Back to Journals » International Journal of Nanomedicine » Volume 14
Liver-targeted liposomes for codelivery of curcumin and combretastatin A4 phosphate: preparation, characterization, and antitumor effects
Authors Jiang H ![]() , Li ZP
, Li ZP ![]() , Tian GX, Pan RY, Xu CM, Zhang B, Wu JL
, Tian GX, Pan RY, Xu CM, Zhang B, Wu JL ![]()
Received 27 September 2018
Accepted for publication 8 February 2019
Published 8 March 2019 Volume 2019:14 Pages 1789—1804
DOI https://doi.org/10.2147/IJN.S188971
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Hong Jiang,1,* Zhi-Peng Li,1,* Gui-Xiang Tian,1,* Rui-Yan Pan,1 Chong-Mei Xu,2 Bo Zhang,2 Jing-liang Wu1
1School of Bioscience and Technology, Weifang Medical University, Weifang, Shandong, China; 2School of Pharmacy, Weifang Medical University, Weifang, Shandong, China
*These authors contributed equally to this work
Background: Recent efforts have been focused on combining two or more therapeutic approaches with different mechanisms to enhance antitumor therapy. Moreover, nanosize drug-delivery systems for codelivering two drugs with proapoptotic and antiangiogenic activities have exhibited great potential in efficient treatment of cancers.
Methods: Glycyrrhetinic acid (GA)–modified liposomes (GA LPs) for liver-targeted codelivery of curcumin (Cur) and combretastatin A4 phosphate (CA4P) were prepared and characterized. In vitro cellular uptake, cytotoxicity, cell migration, in vivo biodistribution, antitumor activity, and histopathological studies were performed.
Results: Compared with unmodified LPs (Cur-CA4P LPs), Cur-CA4P/GA LPs were taken up effectively by human hepatocellular carcinoma cells (BEL-7402) and showed higher cytotoxicity than free drugs. In vivo real-time near-infrared fluorescence–imaging results indicated that GA-targeted LPs increased accumulation in the tumor region. Moreover, Cur-CA4P/GA LPs showed stronger inhibition of tumor proliferation than Cur, Cur + CA4P, and Cur-CA4P LPs in vivo antitumor studies, which was also verified by H&E staining.
Conclusion: GA-modified LPs can serve as a promising nanocarrier for liver-targeted co-delivery of antitumor drugs against hepatocellular carcinoma.
Keywords: liver-targeted, LPs, curcumin, combretastatin A4 phosphate, combination therapy
Introduction
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors, with high incidence and mortality worldwide.1 Chemotherapy is one of the main strategies in the clinical treatment of HCC. However, traditional cytotoxic chemotherapeutic drugs, such as doxorubicin and paclitaxel, exhibit limited clinical efficacy, due to systemic toxicity, lack of selectivity, and drug resistance.2,3 The effectiveness of combination therapy is supported by clinical research, which shows superior antitumor efficacy than single-drug therapy via different antitumor approaches.4–6 Antiangiogenesis, a promising antitumor strategy blocking the development of tumor blood vessels, is widely accepted.7 Therefore, the combination of chemotherapeutics with antiangiogenic drugs is expected to improve therapeutic efficacy for HCC.8,9
Curcumin (Cur), a diphenolic compound derived from the rhizome of turmeric, exhibits potent antitumor efficacy against prostate, breast, and colon cancers and belongs to the third generation of anticancer drugs.10 Cur blocks NFκB signaling to inhibit proliferation, thereby inhibiting cancer-cell growth and inducing apoptosis.11,12 In the clinic, single-drug therapy may increase the occurrence of drug resistance, suggesting the necessity of using other active drugs in combination with Cur to overcome this defect. Hydrophilic combretastatin A4 phosphate (CA4P), isolated from Combretum caffrum, is a new antitumor compound targeting blood vessels and soluble phosphate prodrug derivatives of CA4.13 CA4P acts mainly on the vascular endothelial cells of the tumor, which causes tumor vasoconstriction, dysfunction, and death of neoplastic cells, due to insufficient blood supply.14,15 Few reports about the combination of Cur and CA4P exist.16 Based on the reduced incidence of drug resistance and tumor treatment in different pharmacological pathways by promoting apoptosis and antitumor angiogenesis, we selected the combination of Cur and CA4P to verify the enhanced antitumor efficacy. The combination of Cur and CA4P can improve the effect of chemotherapy via different mechanisms of action and reduce side effects.17 However, chronic single administration is often associated with undesirable side effects or drug resistance, which may cause failure in cancer therapy.18 Possible reasons involve the difficulty in targeted codelivery of the two drugs to tumors, due to their diverse physiochemical properties and mechanisms.
