")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 19
LncRNA AFAP1-AS1 Induces Gefitinib Resistance of Lung Adenocarcinoma Through the miR-653-5p/AGR2 Axis
Authors Zuo T, Jiang P, Fu J, Zhang Y
Received 10 May 2022
Accepted for publication 25 November 2022
Published 5 January 2023 Volume 2023:19 Pages 1—13
DOI https://doi.org/10.2147/TCRM.S374162
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Deyun Wang
Tao Zuo,1,* Ping Jiang,2,* Jun Fu,1 Yongjian Zhang1
1Department of Thoracic Surgery, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 2Department of Ophthalmology, Zhongnan Hospital of Wuhan University, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tao Zuo, Department of Thoracic Surgery, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, No. 26, Shengli Street, Jiang’an District, Wuhan, People’s Republic of China, Tel +86 15002786691, Email [email protected]
Purpose: Gefitinib resistance limits the therapeutic efficacy of gefitinib to lung adenocarcinoma (LUAD). The goal of this research is to learn more about the lncRNA AFAP1-AS1 and how it functions in gefitinib-resistant LUAD cells.
Methods: RT-qPCR was performed to test the expression of AFAP1-AS1, miR-653-5p and AGR2 in LUAD tissues with acquired resistance to gefitinib or not as well as in gefitinib-resistant LUAD cells. Cell proliferation, invasion and apoptosis were measured by CCK8 assays, transwell invasion assays and flow cytometry, respectively. Luciferase reporter assay showed that miR-653-5p and AFAP1-AS1 or AGR2 interactions.
Results: In gefitinib-resistant LUAD cells and tissues, AFAP1-AS1 was overexpressed. Meanwhile, silencing AFAP1-AS1 reduced proliferation and migration while increasing apoptosis and gefitinib sensitivity. Mechanically, AFAP1-AS1 sequestered the miR-653-5p and blocked the inhibition of miR-653-5p to AGR2 and stepwise upregulated AGR2 overexpression in LUAD gefitinib resistant cells, resulting gefitinib resistance in LUAD.
Conclusion: AFAP1-AS1 promotes gefitinib-resistance LUAD cells through a previously unrecognized miR-653-5p/AGR2 axis, suggesting targeting AFAP1-AS1/miR-653-5p/AGR2 axis might be a promising way for LUAD intervention.
Keywords: lncRNA, gefitinib resistance, competing endogenous RNA, lung adenocarcinoma
Introduction
Lung cancer is a common malignant neoplasm originating from the respiratory system. Lung cancer is the second most prevalent neoplasm, barely behind breast cancer, according to worldwide cancer data from 2020, although it is still the main cause of cancer-related death.1 Lung adenocarcinoma (LUAD) is the principal histological subtype of non-small cell lung cancer (NSCLC) and is usually confirmed at a late stage beyond surgical or chemotherapy treatment.2 Mutations in the epidermal growth factor receptor (EGFR) gene that contribute to oncogene addiction to EGFR have been identified in NSCLC.3 EGFR-tyrosine kinase inhibitors (TKI) showed significant effects on EGFR-mutated LUAD, including gefitinib.4,5 Gefitinib is frequently used to treat LUAD patients acquired chemoresistance in clinical practice.6 Hitherto, the application of gefitinib provides notable therapeutic benefits to LUAD patients. However, acquired resistance resulting from the long-termed application of drugs, including gefitinib might limit the drug efficacies and contributes to unsuccessful treatment.7 Several mechanisms have been suggested to contribute to acquired EGFR-TKI resistance, such as secondary EGFR T790M and MET amplification, and activation of the MET/HGF axis.8–10 In addition, conversion from NSCLC to small-cell lung cancer has also been reported as a possible mechanism of acquired resistance to EGFR-TKI in NSCLC.11 However, acquired resistance to gefitinib has not been confirmed. Therefore, exploring the mechanism behind LUAD resistance to gefitinib is highly required.
Long non-coding RNAs (lncRNAs) refer to non-coding transcripts with >200 bp in size. Initially, lncRNAs were regarded as “junk” because of no protein-coding ability.12 However, the advent of high-throughput technology enables us to recognize the significance of lncRNAs in the complexity of higher organisms.13 They can regulate gene expression on various layers and thereby modulate various cellular biofunctions. The dysregulated lncRNAs underscore their multifaceted biofunction of different pathological processes, including tumor acquisition of therapeutic resistance.14 For example, lncRNA MALAT1 is predominantly expressed in gefitinib-resistant A549 cells.15 LncRNA H19 is delivered to normal lung tumor cells via exosomes and confer them gefitinib resistance.16 Additionally, blockade of oncogenic MAPK cascade by lncRNA ZXF1 silence attenuates cisplatin resistance in lung cancer.17
A study found that the antisense version of lncRNA actin filament-associated protein 1 antisense, RNA 1 (lncRNA AFAP1-AS1) is found to be more common in different types of tumors and help cancers grow and spread, including lung cancer. Importantly, increasing resistance to chemotherapeutic drugs, such as cisplatin, fluorouracil, trastuzumab and paclitaxel was witnessed in several cancers, including non-small cell lung cancer (NSCLC) when AFAP1-AS1 upregulation.18–20 And yet, the bifunctional role of AFAP1-AS1 and its underlying mechanism behind gefitinib-resistant LUAD has not been determined.
It is well documented that lncRNAs perform diverse biofunctions by miRNA sponging activity and modulate specific mRNA expression, consequently affecting tumor growth and development.21 AFAP1-AS1 is a critical player during lung malignancy partially through its competing endogenous RNA (ceRNA) activities where it regulates several miRNA/mRNA axes, including miR-139-5p/Ribonucleoside-diphosphate reductase subunit M2,18 miR-545-3p/hepatoma-derived growth factor axis.22 Therefore, we further deciphered its ceRNA activity in gefitinib-resistant LUAD cells. According to bioinformatics analysis, miR-653-5p interested us. Dysregulation of miR-653-5p was observed in various cancers and it performs its oncogenic or anti-oncogenic role.23–25 miR-653-5p, which is overexpressed in NSCLC, is thought to have an anti-oncogenic role in lung cancer because it slows down the growth and spread of lung cancer cells when it is overproduced.26 However, its involvement in LUAD gefitinib resistance remained mysterious.
AGR2 genes located on chromosome 7p21.1 consisting 8 exons. It codes for the endoplasmic reticulum (ER) proteins family member, ie, disulfide isomerase (PDI) that is responsible to catalyze protein folding and interchange reactions of thiol-disulfide. This protein involves in cell migration, cellular transformation and also in metastasis. It acts as a p53 inhibitor in cell processes.27 The tumor-promoting function was detected in different cancers including lung cancer.28,29 Alavi et al also highlighted the prognostic significance of AGR2 in lung cancer.30 Additionally, Xue et al elucidated the oncogenic role of AGR2 in NSCLC in vitro.31 More particularly, high expression abundance of AGR2 was measured in gefitinib-resistant NSCLC cells, and AGR2 silence represses the malignant behavior of gefitinib-resistant NSCLC cells as well as increases the sensitivity of PC9/GR to gefitinib. Therefore, AGR2 contributes to lung cancer gefitinib resistance and thereby promotes lung cancer progression.32 And yet, the behind mechanism is not well recognized.
In our preliminary experiment, we found that a predominant expression of AFAP1-AS1 in LUAD subjects harboring acquired resistance to gefitinib. Through bioinformatics analysis, we established the ceRNA regulatory network involved in lncRNA AFAP1-AS1 to miR-139-5p/AGR2. Furthermore, we further deciphered its functional role in gefitinib resistance to LUAD.
