Barcellini
National Center of Oncological Hadrontherapy (Fondazione CNAO), Strada Campeggi 53, Pavia 27100, Italy
Tel +39 0382078501
Email [email protected]
Abstract: Pancreatic ductal adenocarcinoma (PDAC) is one of the major causes of death in the Western world, and it is estimated to become the second leading cause of tumour-related mortality in the next 10 years. Among pancreatic cancers, ductal adenocarcinomas are by far the most common, characterised by a challenging diagnosis due to the lack of initial and pathognomonic clinical signs. In this scenario, non-metastatic locally advanced pancreatic cancer (LAPC) accounts for a large proportion of all new pancreatic ductal adenocarcinoma diagnoses. There is no consensus on a common definition of LAPC. Still, it usually includes tumours that are not resectable due to vascular involvement. As of today, treatment is limited, and the prognosis is very unfavourable. Curative-intent surgery remains the gold-standard even if often jeopardized by vascular involvement. Continuing progress in our understanding of LAPC genetics and immunology will permit the development of different treatments, targeted or combined, including radiation therapy, hadrontherapy, targeted immunotherapies or new chemotherapies. A multidisciplinary approach combining various fields of expertise is essential in aiming to limit disease progression as well as patient outcome. Using a narrative literature review approach, the manuscript explores the most up-to-date knowledge concerning locally advanced pancreatic ductal adenocarcinoma management.
Keywords: pancreatic cancer, risk factors, treatment, hadrontherapy, surgery
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a deadly malignancy characterised by a somber prognosis. Its incidence is increasing drastically, classed worldwide as the 14th most common cancer and the 7th highest cause of tumour-related mortality. In 2018, 458,918 diagnoses and 432,242 deaths from PDAC had been estimated worldwide1 and by 2030, PDAC is forecasted to be the second leading cause of cancer deaths in the US.2 PDAC is the most frequent type of pancreatic neoplasm, representing 80% to 85% of all malignant pancreatic neoplasms. The absence of specific symptoms and the aggressiveness of the disease are the two main limitations for early diagnosis.3 As it happens today in most clinical disciplines,4–6 a multidisciplinary approach is essential for the management of PDAC, due to the need for primary prevention, importance of an early diagnosis, and the complexity of treatment given that, even combined with radiotherapy, traditional therapeutic strategies have not prolonged the 5-year survival rates (less than 30%) 7. As of today, curative-intent surgical resection remains the only potential for cure. However, at initial presentation, only 15–20% of patients feature a surgically resectable tumour and, additionally, 45–50% of patients experience an openly metastatic disease.8 The remaining 25–30% of patients show either borderline resectable (BRPAC) or locally advanced (LPAC) pancreatic cancer.9 This condition opens up the concept of pancreatic tumour resectability by dividing the progression of pancreatic cancer into BRPAC, LPAC or advanced pancreatic cancer (APAC). As a general principle, the estimation of PDAC resectability should always be driven by the ability to obtain negative resection margins (R0 margins).10 BRPAC has been defined as a condition that encompasses a spectrum of patients ranging from “resectable” to LPAC disease. For these patients, a microscopically positive margin (R1) resection is considered relatively more likely, primarily due to the relationship between the primary pancreatic tumour and the surrounding blood vessels. LPAC is defined as a tumoural condition involving the celiac trunk, or having a tumour-to-artery interface >180° and/or involvement of SMV/PV with no reconstruction options. APAC/LPAC is finally defined as an unresectable pancreatic tumour with metastasis.11 This situation entails to an unmet therapeutic challenge for developing new treatments. This review covers the most up-to-date knowledge of non-metastasized locally advanced pancreatic ductal adenocarcinoma (LPAC) and, in particular, of providing an overview of how new therapies or new therapeutic strategies will guide multidisciplinary disease management. Furthermore, this study also aims to highlight current shortcomings concerning this pathology, which is not yet fully understood.
Epidemiology
PDAC is one of the deadliest malignancies worldwide1 and one of the major concerns is its growing incidence in the Western world. This is supported by the group driven by Saad et al12 which, using data from the USSEER (United States Surveillance, Epidemiology, and End Results Program), found that between 1973 and 2014, PDAC incidence rates increased around 1.03%/year. This is the reason why pancreatic cancer is forecasted to become the second most frequent cause of cancer-related death in the United States by 2030.2,13
Although incidence rates vary considerably in different countries, the highest age-standardised incidence has been detected in industrialised regions such as North America and Europe, and the lowest in less developed areas of Africa and South Central Asia.14 These trends have been confirmed by Wong et al15 who emphasized the positive correlation between the higher human development index and PDAC incidence.
The wide disparity between countries in the incidence of pancreatic cancer indicates how environmental factors may be essential for PDAC development.
PDAC is known as having a complex multistep and multifactorial aetiology linked to the interaction between genetic background as well as environmental susceptibility. The risk of PDAC increases with age in both sexes, the majority of patients who develop PDAC being older than 45 (90% are >55 yrs and 70% >65 yrs), and is diagnosed at a median age of around 70 years old.16
Risk Factors
Although knowledge of the risk factors involved in pancreatic cancer development is poor, some conditions have been associated with an increased risk of pancreatic cancer. Environmental and genetic risk factors have been reported, as already mentioned.17
The most important environmental risk is cigarette smoking,8 with approximately 20% of pancreatic cancers that can be attributed to tobacco use. In 2012 a pooled analysis was performed by Bosetti et al18 reporting a positive association between PDAC and cigarette smoking (OR: 1.2, 95% for former smokers; OR: 2.2, 95% for current smokers). Interestingly, this group has also shown that 20 years of smoking cessation are necessary to reduce the risk of PDAC to the level of never-smokers. These trends have been similarly reported by Lynch et al (OR: 1.8, 95% for current smokers).19 Electronic-cigarette smoke (ECS), meant to provide unburnt nicotine, has been found, in an experimental model, to cause lung cancer in mice.20 Even if today it is too early to have convincing data, it will be of great interest, due to widespread use, to analyse the impact of ECS on PDAC onset in the near future.
Obesity
Large pooled analysis and meta-analyses have confirmed the positive association between obese patients (body mass index [BMI] of 30 or more) and pancreatic cancer risk.21–23 Given that obesity often mirrors an energy imbalance with a caloric overload, three main mechanisms have been proposed to promote pancreas tumourigenesis:
- hormonal and inflammatory effects associated with hyperglycemia, insulin resistance and a chronic inflammatory state.24,25
- diet both due to calory intake26 and a dietary pattern rich in meat and dairy.27,28
- reduced physical activity.29
Overall, studies have concurred that obesity is a risk factor of PDAC (OR:1.33, 95%).30
Diabetes
Type 2 diabetes mellitus (T2DM) is a well-known risk factor for pancreatic cancer, being both a consequence and a prognostic factor for pancreatic cancer. A meta-analysis of 88 cohort studies reported a 94% increase in the risk of pancreatic cancer in individuals with T2DM compared to euglycemic individuals.31 The underlying mechanisms have yet to be fully elucidated. Insulin resistance drives to hyperinsulinemia and high levels of insulin-like growth factor-1 (IGF-1),32,33 exploiting PDAC tumourigenesis by the expression of IGF-1 receptors (IGF-1r) on PDAC cellular membranes.34 As suggested by Li et al35 insulin itself could play an active role in increasing cell proliferation and glucose consumption.36 In parallel, it has been proposed that insulin could activate specific IGF-1r signalling pathways to mediate cell proliferation and inhibition of apoptosis on tumoural cell surface.37
Besides all ongoing studies on PDAC, several trials involve small as well as large animals to investigate the pancreatic parenchyma. Among small animal studies, a variety of experiences emerge on islet transplantation,38 pancreatic transplant39 and ischemic preconditions.40,41 Pancreatic diseases and the possible innovative cure and clinical protocols, including hadrontherapy, stand as a top priority for the scientific community.42
Alcohol Consumption
In the past, principally due to difficulties of investigation, PDAC has been inconsistently associated with alcohol consumption. Nowadays, interesting epidemiological studies have shown a positive association between alcohol intake and pancreatic cancer. In 2018, increased risk for PDAC was reported for heavy drinking habit (>60 g/day), with a hazard ratio of 1.77. It has also been shown that this risk is greater in case of a liquor or beer assumption, but interestingly not with wine. In parallel, this risk was not modified by smoking habits.43
The abovementioned risk factors are summarised in the following Table 1.
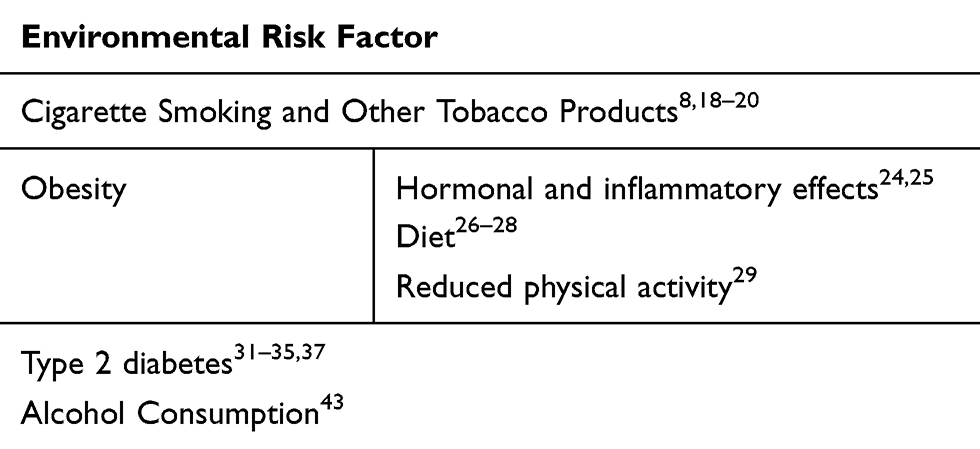 |
Table 1 Risk Factors |
Inherited Pancreatic Cancer Syndromes
Inherited risk factors are responsible for at least 5–10% of pancreatic adenocarcinomas.44 Some genetic variations have been identified as important dominant risk factors, even if not all hereditary pancreatic cancer cases can be tied to a known mutation.45 The most important syndromes increasing risk for pancreatic cancer include the hereditary breast-ovarian cancer syndrome (BRCA1 and BRCA2 genes), familial adenomatous polyposis (APC gene), Lynch syndrome (hereditary non-polyposis colorectal cancer) (MLH1, MSH2, MSH6 and PMS2 genes), and Peutz-Jeghers Syndrome (STK11).