Drug-delivery systems provide a promising opportunity to overcome the dilemma in effective codelivery of multiple cargoes.19,20 Many drug-delivery systems, such as liposomes (LPs), microspheres, nanoemulsions, polymer micelles, polymer–drug conjugates, carbon nanotubes, mesoporous silica nanoparticles, have been widely investigated.21–23 Among the different nanocarriers, LPs have attracted increased attention. LPs contain a hydrophilic core and hydrophobic shell, which can load hydrophilic, hydrophobic, and amphipathic drugs and improve their solubility.24 In addition, the flexibility of the membranal structure in LPs would be beneficial for sustained or controlled drug release. Moreover, LPs <200 nm can improve accumulation in solid tumors, due to the enhanced permeability and retention (EPR) effect, which contributes to enhanced antitumor efficacy and reduced systemic toxicity.25,26 On the basis of these properties, we selected LPs as nanocarriers to improve therapeutic effects against tumors.
Active liver-targeting LPs must improve the accuracy of drug actions on target sites. Glycyrrhetinic acid (GA) is the active aglycone of glycyrrhizin, which belongs to a pentacyclic triterpene derivate.27 Specific receptors for GA on the cytomembrane of hepatocytes have been verified.28,29 Protein kinase Cα is the target binding protein of GA that shows higher expression in HCC cells than that in adjacent nontumor liver cells.30 As such, GA-modified LPs can be expected to exhibit increased active targeting to HCC cells and tissue, which will be beneficial for further enhanced antitumor efficacy than unmodified LPs.
In summary, novel GA-modified LPs for effective codelivery of Cur and CA4P (Cur-CA4P/GA LPs) were developed in this study. Cur-CA4P/GA LPs targeted tumor sites effectively and inhibited tumor-cell proliferation and destroyed tumor neovascularization simultaneously, consequently improving antitumor activity for HCC. In addition to characterization of physicochemical properties of the codelivered LPs, evaluation of in vitro cytotoxicity, cellular uptake, cell migration, and in vivo biodistribution and antitumor efficacy was conducted in this study.
Methods
Materials
Cur (molecular weight [MW] 368.38 Da), L-α-phosphatidylcholine, cholesterol (Chol; MW 386.67 Da), Sephadex TMG50 medium (separation range 1,000–30,000), MTT, DAPI, RPMI-1640 medium, and FBS were purchased from Solarbio (Beijing, China). GA (MW 470.69 Da) was acquired from Fujie Pharmaceutical (Sanyuan, China). 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)–4-methylmorpholinium chloride (DMTMM) was purchased from Medpep (Shanghai, China). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was purchased from Aladdin Reagent (Shanghai, China). DiR (1,1′-dioctadecyl-3,3,3,3-tetramethyl indotricarbocyanine Iodide) was obtained from Pan Bo Biochemistry (Beijing, China). 1,2-Distearyl-sn-glycero-3-phosphoethanolamine (DSPE)–N-hydroxysuccinimide (NHS)–polyethylene glycol 2,000 (PEG2,000) and DSPE-PEG2,000–fluorescein isothiocyanate (FITC) were purchased from Ruixi Technology (Xi’an, China), and CA4P (MW 440.30 kDa) was obtained from Innochem Technology (Beijing, China). All chemical reagents used in the experiments were of analytical reagent grade.
Cell lines
Human HCC cells (BEL7402) and mouse melanoma cells (B16) were purchased from Wuhan University Life Collection Center (Wuhan, China). BEL7402 cells were cultured in RPMI 1640 medium supplemented with 10% FBS and 100 IU/mL penicillin–streptomycin at 37°C in a humidified incubator containing 5% CO2. B16 cells were cultured with DME:F12 (1:1) medium with 10% FBS and 100 IU/mL penicillin–streptomycin in the same environment. Cells were subcultured with trypsin for ~2 minutes, and the culture medium containing FBS was then added to terminate the digestion of trypsin. Suspensions were centrifuged at 1,000 rpm for 5 minutes and the supernatant discarded. Cells were then counted and inoculated. Murine HCC cells (H22) were obtained from KeyGen Biotech (Nanjing, China). H22 cells were cultured under the same conditions as BEL7402 cells and used to establish the tumor-bearing mouse model.
Animals
Male BALB/c mice (20±2 g) aged 4–6 weeks were purchased from Pengyue Experimental Animal Breeding (Jinan, China). Animals were fed a standard diet and allowed water ad libitum. All experiments were performed in compliance with the animal management rules of the Ministry of Health of China (document 55, 2001) and the animal experiment ethics review committee of Weifang Medical University (2017-025).
