")
Back to Journals » Journal of Experimental Pharmacology » Volume 14
Local Anesthetic Like Inhibition of the Cardiac Na+ Channel Nav1.5 by Chloroquine and Hydroxychloroquine
Authors Hage A , de Vries M , Leffler A, Stoetzer C
Received 21 May 2022
Accepted for publication 16 October 2022
Published 8 November 2022 Volume 2022:14 Pages 353—365
DOI https://doi.org/10.2147/JEP.S375349
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Kones
Axel Hage *, Mathis de Vries *, Andreas Leffler, Carsten Stoetzer
Department of Anesthesiology and Intensive Care Medicine, Hannover Medical School, Hanover, Germany
*These authors contributed equally to this work
Correspondence: Axel Hage, Department of Anesthesiology and Intensive Care Medicine, Hannover Medical School, Carl-Neuberg-Strasse 1, Hanover, 30625, Germany, Tel +49 176 1532 2775, Fax +49 511 532 163498, Email [email protected]
Introduction: Chloroquine (CQ) and its derivate hydroxychloroquine (HCQ) are successfully deployed for different diseases beyond the prophylaxis and treatment of malaria. Both substances exhibit antiviral properties and have been proposed for prophylaxis and treatment of COVID-19 caused by SARS-CoV-2. CQ and HCQ cause similar adverse events including life-threatening cardiac arrhythmia generally based on QT-prolongation, which is one of the most reported adverse events for both agents associated with the treatment of COVID-19. Various drugs known to induce QT-prolongation have been proven to exert local anesthetic (LA)-like properties regarding their impact on the cardiac Na+ channel Nav1.5. Inhibition of Nav1.5 is considered as the primary mechanism of cardiotoxicity caused by LAs. However, the mechanism of the arrhythmogenic effects of CQ and HCQ related to Nav1.5 has not yet been fully investigated. Therefore, the exact mechanism of how CQ and HCQ affect the sodium currents generated by Nav1.5 need to be further elucidated.
Objective: This in vitro study aims to investigate the effects of CQ and HCQ on Nav1.5-generated sodium currents to identify possible LA-like mechanisms that might contribute to their arrhythmogenic properties.
Methods: The effects of CQ and HCQ on Nav1.5-generated sodium currents by HEK-293 cells expressing either wild-type human Nav1.5 or mutant Nav1.5 F1760A are measured using the whole-cell patch-clamp technique.
Results: Both agents induce a state-dependent inhibition of Nav1.5. Furthermore, CQ and HCQ produce a use-dependent block of Nav1.5 and a shift of fast and slow inactivation. Results of experiments investigating the effect on the LA-insensitive mutant Nav1.5-F1760A indicate that both agents at least in part employ the proposed LA-binding site of Nav1.5 to induce inhibition.
Conclusion: This study demonstrated that CQ and HCQ exert LA-typical effects on Nav1.5 involving the proposed LA binding site, thus contributing to their arrhythmogenic properties.
Keywords: chloroquine, hydroxychloroquine, patch-clamp, cardiac sodium channel, Nav1.5
Introduction
Chloroquine (CQ) and its hydroxyl analogue hydroxychloroquine (HCQ) have been successfully used in the prophylaxis and treatment of malaria for decades.1–3 Besides their antimalarial effect, CQ and HCQ exhibit immunomodulatory and immunosuppressive characteristics and have been established as disease-modifying antirheumatic drugs in the treatment of rheumatic and immunological diseases.2,4,5 Furthermore, an antiviral effect against several viruses has been demonstrated in vitro and in animal experiments.6–8 Due to the direct antiviral and anti-inflammatory effect, both agents have been proposed for off-label use to treat COVID-19 patients.7,9 Subsequently, after randomized studies were performed without any robust evidence for benefits and uncertainty concerning their safety rose, the FDA retracted an Emergency Use Authorization.10–12 Both agents can cause similar serious side effects ranging from retinopathy, methemoglobinemia or acute kidney injury to life-threatening cardiac arrhythmia.2,11,13,14 QT prolongation, the electrocardiographic equivalent of delayed ventricular repolarization is the most commonly reported adverse event for CQ and HCQ associated with the treatment of COVID-19 pneumonia.15,16 Delayed repolarization facilitates early afterdepolarizations, which can trigger Torsades de Pointes ventricular arrhythmias or even sudden cardiac death. Most of the QT prolongations are induced by drugs and thus classified as acquired long-QT syndrome (LQTS) in contrast to congenital LQTS due to cardiac ion channelopathies. A large proportion of acquired LQTS is triggered by drugs which interact with cardiac hERG K+ channels and thereby affect the rapid component of the delayed rectifier potassium current.17 Indeed, CQ and HCQ inhibit cardiac hERG K+ channels.18,19 In addition, both substances have also been demonstrated to inhibit further cardiac ion channels, such as the inwardly rectifying potassium channel, L-type calcium and sodium channels.20,21 The opioids methadone or buprenorphine, which can also trigger LQTS, inhibit the cardiac-specific isoform of voltage-gated sodium channels, Nav1.5.19,22,23 Vice versa, LAs like bupivacaine, ropivacaine and mepivacaine inhibit hERG K+ channels, suggesting that this may contribute to their cardiotoxic potential.24 However, inhibition of Nav1.5 is suggested to be the primary mechanism of cardiotoxicity caused by LAs.19,25,26 Here we investigated if CQ and HCQ inhibit human Nav1.5 expressed in HEK-293 cells in a manner that may contribute to their cardiotoxicity.
Materials and Methods
Cell Culture and Transfection
HEK-293 cells were purchased commercially (ECACC 96121229, Sigma-Aldrich, Germany) and cultured at 37 °C and 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 100 U/mL penicillin/streptomycin, 25 mM HEPES, 10% heat-inactivated fetal bovine serum (all GIBCO-Invitrogen, Germany), 3 mM taurine (Sigma-Aldrich, Germany), and 0.4% Zeocin (Invitrogen, Carlsbad, CA). For experiments, either HEK-293 cells stably expressing the human Nav1.5 or HEK-293 cells transfected with Nav1.5-F1760A were used. Mutagenesis of Nav1.5-F1760A was carried out according to the instructions of the manufacturer (QuikChange XL Kit; Qiagen GmbH, Hilden, Germany). Transfection of HEK-293 cells were performed with the Nanofectin transfection kit (PAA Laboratories GmbH, Pasching, Austria) as described previously.27
Solutions and Chemicals
The experiments were performed under usage of an external solution that consisted of sterile distilled water and (mmol/l) 70 NaCl, 70 choline chloride, 3 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, and 15 glucoses. The pH value was adjusted to 7.4 with TMA-OH (tetramethylammonium hydroxide). The pipette solution contained (mmol/l) 140 CsF, 10 NaCl, 1 EGTA, and 10 HEPES. The pH value was adjusted to 7.4 with CsOH. Both solutions were prepared sterile, stored protected from light at 4 °C and used up within 1 month as described before.28 Chloroquine was purchased from Hycultec GmbH (Beutelsbach, Germany) and hydroxychloroquine from MedChemExpress (Monmouth Junction, New Jersey, USA). Both agents were prepared as stock solutions to a concentration of 0.1 mol/L according to their data sheets and stored at −80°C. Chloroquine was diluted in dimethyl-sulfoxide (DMSO, Sigma-Aldrich, Munich, Germany) and hydroxychloroquine in distilled water. Directly prior to the experiments, solutions for the patch-clamp recordings were prepared. To achieve the required concentration the appropriate amount of stock solution was titrated with external solution. Solutions were focally applied via a homemade gravity-driven polytetrafluoroethylene-glass application system.