Hereditary Breast-Ovarian Cancer Syndrome (HBOC)
HBOC syndrome is an autosomal dominant disorder associated to an increasing risk for breast cancer (47–55%), ovarian cancer (17–39%) and other cancers, including pancreatic cancer, and is mainly caused by germline mutation in BRCA1 or BRCA2 mutations. Data about prevalence of PDAC among BRCA mutation carriers are heterogeneous, but a study which performed BRCA testing on an unselected collected cohort of 306 patients showed that 4.6% of them had pathogenic BRCA1 or BRCA2 germline variants.46
Familial Adenomatous Polyposis (FAP)
FAP syndrome is an autosomal dominant entity caused by germline mutation of the adenomatous polyposis coli (APC) gene and characterized by the development of numerous adenomatous polyps arising mainly from large intestine epithelium. Patients with FAP have a risk, of almost 100%, of developing colorectal carcinoma by the fourth decade of life,47 and a risk of developing PDAC 4.5 times more than the general population.48
Lynch Syndrome (LS)
LS, also known as hereditary nonpolyposis colorectal cancer (HNPCC) is an autosomal dominant condition caused by germline mutation of genes encoding for mismatch repair (MMR) such as MLH1, MSH2, MSH6 and PMS2.49,50 An alteration of the genes responsible for DNA repair leads to an increase in error rate during replication from 100 to 1000 times, involving areas containing repetitive sequences (microsatellites sequences). These alterations have been associated with the presence of wild-type KRAS and p53 genes, with cumulative risk of PDAC in LS patients around 3.7% (versus 1.5% of general population).51,52
Peutz-Jeghers Syndrome (JPS)
JPS is an autosomal dominant inherited condition deriving from the germline mutation in the STK11 oncosuppressor gene53 and associated with a high risk for developing PDAC. Patients usually exhibit a mucocutaneous hyperpigmentation (oral mucosa, lips and digits) and pathognomonic intestinal hamartomatous polyps. Furthermore, this syndrome puts people at increased risk for developing digestive or genital cancers,54 estimated as high as 93% without specific medical surveillance. PDAC is fully part of this group of neoplasms that can occur in patients with JPS, with a relative risk reported to be as high as 132.55 With such a high relative risk, Peutz-Jeghers syndrome is considered as the hereditary syndrome with the highest risk of developing pancreatic adenocarcinoma.
Genetic Counselling and Risk Assessment
Due to the aggressiveness of this disease, a screening program and genetic counselling is highly recommended for PDAC cases with a suspicion of genetic predisposition (5–10%).56
Histopathology and Molecular Pathways
The World Health Organization (WHO) classification,57 classified pancreatic cancers into different groups based on:
i) the macroscopic features (intraductal, solid, cystic);
ii) type of cell line differentiation (acinar, ductal, endocrine) that is crucial to understand the clinical outcome and biological behaviour of the tumour;
iii) immunophenotypically characteristics, sometimes necessary to define the differentiation line.
By taking into account the most common histotypes, in the following Table 2, we summarise the WHO classification
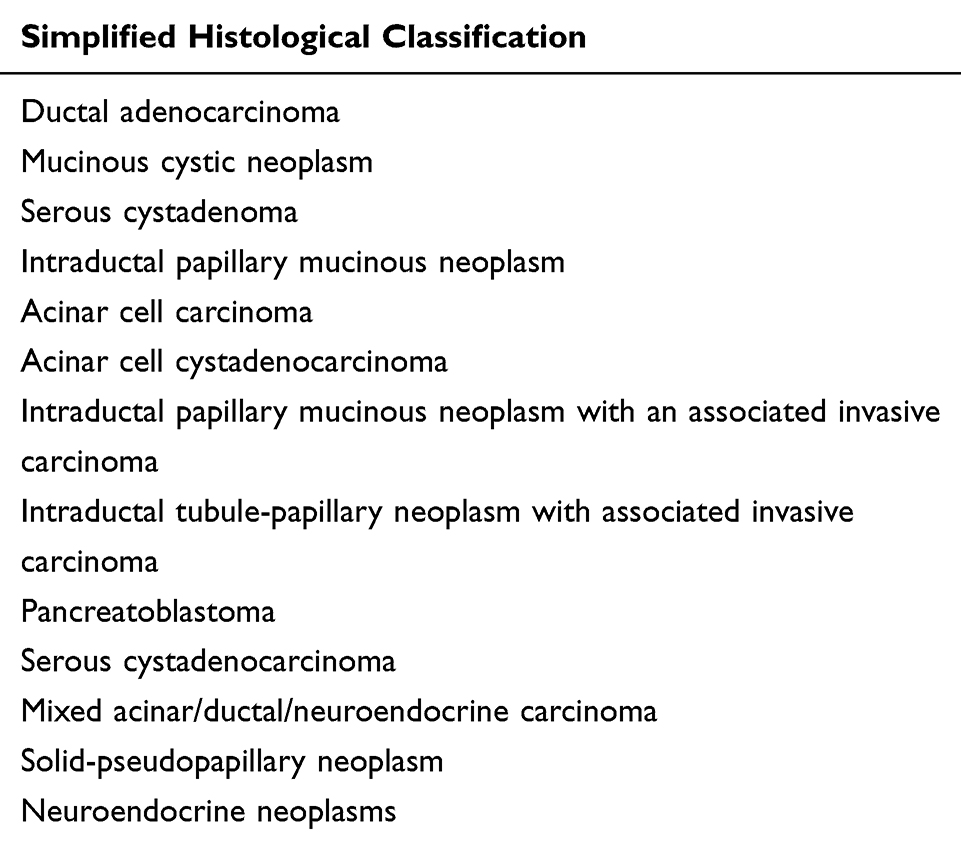 |
Table 2 Common Histotypes of Pancreatic Cancers |
Although in the pancreatic parenchyma, the ductal component is only 20–30%, PDAC is the most frequent pancreatic tumour representing up to 90% of all pancreatic neoplasms.58 Macroscopically, PDCA appears as a solid, hard consistency and shaded margin mass with a colour ranging from yellow to brown encompassing hemorrhagic, necrotic, and/or microcystic areas.59 The anatomical site of presentation (head, body, tail, ampulla, peri-ampullary tissue and inferior third of the common bile duct) influences clinical outcomes. Indeed ampullary carcinomas have generally a better prognosis than those arising in other sites.60 Microscopically, PDAC is characterised by atypical tubular glands with heterogeneous growth patterns including clear-cell or cribriform component that with the tumour grading, which may have an impact on patient survival.61 According to WHO, the histopathological tumour grading for pancreatic cancer considers the architecture (tubular, cribriform and duct-like structures, solid growth), cell shape (cylindrical, cubic, polygonal, pleomorphic, spindle) and amount of mucin (retained, partial loss, complete loss) nuclear polymorphism (slightly, moderately or very polymorphous) and the number of mitoses (1–5/10 HPF, 6–10/10 HPF or >10/10 HPF).57 Immunophenotypically, MUC1, MUC4, MUC5A, CA125, CEA, CA19-9 CK7, CK19, CK18 and sometimes CK20 are usually expressed in PDAC.59
Variants of PDAC
Several variants of PDAC exist distinguished according to the molecular pathogenesis as follow:
- Similar molecular pathogenesis:
- Adenosquamous carcinoma: characterised by a squamous component of up to 30% with a minimum number of glandular ones, an immunohistochemistry positivity for p40 of squamous cells and a worse prognosis than classical PDAC.57
- Anaplastic (undifferentiated) carcinoma: characterised by solid growth, polymorphic cells (including multinuclear giant tumour cells), and positivity for pan-cytokeratin (Pan-CK) and vimentin with an E-cadherin loss. In case of rhabdoid differentiation, it is KRAS wildtype and SMARCB1 mutated.59,62
- Undifferentiated carcinoma with large-duct type carcinoma (resembling non-invasive cystic cancer),63 signet-ring cell carcinoma (expression of nucleus dislocated in the periphery by big cytoplasmatic vacuoles of mucin),64 osteoclastic giant cells (showing histiocytic giant cells positive for CD68) and micropapillary carcinoma (looking like breast carcinoma with an expression of MUC1 in the stroma-facing cell surface and a e-cadherin/galectin-3 cytoplasmatic staining).59
- Different molecular pathogenesis
- Hepatoid adenocarcinoma resembling morphologically and immunocytochemically hepatocellular carcinoma.59
- Colloid carcinoma characterised by tumour cells fitted in great mucin pools and usually associated with high-grade intestinal-type intraductal papillary neoplasm (IPMN).59
- Medullary carcinoma associated with microsatellite instability showed a poor differentiation and an invasive and syncytial pattern of growth.65
Molecular Subtyping of PDAC
Over the years, different molecular classification has been proposed thanks to the introduction of high-throughput techniques and the better knowledge of the pathogenetic role of the PDAC-related genes KRAS, TP53, SMAD4, and CDKN2A.66 Based on the molecular characteristics of PDAC, several authors conducted an interesting molecular classification approach that we summarised below. The molecular classification by Collisson et al67 identified subtypes of PDAC which are different for outcomes and therapeutic response (Table 3): classical, quasi-mesenchymal, and exocrine-like.