Synthesis of DSPE-PEG2,000-GA
DSPE-PEG2,000-GA was synthesized by a two-step reaction.31,32 In the first step, GA solution in methanol was activated by DMTMM to form the intermediate product – GA-ES. At room temperature, GA was added slowly to an ethylene diamine solution to obtain diamine-modified GA (GA-N). In the second step, GA-N and DSPE-PEG2,000-NHS at a molar ratio of 3:1 was dissolved in dimethyl sulfoxide in the presence of EDC (1.58 mg) and 20 μL triethylamine was added to the reaction system to regulate pH. The whole system was reacted for 48 hours under nitrogen protection to prevent bubble formation, followed by dialysis in 1,000 mL double-distilled water for 3 days. The solution was freeze-dried to obtain DSPE-PEG2,000-GA. Chemical structures of DSPE-PEG2,000-NHS, GA-N, and DSPE-PEG2,000-GA were confirmed by 1H nuclear magnetic resonance (JNM ECP-600; Jeol, Tokyo, Japan) by dissolving the target molecule in deuteron chloroform.
Preparation of liver-targeted LPs for coloading Cur and CA4P
Cur-CA4P/GA LPs were prepared via the well-established technique of thin-film evaporation.33 L-α-Phosphatidylcholine (120 mg) and Chol (40 mg) were mixed in 5 mL chloroform at a mass ratio of 3:1 and 20 mg DSPE-PEG2,000-GA was added to this system at a phospholipid:DSPE-PEG2,000-GA mass ratio of 6:1 and mixed with 3 mL of 1 mg/mL Cur anhydrous ethanol solution. The mass ratio of Cur and phospholipids was 1:40. Chol was then removed using a rotary evaporator to form a dry-lipid film at 35°C under uniform speed. CA4P dissolved in PBS (pH 7.4, 5 mL) was added to the lipid film, hydrated for 30–60 minutes, and sonicated for 5 minutes to decrease the particle size. The final concentrations of Cur and CA4P in the system were 0.06 and 0.75 mg/mL, respectively. Finally, 5 mL LP solution was obtained. Unencapsulated Cur was removed using Sephadex G50.34 The chromatographic column was filled with Sephadex G50 medium. After the chromatographic column had reached equilibrium, the sample was added and eluted with PBS. Dialysis was used to remove the unencapsulated CA4P. The sample was sealed in a dialysis bag, and the bag placed in 500 mL PBS solution for 3 days. Finally, the Cur-CA4P/GA LPs were refrigerated (Figure 1). The whole preparation process was conducted without light.
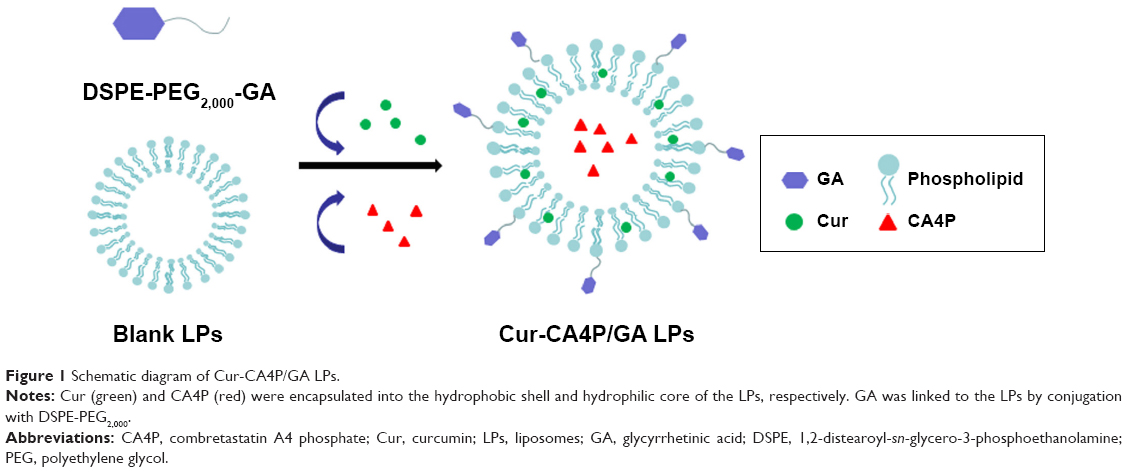 | Figure 1 Schematic diagram of Cur-CA4P/GA LPs. |
Single-load CA4P LPs were prepared through reverse-phase evaporation:35 120 mg L-α-phosphatidylcholine and 15 mg Chol were mixed with 8 mL ether at a mass ratio of 8:1; 10 mg CA4P was dissolved in 5 mL PBS, and the former mix added. Both the ether and PBS solutions were mixed and followed by sonication for 1 minute in an ice bath to form a w:o uniform emulsion. Decompression rotating evaporation removed organic solvents and achieved a colloidal state. Exactly 5 mL PBS was then added for hydration, followed by vortexing to form a uniform suspension. Blank LPs were prepared using the same procedure without drugs. A single-factor method was adopted for prescription screening.