Whole-Cell Patch-Clamp Recordings and Data Acquisition
Membrane Na+ currents were recorded with the whole-cell configuration of the patch-clamp technique and performed at room temperature as described previously.22,28,29 Pipettes for patch-clamp experiments were made of glass capillaries (GB150EFT-10, Science Products, Hofheim, Germany) with a DMZ Universal Puller (Zeitz-Instruments, Germany) and heat polished to achieve a resistance with pipette solution of 2.0 to 2.5 MΩ. Only a single cell was measured per dish and only a single series of measurements was carried out. A patch-clamp amplifier EPC10 amplifier (HEKA Instruments Inc., NY, USA) was operated during all conducted experiments. Its driving program Patchmaster v20 x 60 software (HEKA Instruments Inc., NY, USA) was used for pulse generation but also for data acquisition and to store data obtained from measurement. Currents were filtered at 5 kHz, series-resistance was compensated by at least 60% to minimize voltage errors and the capacitance artifact was cancelled using the amplifier circuitry. Furthermore, linear leak subtraction, based on resistance estimates from four hyperpolarizing pulses applied before the test pulse, was used for all voltage-clamp recordings except for use-dependent block at 10 Hz as described by Stoetzer et al.29 The Pulsefit software (HEKA Instruments Inc., NY, USA) was used for patch-clamp data review and analyzing. Curve fitting and statistical analyses were performed with Origin 7.0 (Microcal Software, Northampton, MA, USA). If applicable, single comparisons of independent groups of data were calculated with the two-tailed Student t-test. Statistical significance was determined at p < 0.05. Data were presented as mean ± S.E.M. or fitted value ± S.E. of the fit. To calculate IC50-values, peak current amplitudes at different drug concentrations were normalized to the value obtained in control solution. Data were fitted with Hill equation y = ymax × {IC50n/ (IC50n × Cn)}, where ymax is the maximal amplitude, IC50 the concentration at which y/ymax = 0.5, and n is the Hill coefficient. To obtain the inactivation curves, peak currents evoked by a test pulse were measured, normalized, and plotted against the conditioning pre-pulse potential. Data were fitted by the Boltzmann equation y = 1/(1 + exp ((EPP − h0.5)/kh)), where EPP is the membrane potential, h0.5 is the voltage at which y = 0.5, and kh is the slope factor.19
Results
Chloroquine and Hydroxychloroquine Inhibit Nav1.5
CQ was shown to block transmembrane Na+ currents in primary feline ventricular cardiomyocytes.20 However, these results were not investigated further regarding the subtype of the voltage-gated Na+ channel nor the effect of HCQ on Na+ currents. Hence, we examined the capability of CQ and HCQ to inhibit recombinant human Nav1.5 expressed in HEK-293 cells. As demonstrated in Figure 1, both CQ and HCQ induce a concentration-dependent tonic block of resting and inactivated Nav1.5 channels. The behavior of resting channels during exposure to CQ or HCQ was explored in cells held at −120 mV by test pulses to 0 mV applied at 0.1 Hz. The IC50 value for CQ was calculated to 69 ± 12 μM (Hill coefficient 0.96 ± 0.04, n = 9) and for HCQ to 446 ± 50 μM (Hill coefficient 0.97 ± 0.02, n = 9). The difference between CQ and HCQ regarding their blocking potency reaches statistical significance (p < 0.001, unpaired t-test). One of the typical properties of LAs is their high-affinity block of inactivated Na+ channels.23 Induction of inactivation by a 10s long pre-pulse at −70 mV followed by 100-ms-long pulse at −120 mV, allowing recovery from fast inactivation, and finally a test pulse to 0 mV leads to a significant stronger tonic block by HCQ (IC50 205 ± 20 μM, Hill coefficient 0.86 ± 0.02, n = 8; p < 0.05, unpaired t-test) in respect to the IC50 of resting channels. Tonic block of inactivated channels induced by CQ also results in a lower IC50 value (IC50 44 ± 9, Hill coefficient 0.97 ± 0.05, n = 12) when compared to resting channels, but this difference did not reach statistical significance (p = 0.62, unpaired t-test).
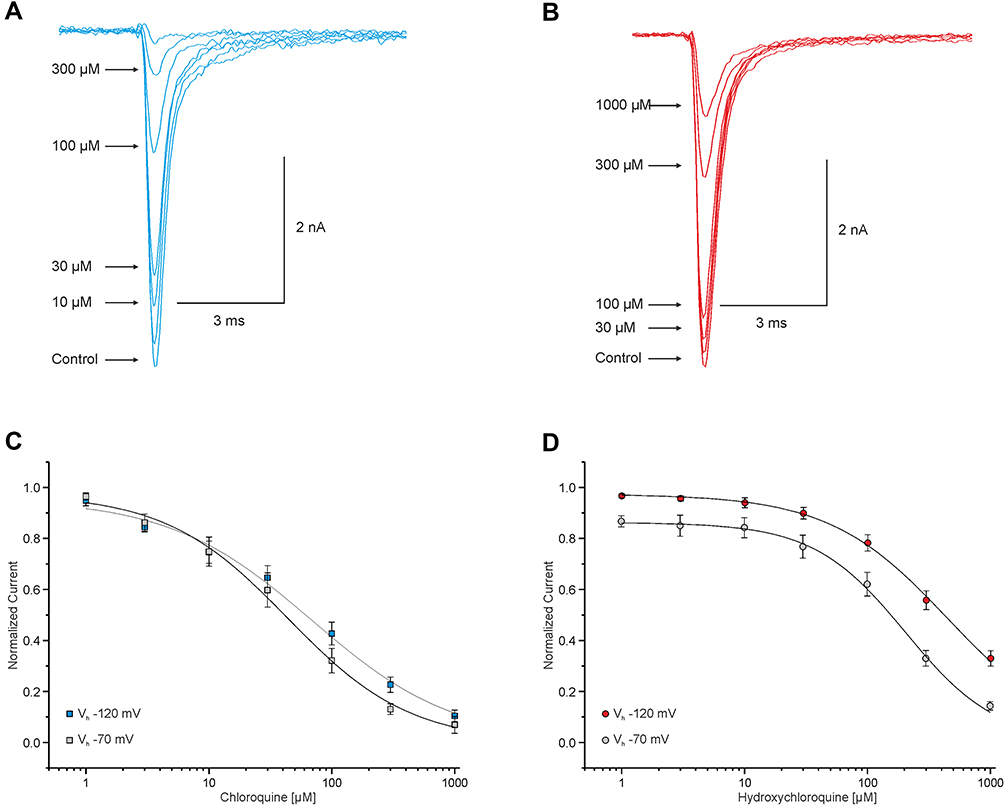 |
Figure 1 Chloroquine and hydroxychloroquine inhibit Nav1.5. (A and B) Representative traces of currents generated by Na+ channels in HEK-293 cells. These currents were triggered through 20 ms test pulses from −120 to 0 mV in intervals of 10s and showing an enhancement of inhibition corresponding to the increasing concentration of CQ (A) and HCQ (B). (C and D) Concentration-dependent block of resting and inactivated Nav1.5 channels. Peak amplitudes of Na+ currents at different concentrations were normalized with respect to the peak amplitude in control solution and plotted against the concentration of CQ (C) and HCQ (D). Data were fitted with the Hill equation as indicated by the solid line. |
Chloroquine and Hydroxychloroquine Induce a Use-Dependent Block of Nav1.5
The use-dependent increase in the affinity of LA upon voltage-gated Na+ channels, leading to a use-dependent block, explains its clinical importance due to the suppression of hyperexcitability, but could also be relevant with regard to its cardiotoxicity.30,31 We questioned if CQ and HCQ induce a use-dependent block of Nav1.5 comparable to LA. Both drugs induce a use-dependent block if Nav1.5 was activated with 60 pulses at 10 Hz in cells held at −120 mV. As it is demonstrated in Figure 2, 10 and 100 µM of CQ induced a current inhibition of 18 ± 0.02% (n = 11) and 33 ± 0.03% (n = 9). HCQ induced a significantly weaker use-dependent block compared to CQ with a reduction of 7 ± 0.01% at 10 µM (n = 10) and 18 ± 0.02% (n = 10) at 100 µM (p < 0.001, unpaired t-test).