Materials and Methods
Clinical Samples
From June 2018 to June 2019, a total of 26 LUAD patients acquired gefitinib-resistance or not were enrolled in The Central Hospital of Wuhan, Tongji Medical College and Huazhong University of Science and Technology. The LUAD clinical tissues were surgically excised and immediately frozen in liquid nitrogen before the next assays. The utilization of the clinical tissues was proved by the ethical committee of The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology (approval number: WHZXKYL2022-063). Each patient signed the written informed consent.
Cell Culture
Human LUAD PC9 and A549 cells, 293T cells were purchased from the American Type Culture Collection, USA. Human LUAD PC9 cells were purchased from BeNa Culture Collection (Beijing, China). A549 cells (F-12K Medium, ThermoFisher, USA), PC9 (RPMI-1640, ThermoFisher, USA) and 293T (DMEM, ThermoFisher, USA) were cultured in medium containing 10% of FBS (ThermoFisher, USA) and 1% penicillin/streptomycin at 37°C with 5% CO2. To establish gefitinib-resistant (GR) PC9 and A549 cells, these cells were continuously exposed to 50 nM gefitinib (Zhejiang East Asia Pharmaceutical Co., Ltd, China). Exposure treatments lasted for 2 months.33
Cell Transfection
MiR-653-5p mimic/inhibitor, mimic/inhibitor NCs, si-lncRNA AFAP1-AS1 (si-AFAP1-AS1), si-AGR2 and si-NCs were obtained from Ribo Bio (Guangzhou, China). Cell transfection was conducted using Lipo2000 (Thermofisher, USA) as instructed. 48h later, the effectiveness of transfection was determined using RT-qPCR.
Assessment of in vitro Proliferation
CCK-8 assay kit (Abcam, USA) was applied to assess cell proliferation. In short, 5 × 103 transfected PC9/GR and A549/GR cells were plated on 96-well plates following indicated time incubation. Cells were continuously exposed to different concentrations of gefitinib ranging from 0.01 µM to 40 µM for 48 h before 10 μL CCK8 reagent for 4h. Plates were read at 450 nm using a microplate reader (Tecan, Männedorf, Switzerland). The IC50 was counted via the online tool IC50 Calculator (https://www.aatbio.com/tools/ic50-calculator) for next cell functional assays.
Transwell Invasion Assay
5 × 103 transfected PC9/GR and A549/GR cells grown in 0.1 µM containing medium upto 24 h and added onto the upper chamber of Millicell insert (Billerica, MA) with Matrigel, where they were supplemented with serum-free medium containing FBS. The prepared transwell invasion inserts were placed onto the lower chamber containing medium with 0.5% FBS. The plates were incubated for 2.5 h to allow cells to invade through the membranes. After the uninvaded cells were removed, the membranes were subjected to the treatment of 5% glutaraldehyde for 10 min prior to staining with 1% crystal violet in 1% ethanol for 10 min. The stained cells were calculated under a microscope (Nikon E800 upright microscope, Nikon Corporation, Tokyo, Japan).
Apoptosis Assay
Apoptosis was detected using the Annexin V-FITC/PI Apoptosis Detection Kit (Vazyme, China). Briefly, PC9/GR and A549/GR cells were grown in 0.1 µM containing medium for 72 h. After being washed in PBS, the cells were exposed to 200 μL of 70% ice-cold ethanol for 1 h. The prepared cells were reacted with 5 μL FITCa before stained with 5 μL PI. Finally, the apoptosis was evaluated by flow cytometry (Becton Dickinson, MD, USA).
RT-qPCR
Total RNAs from tissues and cells were isolated using TRIzol reagent (ThermoFisher, USA). Reverse transcription was conducted with High-Capacity RNA-to-cDNA Kit (Thermofisher, USA) or One Step microRNA cDNA Synthesis Kit (Beijing Baiao Lai Bo Technology Co., Ltd., Beijing, China). Real-time PCR was performed on ABI QuantStudio 6 Pro systems using SYBR Green qPCR Biosharp kit (Biosharp,Co., Ltd, China). Fold changes in transcript are expressed as ΔΔCt value through 2−ΔΔCT method to normalize to U6 or GAPDH. The primers are listed in Table 1.
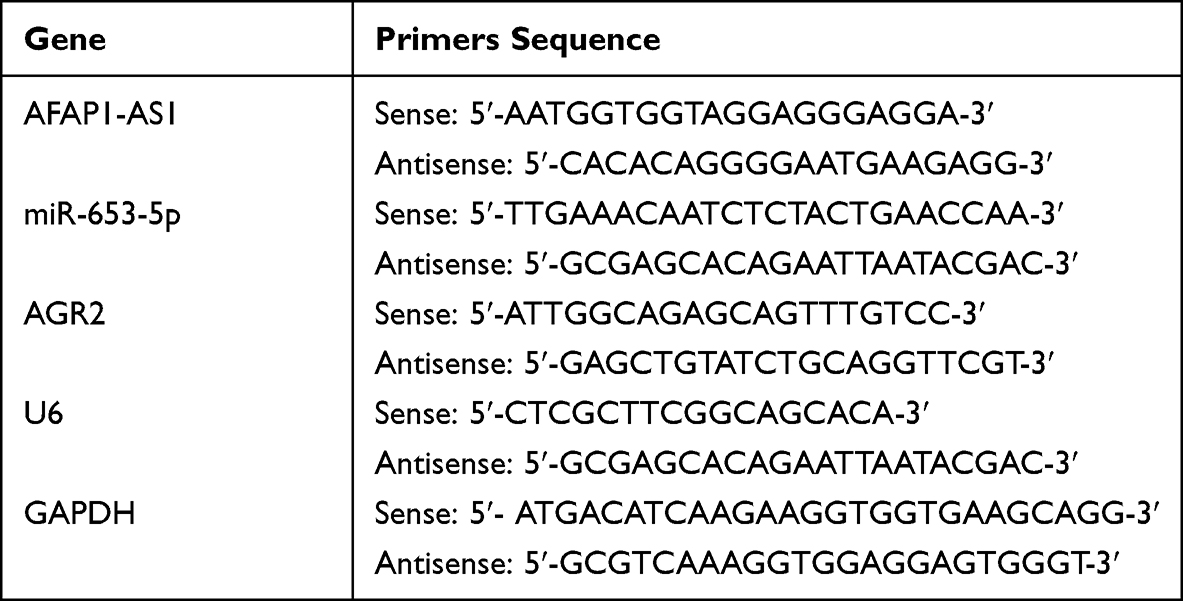 |
Table 1 Primers Used for Quantitative RT-PCR |
Western Blot Assay
Proteins extracted utilizing radioimmunoprecipitation assay (Boster, China), and estimated with a bicinchoninic acid method (Boster) were electrophoresed in 10% sulfate-polyacrylamide gel electrophoresis. Subsequently, proteins were delivered onto polyvinylidene fluoride membranes, washed utilizing TBST, and blocked with 5% BSA blocking buffer at 37°C for 2 h. Then, primary antibody: Bax (ab32503, 1:1000, Abcam, UK), Bcl-2 (ab32124, 1:1000, Abcam) and GAPDH (ab8245, Abcam) were incubated with membranes overnight at 4°C, prior to incubate with secondary antibody (ab6721, Abcam) for 1 h at 37°C. ECL detection kit (Yeasen, China), and ImageJ software applied to visualize and evaluate the protein levels.