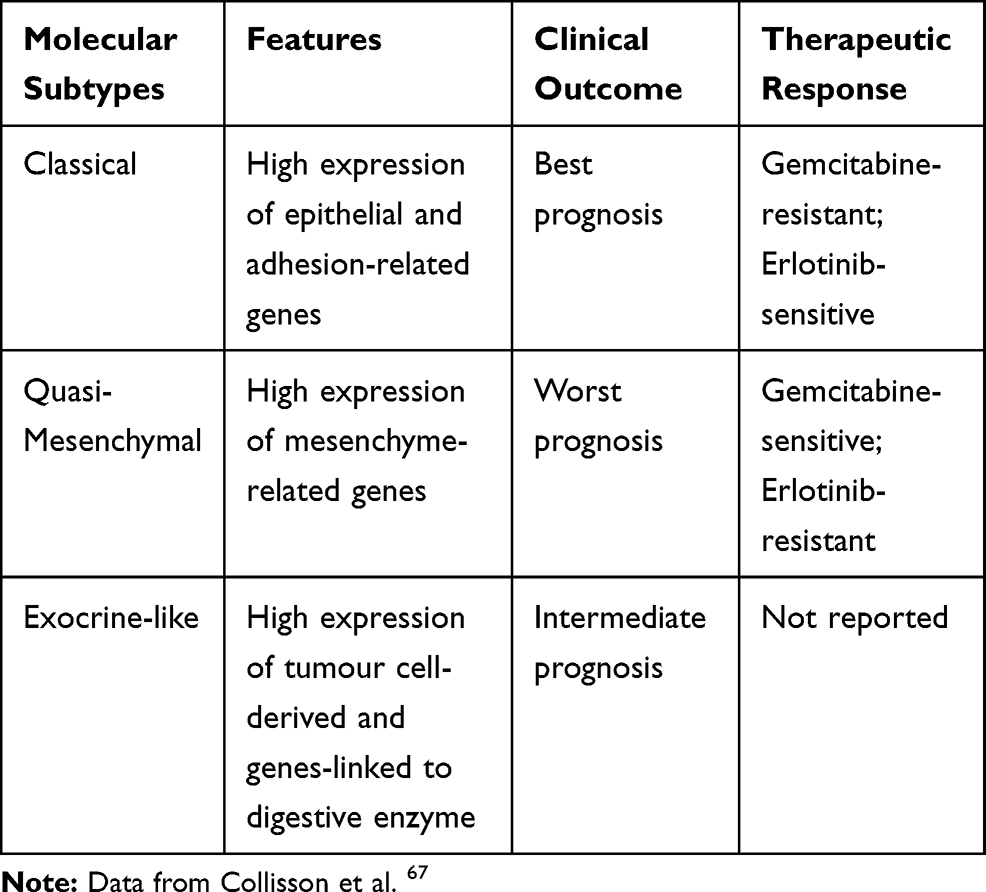 |
Table 3 Summary of Molecular Subtypes |
Another molecular classification is described by Bailey et al68 according to genomic analysis: 1) squamous type, characterised by TP53 and KDM6A mutations, upregulation of the TP63∆N transcriptional network, hypermethylation; 2) pancreatic progenitor type expressing FOXA2/3, PDX1 and MNX1 genes implicated in early pancreatic development 3) immunogenic type displayed upregulation of immune networks; 4) aberrantly differentiated endocrine exocrine (ADEX) type: showed upregulation of genes involved in KRAS activation, exocrine (NR5A2 and RBPJL) and endocrine differentiation (NEUROD1 and NKX2-2). Waddell and colleagues68 proposed a further molecular subtype classification according to genomic stability: 1) termed stable with <50 rearrangements located randomly through the genome; 2) locally rearranged with at least 50 somatic rearrangements clustered on one or few chromosomes; 3) scattered containing 50–200 structural rearrangements spread in genome entirety; 4) unstable with >200 structural rearrangements. Considering the stroma, Moffit et al69 described “normal” and “activate” stroma subtypes with a good and poor prognosis, respectively. In “normal” stroma there is high expression of markers for stellate cells, smooth muscle actin, vimentin, and desmin; in the “activated” stroma there are several genes linked to macrophages (integrin ligand ITGAM and chemokine ligands CCL13- CCL18), associated to tumour progression (released protein SPARC, WNT family members WNT2- WNT5A, gelatinase B MMP9, and stromelysin 3-MMP11) and is also characterised by the presence of fibroblast activation protein FAP. Comparing the gene signatures of the abovementioned classification, it can be observed a non-perfect overlap. As well reported by Primavesi et al66 several working groups used different terminologies as well as different approaches to define biological-similar subtypes and, the definition of consensus genetic models among different classifications is desirable to find new diagnostic and tailored treatment options.
Mutation in Carcinogenesis of Pancreatic Cancer
Early lesion of PDAC can be classified into microscopic (pancreatic intraepithelial neoplasia - PanIN - and atypical flat lesions-AFL) and macroscopic (IPMN-, mucinous cystic neoplasms - MCN - and intraductal tubule-papillary neoplasms - ITPN) precursors whose grading is defined by the importance of cytological atypia. The progression from normal pancreatic tissue to tumour consists of a defined sequence of histopathological and biological events in which the carcinogenesis is the result of a gradual accumulation of multiple and consecutive molecular alterations such as oncogene-activation, inhibition of tumour suppressor genes, gene mutations.59 Since the number of PanIN associated with high-grade dysplasia is increased in patients with a family history of PDAC, these precursor lesions are hypothesized to occur at a very young age.66
K-RAS
The mutation of KRAS (also called K-Ras 2, Ki-Ras, c-K-ras, or c-Ki-ras) is an early event in the carcinogenesis and reported up to 95% of PDAC. KRAS is a small (21KDa) GTP-ase physiologically quiescent and linked to GDP; when GTP replaces GDP, KRAS could active numerous downstream effectors that guide tumour progression: RAF family kinase (RAF1, BRAF, and ARAF) which in turn activate MEK1-MEK2 kinases, that phosphorylate and activate ERK1 and ERK2 kinases. ERKs phosphorylate several proteins of whom ELK1 and c-JUN. KRAS mutation of codon G12, G13, or Q61 makes KRAS constitutively active, typically occurs in early low-grade PanIN-1 lesions, and is associated with minimal cytological architectural atypia.70 Clinical studies show that KRAS mutation is associated with a poor response to Gemcitabine administered as first-line chemotherapy and poorer survival.71 KRAS is mutated in about 95% of advanced PDAC.66
CDKN2A
CDKN2A (p16-INK4a, MTS-1, or CDK4I) is a tumour suppressor that inhibits the progression into the cell cycle by inactivating cyclin D-CDK4 and cyclin D-CDK6 complexes that regulate G1/S phase checkpoint. The inactivation of CDKN2A occurs in 98% of cases of sporadic PDAC and is caused by a different kind of mutation (loss of heterozygosity, homozygous deletion, or promoter silencing). Alteration in CDKN2A is an early event in the pathogenesis and indeed is also described in pre-neoplastic PanIN-2 lesions. Inherited CDKN2A mutations are associated with the familial atypical multiple mole melanoma (FAMMM) syndrome and a greater risk of pancreatic cancer.61,70,72
TP53/SMAD4/BRCA2
TP53 (p53 or antigen NY-CO-13) is a tumour suppressor which modulates the answer to cytotoxic stress by stopping the progression into the cell cycle, arresting growth arrest or inducing apoptosis. TP53 mutation occurs up to 70% of pancreatic cancers and is found in PanIN-3 lesions. Clinical studies observed a worse prognosis in p53 mutated-PDAC73 and a significant improvement of progression-free survival (PFS) in patients with TP53 wild-type.74 SMAD4 (also known as DPC4 or MADH4) is a tumour suppressor protein and a downstream effector of TGF-beta which translocates to the nucleus as heterotrimeric complex promoting the inhibition of growth. Alterations of SMAD4 are reported up to 50% of pancreatic cancer of whom 30% are caused by homozygous deletion. The authors reported a prognostic role of SMAD4 inactivation in terms of overall survival.75 BRCA1 (also known as RNF53) and BRCA2 (also known as FANCD1) are involved in PDAC carcinogenesis. BRCA1 is a tumour suppressor which regulates the answer to DNA damage and the progression in the G2/M cell cycle. BRCA1 mutation occurs in not more than 7% of pancreatic cancer patients, it is associated with familiar cancers and is quite uncommon as sporadic event.76 BRCA2 mutation is found in at most 7–10% of familiar PDAC; few cases of sporadic somatic mutations are reported. BRCA2 is involved in double-strand break repair during the S phase of the cell cycle, centrosome duplication, and cell death.76 Several clinical studies are performed to evaluate the prognostic and predictive role of BRCA2 in PDCA. There are two emblematic cases of an increased sensitivity of BRCA2-mutated-PDCA to DNA-intercalating agents.77,78
Clinical Presentation, Signs and Symptoms
Pancreatic cancer is generally defined as a “silent cancer”; hence, in most cases, symptoms and signs arise when the disease has already progressed to an advanced stage. Here, symptoms are non-specific and vague: patients complain about fatigue, abdominal pain and anorexia, responsible for late diagnosis. However, the clinical presentation is mostly dictated by tumour location within the pancreas and the degree of involvement of surrounding anatomy. In fact, diagnosis is typically made much earlier when the tumour arises in the head of the pancreas with obstruction of the biliary tract rather than in the body or the tail. Sudden and generally painless obstructive jaundice is indeed the most common sign of pancreatic head cancer and pruritus is reported as the most distressing symptom in this subset of patients. Sometimes physical examination can reveal a palpable gallbladder (Courvoisier’s sign) suggesting the presence of mechanical obstruction of the distal common bile duct.79
Instead, the onset of pain may represent a clinical marker of local tumour progression, being it usually related to the extension in the retroperitoneal space with infiltration of the celiac plexus. It is often reported as the first clinical symptom referred from patients with body or tail tumours whose progressive growth has trespassed the pancreatic capsule posteriorly and, with that, spread into the peripancreatic tissues. Direct involvement of major splanchnic vessels is common at this stage. The different setting of clinical presentation between head and body/tail pancreatic tumours account for the different size of the primary lesion that these neoplasms can show at the time of diagnosis; however, tumour size is not the only determinant for resectability and, accordingly, prognosis, as described later. Likewise, body and tail location, cancers of the uncinate process lack early symptoms and bear the worst prognosis because of their close proximity to superior mesenteric vessels which is responsible for a particular dissemination pattern that leads to liver metastasis and lymph node spreading at an earlier course of the disease.80
Pain is located in the mid-epigastric region and radiated to the back. Typically described as relentless, it can be exacerbated in lying position and in some patients by food ingestion too.