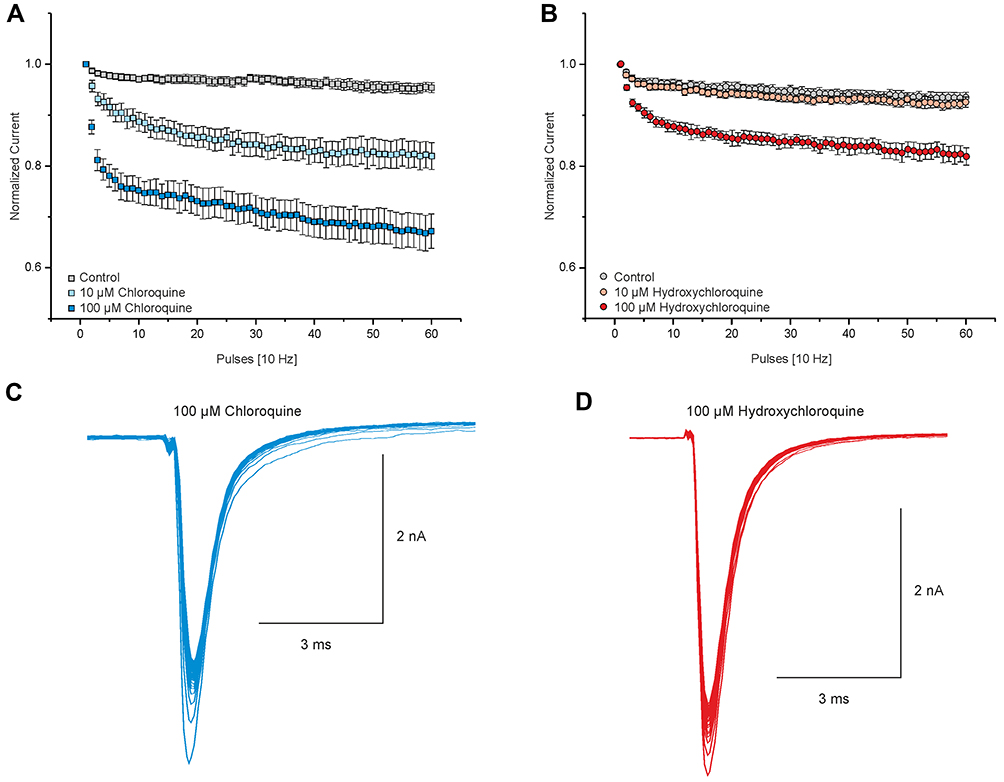 |
Figure 2 Chloroquine and hydroxychloroquine induce a use-dependent block of Nav1.5. (A and B) Displaying the course of the use-dependent block of Nav1.5 by 10 and 100 µM CQ (A) and HCQ (B). Peak currents were normalized to the amplitude of the first pulse and plotted against the pulse number. Currents were induced by 60 test pulses lasting 100 ms, and cells were held at −120 mV. (C and D) Representative current traces of Nav1.5 activated at 10 Hz in the presence of 100 μM CQ (C) and HCQ (D). |
Chloroquine and Hydroxychloroquine Stabilize Fast Inactivation of Nav1.5
Various agents with LA-like effects modify the inactivation properties of Nav1.5 channels.19,28 Here, were characterized the effects of CQ and HCQ on fast inactivation. Fast inactivation was induced by 50 ms pre-pulses ranging from −120 to 0 mV in steps of 5 mV. The remaining fraction of available channels was examined with a 20 ms long pre-pulse to 0 mV. Figure 3 demonstrates that CQ induced a concentration-dependent shift of the steady-state fast inactivation from V0.5 of −85 ± 0.6 mV (n = 9) in control to V0.5 of −96 ± 1 mV (n = 9) with 10 µM and −102 ± 3 mV (n = 11) with 100 µM. The effects induced by HCQ were similar to CQ (control: −86 ± 0.5 mV, n = 9; 10 µM: −95 ± 0.5 mV, n = 9; 100 μM: −100 ± 1 mV, n = 9). The effect of CQ (100 μM) was not significantly different when compared to HCQ (100 μM) (p = 0.59, unpaired t-test).
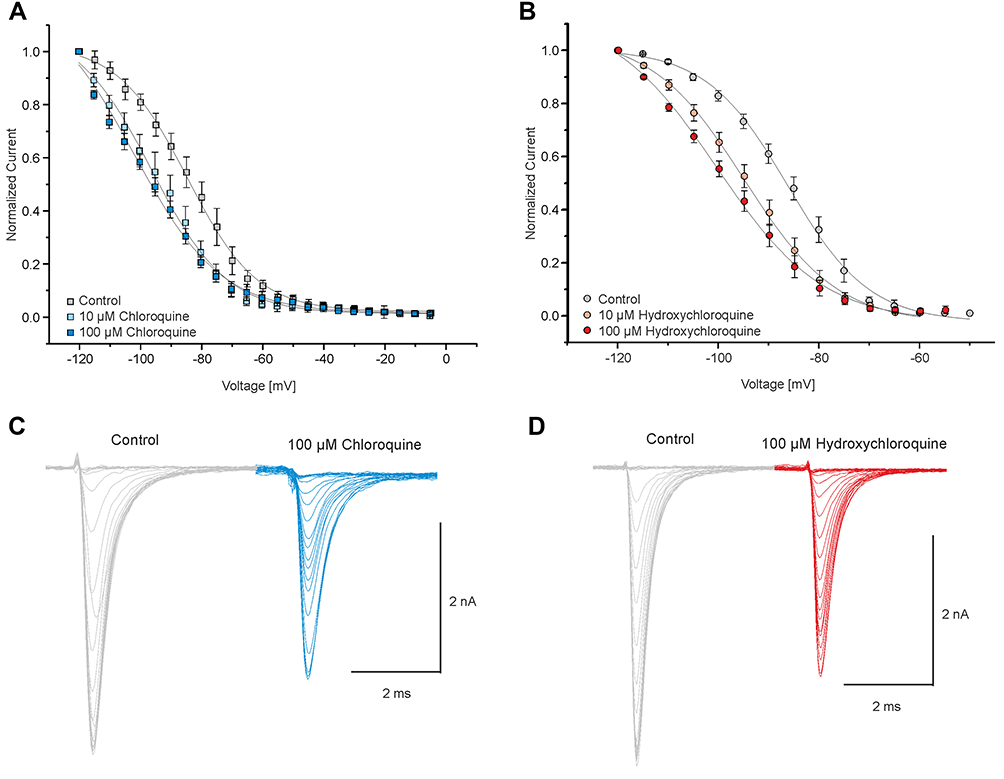 |
Figure 3 Chloroquine and hydroxychloroquine stabilize the fast inactivation state of Nav1.5. To induce the fast inactivation state, cells were exposed to 50-ms-long pre-pulses ranging from −120 to 0 mV in steps of 5 mV and the respective fraction of non-inactivated available channels was tested with a 20-ms-long pre-pulse to 0 mV. (A and B) Voltage-dependency of fast inactivation of Nav1.5 in the presence of control solution and 10 and 100 µM CQ (A) and HCQ (B). Solid lines represent fits obtained with the Boltzmann equation. (C and D) Representative current traces of Nav1.5 each in the presence of 100 μM CQ (C) and HCQ (D) with their corresponding controls. |
Chloroquine and Hydroxychloroquine Do Not Change the Proportion of the Fast and Slow Recovery
LAs are known to exert certain cardiotoxic properties. Previous studies revealed that one of these possible cardiotoxic effects are likely based on a slow dissociation of LAs like bupivacaine and amitriptyline from inactivated channels.26,32,33 Accordingly, we investigated if CQ and HCQ share this potential cardiotoxic property with LAs. The time course of recovery from the inactivated state was explored in the presence of 10 and 100 µM CQ or HCQ. Recovery from inactivation was explored via a two-pulse protocol consisting of a 10-s-long inactivating pulse at −70 mV followed by a test pulse to 0 mV whereby the interval between these pulses varied between 0 and 7.3 s.27,28 As shown in Figure 4, a biphasic course was obtained that was best fitted with a double exponential fit giving two-time constants (τ1 and τ2). In the presence of a drug, τ1 represents recovery of unblocked channels from a fast inactivation state. τ2 represents recovery of inactivated channels that were blocked during the 10-s pre-pulse, including rebinding and dissociation from resting channels and recovery from the slow inactivation state.19,28,33 Application of CQ resulted in a prolongation of both τ1 (control: 9 ± 0.8 ms, n = 10; 10 µM: 37 ± 2 ms; n = 9; 100 µM: 47 ± 5 ms, n = 7) and τ2 (control: 379 ± 125 ms, n = 10; 10 µM: 484 ± 67 ms; n = 9; 100 µM: 633 ± 367 ms, n = 7). Administration of HCQ in the same concentrations resulted in a modest prolongation of τ1 (control: 6 ± 0.4, n = 9 ms; 10 µM: 11 ± 0.8 ms; n = 9; 100 µM: 16 ± 1 ms, n = 8). However, HCQ did not prolong τ2 (control: 368 ± 108 ms, n = 9; 10 µM: 212 ± 63 ms; n = 9; 100 µM: 229 ± 89 ms, n = 8). In contrast to bupivacaine that unbinds slowly from inactivated channels and therefore increases the fraction of channels recovering with a slow time constant in this protocol, CQ and HCQ did not markedly alter the fraction of channels recovering with τ1 (~80–85%) and τ2 (15–20%).
