")
Back to Journals » International Journal of Nanomedicine » Volume 19
Localized Administration of Bcar3 siRNA via Nano-Self-Assembly to Treat Idiopathic Pulmonary Fibrosis by Disrupting Macrophage-Fibroblast Crosstalk
Authors Zeng C, Wang Q, Liu X, Wang K, Wang C , Ju X, Wang T , Zhou Q, Fu X, Yu J, Wang Y
Received 3 November 2023
Accepted for publication 18 February 2024
Published 23 February 2024 Volume 2024:19 Pages 1827—1842
DOI https://doi.org/10.2147/IJN.S444470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Chenxi Zeng,1,* Qi Wang,2,* Xuhan Liu,3,* Kai Wang,1 Congjian Wang,1 Xuetao Ju,1 Tianlai Wang,1 Qing Zhou,4 Xiangning Fu,1 Jun Yu,1 Yi Wang4
1Department of Thoracic Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, People’s Republic of China; 2Department of Geriatrics, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, People’s Republic of China; 3Department of Emergency Medicine, Shenzhen University General Hospital, Shenzhen University Clinical Medical Academy, Shenzhen University, Shenzhen, Guangdong Province, People’s Republic of China; 4Department of Pulmonary and Critical Care Medicine, NHC Key Laboratory of Respiratory Diseases, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yi Wang, Department of Pulmonary and Critical Care Medicine, NHC Key Laboratory of Respiratory Diseases, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, 430030, People’s Republic of China, Email [email protected] Jun Yu, Department of Thoracic Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei Province, 430030, People’s Republic of China, Email [email protected]
Background: Idiopathic pulmonary fibrosis (IPF) is a severe interstitial lung disease characterized by chronic lung injury leading to macrophage infiltration and fibroblast activation. However, there is no effective therapeutic strategy targeting the crucial crosstalk between macrophages and fibroblasts to halt IPF progression.
Methods: Studies were conducted in IPF patients and fibrotic mice models to elucidate the role of Bcar3 in the pathogenesis of pulmonary fibrosis. The effect of Bcar3 on macrophage polarization, fibroblast activation, and related signaling pathways were next investigated to unravel the underlying mechanisms.
Results: Our study elucidates a marked increase in Bcar3 expression in lung tissues from IPF patients and fibrotic mice, recording 1.7 and 7.8-fold increases compared to control subjects, respectively. Additionally, Bcar3 was found to significantly enhance macrophage activation and fibroblast differentiation, observable in both in vivo and in vitro settings. Mechanistically, the upregulation of Bcar3 in macrophages was reliant on Stat6, while in fibroblasts, it depended on TGFβR1/Smad3. Furthermore, Bcar3 augmented IL-4/Stat6 pathway in macrophages and TGF-β/Smad3 pathway in fibroblasts, supporting a synergistic activation loop that expedited lung fibrogenesis. Notably, intratracheal injection of liposomes containing Bcar3 siRNA precisely delivered gene therapeutics to lung macrophages and fibroblasts, effectively reducing Bcar3 expression to 59% of baseline levels. Importantly, this intervention protected mice from lung fibrosis induced by either FITC or bleomycin, as well as human precision-cut lung slices against TGF-β 1 stimulation.
Conclusion: Our study underscores the pivotal role of Bcar3 in orchestrating the macrophage-fibroblast crosstalk during pulmonary fibrosis progression. Targeting Bcar3 emerges as a novel therapeutic avenue to halt IPF progression and enhance patient prognosis.
Keywords: idiopathic pulmonary fibrosis, Bcar3, liposomes, macrophages, fibroblasts
Introduction
Idiopathic pulmonary fibrosis (IPF) is a relentless and life-threatening interstitial lung disease, characterized by persistent cough and exertional dyspnea.1,2 Despite extensive research efforts and a rising incidence of this disorder, there is still a lack of effective treatments that improve survival. Consequently, the 5-year survival rate stands at 70–80% following diagnosis.3 Therefore, development of a safe and effective treatment strategy for IPF is imperative. The disease typically originates from repetitive injury to alveolar epithelial cells, leading to subsequent infiltrations of macrophages and fibroblasts into the fibrotic lesions.4,5 Prior reports have underscored the crucial role of alternatively activated macrophages (M2) in generating fibrotic factors like amphiregulin (AREG), IL-17, and TGF-β, which facilitate fibroblast-to-myofibroblast transition.6,7 These myofibroblasts are central to the deposition and accumulation of the extracellular matrix (ECM) in the lung interstitium during the progression of IPF.8 Although disrupting the interaction between macrophages and fibroblasts presents a theoretical avenue for IPF treatment, the lack of identifiable molecular targets and suitable vehicular systems for drug delivery has impeded its clinical application.
In addition to current therapeutic options, including small-molecule drugs and various biological agents, RNA therapy has emerged as a viable approach for IPF treatment.9 Direct delivery of siRNA into the lungs has beneficial characteristics, such as avoidance of undesired serum interactions and better targeting of pulmonary lesions. However, the structural characteristics (hydrophilicity and negative charge) and instability of siRNA, along with the complex of respiratory tract creates obstacles for effective delivery.10 Recently, positively charged cationic liposomes have emerged as preferred siRNA carriers due to their high loading capacity and significantly enhanced cellular internalization through adsorptive interactions with the cell membrane.11 Furthermore, in our previous studies,12,13 siRNA encapsulated in cationic liposomes shows preferential uptake by macrophages and fibroblasts following intratracheal administration. Therefore, siRNA therapy based on cationic liposomes may represent an optimal strategy for disrupting macrophage-fibroblast crosstalk. Breast Cancer Anti-Estrogen Resistance 3 (Bcar3), initially identified for its role in estrogen resistance in breast cancer, belongs to the novel Src homology 2-containing protein family.14 Functionally, Bcar3 serves as an intermediary adapter protein downstream of diverse growth factor receptors, modulating tumor cell invasion, migration, and adhesion.14 It achieves this by interacting with protein tyrosine phosphatase α and Bcar1 through its N-terminal Src homology 2 domain and C-terminal guanine nucleotide exchange factor (GEF)-like domain, respectively.15,16 Previous studies have revealed that Bcar3 significantly triggers the activation of Cell division cycle 42 (Cdc42) and Src kinases, which are implicated in M2 polarization and the fibroblast-to-myofibroblast transition, respectively.14,17 Additionally, the RNA-sequencing data indicated elevated Bcar3 expression in both IL-4-treated macrophages and TGF-β1-stimulated fibroblasts, suggesting the potential of Bcar3 as an intervention target to disrupt the macrophage-fibroblast crosstalk. However, the roles of Bcar3 in pulmonary fibrosis progression remain to be fully elucidated.
In this investigation, we observed aberrant Bcar3 levels in the lung tissues of IPF patients and pulmonary fibrosis mice. Additionally, Bcar3 expression positively correlated with fibrogenic gene expression. Immunostaining identified Bcar3 predominantly within macrophages and fibroblasts. Moreover, Suppression of Bcar3 in these cell types led to a marked decrease in M2 macrophage polarization and fibroblast differentiation into myofibroblast. Our findings further elucidated the role of Bcar3 in mediating macrophage-fibroblast interactions via the TGF-β pathway. Notably, intratracheal injection of liposomes containing Bcar3 siRNA precisely delivered gene therapeutics to lung macrophages and fibroblasts. Importantly, this intervention protected mice from lung fibrosis induced by either FITC or bleomycin (BLM), as well as human precision-cut lung slices (hPCLS) against TGF-β1 stimulation. Collectively, our research suggests that Bcar3 is intricately responsible for the macrophage-fibroblast crosstalk during IPF progression. Consequently, Therapeutic approaches aimed at targeting Bcar3 show immense potential as promising treatments for IPF.
Materials and Methods
Patient Specimens
Lung specimens were collected from IPF patients (n=5), and non-small cell lung cancer (NSCLC) patients (n=5). The diagnosis of IPF was established following the consensus diagnostic criteria of the American Thoracic Society (ATS)/European Respiratory Society (ERS).18 Additionally, for hPCLS experiments, the lung tissues were collected from NSCLC patients (n=3). All sample were collected in Tongji hospital and informed consent was obtained from all participants. The ethical approval for these studies was granted by the Human Assurance Committee of Tongji Hospital (Reference No. TJ-IRB20220443). Clinical data and pulmonary function tests were provided in Supplementary Table 1.
Animal Experiment
Male C57BL/6 mice (6–8 weeks) were supplied by GemPharmatech (Nanjing, China). The mice were maintained in a specific pathogen-free (SPF) animal facility at Tongji Hospital, with a 12:12-h day/night cycle. After anesthetization with sodium pentobarbital (60 mg/kg), the mice were intratracheally injected with 1.5 mg/kg BLM (MedChemExpress, Shanghai, China) or 14 mg/kg FITC (Sigma-Aldrich, St. Louis, MO, USA) in 50 μL PBS.13 The control group received an equivalent volume of PBS. Subsequently, The mice were intratracheally administered liposomes encapsulating either scrambled (Scr) or Bcar3 siRNA (1 mg/kg) on Days 14 and 17 post-exposure to BLM or FITC. All mice were euthanized on Day 21 for the analysis of pulmonary fibrosis. All animals were cared for in accordance with the “Guidelines for the Ethical Review of Laboratory Animal Welfare (GB/T 35892-2018)”. All experimental protocols received approval from the Animal Care and Use Committee of Tongji Hospital (TJH-202105001).