Weight loss is also seen in the advanced stage of the disease, and its cause may be found in cancer-related anorexia or pancreatic exocrine insufficiency with malabsorption and steatorrhea. When weight loss occurs in the early stage, it may be related to delayed gastric emptying due to larger size tumours predominantly grown on the duodenal side thus determining endoluminal obstruction. These patients often complain of nausea and recurrent vomit too. Moreover, newly onset of diabetes in euglycemic subjects or worsening of known diabetes can often precede the diagnosis of pancreatic cancer. Ascites and a palpable mass in the epigastrium characterise the most advanced clinical pictures of this tumour.
Depression is reported to be more frequent in patients with pancreatic cancer than in patients with other neoplastic diseases. This might be explained with the typical delay of diagnosis of this disease. High incidence of suicide, almost 11 times higher than the remainder of the population, has been reported in male patients with pancreatic adenocarcinoma.81
Migratory thrombophlebitis (Trousseau sign), venous thrombosis, or marantic endocarditis are also reported in patients with pancreatic cancer sometimes as the first clinical presentation.
Diagnostic Investigation
Tumour’s characterization, staging, and determination of resectability, surgical planning, reassessment after neoadjuvant treatment (NAT) are the drivers of proper management for advanced PDAC.82 Contrast-enhanced Multi-Detector Computed Tomography (MDCT) of chest and abdomen is the main, first-line radiological investigation capable of addressing all the above issues when performed by expert radiologists.82,83 Thin-section (<3 mm) multi-Phase Image acquisition based on specific pancreatic protocols and followed by multiplanar reconstructions is recommended to optimize the assessment of the primary lesion, including its local and distant stage, with an overall diagnostic accuracy around 90%.82–84 Being resectability mostly dictated by vascular involvement, MDCT has the highest specificity, sensitivity, positive and negative predictive values (82–100%, 70–96%, 89%, and 100%, respectively) for determining if and to which extent the circumference of critical peripancreatic vessels is affected.82 Despite the persistent lack of consensus on the definition of “borderline resectable” and, to a lesser degree, of “locally advanced” tumours, MDCT remains the preferred modality to address vascular invasion.83 The high-quality distinction between vascular “abutment” (tumour contact ≤180° of vessel circumference) and “encasement” (contact >180°) can be seen on multiplanar-reformatted images, as well as distortion in the contour or shape of vessels indicating possible infiltration by tumoural tissue.85 Contrast-enhanced endoscopic ultrasound (CEUS) may be as much reliable as MDCT for this aim though limited by local availability, operator’s dependency, and presence of anatomical variants in regional vasculature. Therefore, its use is not routinely recommended.85 Arterial variations, if any, should be comprehensively described in MDCT reports. Contact of atypical hepatic or mesenteric arteries with the primary lesion may dictate variations of the standard surgical technique (ie, to allow safe reconstructions, avoid vascular injury) or even rule out resectability in some cases.85 On the other hand, MDCT may fail in detecting tiny liver or peritoneal metastases.82 Current NCCN guidelines still suggest staging laparoscopy, with or without intraoperative ultrasound, when high suspicion of distant disease is raised clinically.83 Magnetic Resonance (MR) is a valuable alternative to MDCT for staging and resectability assessment, especially in the setting of MR angiography, having comparable sensitivity and specificity rates (89% each).85 However, its use is mostly restricted to patients with known allergy to iodinated contrast or with attenuating tumours in which MDCT has lower accuracy.83 Chest X-Ray or non-contrast CT can be used as surrogate tools for chest staging when iodinated contrast medium is contraindicated. Also, MR may help the characterisation of uncertain pancreatic lesions and synchronous small liver nodules appearing indeterminable at MDCT. Specific MR applications, such as Diffusion-weighted Imaging (DWI) and contrast enhancement with Gadoxetic acid, are extremely efficient for these aims.82,85 When a histological diagnosis is needed, EUS with fine-needle aspiration/biopsy (EUS-FNA/FNB) is preferable to US or CT-guided percutaneous approach due to its higher diagnostic yield (over 90% in patients with negative US/CT-guided FNA) and safety profile as to the risk of peritoneal seeding.83,84 Brushing of the common bile/main pancreatic duct can also be performed during ERCP in patients requiring biliary stenting, although the sensitivity rate does not exceed 40% according to the available data.83,86–88 Positron Emission Tomography combined with CT (PET/CT) for diagnosis and staging of PDAC is poorly helpful and should be used selectively in addition to MDCT and/or MR.82–84 However, it may be of help in the evaluation of response to NAT or in the follow-up of resected patients.82–84 Patients receiving NAT should be reassessed with MDCT and/or MR despite morphological criteria indicating clinical response are still unclear. Tumour size and attenuation lack specificity due to the inflammatory reaction causing oedema, fibrosis, and necrosis; instead, reduction in the extent of tumour-vessel contact seems to be more reliable in predicting resectability if evident on MDCT/MR.85 Recently, the combination of PET with MR (PET/MR) has shown better results compared with PET/CT for staging and post-NAT re-evaluation in advanced PDAC patients.82 Reliable, specific biomarkers are still lacking at present. Carbohydrate Antigen 19–9 (CA19-9) has a limited role for diagnosis due to its high false-positive rate and poor specificity for PDAC. Instead, it is useful during post-surgical follow-up and after NAT as a prognostic tool for, respectively, recurrence or resectability.84 Recently, several serum cytokines, proteins, or cancer cell targets have been proposed, alone or combined, as novel potential biomarkers for PDAC that need to be tested on large scale to prove their sensitivity and specificity are higher as expected.84
Medical and Surgical Treatment
In the panorama of locally advanced pancreatic cancer treatments, there are several therapeutic options in the literature. Such therapies may be divided into two groups, the standard ones and those still being tested and validated. This section aims to analyse a variety of the possible therapeutic proposals for the 30% of patients who have locally advanced stage ductal pancreatic cancer without distant metastases.89,90
We may assume that all patients in this stage of disease today would receive induction chemotherapy.91 In 2016 Suker et al demonstrated that LAPC treated with FOLFIRINOX has a very far superior efficacy compared to the previous therapeutic regimens.92 In the last decades, for the first time, new therapeutic regimens have been able to impact on the survival of pancreatic cancer also in its locally advanced form.93 Systematic chemotherapy stands as the standard for patients with LPAC. The evolution of treatments started with fluorouracil (5-FU), which represented the most commonly used treatment until 1996 when Gemcitabine was approved with a modest improvement in survival rate and disease-related symptoms.94
Surgery still stands as the only one potentially curative therapy for pancreatic cancer. New multi-agent chemotherapy regimens, such as Gemcitabine with nab-paclitaxel and FOLFIRINOX, have the aim to be able to determine a downstaging of the disease, allowing surgery to be performed.83 For this reason, as of today, such treatments are widely used to treat LAPC patients. The duration of these systemic therapies is quite well defined in metastatic disease (disease progression or cumulative limiting toxicity), lasting 4–6 months as an average. For LAPC, there seems to be a lack of knowledge on the ideal duration of the treatment. All the recommendations about the duration of treatment are based on quite old studies on single chemotherapy agents.95 Therefore, there is a call to perform new studies on the topic.
As said, the effect of treating these patients with therapy periods of less than 4 months or greater than 6 months is currently unknown. Tuli et al analysed the impact of duration of combination therapy on survival of patients with LAPC, suggesting that a treatment lasting for at least 6 or more months may increase the survival outcomes.96
While such new systemic therapies look more promising and effective than in the past, surgical resection remains the mainstay of cure also for LAPC. In practice, the goal of systemic treatment is to achieve resectability. The resectability rate of LAPC after combination therapies is around 30%.97,98 It is well known that patients with LAPC who undergo surgical resection after chemotherapy show an improved survival outcome compared with those who are not able to achieve resection.99 The role of vascular resection in the treatment of LAPC is well defined by Oba et al in 2020.100 Pancreatic cancer is a systemic disease from the beginning. The development of a new neoadjuvant treatment that improves the survival rate has to be deeply considered. There are probably primary resectable PDACs with bad biology which do not qualify for surgery and locally advanced cancers, with good biology, that should be treated with aggressive surgical operations. The research community should invest in defining better prognostic, patients-related, and biological criteria to select the best ideal candidate for surgery.
Radiotherapy
Historically, locally advanced pancreatic adenocarcinoma (LPAC) has been treated with conformal fractionated radiotherapy (CRT), and conventional fractionation (1.8–2 Gy/fraction) up to a total dose of 50.4 Gy. Treatment target were large fields, including gross tumour volume and regional nodal areas plus further margins, keeping into account the mobility of abdominal structure.
Unsatisfactory results, with reported locoregional control rates between 50 and 70%, have been described with such low doses, not sufficient for a radioresistant tumour, known as one of the “big killer”.101 The intrinsic radio-resistance of PDAC is related to the high percentage of its hypoxic cells. To overcome pancreatic cancer radio-resistance there are two possible modalities:
1. The association of RT to chemo-therapeutic agents to selectively sensitise the tumour to radiations.
2. The dose escalation to the target by increasing the total dose with conventional fractionation (2 Gy per fraction) or the dose per fraction (hypo-fractionated RT regimens). The advantage of associating radiations to chemotherapy has been demonstrated by the LAP07 randomised trial, which showed significantly improved local control in LAPC patients treated by chemoradiotherapy compared to patients treated by chemotherapy alone.102
Dose escalation in pancreatic cancer is limited by its anatomical location and by the proximity to bowel loops, duodenum, and stomach, all organs in motion-sensitive to radiations, so the prescription dose to target volume is necessarily limited by the dose constraints of the surrounding OARs.
Technological advances in radiation delivery, with more conformal dose distribution and lower side effects, allow to partially overcome these limits.
Intensity-modulated RT (IMRT) allows the RT dose to conform to the shape of the target volume by modulating the radiation beam into smaller volumes. The possibility to concentrate higher radiation doses on the tumour, while lowering the dose to surrounding normal critical structures, opened the way to dose escalation.