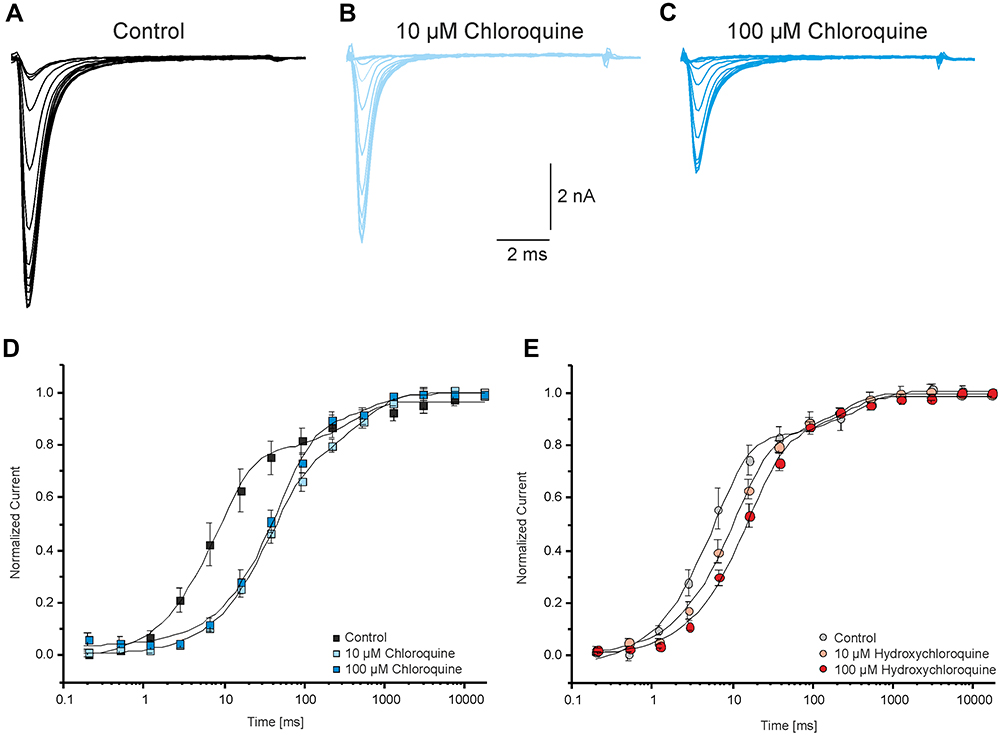 |
Figure 4 Chloroquine and hydroxychloroquine do not change the proportion of the fast and slow recovery. Recovery was explored using a two-pulse paradigm consisting of a 10-s-long inactivating pulse at −70 mV followed by a test pulse to 0 mV. The interval between these pulses varied between 0 and 7.3 s. (A-C) Representative traces of currents generated by Nav1.5 without CQ (A) and in the presence of 10µM (B) and 100µM (C) CQ respectively. (D and E) Time course for the recovery from inactivation in control solution and CQ (D) or HCQ (E). The peak current amplitudes were normalized to the first pulse and plotted against the duration of the interval between both pulses. The time course of recovery from inactivation was best fitted with a double exponential function revealing two-time constants. |
Inhibition of Nav1.5 by Chloroquine and Hydroxychloroquine May Involve the Proposed Binding Sites for Local Anesthetics
Drugs modulate the activity of Nav1.5 by multiple mechanisms. As CQ and HCQ produce a state-dependent inhibition of Nav1.5 which is very similar to the effects of classical LAs, we investigated if inhibition induced by CQ and HCQ involves the established LA binding site of Nav1.5. While multiple residues seem to dictate inhibition by LAs, replacement of F1760 is commonly performed in order to investigate the role of the “LA-binding site”.34–36 Therefore, we investigated the LA-insensitive mutant channel Nav1.5-F1760A as described by Nau et al.33 As displayed in Figure 5, in the presence of CQ Na+ currents generated by Nav1.5-F1760A channels exhibited a significantly weaker concentration-dependent tonic block as compared to wild-type Nav1.5 represented by an approximately fivefold higher IC50 value (IC50 330 ± 14 μM; Hill coefficient 0.96 ± 0.01, n = 9; p < 0.01, unpaired t-test). In contrast to CQ, raising concentrations of HCQ exhibited only a slightly attenuated concentration-dependent tonic block and the corresponding IC50 value was calculated to 666 ± 72 μM (Hill coefficient 0.99 ± 0.02, n = 8; p = 0.17, unpaired t-test). For comparison to the wild-type Nav1.5 channel, the IC50 value for HCQ was calculated to 446 ± 50 μM. Moreover, in the presence of 10 µM and 100 µM CQ the use-dependent block protocol performed with 10 Hz resulted in less inhibition of Nav1.5-F1760A Na+ currents (10 µM: 2.07 ± 0.01%, n=6; 100 μM: 10 ± 0.03%; n = 9; p < 0.01, unpaired t-test) while application of HCQ led to an almost abrogated use-dependent block (100 μM: 0.4 ± 0.02%; n = 8; p < 0.01, unpaired t-test) as compared to wild-type Nav1.5. This experiment indicates that both drugs employ the established LA binding site in order to inhibit Nav1.5.