Histological Analysis
The left lung was excised and fixed in paraformaldehyde (4%) at room temperature for 24 hours. Subsequently, the tissues were embedded in paraffin, followed by sectioning. These sections were then subjected to staining with Sirius red, Masson’s trichrome, and hematoxylin and eosin (H&E), following previously reported methods.3 The degree of fibrosis was assessed using the Ashcroft scoring method on a scale of 0 to 8.7
Immunofluorescence Staining
Paraffin sections or cryosections of lung tissues from IPF patients and pulmonary fibrosis mice were utilized for immunofluorescence staining. The following primary antibodies were employed: Rabbit anti-Bcar3 (1:100, Proteintech, Wuhan, China), mouse anti-PDGFR-β (1:100, Santa Cruz, CA, USA), mouse anti-a-SMA (1:100, Santa Cruz, CA, USA), and mouse anti-Vimentin (1:100, Proteintech, Wuhan, China). To visualize the labeled proteins, Alexa 594- or 488-conjugated anti-rabbit or anti-mouse antibodies (1:400, Abbkine, CA, USA) were utilized as fluorescence secondary antibodies, and DAPI was employed to counterstain the nuclei. Fluorescence images were captured using a fluorescence microscope (Olympus, Shinjuku, Japan).
Isolation and Treatment of Bone Marrow-Derived Macrophages (BMDMs)
BMDMs were isolated from the tibias and femurs of C57BL/6 mice. The BMDMs were cultured in RPMI-1640 medium (Gibco, Thermo Scientific, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Thermo Scientific, MA, USA), 30 ng/mL colony-stimulating factor (M-CSF, PeproTech, MA, USA), and 1% penicillin/streptomycin for a duration of 7 days.19 On Day 7, the cells were stimulated with 10 ng/mL IL-4 (Biolegend, CA, USA), and then collected for protein and mRNA expression analysis.
Isolation and Treatment of Fibroblasts
Mouse lung fibroblasts were extracted from lung tissues of C57BL/6 mice. In brief, the fresh lung tissues were finely minced into small fragments and then evenly distributed in a 10 cm plate with 2 mL of culture medium. After 24 hours, the medium was replaced.12 The cells were grown in DMEM containing 15% FBS and 1% penicillin/streptomycin at 37°C, and passage 3–5 cells were utilized. These cells were then subjected to TGF-β1 (10 ng/mL, MedChemExpress, Shanghai, China) to study the fibroblast responses.
Macrophage-Fibroblast Crosstalk
To investigate the impact of macrophage on fibroblast differentiation, BMDMs were transfected with either Scr or Bcar3 siRNA for 48 hours. Afterward, the supernatant was removed following 10 ng/mL IL-4 induction for 24 hours. Fresh medium was added, followed by incubation for an additional 24 hours. After pretreatment with 10 nM SB431542 or DMSO, the fibroblasts were exposed to macrophage culture medium for 24 hours before harvesting the cells for protein expression analysis.
Western Blot
Cellular and lung tissue lysates were prepared using RIPA lysis buffer (Beyotime, Shanghai, China). Western blot was conducted following established protocols.20 In brief, protein samples were separated through 10% SDS‒PAGE and transferred onto 0.4 μm PVDF membrane. After blocking with 5% milk for 1 hour, the membrane was exposed to anti-Bcar3 (1:1000, Boster, CA, USA), anti-Fibronectin, anti-Collagen 1, anti-GAPDH, and anti-β-actin (1:1000, Proteintech, Wuhan, China) at 4 °C overnight. Horseradish peroxidase (HRP)-labelled anti-rabbit (1:10000, Abbkine, CA, USA) or anti-mouse IgG (1:10000, Abbkine, CA, USA) secondary antibodies were then applied, followed by incubation for 1 hour. Protein bands were examined using a chemiluminescent substrate system (Bio-Rad Laboratories, CA, USA). The gray value was quantified using ImageJ software.
RT-PCR Assays
Total RNA was extracted from cells and lung tissues using TRIzol reagent (Takara, Dalian, China), following established protocols.3 The quality and concentration of the extracted RNA were assessed using a NanoDrop2000 spectrophotometer (Thermo Scientific, MA, USA). cDNA synthesis was performed using a reverse transcriptase kit (Invitrogen, CA, USA). RT-PCR assays were conducted employing SYBR Green mix (Takara, Dalian, China) on a CFX96 RT-PCR system (Bio-Rad Laboratories, CA, USA) with the following conditions: 30 seconds at 95°C, followed by 40 cycles of 95°C for 5 seconds and 60°C for 30 seconds. The mRNA level of Actb was employed as internal controls for normalization. The primer pairs employed for RT-PCR are listed in Supplementary Table 3.
siRNA or Plasmid Transfection
Specific siRNAs targeting Bcar3 (5′-CAACTACATGATCCTTGAT-3′ in Bcar3 mRNA) were procured from RiboBio (Guangzhou, China), followed by transient transfection into fibroblasts with Lipofectamine3000 (Invitrogen, CA, USA). In brief, fibroblasts were cultured in a 12-well plate before siRNA transfection for 48 hours. Subsequently, the cells were exposed to 10 ng/mL TGF-β1 at different time points. Transfection efficiency was assessed by Western blot following 48 hours transfection. To investigate Bcar3 overexpression, a control vector or Bcar3 plasmid (GeneChem, Shanghai, China) was used, and purified plasmid DNA (0.5 μg) was added with Lipofectamine3000 into fibroblasts. The overexpression efficiency of the plasmid was assessed by Western blot following 48 hours transfection.
Preparation of Liposomes Containing siRNAs
The liposomes containing siRNAs were prepared according to previously established methods.21 A lipid solution of mPEG-DMG, DSPC, cholesterol, and lipidoids (C12-200, C70H145N5O5) (1.5:10:38.5:50) was dissolved in sodium citrate (10 mM) and ethanol. Meanwhile, the siRNA was solubilized in a citrate buffer (10 mM). After mixing through vortexing, siRNA: lipidoids (20:1) was obtained. To remove free siRNA, the mixture underwent ultrafiltration centrifugation (3000 rpm, 10 minutes). To assess the liposomes’ properties, dynamic light scattering (DLS) (Malvern Zetasizer Nano-ZS, UK) was employed to measure the stability, zeta potential, polydispersity, and hydrodynamic diameter. The siRNA-entrapment efficiency was calculated using the RiboGreen assay. Additionally, the liposomes were stained with phosphotungstic acid (2%) and examined via transmission electron microscope (TEM, Jeol, Japan).
In vivo Distribution of Liposomes
To assess the in vivo distribution of liposomes, DiR-labeled liposomes were prepared and administered intratracheally to the mice following 14 days of BLM stimulation. After anesthetization using isoflurane (Sigma-Aldrich, St. Louis, MO, USA), the mice were imaged at 0 hour, 4 hours, 3 days, and 6 days using an in vivo imaging system (IVIS Lumina XR, SI Imaging, AZ, USA) with the excitation and emission wavelengths of 745 and 830 nm, respectively. On day 6, the mice were euthanized, and their intestines, kidneys, spleens, livers, hearts, and lungs were collected for ex vivo fluorescence imaging.
Generation and Culture of hPCLS
The para-carcinoma lung tissues (Supplementary Table 2) were gently infused with low-melt agarose (1.5%, Sigma-Aldrich, St. Louis, MO, USA) at 38°C, followed by rapid cooling in ice-cold water for 20 minutes, as previously described.22 The solidified tissues were then sectioned into slices of 400 µm thickness using a Compresstome (VF-300-0Z, Precisionary, CA, USA) at the oscillation frequency and cutting speed of 5 Hz and 6 μm/s, respectively. The generated hPLCS were cultured in a 12-well plate with DMEM-F12 no-phenol red medium (1 mL) containing 1% penicillin/streptomycin and 0.1% FBS at 37°C. Prior to TGF-β (10 ng/mL) stimulation for 24 hours, the hPCLS were exposed to liposomes containing Bcar3 or Scr siRNA for 48 hours.
Statistical Methods
Statistical comparison between groups was performed with GraphPad Prism v8. When comparing two groups with data showing a normal distribution and homogeneity of variance, the two-tailed Student’s t-test was employed. For data without a normal distribution, the two-tailed Mann–Whitney test was utilized for comparisons between two groups. When comparing multiple groups, one-way or two-way ANOVA was used for data with a normal distribution, followed by Tukey’s multiple comparison test for post-hoc analysis. For data without a normal distribution, Kruskal–Wallis test with Dunn’s post-hoc tests was applied. P<0.05 was deemed statistically significant in all cases.
Result
Upregulation of Bcar3 in Pulmonary Fibrosis and Its Localization in Fibroblasts and Macrophages
To elucidate the molecular dynamics underlying macrophage-fibroblast crosstalk, we performed RNA sequencing for transcriptome profiling. This analysis identified 27 genes with abnormal expression in IL-4 treated macrophages and TGF-β1 induced fibroblasts, including 6 genes with upregulation (Figure 1A and B) and 21 genes with downregulation (Supplementary Figure 1A and B). Notably, among the upregulated cohort, Bcar3 emerged as a gene of interest, with its elevated expression levels being confirmed through RT-PCR in both IL-4 induced macrophages and TGF-β1 stimulated fibroblasts. (Figure 1C and D). To further support our results, the expression of Bcar3 was determined in lung tissues from IPF patients and control subjects. Western blot analysis revealed a pronounced increase in Bcar3 expression in the lung tissues of IPF patients (Figure 1E). These results were also validated by coimmunostaining of Bcar3 with CD68 (a marker of macrophages) or PDGFR-β (a marker of fibroblasts) in the lung sections of IPF patients and controls (Figure 1F and G).