Krishnan et al described the clinical benefit of escalating the dose using fractionated IMRT in a population of 200 LAPC patients: who received biological equivalent dose (BED) >70 Gy showed better outcomes in terms of overall survival and locoregional relapse-free survival compared to patients treated with lower BED.103
In a retrospective study on 205 patients treated by IMRT (n=134) and 3D-CRT (n=71) significant lower gastrointestinal toxicity < G2 was found in the IMRT group (16% versus 34%, p< 0.001) compared to the patients treated with conventional 3D conformal technique, while keeping median prescription dose higher in the IMRT group (56 Gy vs 50.4 Gy).104
Further, stereotactic body RT (SBRT), a method of external beam radiotherapy, allows to precisely deliver a high radiation dose to the target, by using a single fraction or few fractions of RT, by prescribing a large dose per fraction, which is known to be biologically more effective on tumour cells response. The potential advantages of hypo-fractionation are based on the assumption that DNA of the healthy tissue easily repairs the damages of the oxidative effects from RT, while tumoural cells, due to their impairment in repairing genomic alterations, cannot.
In contrast to conventional RT, in SBRT dose is delivered only to the primary tumour and involved nodal disease, if, in proximity, no elective regional node regions are irradiated. A further short course of RT by SBRT may bring potential benefits to patients in terms of overall survival by shortening the treatment RT course, reducing the time off of the multi-agents chemotherapy. While the early experience with pancreatic SBRT demonstrated significant gastrointestinal toxicity,105 in more recent studies, SBRT has shown improvement in tolerability by de-escalating dose and decreasing target volumes.
In prospective trials of SBRT delivered alone, median OS range between 5.7 and 19 months, better outcomes are reported when SBRT is delivered after chemotherapy (median OS between 10.3 and 20 months).106,107
Since single fraction, compared to multiple fractions and lower dose/fraction, is related to worst outcomes and side effects, fractionated SBRT is preferred to single-fraction stereotactic RT. Further, fractionated SBRT might be more advantageous because it allows the re-oxygenation of hypoxic tumour cells and redistribution of resistant tumour cells into more radiosensitive cell cycle phases.108 Heavy particles, by protons and carbon ions, are emerging as promising treatment RT modality in radioresistant cancers,109–116 such as pancreatic tumours.117–121
Proton therapy, thanks to its intrinsic physical selectivity, allows delivering dose to the target with no exit dose in the beam path. This translates in a lower dose to surrounding organs and in the possibility of increasing the dose to the target volume with theoretically lower toxicity and better local outcomes.122,123 Clinical data about proton therapy in pancreatic cancer are still scarce.
In locally advanced, unresectable pancreatic cancer, the largest studies with proton radiotherapy are still limited to mono-institutional Japanese experiences. In a retrospective analysis of 42 patients with LAPC treated with proton RT and concurrent chemotherapy, with dose ranging between 50 Gy RBE and 67.5 Gy RBE in 25 fractions, based on the tumour location, after a median follow-up of 14 months (range: 2.4–47.6) no grade 3 or higher late adverse effects were reported. OS at 1 and 2 years was 77.8 and 50.8% with median survival time of 25.6 months, while the LC rate at 1 and 2 years, respectively, of 90.1 and 76.7% with a median time to local recurrence of more than 36 months.124
In opposite the other experiences by proton therapy were less advantageous in terms of gastrointestinal toxicities.
Terashima et al in a Phase I II study treated PDCA with proton radiotherapy with doses up to 70.2 Gy (RBE) in 26 fractions and concomitant Gemcitabine with a relatively high toxicity rate > G3, 8 cases (10%) reported of late gastric ulcer and haemorrhage.125
Such data were supported by Takatori et al in a separate retrospective analysis focalised on gastrointestinal complications of 91 patients treated by proton radiotherapy at Hyogo Medical Center. Acute ulcers were reported in almost 50% of the patients, but late > G3 intestinal side effects in only 3 patients.126
Carbon ions are high linear energy transfer (LET) radiations able to deposit higher energy in the target, compared to conventional photons (low LET radiation), producing significantly more DNA damages to malignant cells, because of their higher relative biological effectiveness (RBE).110,118
Further, the type of damage is different. Whereas photon irradiation leads to indirect DNA damage through the creation of free radicals against the DNA, carbon ion radiotherapy (CIRT) leads to direct DNA damage without an intermediary.
This mechanism leads to the well-known oxygen-dependence of conventional, photon-based radiation, such that hypoxic tumours are radioresistant. Because carbon ions do not require oxygen to damage DNA, CIRT is more effective on hypoxic cells. Thus, because of the superior dose distribution, high RBE, and resistance to hypoxia, CIRT is a promising radiotherapy modality that may improve local control without compromising normal tissues, especially in classically radioresistant tumours.116,118,127
Furthermore, from the physical point of view heavy particles, thanks to their steep dose gradient deposit all the dose in the target volume while keeping a very low dose to the surrounding organs.
Clinical published data about CIRT in pancreatic cancer are limited to Japanese centres.
After a dose-escalation study on 26 patients treated pre-operatively by CIRT (30 to 36.8 Gy[RBE], in 8 fractions, 4 fractions/week),120 Shinoto et al started a dose-escalation trial to treat LAPC patients by CIRT up to 55.2 Gy[RBE], over 3 weeks; concurrent Gemcitabine (1000 mg/m2) was administered. Treatment was generally well tolerated with better local control and overall survival in those patients who received at least 45.6 Gy[RBE], compared to those who received lower doses. In this study, two-year survival at the highest radiation dose levels was 54%, and two-year survival in the cohort of stage III patients treated with at least 45.6 Gy[RBE], was 48%. Median OS was 19.6 months, with 1- and 2-year OS rates in all patients, respectively, of 73% and 35%. Gastrointestinal toxicity greater than G3 (ulcer) was reported in only 1 patient (1%).128
Such promising results in terms of outcomes and toxicities are confirmed by a retrospective multi-institutional study involving Japanese institutions within the study group called “Japan Carbon ion Radiation Oncology Study Group (J-CROS)”. In this study were included 72 LAPC patients treated in three Japanese centres with 52.8 Gy [RBE], or 55.2 Gy [RBE], in 12 fractions. After a median follow-up period of 14.7 months (range, 3.2–37.5), the OS rates were 73% at 1 year, and 46% at 2 years with a median OS of 21.5 months. The three institutions had similar independent results in terms of both LC, OS, and toxicity. Only 1 patient (1%) developed grade 3 duodenal ulcer and no grade 4–5 toxicity was reported.119 Clinical results of particle therapy in LAPC are reported in Table 4.
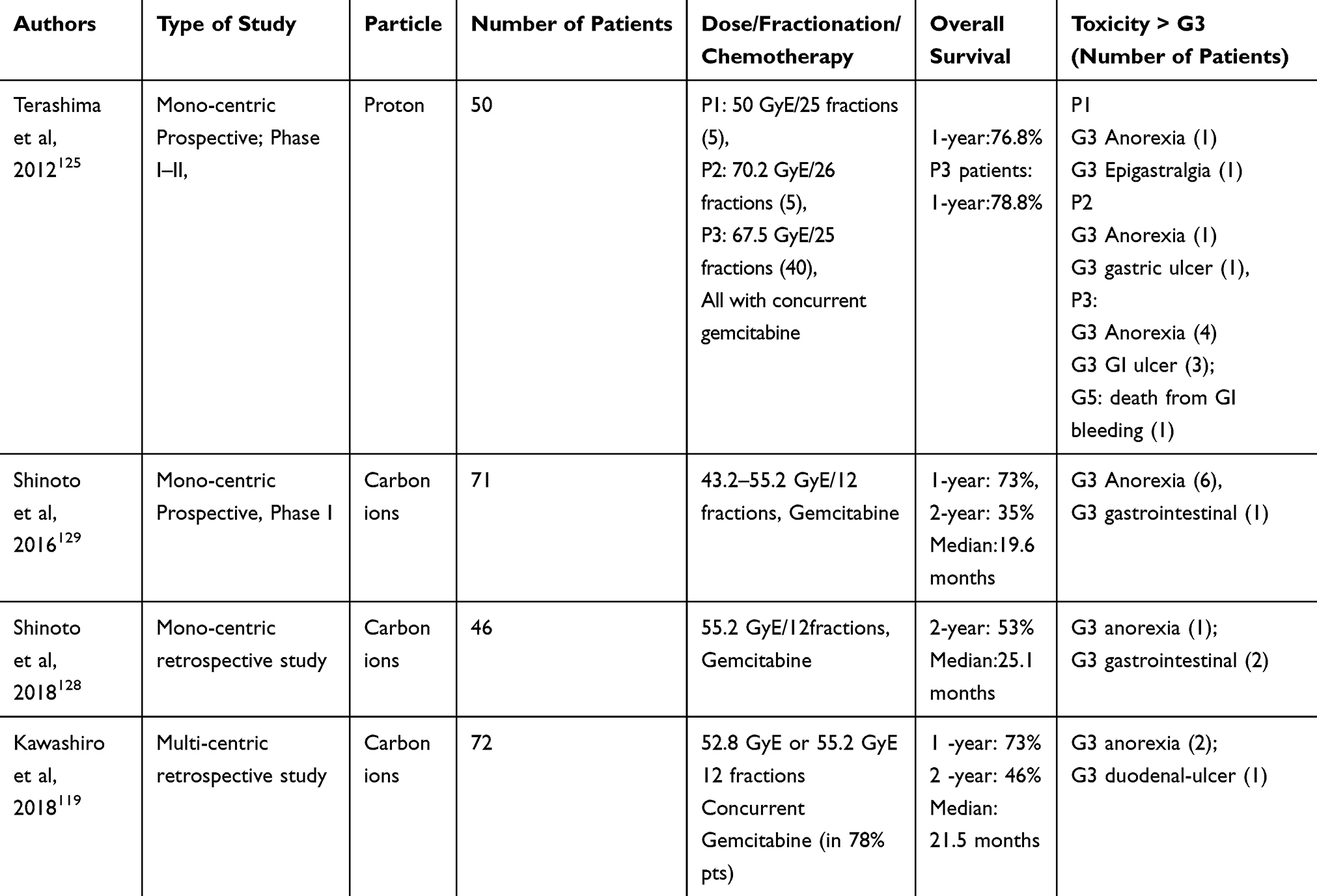 |
Table 4 Particle Therapy Studies |
There are no randomised trials yet showing statistically better outcomes for LAPC of CIRT compared to other conformal RT modalities with photons, but results from Japanese centres are certainly promising and may hopefully represent hope for treatment of disease with such a poor outcome. PIOPPO trial is an ongoing Italian Prospective, Phase II, Multicentre, Single-Arm Study that aims to evaluate the efficacy and the feasibility of 3 cycles of FOLFIRINOX neoadjuvant chemotherapy followed by a short-course CIRT for resectable or borderline resectable pancreatic adenocarcinoma.118
Conclusions
While significant progress in medical knowledge has been made in its management, PDAC is still regarded as one of the deadliest malignancies. This is due to factors such as the lack of early diagnostic markers, delayed detection, diverse genetics and rapid metastasis. In the era of integrated oncological therapies, especially pancreatic cancer, is living and will have a new therapeutic era.42,117 Hadrontherapy appears promising in terms of outcomes and toxicities,122 also in the neoadjuvant schedule.118,120 There is a call for new biological therapies, target treatment as soon as new biotechnological tools for biomarkers that may, in the future, influence the survival of this neoplasm.