Preparation of FITC-labeled LPs
FITC-labeled LPs were prepared through thin-film evaporation similarly to Cur-CA4P/GA LPs. To begin, 120 mg L-α-phosphatidylcholine, 40 mg Chol, and 12 mg DSPE-PEG2,000-FITC were mixed in 5 mL chloroform at a mass ratio of 30:10:3. Chol was evaporated using a rotary evaporator to form a dry-lipid film at 35°C under 18 rpm. Subsequently, 5 mL PBS was added for hydration, lasting for 30–60 minutes. The final step was sonication for 5 minutes to obtain FITC LPs. The preparation of FITC-GA LPs was similar to that of FITC LPs. The only difference was that 20 mg DSPE-PEG2,000-GA was added to the raw materials (mass ratio of L-α-phosphatidylcholine and DSPE-PEG2,000-GA 6:1). The final concentration of FITC was 2.4 mg/mL.
Preparation of DiR-loaded GA LPs
The preparation of DiR-loaded GA LPs was similar to the previous method. First, 120 mg L-α-phosphatidylcholine, 40 mg Chol, 20 mg DSPE-PEG2,000-GA, and 0.2 mg DiR were mixed in 5 mL Chol at a mass ratio of 600:200:100:1 in an eggplant-shaped flask, dried until a thin-lipid film had formed on the rotary evaporator under reduced pressure, and heated in a 35°C water-bath. The film was hydrated with 5 mL PBS, followed by sonication. The obtained DiR-GA LPs were filtered in a 220 nm filter device, and the final concentration of DiR was 40 μg/mL.
Characterization of Cur-CA4P/GA LPs
The morphology of the Cur-CA4P/GA LPs was examined using a transmission electron microscopy. Samples (30 μL) were prepared by placing diluted LPs on a copper grid for 3 minutes, followed by using filter paper to absorb excess samples and staining with 2% phosphotungstic acid (pH 6.5) for 2 minutes. Size distribution and ζ-potential of the LPs were determined by dynamic light scattering at 25°C with a Malvern Zetasizer Nano ZS90. Samples were diluted in PBS and measured in triplicate. For measurement of loading content (LC) and encapsulation efficiency (EE), 1 mL Cur-CA4P/GA LPs was sampled after the removal of free drug and the volume made up with methanol to 10 mL. The absorbance of Cur and CA4P was analyzed with ultraviolet-visible spectrophotometry at 425 and 295 nm.36,37 LC and EE were calculated:
|
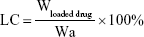
|

where Wloaded drug is drug loaded in LPs, Wtotal drug feeding weight of drug, and Wa weight of all materials.
Stability studies
Cur-CA4P/GA LPs were placed in a 4°C refrigerator for 1, 3, 7, 9, 11, and 14 days. Samples were diluted in PBS to 1 mg/mL for measurement of particle size and ζ-potential, and LC and EE were determined by the sample solution.
In vitro Cur and CA4P release from LPs
In vitro release of Cur and CA4P from LPs was studied by dialysis using PBS (pH 7.4) containing 1% Tween 80 and 20% anhydrous ethanol as release medium.38–40 Cur-CA4P/GA LPs (1 mL) suspension was transferred into the dialysis bag (cutoff 3.5 kDa), and the bag was placed into a conical bottle containing 50 mL release solution. The system was kept in a shaking incubator (THZ-82; Jintan Medical instrument Factory, Jintan, China) with stirring speed of 100 rpm at 37°C. After 0.5, 1, 2, 4, 8, 12, 24, 36, and 48 hours, 4 mL medium was removed and the same volume of release medium added. Concentrations of Cur and CA4P were measured with ultraviolet-visible spectrophotometry at wavelengths of 425 and 295 nm, respectively, and their cumulative release was calculated. Cumulative Cur- and CA4P-release percentage were calculated as follows:
|
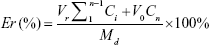
where Md is the amount of drugs in the LPs, V0 the whole volume of the release solution, Ci the concentration of Cur or CA4P in the release solution, and Vr the volume of the replaced solution.