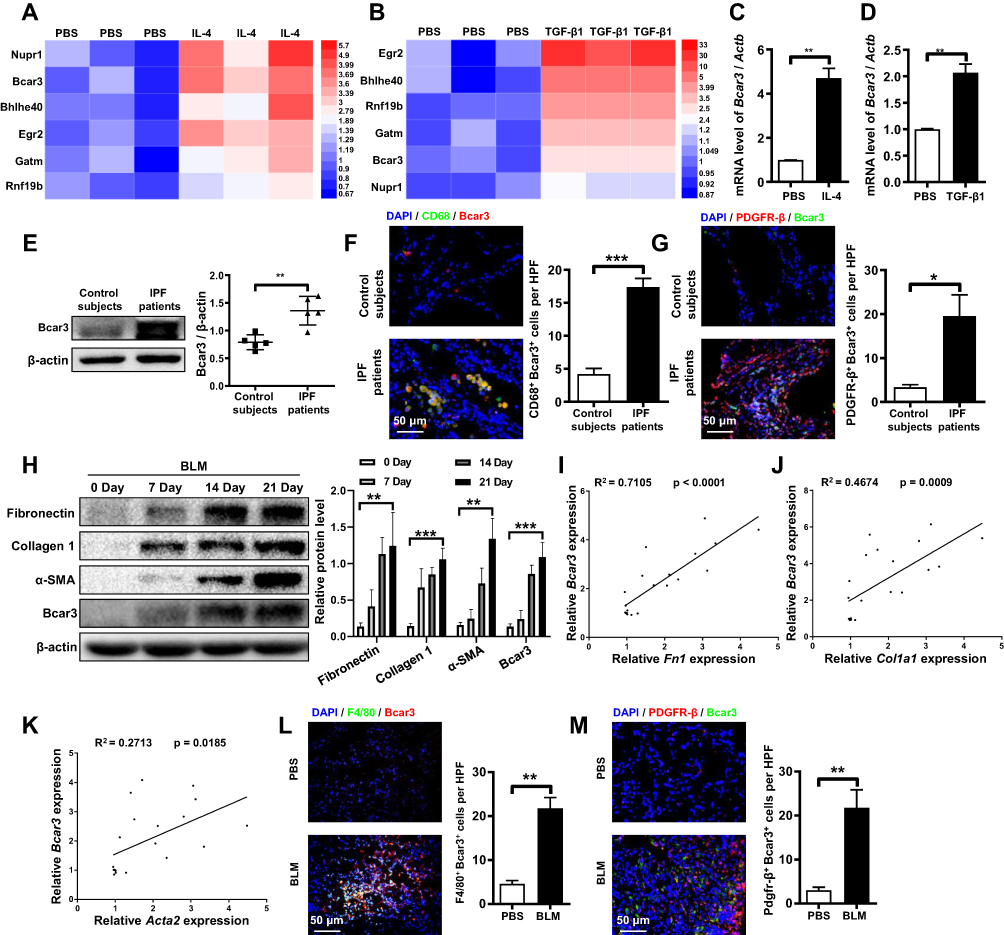 |
Figure 1 Differential expression of Bcar3 in IPF patient samples and pulmonary fibrosis mouse model. (A and B) show heatmaps displaying co-overexpressed genes in IL-4- and TGF-β1-treated macrophage and fibroblast, respectively, with colors representing the fold enrichment. (C and D) present the RT-PCR data of Bcar3 in macrophage and fibroblast after IL-4 and TGF-β1 induction, respectively. (E) depicts the Western blot results of Bcar3 in the lung homogenates of IPF patients (n=5) and control subjects (n=5). (F and G) display representative coimmunostaining data of Bcar3 and CD68 or PDGFR-β in the lung tissues of IPF patients and healthy controls. The nuclei exhibited a blue with DAPI. Magnification ×400. (H) shows the Western blot results of fibronectin, Collagen 1, a-SMA, and Bcar3 in the lung tissues of mice (n=5) at different time points after BLM stimulation. (I–K) demonstrate RT-PCR assays of the correlations among Bcar3, Acta2, Col1a1, and Fn1 upon TGF-β1 stimulation. (L and M) exhibit representative coimmunostaining data of Bcar3 and PDGFR-β or F4/80 in the lung tissues of mice exposed to BLM or PBS. The nuclei exhibited a blue staining with DAPI. Magnification ×400. Mean±SEM. *p<0.05; **p<0.01; ***p<0.001. |
Additionally, we investigated Bcar3 expression in mice lung tissues at different time points following intratracheal administration of BLM to induce pulmonary fibrosis. In line with the results observed in IPF patients, a time-dependent augmentation in Bcar3 expression was observed, accompanied by elevated levels of markers associated with pulmonary fibrosis, including Fibronectin, Collagen 1, and α-SMA (Figure 1H). Furthermore, the mRNA expression of Bcar3 was related to that of Fn1, Col1a1, and Acta2 in fibrotic lungs (Figure 1I–K). We also observed elevated Bcar3 expression in both macrophages and fibroblasts in pulmonary fibrosis mice, as revealed by an elevated number of F4/80+Bcar3+ cells and PDGFR-β+Bcar3+ cells in the lung tissues of mice exposed to BLM compared with control mice (Figure 1L and M). Collectively, these data suggest that overexpression of Bcar3 in macrophages and fibroblasts may be involved in the pathogenesis of pulmonary fibrosis.
The Role of Bcar3 in M2 Macrophage Polarization Through Stat6 Pathway
Macrophages, particularly the M2 subtypes, are pivotal in the development of pulmonary fibrosis.23 Upon IL-4 stimulation, Bcar3 expression in macrophages showed concentration- and time-dependent upregulation (Figure 2A and B), along with increased expression of M2 macrophage markers such as Arg1, Ym1, and CD206. To elucidate the positional role of Bcar3 within the signaling cascade, we employed AS1517499, a specific inhibitor of phosphorylated Stat6 (p-Stat6), which resulted in the attenuation of M2 macrophage polarization and the subsequent downregulation of Ym1 and Arg1 expression (Figure 2C). Notably, this inhibition also significantly reversed the IL-4-induced overexpression of Bcar3, suggesting that Bcar3 expression during M2 macrophage polarization is modulated through the IL-4/p-Stat6 pathway (Figure 2C).
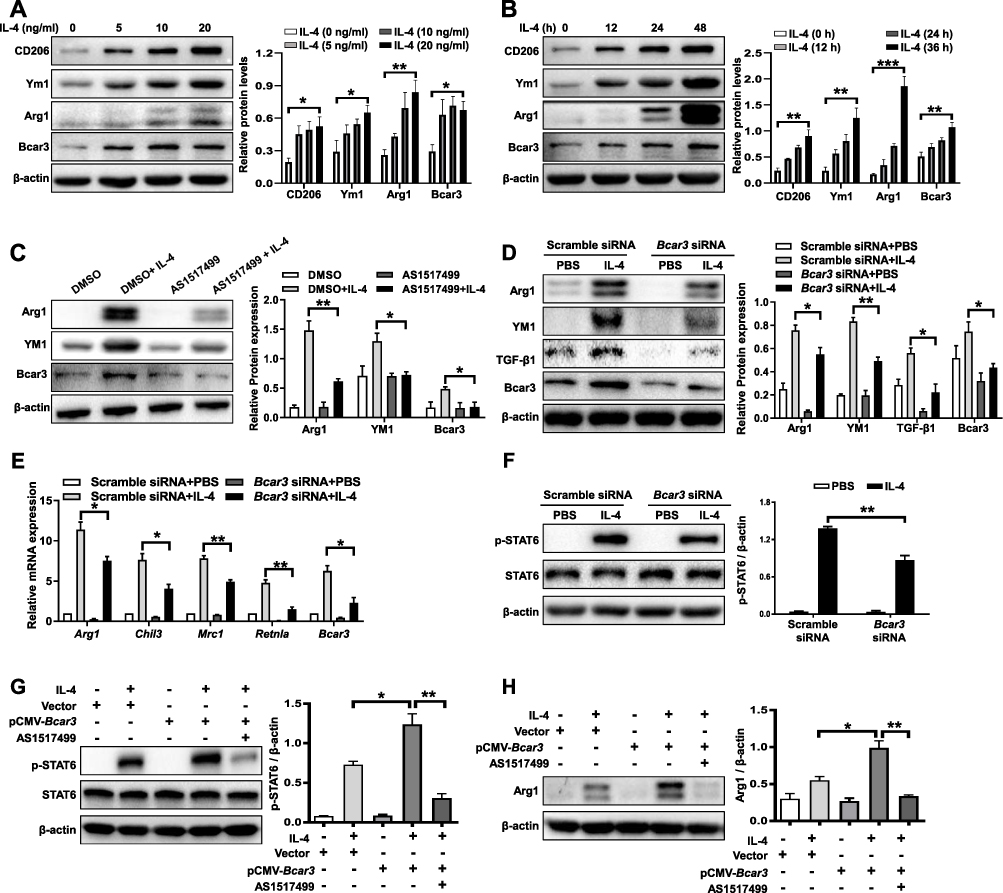 |
Figure 2 The role of Bcar3 in promoting M2 macrophage polarization through enhanced p-Stat6 signaling. (A and B) show the Western blot results of Bcar3, Arg1, Ym1, and CD206 in BMDMs treated with various concentrations of IL-4 (A) and at various time points (B). (C) displays the Western blot results of Bcar3, Arg1, and Ym1 in BMDMs treated with AS1517499 after IL-4 induction. (D) demonstrates the attenuation of M2 macrophage polarization after Bcar3 knockdown, as shown by the Western blot results of Bcar3, Ym1, and Arg1. The corresponding mean data for each group are presented in the bar graph. (E) shows the RT-PCR data of Mrc1, Chil3, Arg1, and Retnla in BMDMs transfected with Bcar3 siRNA following IL-4 induction. (F) illustrates the Western blot results of Stat6 and p-Stat6 in BMDMs transfected with Bcar3 siRNA after 1 hour of IL-4 induction. (G and H) demonstrate the impact of Bcar3 overexpression on p-Stat6 and Arg1 in BMDMs treated with AS1517499 and induced with IL-4 for 1 hour and 24 hours, respectively. Mean±SEM. *p<0.05; **p<0.01; ***p<0.001. |
To examine the role of Bcar3 in macrophage polarization, three Bcar3 siRNAs were designed (Supplementary Figure 2A). Knockdown of Bcar3 led to a marked reduction in M2 macrophage polarization post-IL-4 stimulation, as evidenced by the decreased expression levels of Arg1 and Ym1 (Figure 2D). Additionally, Bcar3 siRNA transfection resulted in diminished TGF-β1 production by M2 macrophages, which is an essential factor involved in fibroblast differentiation. RT-PCR assays further verified these observations, showing reduced expression of Arg1, Chil3, Mrc1, and Retnla following Bcar3 knockdown (Figure 2E).