Luciferase Reporter Assay
The predicted wide-type (WT) complementary sequence of AFAP1-AS1 or AGR2 3’UTR and their corresponding mutant (MUT) sequence were amplified and inserted into pMiR-Report firefly luciferase vectors (Jardin bio, China). The produced luciferase reporter constructs (pmiR-AGR2 3’UTR WT, pmiR-AFAP1-AS1 MUT, pmiR-AFAP1-AS1 WT, and pmiR-AGR2 3’UTR MUT) were delivered into 293T cells with miR-653-5p mimic or mimic NC. 48 h post-transfection, the reporter activities were detected using the Dual-GloLuciferase Assay System (Promega, USA).
Statistical Analysis
GraphPad Prism 9 was adopted to analyze all data presented as mean ± SD. Student’s t-test was done for two group comparison, and one-way ANOVA for multiply group with Bonferroni tests. Correlations between miR-653-5p and AFAP1-AS1 or AGR2 expression were verified using Pearson's correlation. Log rank test was used to analyze the correlation between gene expression and overall survival. P < 0.05 were considered statistically significant.
Results
LncRNA AFAP1-AS1 Silencing Blunts Gefitinib-Resistant Cell Proliferation and Invasion as Well as Induces Cell Apoptosis
We first examine the expression of AFAP1-AS1 in LUAD subjects acquiring gefitinib resistance and underwent no treatment. Elevated AFAP1-AS1 expression was detected in gefitinib acquired resistant LUAD tissues (Figure 1A). In parallel, the highly expressed AFAP1-AS1 was detected in PC9/GR and A549/GR cells (Figure 1B). To further mine the role of AFAP1-AS1 in LUAD gefitinib resistance, we introduced si-AFAP1-AS1 into PC9 and A549 cells and verified the high transfection efficiency and an about four-fold increase in AFAP1-AS1 expression in both LUAD cells (Figure 1C). After gefitinib resistance was acquired, we examined whether AFAP1-AS1 silencing impacted the malignant behaviors of the gefitinib-resistant LUAD cells. The proliferative assessment by CCK-8 assays showed that AFAP1-AS1 silencing lessened the proliferation of PC9/GR and A549/GR cells when exposed to different doses of gefitinib (Figure 1D), and especially the AFAP1-AS1 silencing GR cells present the lower IC50 (half-maximal inhibitory concentration, about 0.10 μg/mL) compared with that of control cells. Corresponding apoptosis activation was also detected in PC9/GR cells and A549/GR cells with AFAP1-AS1 silencing (Figure 1E). Western blot assay showed that the Bax protein expression in the si-AFAP1-AS1 group were higher than that in the si-NC group, while Bcl-2 was lower (Figure 1F). Additionally, the lowered invasive rate of PC9/GR cells and A549/GR cells was also a consequence of AFAP1-AS1 silencing (Figure 1G). Therefore, gefitinib confers resistance to LUAD cells by upregulating AFAP1-AS1 expression.
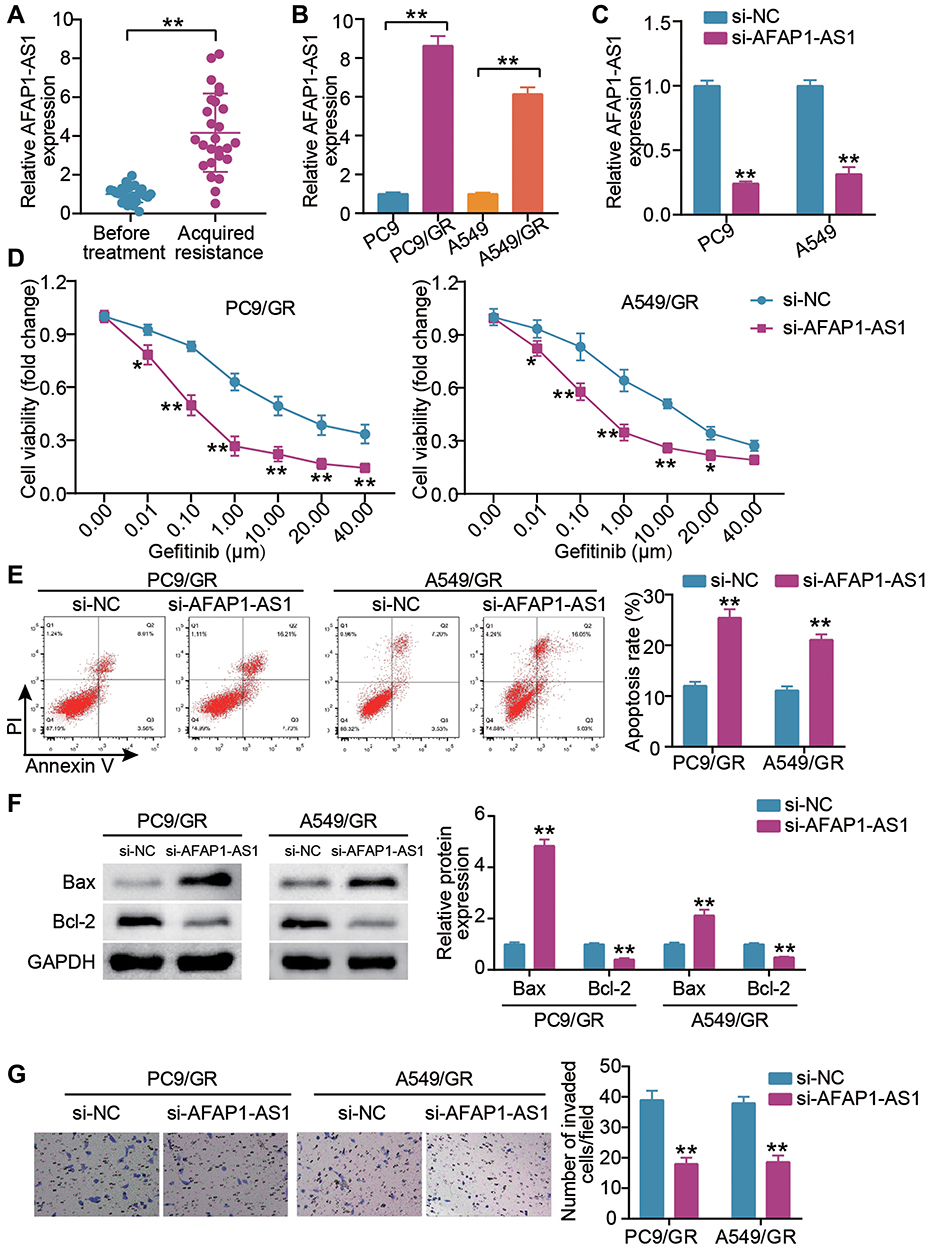 |
Figure 1 LncRNA AFAP1-AS1 silencing blunts gefitinib-resistant cells proliferation, invasion and induces cell apoptosis. (A) AFAP1-AS1 expression levels in LUAD cancer tissues were assessed by qRT-PCR in patients before gefitinib treatment and patients who developed acquired resistance to gefitinib. **P < 0.001 vs Before treatment. (B) AFAP1-AS1 was up-regulated in lung cancer cells with acquired resistance (PC9/GR and A549/GR cells). **P < 0.001 vs PC9 or A549. (C) qRT-PCR analysis for AFAP1-AS1 expression in cells transfected with si-lncRNA AFAP1-AS1(si-AFAP1-AS1) and si-NC for 24 h. **P < 0.001 vs si-NC. (D) CCK-8 assays showed that siRNA knockdown of AFAP1-AS1 remarkably reduced the proliferative capacity of PC9/GR and A549/GR cells. *P < 0.05 and **P < 0.001 vs si-NC. (E) Apoptosis detected by flow cytometry, demonstrated siRNA knockdown of AFAP1-AS1 as observed by a remarkably increase in the apoptosis of PC9/GR and A549/GR cells. **P < 0.001 vs si-NC. (F) Bax and Bcl-2 protein expression were measured by Western blot in PC9/GR and A549/GR cells silenced AFAP1-AS1. **P < 0.001 vs si-NC. (G) Transwell assays showed that the siRNA knockdown of AFAP1-AS1 remarkably reduced the invasion capacity of PC9/GR and A549/GR cells. **P < 0.001 vs si-NC. |
LncRNA AFAP1-AS1 Targets miR-653-5p