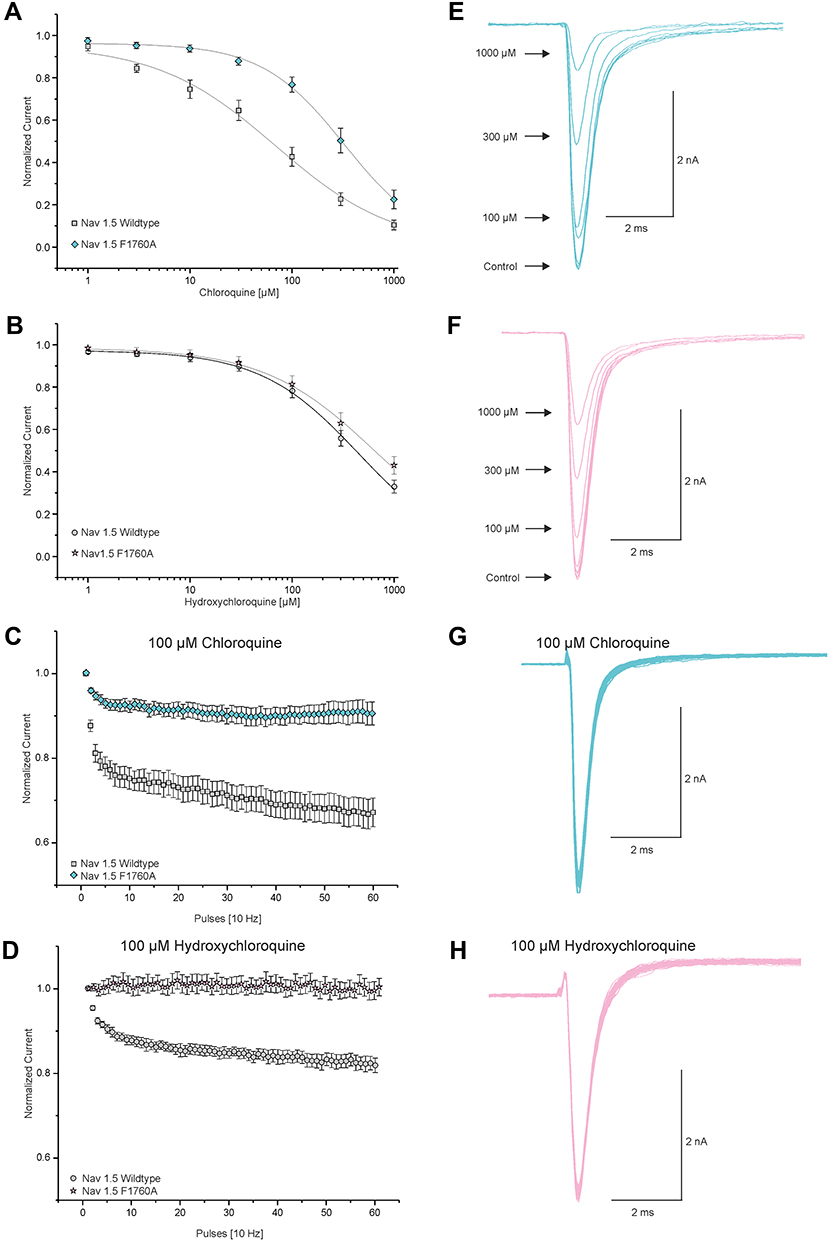 |
Figure 5 Chloroquine and hydroxychloroquine may interact with the proposed LA-binding site of Nav1.5. (A and B) Currents of resting Nav1.5 wild-type and Nav1.5-F1760A channels were triggered by 20 ms test pulses from −120 to 0 mV in intervals of 10s to receive dose–response curves for tonic block by CQ (A) and HCQ (B). Peak amplitudes of Na+ currents at different drug concentrations were normalized with respect to the peak amplitude in control solution and plotted against the concentration of CQ (A) and HCQ (B). Data were fitted with the Hill equation as indicated by the solid line. (C and D) Development of use-dependent block of Nav1.5 wild-type and Nav1.5-F1760A channels by 100 µM CQ (C) and 100 µM HCQ (D). Peak currents were normalized to the amplitude of the first pulse and plotted against the pulse number. (E and F) Representative traces of currents generated by Nav1.5-F1760A for the dose–response curves of CQ (E) and HCQ (F). (G and H) Representative current traces of Nav1.5-F1760A activated at 10 Hz in the presence of 100 μM CQ (G) and HCQ (H). |
Extracellular Alkalinisation Potentiates Inhibition of Nav1.5 by Chloroquine and Hydroxychloroquine
LAs interact with intracellular residues of Na+ channels.37 As LAs are weak bases, alkalinization of the LA-solution increases inhibition of sodium channels due to an increased membrane-permeability.38 To test if this mode of accessibility also applies for the weak bases CQ and HCQ, the pH-value of the extracellular medium was set to 9.0. Indeed, tonic block of resting Nav1.5 channels was increased by alkalization. The IC50 value for CQ was calculated to 6.8 ± 0.9 μM (Hill coefficient 0.9 ± 0.04, n = 9) and for HCQ to 8.3 ± 0.4 μM (Hill coefficient 1.1 ± 0.04, n = 9).
Discussion
CQ and HCQ have been used for the prophylaxis and treatment of malaria for decades. Furthermore, both substances have been established as disease-modifying antirheumatic drugs in the treatment of different rheumatic and immunological diseases due to their immunomodulatory and immunosuppressive characteristics. However, serious side effects accompany usage of CQ and HCQ, ranging from retinopathy, methemoglobinemia or acute kidney injury to life-threatening cardiac arrhythmia.11 Especially cardiovascular side effects linked to their administration are reported and frequently the primary cause of mortality.39–41
CQ and HCQ are considered as Vaughan Williams Class Ia antiarrhythmics because of their structural similarity and comparable antiarrhythmic properties to quinidine.41,42 Class I agents inhibit both peak and late sodium currents by blocking Nav1.5, a feature previously described for CQ and HCQ.21,43,44 Blockade of Nav1.5 is associated with an intracardiac conduction delay and thus, depending on the extent of the propagation delay, with an elongation of the QRS complex in the electrocardiogram.45,46 Drug-induced QRS prolongation due to Nav1.5 inhibition which slows the rate of cardiac depolarization enables life-threatening ventricular arrhythmias and even sudden cardiac death, thus contributing to morbidity and mortality associated with Nav1.5 blockers.46,47 In terms of cardiac toxicity, Nav1.5 is a critical target for LAs.25,28 Previously, for other cardiotoxic drugs with arrhythmogenic properties including buprenorphine, methadone and amitriptyline, LA-like effects on Nav1.5 were described.19,22,29,32,48 In fact, CQ exerts an LA-like pain suppressing effect when injected intradermally or subcutaneously, thus providing immediate analgesia like classical LAs, indicating possible LA-like effects also on Nav1.5.49 This in vitro study was designed to explore whether CQ and HCQ exert LA-like effects on the cardiac voltage-gated Na+ channel Nav1.5 as a feasible target for their proarrhythmic properties. We were able to demonstrate that both drugs are concentration- and state-dependent inhibitors of Nav1.5. Furthermore, the LA-insensitive mutant Nav1.5-F1760A displayed a reduced sensitivity to both substances. Thus, our data suggest that CQ and HCQ indeed have LA-like properties and even employ the proposed intracellular LA-binding site. However, prolongation of the QT interval leading to acquired LQTS has been primarily explained by the interaction between CQ and HCQ with cardiac hERG K+ channels and not linked to Nav1.5.41,42,46 Furthermore, various sodium channel blockers like the LA lidocaine, the antiarrhythmics mexiletine and flecainide, but also CQ, have been shown to suppress persistent sodium currents which are associated with congenital LQTS-type-3 and thereby may partially offset the QT prolongation that is induced through blockade of hERG K+ channels.42,50 In addition, in an analysis of pooled congenital LQTS patients by Yang et al mexiletine and flecainide as representatives of class Ib and Ic antiarrhythmics, significantly shortened the corrected QT time.51 Although this property of various sodium channel blockers mediate an antiarrhythmic effect, the use of CQ and HCQ is accompanied by a widened QRS complex and a prolongation of the QT interval as a commonly reported side effect, as recently reported in patients with COVID-19 and concomitant use of CQ or HCQ.52,53 Accordingly, in contrast to class Ib and Ic antiarrhythmics, the clinical significance of blocking late sodium currents in terms of QT interval shortening by both agents seems to be negligible. Moreover, it should be considered that Nav1.5 blockade by CQ and HCQ could secondarily affect repolarization via a delay in the course of depolarization, thus rather contributing to a prolonged QT interval.45 Hence, our results provide a more detailed understanding of how modulation of Nav1.5 channels by CQ and HCQ might contribute to their arrhythmogenic effects.