Acknowledgments
We would like to thank Francesca Dal Mas, MSc, JD, PhD (University of Lincoln) for her valuable contribution in the review and editing of the draft.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. doi:10.1158/0008-5472.CAN-14-0155
3. Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318–348. doi:10.3322/caac.21190
4. Cobianchi L, Dal Mas F, Piccolo D, et al. Digital transformation in healthcare. The challenges of translating knowledge in a primary research, educational and clinical centre. In: Soliman KS (Ed.),
5. Dal Mas F, Biancuzzi H, Massaro M, Barcellini A, Cobianchi L, Miceli L. Knowledge translation in oncology. A case study. Electron J Knowl Manag. 2020;18(3):212–223, doi:10.34190/EJKM.18.03.002
6. Bednarova R, Biancuzzi H, Rizzardo A, et al. Cancer rehabilitation and physical activity: the ‘Oncology in Motion’ project. J Cancer Educ. 2020. doi:10.1007/s13187-020-01920-0
7. Howlader N, Noone AM, Krapcho M, et al. Bethesda, MD: 2016. Apr, SEER Cancer Statistics Review, 1975–2013. National Cancer Institute. Based on November 2015 SEER data submission, posted to the SEER web site. Available from: http://seer.cancer.gov/csr/1975_2013/.
8. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–620. doi:10.1016/S0140-6736(10)62307-0
9. Varadhachary GR, Tamm EP, Abbruzzese JL, et al. Borderline resectable pancreatic cancer: definitions, management, and role of preoperative therapy. Ann Surg Oncol. 2006;13(8):1035–1046. doi:10.1245/ASO.2006.08.011
10. Loehrer AP, Ferrone CR. Treatment of locally advanced pancreatic ductal adenocarcinoma. Dig Surg. 2016;33(4):343–350. doi:10.1159/000445020
11. Soweid AM. The borderline resectable and locally advanced pancreatic ductal adenocarcinoma: definition. Endosc Ultrasound. 2017;6(Suppl 3):S76–S78. doi:10.4103/eus.eus_66_17
12. Saad AM, Turk T, Al-Husseini MJ, Abdel-Rahman O. Trends in pancreatic adenocarcinoma incidence and mortality in the United States in the last four decades; a SEER-based study. BMC Cancer. 2018;18(1):688. doi:10.1186/s12885-018-4610-4
13. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi:10.3322/caac.21387
14. Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22(44):9694–9705. doi:10.3748/wjg.v22.i44.9694
15. Wong MCS, Jiang JY, Liang M, Fang Y, Yeung MS, Sung JJY. Global temporal patterns of pancreatic cancer and association with socioeconomic development. Sci Rep. 2017;7(1):3165. doi:10.1038/s41598-017-02997-2
16. McWilliams RR, Maisonneuve P, Bamlet WR, et al. Risk factors for early-onset and very-early-onset pancreatic adenocarcinoma. Pancreas. 2016;45(2):311–316. doi:10.1097/MPA.0000000000000392
17. Becker AE. Pancreatic ductal adenocarcinoma: risk factors, screening, and early detection. World J Gastroenterol. 2014;20(32):11182. doi:10.3748/wjg.v20.i32.11182
18. Bosetti C, Lucenteforte E, Silverman DT, et al. Cigarette smoking and pancreatic cancer: an analysis from the International Pancreatic Cancer Case-Control Consortium (Panc4). Ann Oncol. 2012;23(7):1880–1888. doi:10.1093/annonc/mdr541
19. Lynch SM, Vrieling A, Lubin JH, et al. Cigarette smoking and pancreatic cancer: a pooled analysis from the Pancreatic Cancer Cohort Consortium. Am J Epidemiol. 2009;170(4):403–413. doi:10.1093/aje/kwp134
20. Tang M, Wu X-R, Lee H-W, et al. Electronic-cigarette smoke induces lung adenocarcinoma and bladder urothelial hyperplasia in mice. Proc Natl Acad Sci. 2019;116(43):21727–21731. doi:10.1073/pnas.1911321116
21. Bracci PM. Obesity and pancreatic cancer: overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog. 2012;51(1):53–63. doi:10.1002/mc.20778
22. Jiao L, Berrington de Gonzalez A, Hartge P, et al. Body mass index, effect modifiers, and risk of pancreatic cancer: a pooled study of seven prospective cohorts. Cancer Causes Control. 2010;21(8):1305–1314. doi:10.1007/s10552-010-9558-x
23. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet. 2008;371(9612):569–578. doi:10.1016/S0140-6736(08)60269-X
24. Douglas JB, Silverman DT, Pollak MN, Tao Y, Soliman AS, Stolzenberg-Solomon RZ. Serum IGF-I, IGF-II, IGFBP-3, and IGF-I/IGFBP-3 molar ratio and risk of pancreatic cancer in the prostate, lung, colorectal, and ovarian cancer screening trial. Cancer Epidemiol Biomarkers Prev. 2010;19(9):2298–2306. doi:10.1158/1055-9965.EPI-10-0400
25. Ramos-Nino ME. The role of chronic inflammation in obesity-associated cancers. ISRN Oncol. 2013;2013:1–25. doi:10.1155/2013/697521
26. Sousa CM, Kimmelman AC. The complex landscape of pancreatic cancer metabolism. Carcinogenesis. 2014;35(7):1441–1450. doi:10.1093/carcin/bgu097
27. Chan JM, Gong Z, Holly EA, Bracci PM. Dietary patterns and risk of pancreatic cancer in a large population-based case-control study in the San Francisco Bay Area. Nutr Cancer. 2013;65(1):157–164. doi:10.1080/01635581.2012.725502
28. Bosetti C, Bravi F, Turati F, et al. Nutrient-based dietary patterns and pancreatic cancer risk. Ann Epidemiol. 2013;23(3):124–128. doi:10.1016/j.annepidem.2012.12.005
29. Behrens G, Jochem C, Schmid D, Keimling M, Ricci C, Leitzmann MF. Physical activity and risk of pancreatic cancer: a systematic review and meta-analysis. Eur J Epidemiol. 2015;30(4):279–298. doi:10.1007/s10654-015-0014-9
30. Arslan AA. Anthropometric measures, body mass index, and pancreatic cancer. Arch Intern Med. 2010;170(9):791. doi:10.1001/archinternmed.2010.63
31. Batabyal P, Vander Hoorn S, Christophi C, Nikfarjam M. Association of diabetes mellitus and pancreatic adenocarcinoma: a meta-analysis of 88 studies. Ann Surg Oncol. 2014;21(7):2453–2462. doi:10.1245/s10434-014-3625-6
32. Ireland L, Santos A, Ahmed MS, et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016;76(23):6851–6863. doi:10.1158/0008-5472.CAN-16-1201
33. Il Jang W, Kim M-S, Kang SH, et al. Association between metformin use and mortality in patients with type 2 diabetes mellitus and localized resectable pancreatic cancer: a nationwide population-based study in korea. Oncotarget. 2017;8(6):9587–9596. doi:10.18632/oncotarget.14525
34. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30(6):586–623. doi:10.1210/er.2008-0047
35. Li Y, Bian X, Wei S, He M, Yang Y. The relationship between pancreatic cancer and type 2 diabetes: cause and consequence. Cancer Manag Res. 2019;11:8257–8268. doi:10.2147/CMAR.S211972
36. Goodwin PJ. Insulin in the adjuvant breast cancer setting: a novel therapeutic target for lifestyle and pharmacologic interventions? J Clin Oncol. 2008;26(6):833–834. doi:10.1200/JCO.2007.14.7132
37. Cui Y, Andersen DK. Diabetes and pancreatic cancer. Endocr Relat Cancer. 2012;19(5):F9–F26. doi:10.1530/ERC-12-0105
38. Pileggi A, Cobianchi L, Inverardi L, Ricordi C. Overcoming the challenges now limiting islet transplantation: a sequential, integrated approach. Ann N Y Acad Sci. 2006;1079:383–398. doi:10.1196/annals.1375.059
39. Merani S, Toso C, Emamaullee J, Shapiro AMJ. Optimal implantation site for pancreatic islet transplantation. Br J Surg. 2008;95(12):1449–1461. doi:10.1002/bjs.6391
40. Hogan AR, Doni M, Molano RD, et al. Beneficial effects of ischemic preconditioning on pancreas cold preservation. Cell Transplant. 2012;21(7):1349–1360. doi:10.3727/096368911X623853
41. Hogan AR, Doni M, Ribeiro MM, et al. Ischemic preconditioning improves islet recovery after pancreas cold preservation. Transplant Proc. 2009;41(1):354–355. doi:10.1016/j.transproceed.2008.11.003
42. Cobianchi L, Dal Mas F, Barcellini A, et al. Knowledge translation in challenging healthcare environments: The PIOPPO experience at the National Centre of Oncological Hadrontherapy (CNAO Foundation), In: Garcia-Perez A, Simkin L, Editors. Proceedings of the 21st European Conference on Knowledge Management - ECKM2020, 3-4 December Coventry, Academic Conferences and Publishing International Limited, Reading; 124–132 doi:10.34190/EKM.20.039
43. Naudin S, Li K, Jaouen T, et al. Lifetime and baseline alcohol intakes and risk of pancreatic cancer in the European prospective investigation into cancer and nutrition study. Int J Cancer. 2018;143(4):801–812. doi:10.1002/ijc.31367
44. Brand RE, Lerch MM, Rubinstein WS, et al. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56(10):1460–1469. doi:10.1136/gut.2006.108456
45. Klein AP, Beaty TH, Bailey-Wilson JE, Brune KA, Hruban RH, Petersen GM. Evidence for a major gene influencing risk of pancreatic cancer. Genet Epidemiol. 2002;23(2):133–149. doi:10.1002/gepi.1102
46. Holter S, Borgida A, Dodd A, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol. 2015;33(28):3124–3129. doi:10.1200/JCO.2014.59.7401
47. Waller A, Findeis S, Lee MJ. Familial adenomatous polyposis. J Pediatr Genet. 2016;5(2):78–83. doi:10.1055/s-0036-1579760
48. Moussata D, Senouci L, Berger F, et al. Familial adenomatous polyposis and pancreatic cancer. Pancreas. 2015;44(3):512–513. doi:10.1097/MPA.0000000000000295
49. Ligtenberg MJL, Kuiper RP, Chan TL, et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3ʹ exons of TACSTD1. Nat Genet. 2009;41(1):112–117. doi:10.1038/ng.283
50. Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66(15):7810–7817. doi:10.1158/0008-5472.CAN-06-1114
51. Leoz ML, Sánchez A, Carballal S, et al. [Hereditary gastric and pancreatic cancer predisposition syndromes]. Gastroenterol Hepatol. 2016;39(7):481–493. Spanish. doi:10.1016/j.gastrohep.2015.11.009
52. Bujanda L, Herreros-Villanueva M. Pancreatic cancer in lynch syndrome patients. J Cancer. 2017;8(18):3667–3674. doi:10.7150/jca.20750
53. Beggs AD, Latchford AR, Vasen HFA, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986. doi:10.1136/gut.2009.198499
54. Latchford A, Greenhalf W, Vitone LJ, Neoptolemos JP, Lancaster GA, Phillips RKS. Peutz-Jeghers syndrome and screening for pancreatic cancer. Br J Surg. 2006;93(12):1446–1455. doi:10.1002/bjs.5609
55. Hearle NCM, Rudd MF, Lim W, et al. Exonic STK11 deletions are not a rare cause of Peutz-Jeghers syndrome. J Med Genet. 2006;43(4):e15. doi:10.1136/jmg.2005.036830
56. Amundadottir LT. Pancreatic Cancer Genetics. Int J Biol Sci. 2016;12(3):314–325. doi:10.7150/ijbs.15001
57. Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumours of the Digestive System. World Health Organization; 2010.