The top 50 unregulated genes in LUAD samples were screened from GEPIA databases (Supplementary Table 1). Uploading these unregulated genes to Metascape website (https://metascape.org/) for GO enrichment, lung development including three genes (MMP12, AGR2, SPDEF) were enriched (Figure 2A). According to the data from GEPIA, it was found that AGR2 having high expression was related to the poor prognosis of LUAD (Figure 2B). Hence, AGR2 was selected as our interested gene, and its upstream miRNAs were predicted by TargetScan. Meanwhile, the downstream of AFAP1-AS1 was predicted by starBase. The result showed that only miR-653-5p was the common miRNAs via TargetScan and starBase (Figure 2C). Through StarBase (http://starbase.sysu.edu.cn/), we found that the AFAP1-AS1 contained the miR-653-5p putative binding sits (Figure 2D). Subsequently, AFAP1-AS1 WT/MUT luciferase reporter vectors were constructed. When these vectors were transfected into 293T with miR-653-5p mimic, results depicted AFAP1-AS1 WT-vectors exhibited lowered luciferase activity, while no change occurred in AFAP1-AS1 MUT vectors (Figure 2E), suggesting the ceRNA activity of AFAP1-AS1 to miR-653-5p. Hence, we mined the clinical data of the miR-653-5p expression in subjects acquired resistance or not. A clear downregulation of miR-653-5p expression was shown in gefitinib-resistant LUAD patient samples (Figure 2F). More interesting, miR-653-5p negatively correlated with AFAP1-AS1 (Figure 2G). Collectively, miR-653-5p is a target of AFAP1-AS1 in LUAD.
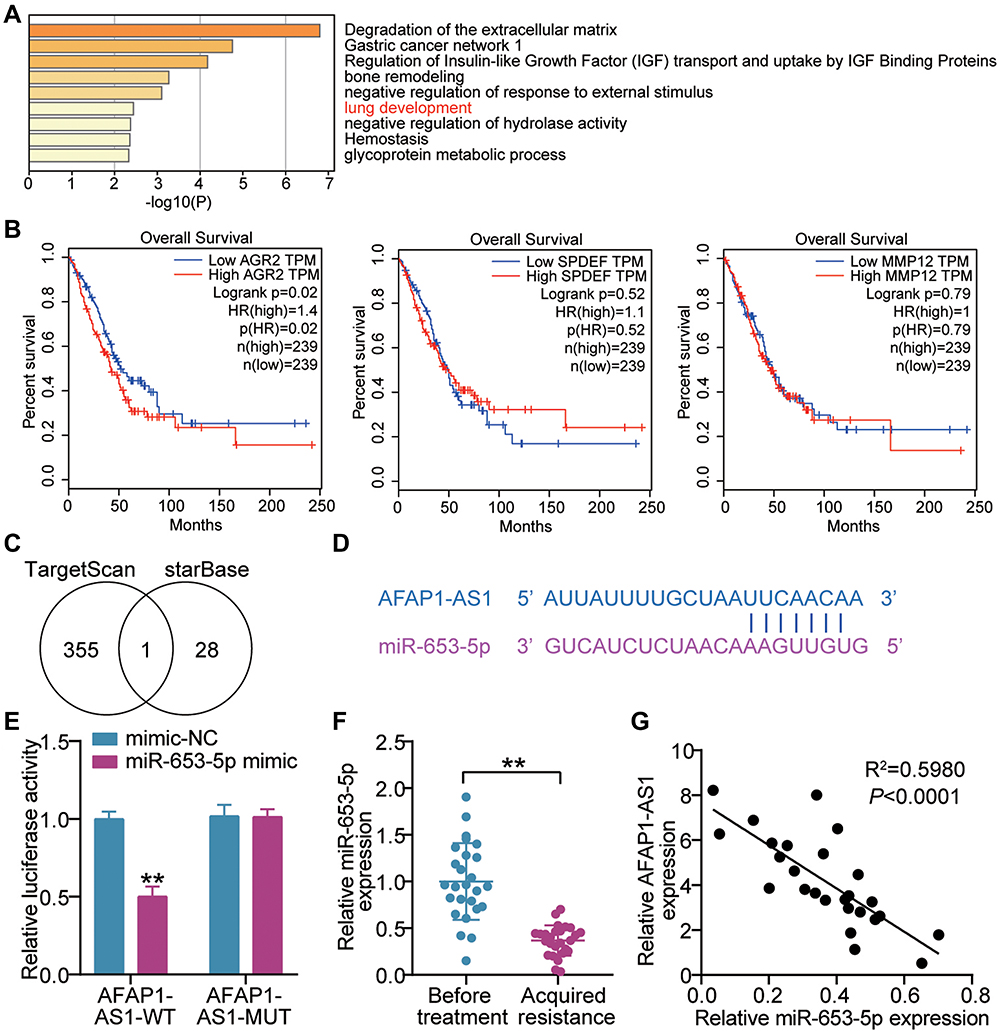 |
Figure 2 LncRNA AFAP1-AS1 targets miR-653-5p. (A) Lung development was enriched by Metascape. (B) The correlation between the prognosis of LUAD patients and three genes (AGR2, SPDEF and MMP12). (C) MiR-653-5p was the miRNA binding to AFAP1-AS1 and AGR2. TargetScan and starBase were used to predict the miRNAs binding to AGR2 and AFAP1-AS1, respectively. (D) Bioinformatics was used to predict the binding sites between AFAP1-AS1 and miR-653-5p. (E) Luciferase reporter assay showed that miR-653-5p mimic significantly inhibited the luciferase activity of pGL3 plasmids harbouring seed region of AFAP1-AS1 WT in 293T cells. **P < 0.001 vs mimic-NC. (F) RT-qPCR showing a clear miR-653-5p downregulation in the tumor tissues from gefitinib-resistant LUAD patients compared with that from gefitinib-resistant was observed. **P < 0.001 vs Before treatment. (G) Pearson analysis of miR-653-5p and AFAP1-AS1. |
MiR-653-5p Blockade Neutralizes the Effect of lncRNA AFAP1-AS1 Silencing on Gefitinib-Resistant LUAD Cell Growth
Since the target relationship between AFAP1-AS1 and miR-653-5p was determined, we next mined the interaction in PC9 and A549 cells. By co-delivering si-AFAP1-AS1 and miR-653-5p inhibitor into A549 and PC9 cells, we found that AFAP1-AS1 silencing successfully caused the enhanced expression of miR-653-5p, and yet this upregulation was blocked by additional transfection of miR-653-5p inhibitor (Figure 3A). In gefitinib resistance acquired PC9 and A549 cells (PC9/GR and A549/GR), a pronounced increment of cell viability was observed along with alone transfection of miR-653-5p inhibitor, more interesting, accompanied transfection of miR-653-5p inhibitor and si-AFAP1-AS1 enabled the increased viability regress to the levels comparable to that of the control group (Figure 3B). Furthermore, parallel analysis of apoptosis showed that miR-653-5p silencing lessened cellular apoptosis rate in PC9/GR and A549/GR cells, while this effect was offset by the resistant apoptosis of LUAD gefitinib-resistant cells resulting from AFAP1-AS1 silencing (Figure 3C). Western blot showed that Bcl-2 was facilitated while Bax was restrained in the inhibitor group compared with the si-AFAP1-AS1 + inhibitor group (Figure 3D). Additional analysis of cell invasion showed that anti-miR-653-5p treatment aggravated the invasive phenotypes of gefitinib-resistant LUAD cells, while the aggravated malignant phenotypes almost diminished along with additional transfection of si- AFAP1-AS1 (Figure 3E). These findings validate that AFAP1-AS1 modulate LUAD gefitinib resistance by sponging miR-653-5p.