Given the pivotal role of p-Stat6 in the polarization of IL-4-stimulated M2 macrophages,24 we investigated the effects of Bcar3 on p-Stat6 expression after IL-4 induction. IL-4 exposure led to a significant increase in p-Stat6 levels, which was effectively abrogated by Bcar3 siRNA, demonstrating Bcar3’s involvement in the regulation of p-Stat6 expression (Figure 2F). Overexpression studies in BMDMs confirmed that Bcar3 significantly enhances Arg1 and p-Stat6 levels (Supplementary Figure 2B and Figure 2G and H), reinforcing the notion of Bcar3’s regulatory effect on p-Stat6. Moreover, inhibition of p-Stat6 with AS1517499 effectively negated the Bcar3-induced upregulation of Arg1 and p-Stat6, further substantiating the dependency of Bcar3-regulated M2 macrophage polarization on the p-Stat6 pathway (Figure 2G and H).
A Positive Feedback Loop Between Bcar3 and TGFβR1/Smad3 Pathway During the Transition of Fibroblast to Myofibroblast
To explore the functions of Bcar3 in fibroblasts, we cultured fibroblasts and subjected them to TGF-β1 stimulation. Intriguingly, TGF-β1 induction led to a dose- and time-dependent upregulation of Bcar3 expression, coinciding with increased levels of fibrotic biomarkers (Figure 3A and B). These results indicate that TGF-β pathway contributes to the increased expression of Bcar3 during fibroblast differentiation. To explore this further, we investigated the impact of TGF-β pathway on Bcar3 expression. Inhibition of TGFβR1 with SB-431542 and p-Smad3 with SIS3-HCl effectively abrogated the TGF-β1-induced upregulation of Bcar3 (Figure 3C and D), underscoring the critical role of TGFβR1 and Smad3 in mediating Bcar3 expression during myofibroblast formation. Additionally, silencing Bcar3 expression significantly mitigated the formation of myofibroblasts and reduced the expression of fibrotic biomarkers in response to TGF-β1, suggesting that Bcar3 is essential for fibroblast differentiation (Figure 3E). In contrast, upregulation of Bcar3 could facilitate fibroblast-to-myofibroblast differentiation (Figure 3F).
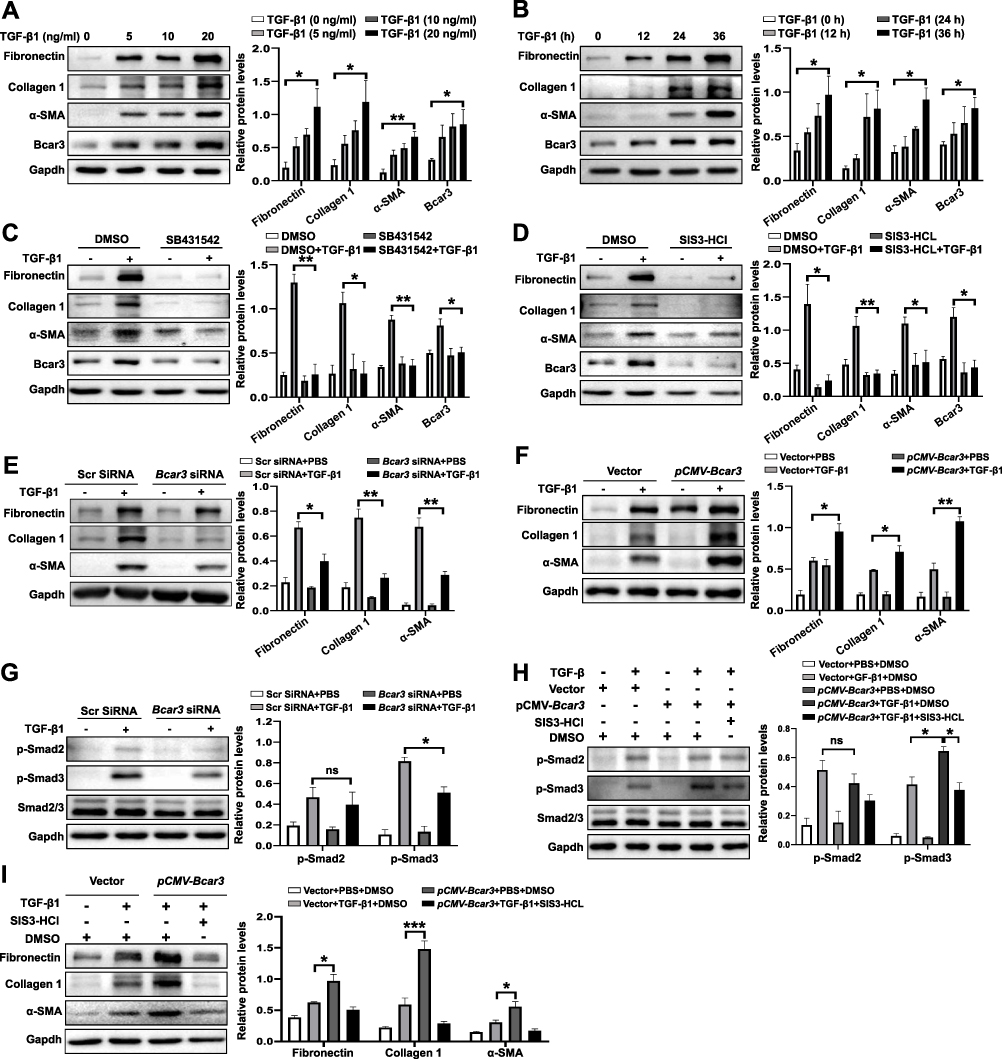 |
Figure 3 The involvement of Bcar3 in promoting fibroblast-to-myofibroblast differentiation through the TGF-β1/Smad3 pathway. (A and B) show the Western blot results of Bcar3, fibronectin, Collagen 1, and a-SMA in fibroblasts treated with different doses of TGF-β1 (A) and at various time points (B). (C and D) depict Western blot results of Bcar3, fibronectin, Collagen 1, and a-SMA in fibroblasts treated with SB431542 (C) or SIS3-HCl (D) after TGF-β1 induction for 24 hours. (E) shows the Western blot results of Bcar3, fibronectin, Collagen 1, TGF-β1, and a-SMA in fibroblasts transfected with Bcar3 siRNA after 24 hours of TGF-β1 stimulation. (F) presents the Western blot results of fibronectin, Collagen 1, and a-SMA in Bcar3-overexpressing fibroblasts. (G and H) display Western blot results of Bcar3, Smad2/3 and p-Smad2/3 in fibroblasts transfected with Bcar3 siRNA after 1 hour of TGF-β1 stimulation, and in Bcar3-overexpressing fibroblasts exposed to SIS3-HCl and induced with TGF-β1 for 1 hour, respectively. (I) presents the Western blot results of fibronectin, Collagen 1, and a-SMA in Bcar3-overexpressing fibroblasts treated with SIS3-HCl after TGF-β1 induction for 24 hours. Mean±SEM. *p<0.05; **p<0.01; ***p<0.001. |
The impact of Bcar3 on TGF-β signaling pathway was also evaluated. Exposure to TGF-β1 significantly increased the phosphorylation of Smad2 and Smad3, while suppressing Bcar3 expression selectively impeded the phosphorylation of Smad3 without significantly affecting Smad2 phosphorylation (Figure 3G). Conversely, overexpressing Bcar3 prominently elevated p-Smad3 levels (Figure 3H), providing consistent evidence for the regulatory role of Bcar3 in modulating p-Smad3 during this process. In addition, inhibition of p-Smad3 abolished the fibroblast transition induced by Bcar3 (Figure 3I). These results demonstrate the existence of a positive feedback loop between Bcar3 and TGFβR1/Smad3 pathway during fibroblast to myofibroblast transition.