58. Cascinu S, Falconi M, Valentini V, Jelic S. Pancreatic cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v55–58. doi:10.1093/annonc/mdq165
59. Haeberle L, Esposito I. Pathology of pancreatic cancer. Transl Gastroenterol Hepatol. 2019;4:50. doi:10.21037/tgh.2019.06.02
60. Carter JT, Grenert JP, Rubenstein L, Stewart L, Way LW. Tumors of the ampulla of vater: histopathologic classification and predictors of survival. J Am Coll Surg. 2008;207(2):210–218. doi:10.1016/j.jamcollsurg.2008.01.028
61. Schlitter AM, Segler A, Steiger K, et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): identification of prognostic subtypes. Sci Rep. 2017;7:41064. doi:10.1038/srep41064
62. Komatsu H, Egawa S, Motoi F, et al. Clinicopathological features and surgical outcomes of adenosquamous carcinoma of the pancreas: a retrospective analysis of patients with resectable stage tumors. Surg Today. 2015;45(3):297–304. doi:10.1007/s00595-014-0934-0
63. Bagci P, Andea AA, Basturk O, Jang K-T, Erbarut I, Adsay V. Large duct type invasive adenocarcinoma of the pancreas with microcystic and papillary patterns: a potential microscopic mimic of non-invasive ductal neoplasia. Mod Pathol. 2012;25(3):439–448. doi:10.1038/modpathol.2011.181
64. Yepuri N, Naous R, Richards C, Dhir M, Jain A. Poorly differentiated signet ring cell carcinoma of pancreas masquerading as chronic pancreatitis. J Surg Case Rep. 2018;2018(8):rjy218. doi:10.1093/jscr/rjy218
65. Wilentz RE, Goggins M, Redston M, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000;156(5):1641–1651. doi:10.1016/S0002-9440(10)65035-3
66. Primavesi F, Stättner S, Schlick K, et al. Pancreatic cancer in young adults: changes, challenges, and solutions. Onco Targets Ther. 2019;12:3387–3400. doi:10.2147/OTT.S176700
67. Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17(4):500–503. doi:10.1038/nm.2344
68. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52. doi:10.1038/nature16965
69. Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47(10):1168–1178. doi:10.1038/ng.3398
70. Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte-Kausiliene E, Skeberdyte A, Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers (Basel). 2017;9(12):42. doi:10.3390/cancers9050042
71. Boeck S, Jung A, Laubender RP, et al. KRAS mutation status is not predictive for objective response to anti-EGFR treatment with erlotinib in patients with advanced pancreatic cancer. J Gastroenterol. 2013;48(4):544–548. doi:10.1007/s00535-013-0767-4
72. Hayashi H, Kohno T, Ueno H, et al. Utility of assessing the number of mutated KRAS, CDKN2A, TP53, and SMAD4 genes using a targeted deep sequencing assay as a prognostic biomarker for pancreatic cancer. Pancreas. 2017;46(3):335–340. doi:10.1097/MPA.0000000000000760
73. Grochola LF, Taubert H, Greither T, Bhanot U, Udelnow A, Würl P. Elevated transcript levels from the MDM2 P1 promoter and low p53 transcript levels are associated with poor prognosis in human pancreatic ductal adenocarcinoma. Pancreas. 2011;40(2):265–270. doi:10.1097/mpa.0b013e3181f95104
74. Ormanns S, Siveke JT, Heinemann V, et al. pERK, pAKT and p53 as tissue biomarkers in erlotinib-treated patients with advanced pancreatic cancer: a translational subgroup analysis from AIO-PK0104. BMC Cancer. 2014;14:624. doi:10.1186/1471-2407-14-624
75. Blackford A, Serrano OK, Wolfgang CL, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15(14):4674–4679. doi:10.1158/1078-0432.CCR-09-0227
76. Stadler ZK, Salo-Mullen E, Patil SM, et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer. 2012;118(2):493–499. doi:10.1002/cncr.26191
77. Sonnenblick A, Kadouri L, Appelbaum L, et al. Complete remission, in BRCA2 mutation carrier with metastatic pancreatic adenocarcinoma, treated with cisplatin based therapy. Cancer Biol Ther. 2011;12(3):165–168. doi:10.4161/cbt.12.3.16292
78. James E, Waldron-Lynch MG, Saif MW. Prolonged survival in a patient with BRCA2 associated metastatic pancreatic cancer after exposure to camptothecin: a case report and review of literature. Anticancer Drugs. 2009;20(7):634–638. doi:10.1097/CAD.0b013e32832b511e
79. Murr MM, Sarr MG, Oishi AJ, van Heerden JA. Pancreatic cancer. CA Cancer J Clin. 1994;44(5):304–318. doi:10.3322/canjclin.44.5.304
80. Beger HG, Rau B, Gansauge F, Poch B, Link K-H. Treatment of pancreatic cancer: challenge of the facts. World J Surg. 2003;27(10):1075–1084. doi:10.1007/s00268-003-7165-7
81. Turaga KK, Malafa MP, Jacobsen PB, Schell MJ, Sarr MG. Suicide in patients with pancreatic cancer. Cancer. 2011;117:642–647. doi:10.1002/cncr.25428
82. Lee ES, Lee JM. Imaging diagnosis of pancreatic cancer: a state-of-the-art review. World J Gastroenterol. 2014;20(24):7864–7877. doi:10.3748/wjg.v20.i24.7864
83. Tempero MA, Malafa MP, Chiorean EG, et al. Pancreatic adenocarcinoma, version 1.2019. J Natl Compr Canc Netw. 2019;17(3):202–210. doi:10.6004/jnccn.2019.0014
84. Zhang L, Sanagapalli S, Stoita A. Challenges in diagnosis of pancreatic cancer. World J Gastroenterol. 2018;24(19):2047–2060. doi:10.3748/wjg.v24.i19.2047
85. Elbanna KY, Jang H-J, Kim TK. Imaging diagnosis and staging of pancreatic ductal adenocarcinoma: a comprehensive review. Insights Imaging. 2020;11(1):58. doi:10.1186/s13244-020-00861-y
86. Burnett AS, Calvert TJ, Chokshi RJ. Sensitivity of endoscopic retrograde cholangiopancreatography standard cytology: 10-y review of the literature. J Surg Res. 2013;184(1):304–311. doi:10.1016/j.jss.2013.06.028
87. Pereira P, Morais R, Vilas-Boas F, et al. Brush cytology performance for the assessment of biliopancreatic strictures. Acta Cytol. 2020;64(4):344–351. doi:10.1159/000502791
88. Aly FZ, Mostofizadeh S, Jawaid S, Knapik J, Mukhtar F, Klein R. Effect of single operator cholangioscopy on accuracy of bile duct cytology. Diagn Cytopathol. 2020;48:1230–1236. doi:10.1002/dc.24553
89. Weniger M, Moir J, Damm M, et al. Respect - A multicenter retrospective study on preoperative chemotherapy in locally advanced and borderline resectable pancreatic cancer. Pancreatology. 2020;20(6):1131–1138. doi:10.1016/j.pan.2020.06.012
90. Chauffert B, Mornex F, Bonnetain F, et al. Phase III trial comparing intensive induction chemoradiotherapy (60 Gy, infusional 5-FU and intermittent cisplatin) followed by maintenance gemcitabine with gemcitabine alone for locally advanced unresectable pancreatic cancer. Ann Oncol. 2008;19(9):1592–1599. doi:10.1093/annonc/mdn281
91. Hackert T, Sachsenmaier M, Hinz U, et al. Locally advanced pancreatic cancer: neoadjuvant therapy with folfirinox results in resectability in 60% of the patients. Ann Surg. 2016;264(3):457–463. doi:10.1097/SLA.0000000000001850
92. Suker M, Beumer BR, Sadot E, et al. FOLFIRINOX for locally advanced pancreatic cancer: a systematic review and patient-level meta-analysis. Lancet Oncol. 2016;17(6):801–810. doi:10.1016/S1470-2045(16)00172-8
93. Kieler M, Unseld M, Bianconi D, et al. Impact of new chemotherapy regimens on the treatment landscape and survival of locally advanced and metastatic pancreatic cancer patients. J Clin Med. 2020;9(3):648. doi:10.3390/jcm9030648
94. Burris HA, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–2413. doi:10.1200/JCO.1997.15.6.2403
95. Hammel P, Huguet F, van Laethem J-L, et al. Effect of chemoradiotherapy vs chemotherapy on survival in patients with locally advanced pancreatic cancer controlled after 4 months of gemcitabine with or without erlotinib: the LAP07 randomized clinical trial. JAMA. 2016;315(17):1844–1853. doi:10.1001/jama.2016.4324
96. Tuli R, David J, Lobaugh S, Zhang Z, O’Reilly EM. Duration of therapy for locally advanced pancreatic cancer: does it matter? Cancer Med. 2020;9(13):4572–4580. doi:10.1002/cam4.3081