LAs inhibit Na+ channels by locking the pore from the intracellular side during the open state and are therefore described as use-dependent open state block.37 At physiological pH-values, most LAs are predominantly positively charged due to protonation and have to be deprotonated into their neutral form before crossing the cell membrane. Alkalization of the extracellular medium changes the ratio between the two states in favor of the deprotonated form leading to an increased membrane permeability and pharmacological effect.38,54 The IC50 values for inhibition of Nav1.5 by CQ and HCQ were reduced as well, supporting our notion that both substances inhibit Nav1.5 by binding to an intracellular site. Along with the findings that CQ and HCQ induce a use-dependent block and may bind to the proposed binding sites for LA, these results suggest that both agents have to be deprotonated to pass the cell membrane before reaching their binding site.
CQ and HCQ have similar pharmacokinetics. After application, sequestration across all tissues is an important attribute, whereby only clinically unimportant differences in tissue distribution of CQ and HCQ were observed.55,56 Preclinical experiments revealed that the tissue/plasma CQ-concentration ratio in the heart after an intraperitoneal dose vary between 6.8 after 1 hour and 20 after 4 hours, indicating that CQ-concentrations are higher in heart tissue than in the plasma.57 These results were confirmed in different animal and human studies.56 Steady-state plasma concentration of CQ during therapy of rheumatoid arthritis generates plasma concentrations of 1–3μmol/L.58 In patients with long-term use of CQ, serum concentrations above 2.5μmol/L are frequently associated with typical side effects.59 HCQ was reported to be less toxic but safety concerns have also been raised for cardiac toxicity.7 However, a meta-analysis of randomized trials found an increased mortality in HCQ-treated groups for therapy of COVID-19.60 Due to their comparable tissue distribution and reported cardiac toxicity in prevention and treatment of COVID-19, clinically relevant levels of CQ and HCQ seems to be close to the concentration range of CQ and HCQ which may cause effects on Nav1.5.
In general, the IC50 value is an accepted value to describe the inhibitory potency of a substance. The IC50 values for resting Nav1.5 channels yielded from our experiments were about 69 μM for CQ and 446 μM for HCQ. Jordaan et al found somewhat divergent IC50 values for CQ (8 µM) and HCQ (>300 µM).52 In contrast, Thomet et al stated that on ATX-II-induced non-inactivating persistent currents of Nav1.5, inhibition by CQ and HCQ gave IC50 values of 159 μM and 96 μM respectively.21 The IC50 values for the rapid component of the delayed rectifier potassium current mediated by hERG K+ channels are considerably lower (2–8 μM for CQ and 3–10 μM for HCQ).21,52,61,62 However, the sole validity of IC50 values for predicting proarrhythmic risks due to inhibition of hERG K+ channels have been challenged, noting that they do not account for dynamic time- and state-dependent drug–channel interactions. Furthermore, the IC50 value is not necessarily related to the arrhythmogenic potential.63,64 Both CQ and HCQ also inhibit L-type calcium currents with IC50 values of 3–30 μM and 8–90 μM, respectively.21,52 Kir2.1, generating the rectifying potassium current, is also blocked by both agents with IC50 values of 6 μM for CQ and 30 μM for HCQ.21 Taken together, different studies confirm that both CQ and HCQ inhibit several cardiac ion channels with potential significance for cardiac arrhythmias.
Resting Nav1.5 channels appear to require high concentrations of both substances to get inhibited, thus the relevance for their arrhythmogenic potential remains unclear. Resting potentials in mammalian hearts depend on the type of cells and varies between −50 mV in sinuatrial node cells and −90 mV in ventricular cells.65 Thus, recording currents of resting channels held at hyperpolarized potentials of −120 mV as applied in this study is artificial and not representative for the membrane properties of cardiomyocytes in vivo.28 Since the physiological transmembrane resting potential of mammalian hearts lays around −90 mV in ventricular cells and, due to an ongoing cardiac activity, different conformational states of Nav1.5, the clinical relevance of inhibitors should also be measured from recordings on inactivated channels.19,23,28 Indeed, our data show that both CQ and HCQ exert an LA-like high-affinity block of Nav1.5 channels due to an enhanced binding to the inactivated state. Another important property of LA regarding their cardiotoxicity may be use-dependent inhibition. For example, bupivacaine known for its high cardiotoxic potential induces a very strong use-dependent block.28,33 Sánchez-Chapula et al demonstrated that CQ blocked several ion currents in primary feline ventricular myocytes, including Na+ currents. However, 10 µM CQ did not induce a use-dependent block.20 In contrast, our experiments suggest that CQ and HCQ induce a modest use-dependent block of Nav1.5 using a frequency concordant to previous studies.19,66 Furthermore, CQ induced a stronger use-dependent block as compared to HCQ. This difference between CQ and HCQ correlates with their different cardiotoxicities. Nevertheless, both agents induce less use-dependent block compared to bupivacaine and other LAs but showing similar effects to methadone which was reported to have relevant LA-like properties.28,29,67 In addition to the use-dependent block as a surrogate for an increasing affinity of Nav1.5 channels towards an agent as a response to repetitive depolarizations, substance-specific kinetics are responsible for the duration of their blocking capabilities. Fast onset of inhibition and slow dissociation from inactivated cardiac Na+ channels of bupivacaine and amitriptyline led to the generally used classification of “fast in” and “slow out” blockers.26,28,32 Due to slow dissociation of agents with inhibiting properties from inactivated Na+ channels, the vast majority of channels may remain in an inactive state between two action potentials, resulting in an accumulation of agent-bound inactivated channels and finally to cardiac arrhythmias.19 However, neither CQ nor HCQ exhibit effects similar to those of bupivacaine or amitriptyline. CQ and HCQ only exhibit a modest change of the proportion of the fast and slow recovery components. This indicates that both CQ and HCQ appear to dissociate quite fast from inactivated channels, a property they share for example with lidocaine and therefore do not readily explain their cardiotoxicity.
Conclusion
Both CQ and HCQ can cause ventricular arrhythmias and sudden cardiac death by affecting ventricular depolarization and repolarization, which manifest as prolongation of the QRS complex and the QT interval, latter classified as acquired LQTS. Furthermore, several studies suggest that these mechanisms are caused by a multiple ion channel block in cardiac cells. In this study, it was demonstrated that both agents modify Nav1.5 and exhibit LA-like properties. Our experiments on LA-insensitive Nav1.5-F1760A channels suggest that CQ and HCQ bind to the proposed LA-binding site to inhibit Nav1.5. We conclude that this study identified an additional molecular mode of action for the proarrhythmic properties of CQ and HCQ.
Abbreviations
LA, local anesthetic; hERG, human ether-à-go-go-related gene; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; COVID-19, coronavirus disease 2019; FDA, US Food and Drug Administration; LQTS, long-QT syndrome; HEK-293 cells, human embryonic kidney-293 cells.