Bcar3 Impedes Reciprocal Macrophage-Fibroblast Crosstalk
M2 macrophages are known to be one of main sources of TGF-β1, a critical driver of fibroblast differentiation in the development of IPF,25 Based on these understanding, we formulated a hypothesis that Bcar3 facilitates the progression of pulmonary fibrosis via TGF-β1-mediated macrophage-fibroblast crosstalk. To investigate this hypothesis, we induced fibroblasts treated with SB431542 or DMSO with supernatants from M2 macrophages transfected with either Scr or Bcar3 siRNA (Figure 4A).
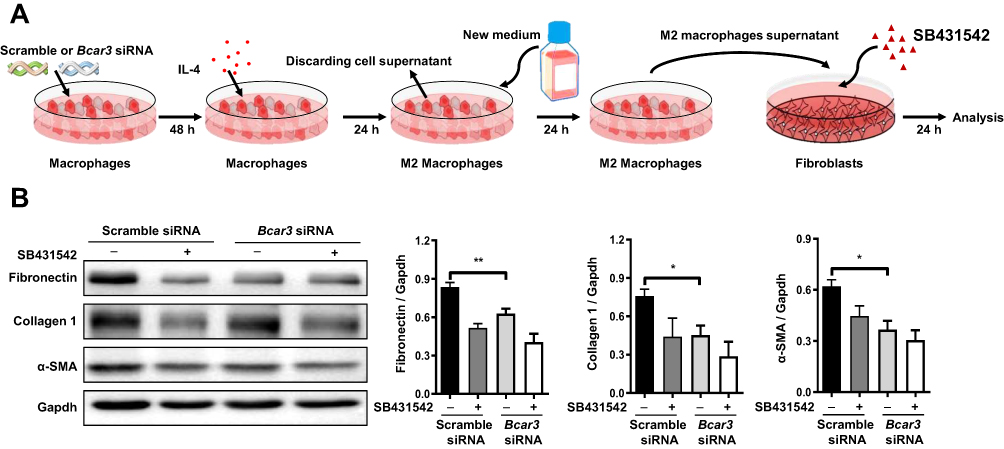 |
Figure 4 The inhibitory effect of Bcar3 on reciprocal macrophage-fibroblast crosstalk. (A) presents a diagram illustrating the experimental setup of macrophage induction of fibroblast differentiation. (B) shows the Western blot results of fibronectin, Collagen 1, and a-SMA in fibroblasts treated with BMDM supernatant. Mean±SEM. *p<0.05; **p<0.01. |
Consistent with our hypothesis, the application of SB431542 markedly suppressed fibroblast differentiation stimulated by supernatants from M2 macrophages transfected with Scr siRNA, implying that TGF-β1 is a key profibrotic factor in this cellular interaction (Figure 4B). Notably, when fibroblasts were exposed to supernatants from M2 macrophages transfected with Bcar3 siRNA, a pronounced reduction in the expression of fibrotic biomarkers was observed, compared to those exposed to supernatants from Scr siRNA-transfected M2 macrophages (Figure 4B), implying that Bcar3 impedes reciprocal macrophage-fibroblast crosstalk.
Analysis of Liposomes Containing Bcar3 siRNA and in vivo Distribution Following Intratracheal Administration
To explore potential therapeutic benefits for pulmonary fibrosis, we developed cationic lipid (C12-200)-based nanoparticles containing Bcar3 siRNA (Figure 5A). These liposomes exhibited high entrapment efficiency (>95%) for siRNA, with a zeta potential of 4.9 mV (Figure 5B). Additionally, the liposomes had a mean diameter of approximately 100 nm (Figure 5C) and displayed an even spherical morphology (Figure 5D). Moreover, the liposomes remained stable for over 24 hours (Figure 5E).
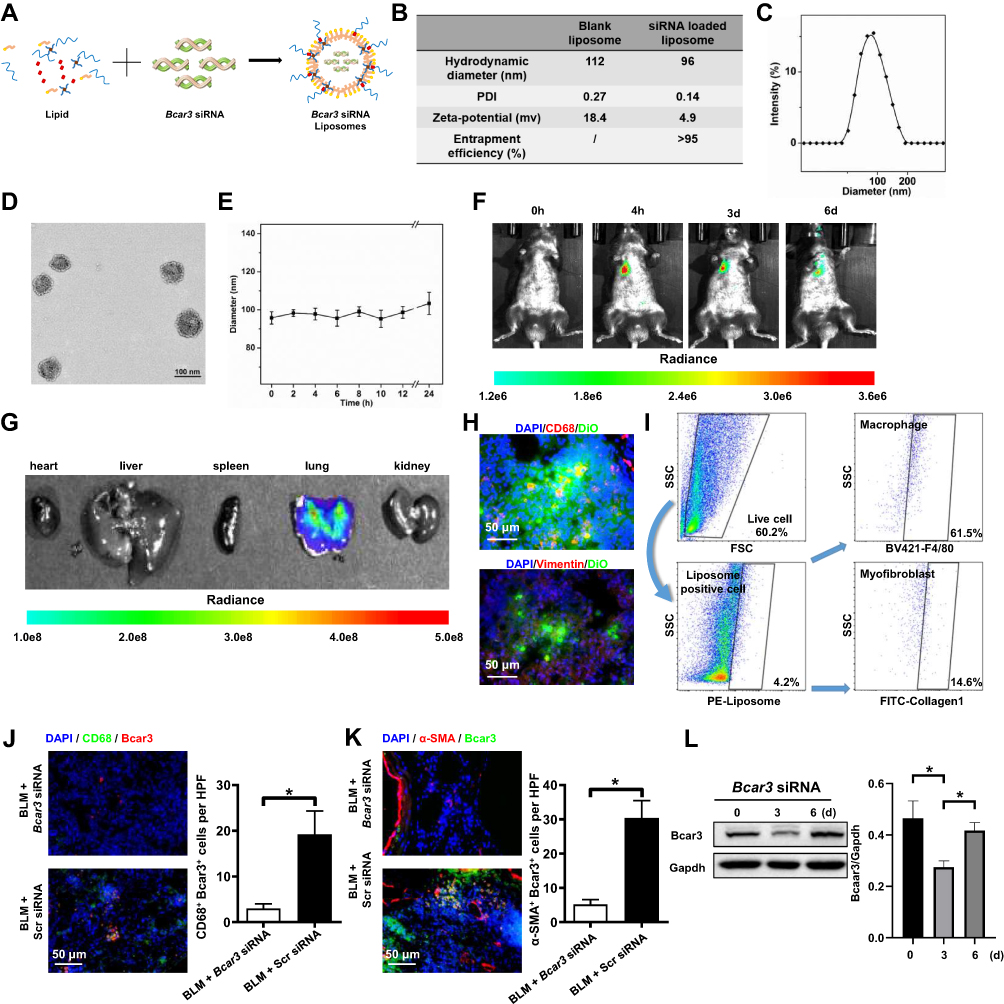 |
Figure 5 The distribution pattern of Bcar3 siRNA liposomes following intratracheal administration. (A) shows the preparation of liposomes loaded with Bcar3 siRNA. (B) presents the zeta potential, PDI, hydrodynamic diameter, and entrapment efficiency of liposomes (Bcar3 siRNA-loaded or empty). (C) displays a representative TEM image of liposomes loaded with Bcar3 siRNA. (D) shows the hydrodynamic diameter distribution of liposomes loaded with Bcar3 siRNA. (E) demonstrates the colloidal stability of liposomes loaded with Bcar3 siRNA in PBS. (F) shows the time-lapse IVIS imaging depicting the mouse’s response to intratracheally injected liposomes labeled with DiR at different time points. (G) presents ex vivo IVIS images of the main organs of mice. (H) displays immunofluorescence images illustrating the distribution of Vimentin+ cells (red), CD68+ cells (red), and DiO-labeled liposomes (green) in the mouse lungs. (I) presents the flow cytometry of liposome distribution in the lungs. (J and K) show immunofluorescent staining of CD68 (green) and Bcar3 (red), and Bcar3 (green) and a-SMA (red) in mice exposed to BLM followed by intravenous injection of liposomes containing Bcar3 siRNA. (L) presents the Western blot results of Bcar3 in mice exposed to BLM followed by intravenous injection of liposomes containing Bcar3 siRNA. Mean±SEM. *p<0.05. |
To investigate the in vivo distribution of the liposomes, mice were intratracheally administered with DiR-labeled liposomes, and then imaged at 0 hour, 4 hours, 3 days, and 6 days using IVIS. The fluorescent signals were predominantly localized in the lungs, with a gradual decrease over time (Figure 5F). Ex vivo imaging corroborated these results, further validating the targeted distribution of the liposomes (Figure 5G). Additionally, to investigate the cellular localization of liposomes in fibrotic lungs, DiO-labeled liposomes were administered intratracheally to pulmonary fibrosis mice for 48 hours. Immunostaining revealed that DiO (green) predominantly accumulated in macrophages (CD68+ cells) and fibroblasts (Vimentin+ cells) (Figure 5H). Flow cytometry analysis yielded similar findings (Figure 5I). Importantly, lung tissues from mice treated with Bcar3 siRNA-loaded liposomes showed a marked reduction in Bcar3 expression in both macrophages (Figure 5J) and fibroblasts (Figure 5K). A significant decrease in Bcar3 expression was observed by Day 3 post-intratracheal injection, with a subsequent gradual restoration by Day 6 (Figure 5L and Supplementary Figure 3A and B), highlighting the efficacy and temporal dynamics of gene silencing achieved through liposomal delivery. In contrast, direct administration of free Bcar3 siRNA failed to achieve similar downregulation, likely due to rapid degradation or insufficient cellular uptake (Supplementary Figure 3A–C). Based on these insights, we treated the mice with liposomes at Days 14 and 17 following BLM stimulation.