97. Gillen S, Schuster T, Meyer Zum Büschenfelde C, Friess H, Kleeff J, Seiler C. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7(4):e1000267. doi:10.1371/journal.pmed.1000267
98. Assifi MM, Lu X, Eibl G, Reber HA, Li G, Hines OJ. Neoadjuvant therapy in pancreatic adenocarcinoma: a meta-analysis of Phase II trials. Surgery. 2011;150(3):466–473. doi:10.1016/j.surg.2011.07.006
99. Wo JY, Niemierko A, Ryan DP, et al. Tolerability and long-term outcomes of dose-painted neoadjuvant chemoradiation to regions of vessel involvement in borderline or locally advanced pancreatic cancer. Am J Clin Oncol. 2018;41(7):656–661. doi:10.1097/COC.0000000000000349
100. Oba A, Bao QR, Barnett CC, et al. Vascular resections for pancreatic ductal adenocarcinoma: vascular resections for PDAC. Scand J Surg. 2020;109(1):18–28. doi:10.1177/1457496919900413
101. Bruynzeel AME, Lagerwaard FJ. The role of biological dose-escalation for pancreatic cancer. Clin Transl Radiat Oncol. 2019;18:128–130. doi:10.1016/j.ctro.2019.04.020
102. Hammel P, Huguet F, Van Laethem J-L, et al. Comparison of chemoradiotherapy (CRT) and chemotherapy (CT) in patients with a locally advanced pancreatic cancer (LAPC) controlled after 4 months of gemcitabine with or without erlotinib: final results of the international phase III LAP 07 study. J Clin Oncol. 2013;31:LBA4003–LBA4003. doi:10.1200/jco.2013.31.18_suppl.lba4003
103. Krishnan S, Chadha AS, Suh Y, et al. Focal radiation therapy dose escalation improves overall survival in locally advanced pancreatic cancer patients receiving induction chemotherapy and consolidative chemoradiation. Int J Radiat Oncol Biol Phys. 2016;94(4):755–765. doi:10.1016/j.ijrobp.2015.12.003
104. Prasad S, Cambridge L, Huguet F, et al. Intensity modulated radiation therapy reduces gastrointestinal toxicity in locally advanced pancreas cancer. Pract Radiat Oncol. 2016;6(2):78–85. doi:10.1016/j.prro.2015.09.006
105. Schellenberg D, Goodman KA, Lee F, et al. Gemcitabine chemotherapy and single-fraction stereotactic body radiotherapy for locally advanced pancreatic cancer. Int J Radiat Oncol Biol Phys. 2008;72(3):678–686. doi:10.1016/j.ijrobp.2008.01.051
106. Koong AC, Christofferson E, Le QT, et al. Phase II study to assess the efficacy of conventionally fractionated radiotherapy followed by a stereotactic radiosurgery boost in patients with locally advanced pancreatic cancer. Int J Radiat Oncol Biol Phys. 2005;63:320–323. doi:10.1016/j.ijrobp.2005.07.002
107. Goyal K, Einstein D, Ibarra RA, et al. Stereotactic body radiation therapy for nonresectable tumors of the pancreas. J Surg Res. 2012;174:319–325. doi:10.1016/j.jss.2011.07.044
108. Pollom EL, Alagappan M, Von Eyben R, et al. Single- versus multifraction stereotactic body radiation therapy for pancreatic adenocarcinoma: outcomes and toxicity. Int J Radiat Oncol Biol Phys. 2014;90:918–925. doi:10.1016/j.ijrobp.2014.06.066
109. Vitolo V, Fiore MR, Barcellini A, et al. Carbon ion radiotherapy in the management of the tumors of the peripheral nervous system. Anticancer Res. 2019;39(2):909–913. doi:10.21873/anticanres.13193
110. Barcellini A, Vitolo V, Mastella E, Mirandola A, Valvo F. Letter to the Editor concerning “Re-irradiation in gynaecological cancers, present experiences and future hopes”. J Radiat Oncol. 2019;8(3):355–356. doi:10.1007/s13566-019-00396-w
111. Barcellini A, Roccio M, Laliscia C, et al. Endometrial cancer: when upfront surgery is not an option. Oncol. 2020;1–7. doi:10.1159/000510690.
112. Barcellini A, Gadducci A, Laliscia C, et al. Adenoid cystic carcinoma of Bartholin’s gland: what is the best approach? Oncology. 2020;1–7. doi:10.1159/000506485.
113. Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol. 2010;7(1):37–43. doi:10.1038/nrclinonc.2009.183
114. Barcellini A, Vitolo V, Cobianchi L, et al. Re-irradiation with carbon ion radiotherapy for pelvic rectal cancer recurrences in patients previously irradiated to the pelvis. In Vivo (Brooklyn). 2020;34(3):1547–1553. doi:10.21873/invivo.11944
115. Wang L, Wang X, Zhang Q, et al. Is there a role for carbon therapy in the treatment of gynecological carcinomas? A systematic review. Future Oncol. 2019;15(26):3081–3095. doi:10.2217/fon-2019-0187
116. Vitolo V, Barcellini A, Fossati P, et al. Carbon ion radiotherapy in the management of unusual liposarcomas: a case report. In Vivo (Brooklyn). 2019;33(2). doi:10.21873/invivo.11506
117. Barcellini A, Vitolo V, Cobianchi L, et al. Pancreatic cancer: does a short course of carbon ion radiotherapy worth during COVID-19 outbreak? Pancreatology. 2020;20:1004–1005. doi:10.1016/j.pan.2020.05.007
118. Vitolo V, Cobianchi L, Brugnatelli S, et al. Preoperative chemotherapy and carbon ions therapy for treatment of resectable and borderline resectable pancreatic adenocarcinoma: a prospective, phase II, multicentre, single-arm study. BMC Cancer. 2019;19(1). doi:10.1186/s12885-019-6108-0
119. Kawashiro S, Yamada S, Okamoto M, et al. Multi-institutional study of carbon-ion radiotherapy for locally advanced pancreatic cancer: Japan Carbon-ion Radiation Oncology Study Group (J-CROS) study 1403 pancreas. Int J Radiat Oncol Biol Phys. 2018;101(5):1212–1221. doi:10.1016/j.ijrobp.2018.04.057
120. Shinoto M, Yamada S, Yasuda S, et al. Phase 1 trial of preoperative, short-course carbon-ion radiotherapy for patients with resectable pancreatic cancer. Cancer. 2013;119(1):45–51. doi:10.1002/cncr.27723
121. Kawashiro S, Mori S, Yamada S, et al. Dose escalation study with respiratory-gated carbon-ion scanning radiotherapy using a simultaneous integrated boost for pancreatic cancer: simulation with four-dimensional computed tomography. Br J Radiol. 2017;90(1072):20160790. doi:10.1259/bjr.20160790
122. Facoetti A, Barcellini A, Valvo F, Pullia M. The role of particle therapy in the risk of radio-induced second tumors: a review of the literature. Anticancer Res. 2019;39(9):4613–4617. doi:10.21873/anticanres.13641
123. Vitolo V, Barcellini A, Mirandola A, et al. Is proton beam radiotherapy worthwhile in the management of angiosarcoma of the scalp? Anticancer Res. 2020;40(3):1645–1649. doi:10.21873/anticanres.14114
124. Hiroshima Y, Fukumitsu N, Saito T, et al. Concurrent chemoradiotherapy using proton beams for unresectable locally advanced pancreatic cancer. Radiother Oncol. 2019;136:37–43. doi:10.1016/j.radonc.2019.03.012
125. Terashima K, Demizu Y, Hashimoto N, et al. A phase I/II study of gemcitabine-concurrent proton radiotherapy for locally advanced pancreatic cancer without distant metastasis. Radiother Oncol. 2012;103(1):25–31. doi:10.1016/j.radonc.2011.12.029
126. Takatori K, Terashima K, Yoshida R, et al. Upper gastrointestinal complications associated with gemcitabine-concurrent proton radiotherapy for inoperable pancreatic cancer. J Gastroenterol. 2014;49(6):1074–1080. doi:10.1007/s00535-013-0857-3
127. Cuccia F, Fiore MR, Barcellini A, et al. Outcome and toxicity of carbon ion radiotherapy for axial bone and soft tissue sarcomas. Anticancer Res. 2020;40(5):2853–2859. doi:10.21873/anticanres.14260
128. Shinoto M, Terashima K, Suefuji H, et al. A single institutional experience of combined carbon-ion radiotherapy and chemotherapy for unresectable locally advanced pancreatic cancer. Radiother Oncol. 2018;129(2):333–339. doi:10.1016/j.radonc.2018.08.026
129. Shinoto M, Yamada S, Terashima K, et al. Carbon ion radiation therapy with concurrent gemcitabine for patients with locally advanced pancreatic cancer. Int J Radiat Oncol Biol Phys. 2016;95(1):498–504. doi:10.1016/j.ijrobp.2015.12.362
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.