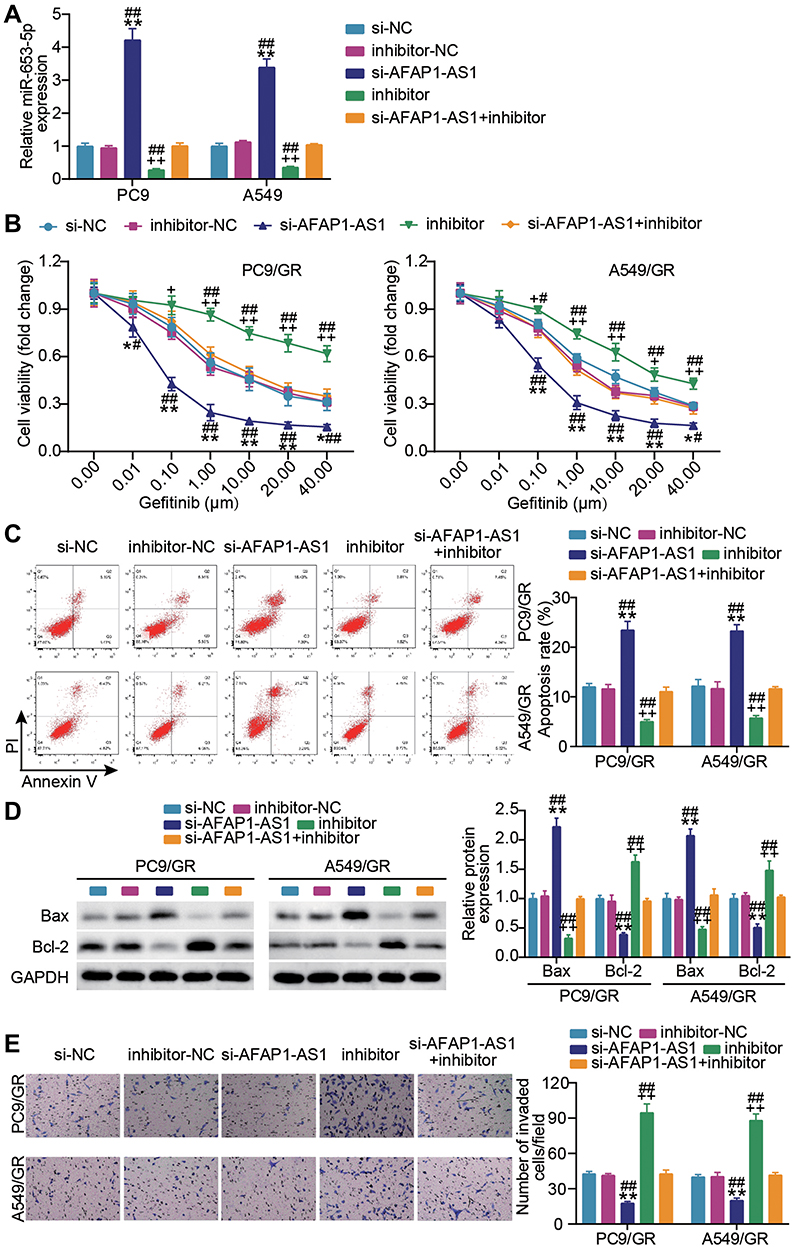 |
Figure 3 MiR-653-5p blockade neutralizes the effect of lncRNA AFAP1-AS1 silencing on gefitinib-resistant LUAD cell growth. (A) Verification of transfection efficiency. PC9 and A549 cells were transfected with si-AFAP1-AS1, si-NC, miR-653-5p inhibitor (inhibitor), inhibitor-NC, si-AFAP1-AS1 + inhibitor. After 48 h, RT-qPCR was implemented to test the miR-653-5p expression. (B–D) PC9/GR and A549/GR cells were transfected with si-AFAP1-AS1, si-NC, miR-653-5p inhibitor (inhibitor), inhibitor NC, si-AFAP1-AS1 + inhibitor. 48h post-transfection. Proliferation curves were obtained using CCK8 assays (B). Cell apoptosis was detected by flow cytometry (C) and Western blot (D). Cell invasion was determined by Transwell invasion assay (E). *P < 0.05 and **P < 0.001 vs si-NC; +P < 0.05, ++P < 0.001 vs inhibitor-NC; #P < 0.05 and ##P < 0.001 vs si-AFAP-AS1+inhibitor. |
MiR-653-5p Targets AGR2
According to the above bioinformatics analysis, we continuously interrogated the interaction of miR-653-5p and AGR2. In agreement with these above mentioned results, AGR2 3’ UTR could recognize the miR-653-5p by complementary sequences (Figure 4A). The data were further supported by reporter assays of luciferase demonstrating that miR-653-5p mimic strongly weakened the AGR2 3’ UTR WT-mediated luciferase activity (Figure 4B). We next asked whether there was clinical relevance between miR-653-5p and AGR2. Analysis of RT-qPCR demonstrated that a robust expression of AGR2 was observed in tumor tissues acquire gefitinib resistance (Figure 4C). The negative relevance was shown between the expression of miR-653-5p and AGR2 (Figure 4D). To sum, miR-653-5p could recognize the 3’UTR AGR2 and then edit AGR2 expression.