Liposomes Containing Bcar3 siRNA Exert Protective Effects Against Pulmonary Fibrosis and Fibrosis-Related Phenotypes in hPCLS
To assess the therapeutic potential of liposomes containing Bcar3 siRNA for pulmonary fibrosis, we initially established a mouse model of FITC-stimulated pulmonary fibrosis. Mice exposed to liposomes containing Bcar3 siRNA showed markedly reduced fluorescence signal of FITC compared with control group (Figure 6A). Histological assessment indicated substantial lung damage and excessive collagen accumulation in the lungs of mice exposed to FITC and liposomes containing FITC+Scr siRNA, whereas those exposed to liposomes containing Bcar3 siRNA demonstrated significant mitigation in lung damage and fibrosis (Figure 6A). Moreover, the levels of hydroxyproline were dramatically lower in the lungs of mice exposed to liposomes containing Bcar3 siRNA (Figure 6B). Additionally, Western blot and RT-PCR analyses further validated the therapeutic effects by showing downregulation of myofibroblast markers (Figure 6C and D). Moreover, a significant reduction in Arg1 and CD206 was observed in the Bcar3 siRNA treatment group, highlighting the role of Bcar3 in M2 macrophage polarization and fibrosis progression (Figure 6E). These findings were further supported by CD206 immunostaining (Supplementary Figure 4A).
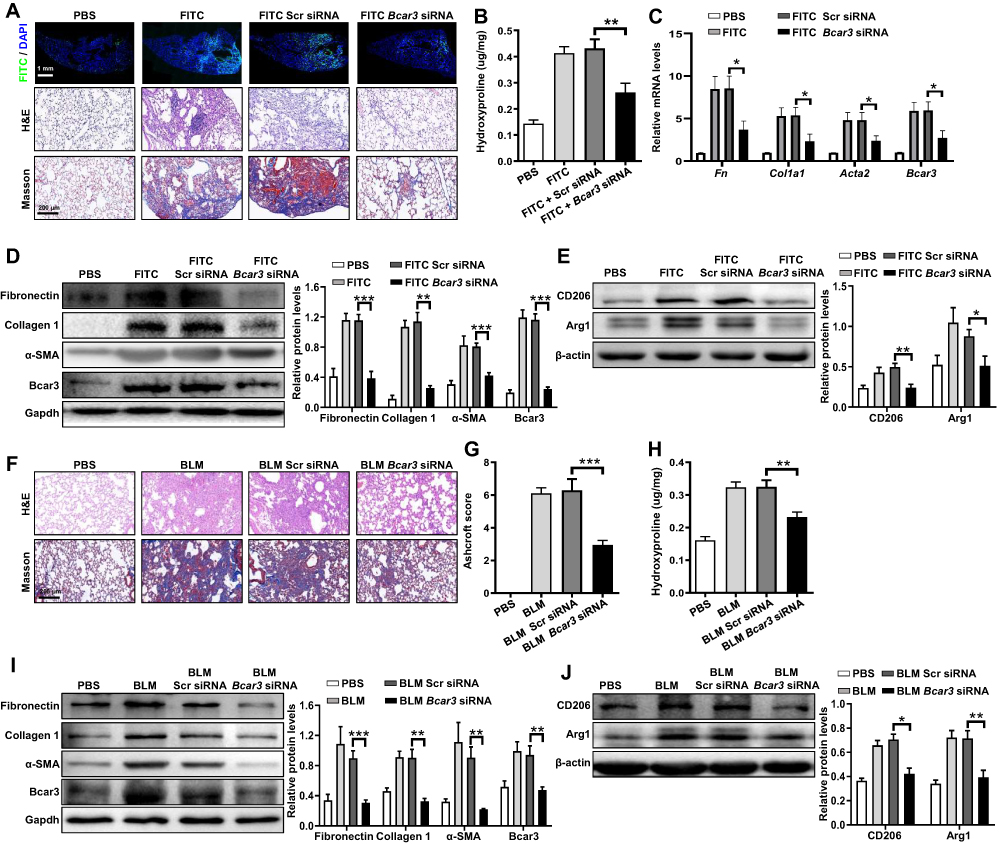 |
Figure 6 The protective effects of intratracheal injection of liposomes containing Bcar3 siRNA in pulmonary fibrosis mice. (A) shows the FITC (top), H&E (middle), and Masson (bottom) staining of the lung tissues following FITC simulation. Magnification 200×. (B) presents a bar graph illustrating hydroxyproline levels in following FITC stimulation. (C) displays the RT-PCR data of Bcar3, Acta2, Col1a1, and Fn1 following FITC stimulation. (D) shows the Western blot results of fibronectin, Collagen 1, a-SMA, and Bcar3 in mice exposed to FITC. (E) illustrates Western blot results of Arg1 and CD206 in mice exposed to FITC. (F) shows H&E (up) and Masson (down) staining of the lungs in after BLM stimulation. (G) presents a bar graph revealing the semi-quantitative Ashcroft scores of fibrosis severity. Magnification 200×. (H) shows a bar graph of hydroxyproline levels in the lungs following BLM stimulation. (I) presents the Western blot results of fibronectin, Collagen 1, a-SMA, and Bcar3 in mice exposed to BLM. (J) illustrates the Western blot results of Arg1 and CD206 following BLM stimulation. Mean±SEM. *p<0.05; **p<0.01; ***p<0.001. |
Similarly, in the BLM-induced pulmonary fibrosis mouse model, treatment with liposomes containing Bcar3 siRNA markedly reduced fibrotic lesions, as demonstrated by histopathological staining (Figure 6F), reduced Ashcroft scores (Figure 6G), decreased hydroxyproline levels (Figure 6H), and downregulated expression of fibrotic biomarkers (Figure 6I). Additionally, intratracheal injection of liposomes containing Bcar3 siRNA remarkably suppressed M2 macrophage polarization, as evidenced by downregulation of CD206 and Arg1 (Figure 6J). Consistent findings were obtained from CD206 immunostaining (Supplementary Figure 4B).
In addition, we explored the effect of liposomes containing Bcar3 siRNA on hPCLS. Exposure to liposomes containing Bcar3 siRNA markedly decreased collagen deposition and the expression of fibronectin and collagen 1 upon TGF-β1 induction (Figure 7A and B). RT-PCR assays confirmed these observations (Supplementary Figure 5). Furthermore, liposomes containing Bcar3 siRNA demonstrated effective inhibition of M2 macrophage polarization in hPCLS (Figure 7C and D). Taken together, these findings demonstrate the promise of liposomes containing Bcar3 siRNA as a novel therapeutic approach for the treatment of pulmonary fibrosis.