Acknowledgment
The authors gratefully acknowledge Heike Buerger (Anesthesiology, Hannover) for the excellent assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tanenbaum L.; Antimalarial agents. Antimalarial agents. Arch Dermatol. 1980;116(5):587–591. doi:10.1001/archderm.1980.01640290097026
2. Al-Bari MAA. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J Antimicrob Chemother. 2015;70(6):1608–1621. doi:10.1093/jac/dkv018
3. Naarding MA, Baan E, Pollakis G, Paxton WA. Effect of chloroquine on reducing HIV-1 replication in vitro and the DC-SIGN mediated transfer of virus to CD4+ T-lymphocytes. Retrovirology. 2007;4(1):6. doi:10.1186/1742-4690-4-6
4. Dos Reis Neto ET, Kakehasi AM, Medeiros Pinheiro M, et al. Revisiting hydroxychloroquine and chloroquine for patients with chronic immunity-mediated inflammatory rheumatic diseases. Adv Rheumatol. 2020;60(1):32. doi:10.1186/s42358-020-00134-8
5. Plantone D, Koudriavtseva T. Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin Drug Investig. 2018;38(8):653–671. doi:10.1007/s40261-018-0656-y
6. Kumar A, Liang B, Aarthy M, et al. Hydroxychloroquine inhibits zika virus NS2B-NS3 protease. ACS Omega. 2018;3(12):18132–18141. doi:10.1021/acsomega.8b01002
7. Oscanoa TJ, Romero-Ortuno R, Carvajal A, Savarino A. A pharmacological perspective of chloroquine in SARS-CoV-2 infection: an old drug for the fight against a new coronavirus? Int J Antimicrob Agents. 2020;56(3):106078. doi:10.1016/j.ijantimicag.2020.106078
8. Hashem AM, Alghamdi BS, Algaissi AA, et al. Therapeutic use of chloroquine and hydroxychloroquine in COVID-19 and other viral infections: a narrative review. Travel Med Infect Dis. 2020;35:101735. doi:10.1016/j.tmaid.2020.101735
9. Khuroo MS. Chloroquine and hydroxychloroquine in coronavirus disease 2019 (COVID-19). Facts, fiction and the hype: a critical appraisal. Int J Antimicrob Agents. 2020;56(3):106101. doi:10.1016/j.ijantimicag.2020.106101
10. Claudia E, Fabio C, Sarah E, Mantovani A, Maurizio C. Potential harm caused by physicians’ a-priori beliefs in the clinical effectiveness of hydroxychloroquine and its impact on clinical and economic outcome – a simulation approach. J Crit Care. 2020;1:85.
11. Perez J, Roustit M, Lepelley M, Revol B, Cracowski J-L, Khouri C. Reported adverse drug reactions associated with the use of hydroxychloroquine and chloroquine during the COVID-19 pandemic. Ann Intern Med. 2021. doi:10.7326/M20-7918
12. Food and Drug Administration. Memorandum explaining basis for revocation of emergency use authorization for emergency use of chloroquine phosphate and hydroxychloroquine Sulfate. Available from: https://www.fda.gov/media/138945/download.
13. Singh AP, Tousif S, Umbarkar P, Lal H. A pharmacovigilance study of hydroxychloroquine cardiac safety profile: potential implication in COVID-19 mitigation. J Clin Med. 2020;9(6). doi:10.3390/jcm9061867
14. Cohen IV, Makunts T, Moumedjian T, Issa MA, Abagyan R. Cardiac adverse events associated with chloroquine and hydroxychloroquine exposure in 20 years of drug safety surveillance reports. Sci Rep. 2020;10(1):19199. doi:10.1038/s41598-020-76258-0
15. Food and Drug Administration. FDA cautions against use of hydroxychloroquine or chloroquine for COVID-19 outside of the hospital setting or a clinical trial due to risk of heart rhythm problems. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/OSE%20Review_Hydroxychloroquine-Cholorquine%20-%2019May2020_Redacted.pdf.
16. Jankelson L, Karam G, Becker ML, Chinitz LA, Tsai M-C. QT prolongation, torsades de pointes, and sudden death with short courses of chloroquine or hydroxychloroquine as used in COVID-19: a systematic review. Heart Rhythm. 2020;17(9):1472–1479. doi:10.1016/j.hrthm.2020.05.008
17. El-Sherif N, Turitto G, Boutjdir M. Acquired long QT syndrome and electrophysiology of Torsade de Pointes. Arrhythm Electrophysiol Rev. 2019;8(2):122–130. doi:10.15420/aer.2019.8.3
18. Traebert M, Dumotier B, Meister L, Hoffmann P, Dominguez-Estevez M, Suter W. Inhibition of hERG K+ currents by antimalarial drugs in stably transfected HEK293 cells. Eur J Pharmacol. 2004;484(1):41–48. doi:10.1016/j.ejphar.2003.11.003
19. Schulze V, Stoetzer C, O’Reilly AO, et al. The opioid methadone induces a local anaesthetic-like inhibition of the cardiac Na+ channel, Na(v)1.5. Br J Pharmacol. 2014;171(2):427–437. doi:10.1111/bph.12465
20. Sánchez-Chapula JA, Salinas-Stefanon E, Torres-Jácome J, Benavides-Haro DE, Navarro-Polanco RA. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J Pharmacol Exp Ther. 2001;297(1):437–445.
21. Thomet U, Amuzescu B, Knott T, Mann SA, Mubagwa K, Radu BM. Assessment of proarrhythmogenic risk for chloroquine and hydroxychloroquine using the CiPA concept. Eur J Pharmacol. 2021;913:174632. doi:10.1016/j.ejphar.2021.174632
22. Leffler A, Frank G, Kistner K, et al. Local anesthetic-like inhibition of voltage-gated Na(+) channels by the partial μ-opioid receptor agonist buprenorphine. Anesthesiology. 2012;116(6):1335–1346. doi:10.1097/ALN.0b013e3182557917
23. Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201(1):1–8. doi:10.1007/s00232-004-0702-y
24. Siebrands CC, Schmitt N, Friederich P. Local anesthetic interaction with human ether-A-go-go-related gene (HERG) channels: role of aromatic amino acids Y652 and F656. Anesthesiology. 2005;103(1):102–112. doi:10.1097/00000542-200507000-00017
25. Butterworth JF. Models and mechanisms of local anesthetic cardiac toxicity: a review. Reg Anesth Pain Med. 2010;35(2):167–176. doi:10.1097/aap.0b013e3181d231b9
26. Clarkson CW, Hondeghem LM. Mechanism for bupivacaine depression of cardiac conduction. Anesthesiology. 1985;62(4):396–405. doi:10.1097/00000542-198504000-00006
27. Stoetzer C, Papenberg B, Doll T, et al. Differential inhibition of cardiac and neuronal Na(+) channels by the selective serotonin-norepinephrine reuptake inhibitors duloxetine and venlafaxine. Eur J Pharmacol. 2016;783:1–10. doi:10.1016/j.ejphar.2016.04.051
28. Stoetzer C, Reuter S, Doll T, Foadi N, Wegner F, Leffler A. Inhibition of the cardiac Na+ channel α-subunit Nav1.5 by propofol and dexmedetomidine. Naunyn Schmiedebergs Arch Pharmacol. 2016;389(3):315–325. doi:10.1007/s00210-015-1195-1
29. Stoetzer C, Kistner K, Stüber T, et al. Methadone is a local anaesthetic-like inhibitor of neuronal Na+ channels and blocks excitability of mouse peripheral nerves. Br J Anaesth. 2015;114(1):110–120. doi:10.1093/bja/aeu206
30. Huang C-J, Schild L, Moczydlowski EG. Use-dependent block of the voltage-gated Na(+) channel by tetrodotoxin and saxitoxin: effect of pore mutations that change ionic selectivity. J General Physiol. 2012;140(4):435–454. doi:10.1085/jgp.201210853
31. Nadrowitz F, Stoetzer C, Foadi N, et al. The distinct effects of lipid emulsions used for “lipid resuscitation” on gating and bupivacaine-induced inhibition of the cardiac sodium channel Nav1.5. Anesth Analg. 2013;117(5):1101–1108. doi:10.1213/ANE.0b013e3182a1af78
32. Nau C, Seaver M, Wang SY, Wang GK. Block of human heart hH1 sodium channels by amitriptyline. J Pharmacol Exp Ther. 2000;292(3):1015–1023.