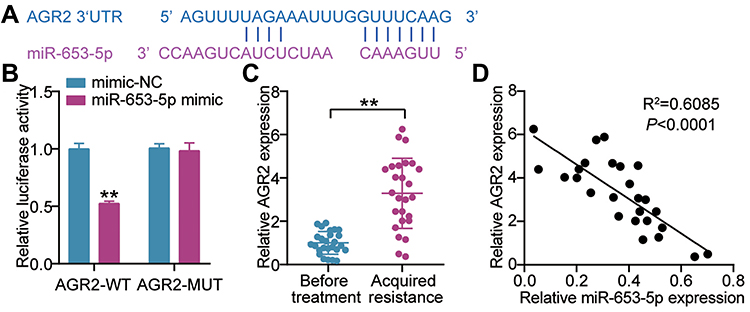 |
Figure 4 miR-653-5p targets AGR2. (A) Seed sequences of wild-type AGR2 3’UTR were predicted by Starbase (http://starbase.sysu.edu.cn/). (B) Normalized luciferase activity of 293T cells nucleofected with the miR-653-5p mimics and AGR2 3’UTR WT and AGR2 3’UTR MUT luciferase reporter vectors. **P < 0.001 vs mimic-NC. (C) mRNA expression level of AGR2 in LUAD subjects before treatment and after gefitinib resistance was acquired by RT-qPCR. **P < 0.001 vs Before treatment. (D) Pearson's correlation analysis of miR-653-5p expression with AGR2 expression. |
AGR2 Knockdown Compromises miR-653-5p Inhibitor Effect on Gefitinib-Resistant LUAD Cell Growth
We next asked whether AGR2 impacted the miR-653-5p-mediated malignant behaviors of gefitinib-resistant LUAD cells. In congruence with the above-mentioned interaction between miR-653-5p and AGR2, we found that a notable increment of AGR2 was induced by A549 and PC9 cells alone transfection of miR-653-5p inhibitor while compromised by together transfection with miR-653-5p inhibitor and si-AGR2 (Figure 5A). Subsequently, we captured the cell phenotype alternation in PC9/GR cells and A549/GR cells upon co-transfection with si-AGR2 and miR-653-5p inhibitor. As shown in Figure 5B, the AGR2 silence lessened the proliferation of PC9/GR and A549/GR cells, and while the notably decreased proliferation was abrogated by miR-653-5p inhibitor. Conversely, an evident apoptosis inactivation was found in PC9/GR and A549/GR cells when AGR2 expression was silenced, and while restored by together transfection with miR-653-5p inhibitor and si-AGR2 (Figure 5C). Western blot manifested that contrast to the si-NC group, Bcl-2 protein level in the si-AGR2 group was inhibited while Bax was promoted, and the apoptosis inhibited by miR-653-5p inhibitor was partially eliminated by AGR2 silencing (Figure 5D). Likewise, the invasion of AGR2 silencing C9GR and A549/GR cells was lower than that of C9GR and A549/GR cells transfected si-NC, and yet the lower invasion was almost overturned by miR-653-5p inhibitor (Figure 5E). Based on these data, miR-653-5p promotes the malignant behaviors of PC9/GR and A549/GR cells by targeting AGR2.
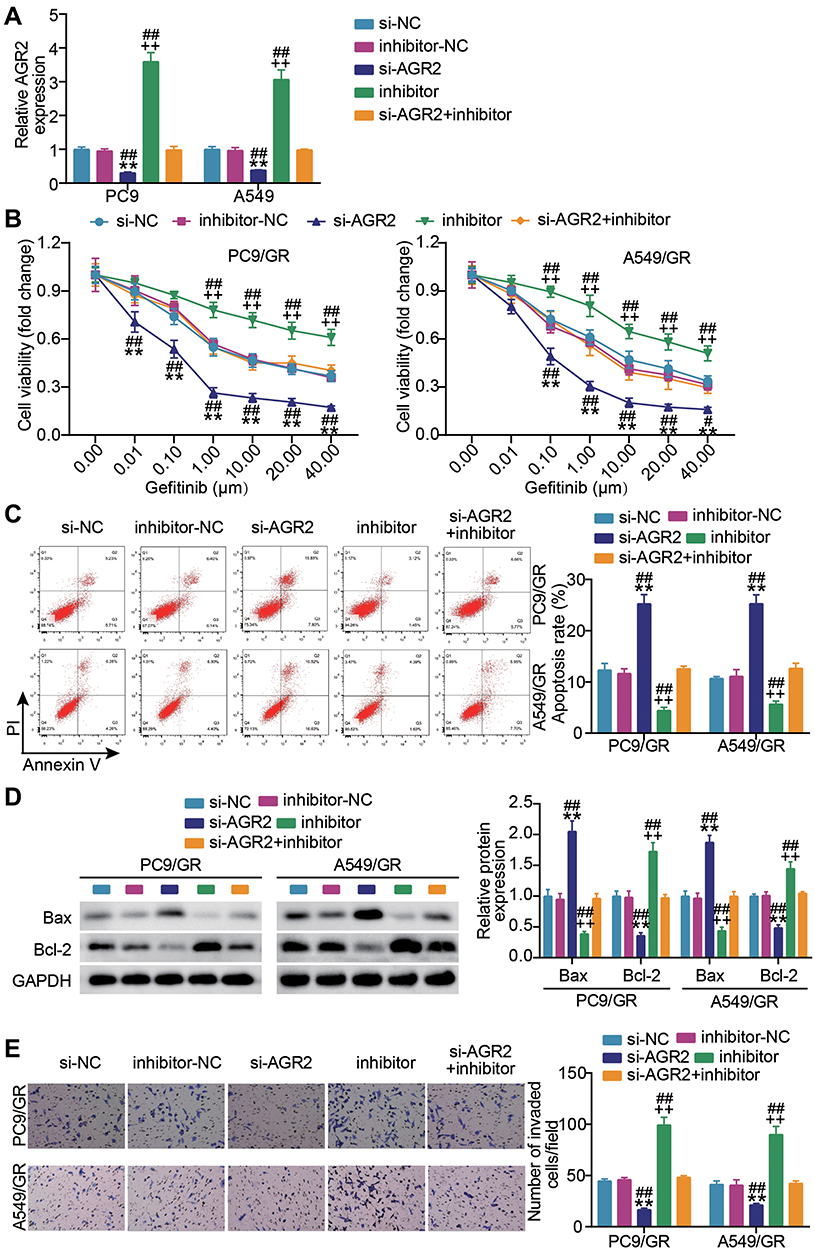 |
Figure 5 AGR2 knockdown compromises the effect of miR-653-5p inhibitor on gefitinib-resistant LUDA cell growth. (A) Verification of transfection efficiency. PC9 and A549 cells were transfected with si-NC, si-AGR2, miR-653-5p inhibitor, inhibitor-NC and miR-653-5p inhibitor + si-AGR2. After 48h, RT-qPCR was performed to test the miR-653-5p expression. (B–D) PC9/GR and A549/GR cells were transfected with si-NC, si-AGR2, miR-653-5p inhibitor, inhibitor-NC and miR-653-5p inhibitor + si-AGR2. 48h post-transfection. Proliferation curves were obtained using CCK-8 assays (B). Cell apoptosis was detected by flow cytometry (C) and Western blot (D). Cell invasion was determined by Transwell invasion assay (E). **P < 0.001 vs si-NC; ++P < 0.001 vs inhibitor-NC; ##P < 0.001 vs si-AFAP-AS1+inhibitor. |
Discussion
EGFR mutations and ALK translocation have been found to be associated with acquired resistance in NSCLC including gefitinib.4 A previous study revealed that lncRNA-ABCA12-8 was significantly upregulated in NSCLC patients with acquired resistance to gefitinib and facilitates cancer cell malignancy.34 In the present work, we found another lncRNA, AFAP1-AS1, is predominantly expressed in LUAD patients reporting gefitinib resistance, AFAP1-AS1 silence attenuates the oncogenic behaviors of PC9/GR and A549/GR cells. Furthermore, the AFAP1-AS1 specifically sequesters miR-653-5p and stepwise upregulates the expression of AGR2 targeted by miR-653-5p. The subsequent upregulation of AGR2 protein strengthens the proliferation and invasion of PC9/GR and A549/GR cells and blunted cell apoptosis. The antagonistic expression of miR-653-5p to AFAP1-AS1 or AGR2 further supports the ceRNA activity of AFAP1-AS1 to miR-653-5p/AGR2 axis. Our findings indicate that AFAP1-AS1 might be a candidate target for LUAD therapy.
Previously, AFAP1-AS1 contributes to drug resistance in several cancers. For example, AFAP1-AS1 inducing Wnt/β-Catenin Signaling Pathway elicits trastuzumab resistance of triple-negative breast cancer.35 AFAP1-AS1 knockdown downregulates the paclitaxel sensitivity in prostate cancer.19 In NSCLC, activation of the PI3K/AKT pathway by AFAP1-AS1 results in cisplatin-resistance.36 However, its role in LUAD gefitinib resistance is poorly understood. Our data demonstrated that AFAP1-AS1 was robustly expressed in gefitinib acquired resistant LUAD tissues as well as gefitinib resistant LUAD cells. AFAP1-AS1 silence attenuated the viability and invasion of gefitinib resistant LUAD cells and induced cell apoptosis. Accordingly, our findings indicate the previously unappreciated importance of AFAP1-AS1 in LUAD gefitinib resistance.