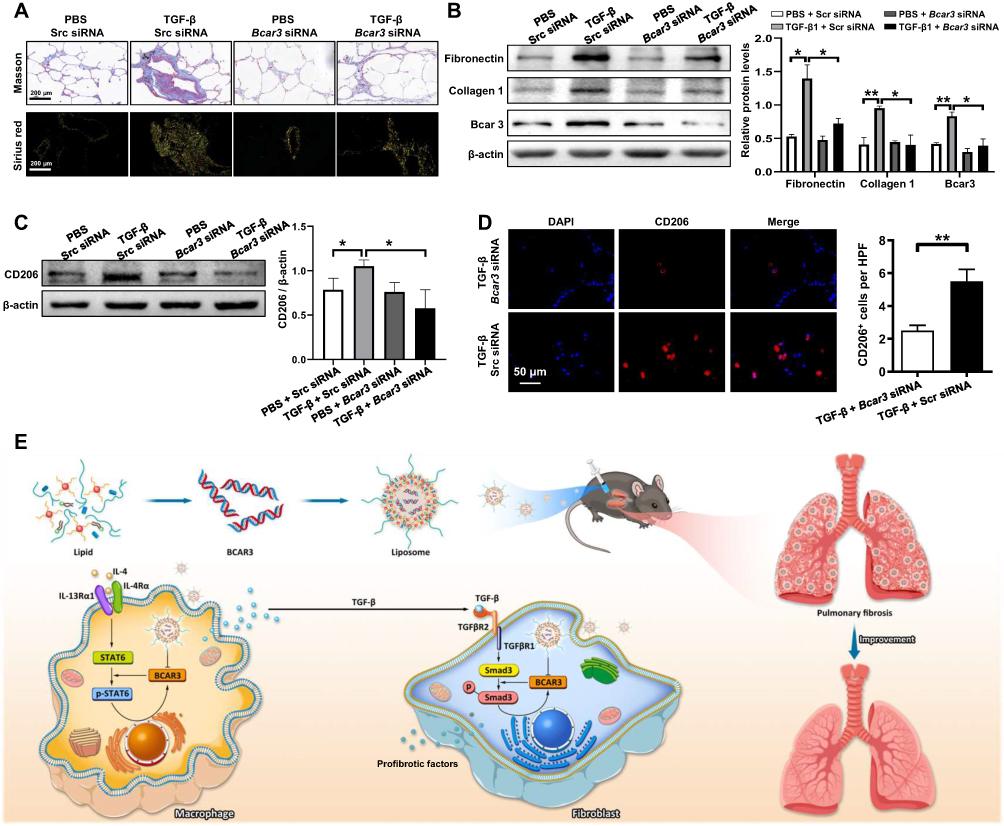 |
Figure 7 The attenuation of fibrosis-related phenotypes in TGF-β-exposed hPCLS after treatment with liposomes containing Bcar3 siRNA. (A) shows Sirius red staining (bottom) and Masson staining (top) of fibrosis-related phenotypes in TGF-β-exposed hPCLS after treatment with liposomes containing Bcar3 siRNA. (B) presents the Western blot results of Bcar3, fibronectin, and Collagen 1 in hPCLS after treatment with TGF-β and liposomes containing Bcar3 siRNA. The left panel shows representative Western blot data, and the right panel presents the bar graph of Western blot data. (C) displays the Western blot results of CD206 in hPCLS after treatment with TGF-β and liposomes containing Bcar3 siRNA. The left panel shows representative Western blot data, and the right panel presents the bar graph of Western blot data. (D) shows the immunofluorescence of CD206 in hPCLS after treatment with TGF-β and liposomes containing Bcar3 siRNA. (E) depicts a schematic of the mechanism by which Bcar3 regulates pulmonary fibrosis. Mean±SEM. *p<0.05; **p<0.01. |
Discussion
Herein, the effect of Bcar3 on the macrophage-fibroblast crosstalk during pulmonary fibrosis progression was investigated. Our results indicated that the expression of Bcar3 was increased in fibroblasts and macrophages, and this upregulated expression was also detected in the lungs of IPF patients and pulmonary fibrosis mice. Additionally, the increased expression of Bcar3 could facilitate M2 macrophage polarization and fibroblast-to-myofibroblast differentiation. Mechanistic research further determined positive feedback loops involving IL-4/Stat6/Bcar3 in macrophages and TGF-β1/Smad3/Bcar3 in fibroblasts. Notably, inhibition of Bcar3 effectively disrupted the macrophage-fibroblast crosstalk mediated by TGF-β1. Our study demonstrated that intratracheal injection of liposomes containing Bcar3 siRNA led to a significant reversal of pulmonary fibrosis induced by BLM or FITC, and in hPCLS induced with TGF-β1 (Figure 7E). These findings highlight the importance of Bcar3 in the macrophage-fibroblast crosstalk during pulmonary fibrosis progression. Moreover, our study demonstrated that intratracheal injection of liposomes containing Bcar3 siRNA can effectively reverse pulmonary fibrosis, suggesting a potential reliable treatment strategy for this condition.
Growing evidence indicates that fibroblasts and macrophages communicate to promote pulmonary fibrosis progression.6,8 Activated macrophages generate various factors, including AREG, PDGF, and TGF-β, which modulate fibroblast proliferation and differentiation. Targeting the macrophage-fibroblast crosstalk has been regarded as a promising treatment strategy for IPF. Current first-line drugs, nintedanib and pirfenidone, partially act on this strategy by inhibiting macrophage and fibroblast activation.26 Nonetheless, their multiple targets often lead to serious adverse events in IPF patients, thus impeding their clinical implementation. Herein, we aimed to determine key regulatory factors of macrophage activation and fibroblast differentiation. Through RNA-seq analysis, 27 genes were identified to have differential expression in fibroblasts and macrophages after induction, with 6 genes with up-regulation and 21 genes with down-regulation. Among these, Bcar3 is the member of novel Src homology 2 containing protein family, which is mainly reported in cancer progression. Previous study showed that Bcar3/Cas/c-Src signaling axis is a prominent regulator of c-Src activity in breast tumor cells.27 While, Src activity is associated with M2 macrophage polarization28 and fibroblast differentiation.29 Consequently, we focused on investigating the roles of Bcar3 in fibroblasts and macrophages during pulmonary fibrosis progression.
To investigate the role of Bcar3 in macrophages, we detected its expression in macrophages treated with IL-4, which is a strong inducer for alternatively activated macrophages. It was observed that Bcar3 was overexpressed during M2 macrophage polarization, and this effect was significantly reversed by a Stat6 inhibitor, indicating that the expression of Bcar3 in macrophages is regulated by IL-4/p-Stat6 pathway. Previous data showed that Bcar3 could binds to Bcar1 via its COOH-terminal domain and activate Cdc42, a small GTPase of the Rho family, which facilitated Stat6 activation during differentiation of Th2 cells.30,31 Therefore, we detected the effect of Bcar3 on IL-4/Stat6 pathway. Consistently, Bcar3 was found to promote the IL-4 stimulated M2 program by inducing Stat6 activation, as evidenced by suppressing p-Stat6 with AS1517499 markedly abolished the increased expression of Arg1 and p-Stat6 induced by Bcar3. Together, our research has identified a positive feedback loop centered around Bcar3 that promotes M2 macrophage polarization.
In fibroblasts, Bcar3 had similar expression patterns in a concentration- and time-dependent fashion upon TGF-β1 induction. The findings demonstrated that Bcar3 expression in fibroblasts exposed to TGF-β1 was dependent on TGFβR1 and Smad3, as revealed by the decreased Bcar3 expression in fibroblasts treated with SIS3-HCl or SB-431542 upon TGF-β1 induction. Importantly, gain-loss assays indicated that Bcar3 expression in fibroblasts promotes their transition into myofibroblasts after TGF-β1 exposure in a Smad3-dependent manner. However, in contrast to our observation, Guo et al reported that Bcar3 antagonizes TGFβ-induced Smad phosphorylation.32 This discrepancy is most likely caused by the differences of experimental approaches employed. Specifically, they conducted the experiment in MDA-MB-231 cells, a breast cancer cell line, with TGF-β (200 pM) induction, while we stimulated the primary lung fibroblasts with TGF-β1 (10 ng/mL). Nevertheless, regardless of which point of view is more persuasive, all of these findings suggest the implication of Bcar3 could modulate TGF-β/Smad signaling pathway.
Due to the limited therapeutic options available for IPF, we aimed to extrapolate these data into a potential treatment strategy. RNA therapy, utilizing specific exogenous nucleotide fragments such as siRNAs, offers the advantage of precise modulation of target cells, even those previously considered “undruggable”.33 The utilization of siRNA for disease treatment faces a crucial challenge, attributed to the structural characteristics (hydrophilicity and negative charge) and instability of siRNA. An effective and safe delivery system is imperative in overcoming this challenge. While both viral and non-viral vector systems can efficiently deliver RNA, viral vectors are limited by high immunogenicity, the risk of chromosomal integration, and poor reproducibility. In contrast, non-viral vector systems, such as biodegradable polymeric nanoparticles (NPs), are gaining popularity among researchers. Notably, positively charged cationic liposomes have emerged as preferred siRNA carriers due to their high loading capacity and significantly enhanced cellular internalization through adsorptive interactions with the cell membrane.34 For instance, patisiran (Onpattro), the first lipid nanoparticle-based siRNA drug for hereditary transthyretin-mediated amyloidosis, received approval from the Food and Drug Administration (FDA) in 2018, underscoring the translation of such concepts into clinical practice.35 Herein, we employed novel liposomes that are easily prepared and suitable for clinical implementation. Intratracheal injection of these liposomes demonstrated exceptional biocompatibility, as they predominantly accumulated at the fibrosis site and selectively targeted macrophages and fibroblasts. Liposome concentrations spanning from 25–400nM exhibited no discernible impact on cell viability (Supplementary Figure 6C and D). Above all, intratracheal administration of liposomes containing Bcar3 siRNA effectively reversed pulmonary fibrosis without inducing any adverse events (Supplementary Figure 6A and B).
This research has certain limitations that need to be addressed. As this was the first study to explore the role of Bcar3 in fibroblast differentiation and macrophage polarization, the exact mechanism remains unclear. While we demonstrated that Bcar3 can promote Smad3 and Stat6 pathways in macrophages and fibroblasts, respectively, a detailed mechanistic investigation is warranted. Furthermore, while we elucidated the functions of Bcar3 in both macrophages and fibroblasts, it is essential to consider that Bcar3 is also expressed in other cell types (eg, T cells and epithelial cells). Investigating the role of Bcar3 in these cell types will be necessary for a comprehensive understanding. Additionally, our study revealed that TGF-β1 may play a significant role in the Bcar3-mediated macrophage-fibroblast interaction during pulmonary fibrosis progression. However, it is plausible that other signaling pathways might also be involved in this interaction. Exploring the contribution of these alternative pathways is essential to gain a comprehensive understanding of the underlying mechanisms.