33. Nau C, Wang SY, Strichartz GR, Wang GK. Block of human heart hH1 sodium channels by the enantiomers of bupivacaine. Anesthesiology. 2000;93(4):1022–1033. doi:10.1097/00000542-200010000-00026
34. Beyder A, Strege PR, Bernard C, Farrugia G. Membrane permeable local anesthetics modulate Na(V)1.5 mechanosensitivity. Channels. 2012;6(4):308–316. doi:10.4161/chan.21202
35. Mike A, Lukacs P. The enigmatic drug binding site for sodium channel inhibitors. CMP. 2010;3(3):129–144. doi:10.2174/1874467211003030129
36. Carboni M, Zhang Z-S, Neplioueva V, Starmer CF, Grant AO. Slow sodium channel inactivation and use-dependent block modulated by the same domain IV S6 residue. J Membr Biol. 2005;207(2):107–117. doi:10.1007/s00232-005-0805-0
37. van Theemsche KM, van de Sande DV, Snyders DJ, Labro AJ. Hydrophobic drug/toxin binding sites in voltage-dependent K+ and Na+ channels. Front Pharmacol. 2020;11:735. doi:10.3389/fphar.2020.00735
38. Chernoff DM, Strichartz GR. Kinetics of local anesthetic inhibition of neuronal sodium currents. pH and hydrophobicity dependence. Biophys J. 1990;58(1):69–81. doi:10.1016/S0006-3495(90)82354-7
39. Chen C-Y, Wang F-L, Lin -C-C. Chronic hydroxychloroquine use associated with QT prolongation and refractory ventricular arrhythmia. Clin Toxicol. 2006;44(2):173–175. doi:10.1080/15563650500514558
40. Morgan ND, Patel SV, Dvorkina O. Suspected hydroxychloroquine-associated QT-interval prolongation in a patient with systemic lupus erythematosus. J Clin Rheumatol. 2013;19(5):286–288. doi:10.1097/RHU.0b013e31829d5e50
41. Lebin JA, LeSaint KT. Brief review of chloroquine and hydroxychloroquine toxicity and management. West J Emerg Med. 2020;21(4):760–763. doi:10.5811/westjem.2020.5.47810
42. Mubagwa K. Cardiac effects and toxicity of chloroquine: a short update. Int J Antimicrob Agents. 2020;56(2):106057. doi:10.1016/j.ijantimicag.2020.106057
43. Osadchii OE. Effects of Na+ channel blockers on the restitution of refractory period, conduction time, and excitation wavelength in perfused Guinea-pig heart. PLoS One. 2017;12(2):e0172683. doi:10.1371/journal.pone.0172683
44. Burashnikov A, Antzelevitch C. Role of late sodium channel current block in the management of atrial fibrillation. Cardiovasc Drugs Ther. 2013;27(1):79–89. doi:10.1007/s10557-012-6421-1
45. Colunga S, Padrón R, García-Iglesias D, et al. The QT interval dynamic in a human experimental model of controlled heart rate and QRS widening. J Clin Med. 2019;8(9):845. doi:10.3390/jcm8091417
46. Harmer AR, Valentin J-P, Pollard CE. On the relationship between block of the cardiac Na+ channel and drug-induced prolongation of the QRS complex. Br J Pharmacol. 2011;164(2):260–273. doi:10.1111/j.1476-5381.2011.01415.x
47. Heist EK, Ruskin JN. Drug-induced arrhythmia. Circulation. 2010;122(14):1426–1435. doi:10.1161/CIRCULATIONAHA.109.894725
48. O’Reilly AO, Eberhardt E, Weidner C, Alzheimer C, Wallace BA, Lampert A. Bisphenol A binds to the local anesthetic receptor site to block the human cardiac sodium channel. PLoS One. 2012;7(7):e41667. doi:10.1371/journal.pone.0041667
49. Mandel EH. A new local anesthetic with anticoagulant properties, chloroquine (aralen) dihydrochloride. Arch Dermatol. 1960;81:260–263. doi:10.1001/archderm.1960.03730020096015
50. Johannesen L, Vicente J, Mason JW, et al. Late sodium current block for drug-induced long QT syndrome: results from a prospective clinical trial. Clin Pharmacol Ther. 2016;99(2):214–223. doi:10.1002/cpt.205
51. Yang Y, Lv -T-T, Li SY, Zhang P. Sodium channel blockers in the management of long QT syndrome types 3 and 2: a system review and meta-analysis. J Cardiovasc Electrophysiol. 2021;32(11):3057–3067. doi:10.1111/jce.15223
52. Jordaan P, Dumotier B, Traebert M, et al. Cardiotoxic potential of hydroxychloroquine, chloroquine and azithromycin in adult human primary cardiomyocytes. Toxicol Sci. 2021;180(2):356–368. doi:10.1093/toxsci/kfaa194
53. Becker ML, Snijders D, van Gemeren CW, Kingma HJ, van Lelyveld SFL, Giezen TJ. QTc Prolongation in COVID-19 patients using chloroquine. Cardiovasc Toxicol. 2021;21(4):314–321. doi:10.1007/s12012-020-09621-2
54. Lenkey N, Karoly R, Kiss JP, Szasz BK, Vizi ES, Mike A. The mechanism of activity-dependent sodium channel inhibition by the antidepressants fluoxetine and desipramine. Mol Pharmacol. 2006;70(6):2052–2063. doi:10.1124/mol.106.026419
55. Schultz KR, Gilman AL. The lysosomotropic amines, chloroquine and hydroxychloroquine: a potentially novel therapy for graft-versus-host disease. Leuk Lymphoma. 1997;24(3–4):201–210. doi:10.3109/10428199709039008
56. Browning DJ. Hydroxychloroquine and Chloroquine Retinopathy. New York, NY: Springer New York; 2014.
57. Adelusi SA, Salako LA. Kinetics of the distribution and elimination of chloroquine in the rat. General Pharmacol. 1982;13(5):433–437. doi:10.1016/0306-3623(82)90110-0
58. Wollheim FA, Hanson A, Laurell CB. Chloroquine treatment in rheumatoid arthritis. Correlation of clinical response to plasma protein changes and chloroquine levels. Scand J Rheumatol. 1978;7(3):171–176. doi:10.3109/03009747809095649
59. Frisk-Holmberg M, Bergkvist Y, Domeij-Nyberg B, Hellström L, Jansson F. Chloroquine serum concentration and side effects: evidence for dose-dependent kinetics. Clin Pharmacol Ther. 1979;25(3):345–350. doi:10.1002/cpt1979253345
60. Axfors C, Schmitt AM, Janiaud P, et al. Mortality outcomes with hydroxychloroquine and chloroquine in COVID-19 from an international collaborative meta-analysis of randomized trials. Nat Commun. 2021;12(1):2349. doi:10.1038/s41467-021-22446-z
61. Sánchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J Biol Chem. 2002;277(26):23587–23595. doi:10.1074/jbc.M200448200
62. Yu R, Li P. Computational and experimental studies on the inhibitory mechanism of hydroxychloroquine on hERG. Toxicology. 2021;458:152822. doi:10.1016/j.tox.2021.152822
63. Li Z, Dutta S, Sheng J, et al. Improving the in silico assessment of proarrhythmia risk by combining hERG (human ether-à-go-go-related gene) channel-drug binding kinetics and multichannel pharmacology. Circ Arrhythm Electrophysiol. 2017;10(2):e004628. doi:10.1161/CIRCEP.116.004628
64. Kowalska M, Nowaczyk J, Nowaczyk A. KV11.1, NaV1.5, and CaV1.2 transporter proteins as antitarget for drug cardiotoxicity. IJMS. 2020;21(21):8099. doi:10.3390/ijms21218099
65. Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J. 1994;21(1):30–41.
66. Meents JE, Juhasz K, Stölzle-Feix S, Peuckmann-Post V, Rolke R, Lampert A. The opioid oxycodone use-dependently inhibits the cardiac sodium channel NaV 1.5. Br J Pharmacol. 2018;175:3007–3020.
67. Zhang H, Ji H, Liu Z, et al. Voltage-dependent blockade by bupivacaine of cardiac sodium channels expressed in Xenopus oocytes. Neurosci Bull. 2014;30(4):697–710. doi:10.1007/s12264-013-1449-1
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.