LncRNAs reportedly performed their ceRNA activity to antagonize the inhibition of miRNAs to mRNAs. MiRNAs, intrinsic non-coding RNAs with about 20 nucleotides, are found to be active participants of a myriad of cellular process, including tumorigenesis. Our data showed that AFAP1-AS1 targeted miR-653-5p. This miRNA was found as a pro-oncogenic gene or tumor suppressor in different cancers. An example of its oncomir activity is the stimulation of miR-653-5p to the oncogenic Wnt/β-catenin signaling during prostate cancerigenesis.37 And yet, its tumor-suppressive role was also depicted in breast cancer, showing overexpression of miR‑653‑5p contributes to breast cancer cells in vitro proliferation and migration.38 In lung cancer, miR-653-5p silence favors cellular growth and invasion in vitro.26 However, its role in LUAD gefitinib resistance remained elusive. Our data found that miR-653-5p inhibitor increased the viability as IC50 value of gefitinib and invasion of PC9/GR and A549/GR cells, in support of the tumor-suppressing role in lung cancer. On the other hand, we also for the first time uncovered the interaction between miR-653-5p and AFAP1-AS1. This interaction was further supported by a contrary expression relationship of miR-653-5p and AFAP1-AS1. More importantly, miR-653-5p inhibitor could abrogate the tumor-suppressing effect of AFAP1-AS1 silence in gefitinib-resistant LUAD cells. All these data suggest that AFAP1-AS1 perform its oncogenic biofunction by targeting miR-653-5p. In addition, one previous study reported that miR-653-5p targeted by circ-RAD23B also play a role during lung cancer progression.26 Therefore, miR-653-5p is likely to mediate lung cancer by various regulatory network crosstalk. The complex regulatory network d warrants further investigation.
AGR2 is essential for maintaining endoplasmic reticulum (ER) protein homeostasis which disruption enables tumor heterogeneity and initiates tumor formation.39 Hence, AGR2 is dissected as a pro-oncogenic gene in cancers.28 Indeed, AGR2 is amplified in lung cancer tissues and related to dismal clinical outcomes.30 Furthermore, overexpressing AGR2 mitigates the suppression of miR-200c on LUAD cell malignant action.40 More importantly, the AGR2 deficiency causes the sensitivity of PC9-gefitinib cells to gefitinib.32 Consistently, our data showed that AGR2 overexpressed in gefitinib acquired resistant LUAD tissue, and AGR2 silence inhibited the sustaining proliferation and invasion of PC9/GR and A549/GR cells as well restored the sensitivity to both cells to gefitinib. However, its underlying mechanism remained elusive. Our data for the first time elucidated the interaction of miR-653-5p and AGR2. More importantly, the oncogenic role of miR-653-5p was neutralized by AGR2 silence. All findings suggest miR-653-5p downregulates AGR2 expression and attenuates its oncogenicity in lung cancer.
However, there were non-negligible limitations. First, we have not validated the role of AFAP1-AS1 in lung cancer in vivo and this merits further investigation. Whether other miRNAs or mRNA are involved in the ceRNA activity of AFAP1-AS1 to modulate LUAD gefitinib resistance would be sought in further work. Additionally, more patient samples with LUAD would be required for further verification.
Conclusion
In conclusion, our work found that the novel role of AFAP1-AS1 in modulating miR-653-5p/AGR2 axis and contributing gefitinib resistance. Targeting AFAP1-AS1 or antagonizing miR-653-5p/AGR2 axis might be a druggable approach to attenuate the gefitinib resistance during cancer treatment.
Ethics Approval
The present study was approved by the Ethics Committee of The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology (Wuhan, China). The clinical tissue samples processing in accordance with strict compliance with the ethical standards of the Declaration of Helsinki. Written informed consent was signed by all patients.
Consent to Participate
Written informed consent was signed by all patients.
Consent for Publication
Consent for publication was taken from the participants.
Data Sharing Statement
All data generated or analyzed during this study can be found in below websites. The top 50 unregulated genes in LUAD samples were screened from GEPIA databases(http://gepia.cancer-pku.cn/). Lung development was enriched by Metascape(https://metascape.org/). TargetScan(http://www.targetscan.org/vert_80/) and starBase(https://starbase.sysu.edu.cn/starbase2/) were used to predict the binding sites.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article. This work was supported by the Wuhan Health Science Foundation (WZ20Y05).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Sung H, Ferlay J, Siegel RL., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Sun GZ, Zhao TW. Lung adenocarcinoma pathology stages related gene identification. Math Biosci Eng. 2019;17(1):737–746. doi:10.3934/mbe.2020038
3. Chen Y, Chen Z, Chen R, et al. Immunotherapy-based combination strategies for treatment of EGFR-TKI-resistant non-small-cell lung cancer. Future Oncol. 2022;18(14):1757–1775. doi:10.2217/fon-2021-0862
4. Sini C, Tuzi A, Rossi G, Russo A, Pezzuto A. Acquired resistance in oncogene-addicted non-small-cell lung cancer. Future Oncol. 2018;14(13s):29–40. doi:10.2217/fon-2018-0097
5. Noronha V, Patil VM, Joshi A, et al. Gefitinib versus gefitinib plus pemetrexed and carboplatin chemotherapy in EGFR-mutated lung cancer. J Clin Oncol. 2020;38(2):124–136. doi:10.1200/JCO.19.01154
6. Hosomi Y, Morita S, Sugawara S, et al. Gefitinib alone versus gefitinib plus chemotherapy for non-small-cell lung cancer with mutated epidermal growth factor receptor: NEJ009 study. J Clin Oncol. 2020;38(2):115–123. doi:10.1200/JCO.19.01488
7. Mok TS, Cheng Y, Zhou X, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. J Clin Oncol. 2018;36(22):2244–2250. doi:10.1200/JCO.2018.78.7994
8. Liao BC, Griesing S, Yang JC. Second-line treatment of EGFR T790M-negative non-small cell lung cancer patients. Ther Adv Med Oncol. 2019;11:1758835919890286. doi:10.1177/1758835919890286
9. Yang JJ, Fang J, Shu YQ, et al. A phase Ib study of the highly selective MET-TKI savolitinib plus gefitinib in patients with EGFR-mutated, MET-amplified advanced non-small-cell lung cancer. Invest New Drugs. 2021;39(2):477–487. doi:10.1007/s10637-020-01010-4