Conclusion
In summary, we provide evidence that Bcar3 can contribute to the pathogenesis of IPF by promoting M2 macrophage polarization and fibroblast differentiation. Notably, Bcar3 participates in two positive feedback loops in macrophages and fibroblasts, contributing to the disease progression. Crucially, inhibition of Bcar3 disrupts the macrophage-fibroblast crosstalk mediated by TGF-β1. Thus, our findings suggest that intratracheal injection of liposomes containing Bcar3 siRNA holds promise for effectively reversing pulmonary fibrosis stimulated by BLM and FITC without any adverse events. Consequently, targeting Bcar3 can be an effective strategy for treating pulmonary fibrosis. Nevertheless, further research is warranted to fully understand the molecular mechanisms and potential applications of this strategy.
Data Sharing Statement
The data and materials used in the current study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
The study was conducted in compliance with the Declaration of Helsinki and was approved by the Human Assurance Committee of Tongji Hospital (TJ-IRB20220443). All experimental procedures were approved by the Animal Care and Use Committee of Tongji Hospital (TJH-202105001).
Acknowledgment
We are grateful to those patients for donating their pulmonary tissue for the studies.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The present study was supported by the National Natural Science Foundation of China (82170078). The Tongji Hospital (HUST) Foundation for Excellent Young Scientists (2020YQ03) and Wuhan Science and Technology Bureau “Dawn Light Plan Talent Project” (2022020801020450).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Prele CM, Miles T, Pearce DR, et al. Plasma cell but not CD20-mediated B-cell depletion protects from bleomycin-induced lung fibrosis. Eur Respir J. 2022;60(5):2101469. doi:10.1183/13993003.01469-2021
2. Wang Y, Zhao J, Zhang H, Wang CY. Arginine is a key player in fibroblasts during the course of IPF development. Mol Ther. 2021;29(4):1361–1363. doi:10.1016/j.ymthe.2021.02.023
3. Hu Y, Wang Q, Yu J, et al. Tartrate-resistant acid phosphatase 5 promotes pulmonary fibrosis by modulating beta-catenin signaling. Nat Commun. 2022;13(1):114. doi:10.1038/s41467-021-27684-9
4. Wang Y, Zhang L, Wu GR, et al. MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci Adv. 2021;7(1):eabb6075.
5. Wang Y, Zhang L, Huang T, et al. The methyl-CpG-binding domain 2 facilitates pulmonary fibrosis by orchestrating fibroblast to myofibroblast differentiation. Eur Respir J. 2022;60(3):2003697. doi:10.1183/13993003.03697-2020
6. Buechler MB, Fu W, Turley SJ. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity. 2021;54(5):903–915. doi:10.1016/j.immuni.2021.04.021
7. Rao LZ, Wang Y, Zhang L, et al. IL-24 deficiency protects mice against bleomycin-induced pulmonary fibrosis by repressing IL-4-induced M2 program in macrophages. Cell Death Differ. 2021;28(4):1270–1283. doi:10.1038/s41418-020-00650-6
8. Prasse A, Pechkovsky DV, Toews GB, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173(7):781–792. doi:10.1164/rccm.200509-1518OC
9. Doroudian M, O’Neill A, Mac Loughlin R, Prina-Mello A, Volkov Y, Donnelly SC. Nanotechnology in pulmonary medicine. Curr Opin Pharmacol. 2021;56:85–92. doi:10.1016/j.coph.2020.11.002
10. Ding L, Tang S, Tang W, et al. Perfluorocarbon nanoemulsions enhance therapeutic siRNA delivery in the treatment of pulmonary fibrosis. Adv Sci. 2022;9(8):e2103676.
11. Ghasemzad M, Hashemian SMR, Memarnejadian A, Akbarzadeh I, Hossein-Khannazer N, Vosough M. The nano-based theranostics for respiratory complications of COVID-19. Drug Dev Ind Pharm. 2021;47(9):1353–1361. doi:10.1080/03639045.2021.1994989
12. Wang Q, Liu J, Hu Y, et al. Local administration of liposomal-based Srpx2 gene therapy reverses pulmonary fibrosis by blockading fibroblast-to-myofibroblast transition. Theranostics. 2021;11(14):7110–7125. doi:10.7150/thno.61085
13. Pan T, Zhou Q, Miao K, et al. Suppressing Sart1 to modulate macrophage polarization by siRNA-loaded liposomes: a promising therapeutic strategy for pulmonary fibrosis. Theranostics. 2021;11(3):1192–1206. doi:10.7150/thno.48152
14. Arras J, Thomas KS, Myers PJ, et al. Breast Cancer Antiestrogen Resistance 3 (BCAR3) promotes tumor growth and progression in triple-negative breast cancer. Am J Cancer Res. 2021;11(10):4768–4787.
15. Steenkiste EM, Berndt JD, Pilling C, Simpkins C, Cooper JA. A Cas-BCAR3 co-regulatory circuit controls lamellipodia dynamics. Elife. 2021;10:e67078.
16. Zhang W, Lin Y, Liu X, et al. Prediction and prognostic significance of BCAR3 expression in patients with multiple myeloma. J Transl Med. 2018;16(1):363. doi:10.1186/s12967-018-1728-8
17. Riggins RB, Quilliam LA, Bouton AH. Synergistic promotion of c-Src activation and cell migration by Cas and AND-34/BCAR3. J Biol Chem. 2003;278(30):28264–28273. doi:10.1074/jbc.M303535200
18. Raghu G, Remy-Jardin M, Myers J, Richeldi L, Wilson KC. The 2018 diagnosis of idiopathic pulmonary fibrosis guidelines: surgical lung biopsy for radiological pattern of probable usual interstitial pneumonia is not mandatory. Am J Respir Crit Care Med. 2019;200(9):1089–1092. doi:10.1164/rccm.201907-1324ED
19. Mou Y, Wu GR, Wang Q, et al. Macrophage-targeted delivery of siRNA to silence Mecp2 gene expression attenuates pulmonary fibrosis. Bioeng Transl Med. 2022;7(2):e10280. doi:10.1002/btm2.10280
20. Wang Q, Yu J, Hu Y, et al. Indirubin alleviates bleomycin-induced pulmonary fibrosis in mice by suppressing fibroblast to myofibroblast differentiation. Biomed Pharmacother. 2020;131:110715. doi:10.1016/j.biopha.2020.110715
21. Love KT, Mahon KP, Levins CG, et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc Natl Acad Sci U S A. 2010;107(5):1864–1869. doi:10.1073/pnas.0910603106
22. Ahangari F, Becker C, Foster DG, et al. Saracatinib, a selective src kinase inhibitor, blocks fibrotic responses in preclinical models of pulmonary fibrosis. Am J Respir Crit Care Med. 2022;206(12):1463–1479. doi:10.1164/rccm.202010-3832OC
23. Huang J, Maier C, Zhang Y, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. 2017;76(11):1941–1948. doi:10.1136/annrheumdis-2016-210823
24. Yao Y, Wang Y, Zhang Z, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24(5):915–925. doi:10.1038/mt.2016.36
25. Lv Q, Wang J, Xu C, Huang X, Ruan Z, Dai Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3beta/beta-catenin and TGF-beta1/Smad2/3 signaling pathways. Mol Med. 2020;26(1):49. doi:10.1186/s10020-020-00173-3
26. Zhang L, Wang Y, Wu G, Xiong W, Gu W, Wang CY. Macrophages: friend or foe in idiopathic pulmonary fibrosis? Respir Res. 2018;19(1):170. doi:10.1186/s12931-018-0864-2
27. Schuh NR, Guerrero MS, Schrecengost RS, Bouton AH. BCAR3 regulates Src/p130 Cas association, Src kinase activity, and breast cancer adhesion signaling. J Biol Chem. 2010;285(4):2309–2317. doi:10.1074/jbc.M109.046631
28. Hu X, Wang H, Han C, Cao X. Src promotes anti-inflammatory (M2) macrophage generation via the IL-4/STAT6 pathway. Cytokine. 2018;111:209–215. doi:10.1016/j.cyto.2018.08.030
29. Hu M, Che P, Han X, et al. Therapeutic targeting of SRC kinase in myofibroblast differentiation and pulmonary fibrosis. J Pharmacol Exp Ther. 2014;351(1):87–95. doi:10.1124/jpet.114.216044
30. Cai D, Iyer A, Felekkis KN, et al. AND-34/BCAR3, a GDP exchange factor whose overexpression confers antiestrogen resistance, activates Rac, PAK1, and the cyclin D1 promoter. Cancer Res. 2003;63(20):6802–6808.
31. Fu J, Liu B, Zhang H, et al. The role of cell division control protein 42 in tumor and non-tumor diseases: a systematic review. J Cancer. 2022;13(3):800–814. doi:10.7150/jca.65415
32. Guo J, Canaff L, Rajadurai CV, et al. Breast cancer anti-estrogen resistance 3 inhibits transforming growth factor beta/Smad signaling and associates with favorable breast cancer disease outcomes. Breast Cancer Res. 2014;16(6):476. doi:10.1186/s13058-014-0476-9
33. Paunovska K, Loughrey D, Dahlman JE. Drug delivery systems for RNA therapeutics. Nat Rev Genet. 2022;23(5):265–280. doi:10.1038/s41576-021-00439-4
34. Kedmi R, Veiga N, Ramishetti S, et al. A modular platform for targeted RNAi therapeutics. Nat Nanotechnol. 2018;13(3):214–219. doi:10.1038/s41565-017-0043-5
35. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18(6):421–446. doi:10.1038/s41573-019-0017-4
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.