10. Titmarsh HF, O’Connor R, Dhaliwal K, Akram AR. Corrigendum: the emerging role of the c-MET-HGF axis in non-small cell lung cancer tumor immunology and immunotherapy. Front Oncol. 2020;10:1516. doi:10.3389/fonc.2020.01516
11. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. doi:10.1056/NEJMoa040938
12. Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Adv Exp Med Biol. 2017;1008:1–46. doi:10.1007/978-981-10-5203-3_1
13. Puvvula PK. LncRNAs regulatory networks in cellular senescence. Int J Mol Sci. 2019;20:11. doi:10.3390/ijms20112615
14. Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77(15):3965–3981. doi:10.1158/0008-5472.CAN-16-2634
15. Feng C, Zhao Y, Li Y, Zhang T, Ma Y, Liu Y. LncRNA MALAT1 promotes lung cancer proliferation and gefitinib resistance by acting as a miR-200a sponge. Arch Bronconeumol. 2019;55(12):627–633. doi:10.1016/j.arbr.2019.03.018
16. Lei Y, Guo W, Chen B, Chen L, Gong J, Li W. Tumor‑released lncRNA H19 promotes gefitinib resistance via packaging into exosomes in non‑small cell lung cancer. Oncol Rep. 2018;40(6):3438–3446. doi:10.3892/or.2018.6762
17. Yu T, Bai W, Su Y, Wang Y, Wang M, Ling C. Enhanced expression of lncRNA ZXF1 promotes cisplatin resistance in lung cancer cell via MAPK axis. Exp Mol Pathol. 2020;116:104484. doi:10.1016/j.yexmp.2020.104484
18. Huang N, Guo W, Ren K, et al. LncRNA AFAP1-AS1 Supresses miR-139-5p and promotes cell proliferation and chemotherapy resistance of non-small cell lung cancer by competitively upregulating RRM2. Front Oncol. 2019;9:1103. doi:10.3389/fonc.2019.01103
19. Leng W, Liu Q, Zhang S, Sun D, Guo Y. LncRNA AFAP1-AS1 modulates the sensitivity of paclitaxel-resistant prostate cancer cells to paclitaxel via miR-195-5p/FKBP1A axis. Cancer Biol Ther. 2020;21(11):1072–1080. doi:10.1080/15384047.2020.1829266
20. Han M, Gu Y, Lu P, et al. Exosome-mediated lncRNA AFAP1-AS1 promotes trastuzumab resistance through binding with AUF1 and activating ERBB2 translation. Mol Cancer. 2020;19(1):26. doi:10.1186/s12943-020-1145-5
21. Robinson EK, Covarrubias S, Carpenter S. The how and why of lncRNA function: an innate immune perspective. Biochim Biophys Acta - Gene Regul Mech. 2020;1863(4):194419. doi:10.1016/j.bbagrm.2019.194419
22. Sun J, Min H, Yu L, Yu G, Shi Y, Sun J. The knockdown of LncRNA AFAP1-AS1 suppressed cell proliferation, migration, and invasion, and promoted apoptosis by regulating miR-545-3p/hepatoma-derived growth factor axis in lung cancer. Anticancer Drugs. 2021;32(1):11–21. doi:10.1097/CAD.0000000000001003
23. Chi R, Chen X, Liu M, et al. Role of SNHG7-miR-653-5p-STAT2 feedback loop in regulating neuroblastoma progression. J Cell Physiol. 2019;234(8):13403–13412. doi:10.1002/jcp.28017
24. Lian LP, Xi XY. Long non-coding RNA XIST protects chondrocytes ATDC5 and CHON-001 from IL-1β-induced injury via regulating miR-653-5p/SIRT1 axis. J Biol Regul Homeost Agents. 2020;34(2):379–391. doi:10.23812/19-549-A-65
25. Liu F, Hu L, Pei Y, et al. Long non-coding RNA AFAP1-AS1 accelerates the progression of melanoma by targeting miR-653-5p/RAI14 axis. BMC Cancer. 2020;20(1):258. doi:10.1186/s12885-020-6665-2
26. Han W, Wang L, Zhang L, Wang Y, Li Y. Circular RNA circ-RAD23B promotes cell growth and invasion by miR-593-3p/CCND2 and miR-653-5p/TIAM1 pathways in non-small cell lung cancer. Biochem Biophys Res Commun. 2019;510(3):462–466. doi:10.1016/j.bbrc.2019.01.131
27. Obacz J, Takacova M, Brychtova V, et al. The role of AGR2 and AGR3 in cancer: similar but not identical. Eur J Cell Biol. 2015;94(3–4):139–147. doi:10.1016/j.ejcb.2015.01.002
28. Moidu NA, Ns AR, Syafruddin SE, Low TY, Mohtar MA. Secretion of pro-oncogenic AGR2 protein in cancer. Heliyon. 2020;6(9):e05000. doi:10.1016/j.heliyon.2020.e05000
29. Tian S, Hu J, Tao K, et al. Secreted AGR2 promotes invasion of colorectal cancer cells via Wnt11-mediated non-canonical Wnt signaling. Exp Cell Res. 2018;364(2):198–207. doi:10.1016/j.yexcr.2018.02.004
30. Alavi M, Mah V, Maresh EL, et al. High expression of AGR2 in lung cancer is predictive of poor survival. BMC Cancer. 2015;15:655. doi:10.1186/s12885-015-1658-2
31. Xue X, Fei X, Hou W, Zhang Y, Liu L, Hu R. miR-342-3p suppresses cell proliferation and migration by targeting AGR2 in non-small cell lung cancer. Cancer Lett. 2018;412:170–178. doi:10.1016/j.canlet.2017.10.024
32. Luu TT, Bach DH, Kim D, Hu R, Park HJ, Lee SK. Overexpression of AGR2 Is associated with drug resistance in mutant non-small cell lung cancers. Anticancer Res. 2020;40(4):1855–1866. doi:10.21873/anticanres.14139
33. Huang Z, Ma Y, Zhang P, Si J, Xiong Y, Yang Y. Long non-coding RNA H19 confers resistance to gefitinib via miR-148b-3p/DDAH1 axis in lung adenocarcinoma. Anticancer Drugs. 2020;31(1):44–54. doi:10.1097/CAD.0000000000000831
34. He S, Shi J, Zhou H, Li Q, Wu L. Lnc-ABCA12-8 confers acquired resistance to gefitinib in non-small cell lung cancer by regulating the alternative splicing of fibronectin 1 in the IIICS region. Cancer Gene Ther. 2022;29:1686–1696. doi:10.1038/s41417-022-00483-0
35. Bi Z, Li Q, Dinglin X, et al. Nanoparticles (NPs)-Meditated LncRNA AFAP1-AS1 silencing to block Wnt/β-catenin signaling pathway for synergistic reversal of radioresistance and effective cancer radiotherapy. Adv Sci. 2020;7(18):2000915. doi:10.1002/advs.202000915
36. Liu Y, Hu Q, Wang X. AFAP1-AS1 induces cisplatin resistance in non-small cell lung cancer through PI3K/AKT pathway. Oncol Lett. 2020;19(1):1024–1030. doi:10.3892/ol.2019.11175
37. Fu Q, Sun Z, Yang F, Mao T, Gao Y, Wang H. SOX30, a target gene of miR-653-5p, represses the proliferation and invasion of prostate cancer cells through inhibition of Wnt/β-catenin signaling. Cell Mol Biol Lett. 2019;24:71. doi:10.1186/s11658-019-0195-4
38. Zhang M, Wang H, Zhang X, Liu F. miR‑653‑5p suppresses the growth and migration of breast cancer cells by targeting MAPK6. Mol Med Rep. 2021;23:3. doi:10.3892/mmr.2020.11641
39. Gong W, Ekmu B, Wang X, Lu Y, Wan L. AGR2-induced glucose metabolism facilitated the progression of endometrial carcinoma via enhancing the MUC1/HIF-1α pathway. Hum Cell. 2020;33(3):790–800. doi:10.1007/s13577-020-00356-4
40. Sommerova L, Ondrouskova E, Martisova A, Zoumpourlis V, Galtsidis S, Hrstka R. ZEB1/miR-200c/AGR2: a new regulatory loop modulating the epithelial-mesenchymal transition in lung adenocarcinomas. Cancers. 2020;12:6. doi:10.3390/cancers12061614
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.