In vitro cellular uptake assays
Cellular uptake of liposomal formulations was evaluated in BEL7402 cell by qualitative methods.41 Cells (5×103) were seeded onto a glass-bottom cell-culture dish (ϕ20 mm) and incubated for 24 hours at 37°C until adherent growth was observed. After incubation with FITC LPs or FITC-GA LPs (2.4 mg/mL) for 30 minutes, all reagents were removed and cells washed in cold PBS (pH 7.4) three times, fixed with 4% polyformaldehyde for 10 minutes, and stained with DAPI (0.1 μg/mL, diluted with PBS) for 5 minutes in the CO2 incubator. Finally, cells were washed three times with PBS and photographed under confocal laser-scanning microscopy (TCSSP8; Leica, Wetzlar, Germany). Excitation and emission wavelengths of DAPI and FITC were 359 and 461 nm and 490 and 520 nm, respectively. All operations were performed in the dark. Images were merged with Image-Pro Plus 6.0.
In vitro cytotoxicity assays
The in vitro cytotoxicity of different formulations was assessed using MTT assays.42 Approximately 5×103 B16 or BEL7402 cells were seeded in 96-well plates and incubated for 24 hours at 37°C. Cell-compatibility tests had to be performed before cells were treated with drugs. Cells were treated with different concentrations of blank GA LPs for 24 or 48 hours at 37°C. MTT reagent (10 μL, 5 mg/mL) was added to each well and incubated for 4 hours at 37°C in the dark. Approximately 150 μL dimethyl sulfoxide was then added to each well and shaken for 10 minutes to dissolve the formazan crystals. The absorbance value of each hole was detected at 490 nm using an enzyme marker (ELX800; BioTek Instruments, Winooski, VT, USA). Cells were treated with various concentrations of free Cur and CA4P, Cur + CA4P, Cur-CA4P LPs, and Cur-CA4P/GA LPs, and incubated for 24, 48, and 72 hours.
Cell-migration assays
Cell migration of free Cur and CA4P, Cur + CA4P (1:5), Cur-CA4P LPs, and Cur-CA4P/GA LPs were assessed through wound-healing assays.43 In brief, 104 B16 or BEL7402 cells were seeded in six-well plates and grown for 24 hours at 37°C in a CO2 incubator. When cell density reached 90%, cells were scratched with a sterile pipette tip with at least three lines per well. Scratched cells were washed off with PBS and the different drug formulations added. Images of cells invading the scratch were captured at 0, 24, and 48 hours using microscopy. The effect of drugs on cell migration was evaluated by calculating the migration rate:
|
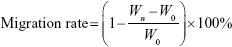
where Wn and W0 represent average scratch widths at n and 0 hours, respectively.
In vivo real-time near-infrared fluorescence imaging
Near-infrared fluorescence imaging (NIRF) was used to observe the biodistribution of GA LPs formulation in vivo, and DiR-GA LPs (40 μg/mL) were prepared to monitor the fate of LPs. When tumors were 100 mm3, DiR-GA LPs were injected into tail veins of mice. Free DiR was used as control. Precisely 10 mg DiR iodide in a 10 mL volumetric bottle was dissolved in methanol at a constant volume and prepared into a 1 mg/mL drug-reserve solution, which was stored at 4°C in the refrigerator for later use. DiR iodide reserve solution diluted with normal saline to the concentration of 40 μg/mL. Mice were anesthetized with 10% chloral hydrate and real-time NIRF images obtained with excitation and emission at 745 and 835 nm, respectively.44 Results were analyzed using the Living Image 3.1 software (PerkinElmer, Waltham, MA, USA).
In vivo antitumor efficacy
To evaluate the in vivo antitumor efficacy of the Cur-CA4P/GA LPs, we established the H22 tumor-bearing BALB/c male mice model. Establishment of this model was divided into two steps. First, 0.3 mL hepatoma H22 cells was injected into the abdominal cavity of five mice. After 2 weeks, the mice had developed ascites tumors. Second, cells from the H22 liver cancer ascites-tumor sources were collected in sterile conditions and diluted to 4×106 tumor cells/mL with normal saline. Approximately 0.2 mL of the suspension was subcutaneously inoculated into the flanks of mice. When tumor volume had reached ~100 mm3, mice were divided into five groups: normal saline (control group), free Cur, Cur + CA4P, Cur-CA4P LPs, and Cur-CA4P/GA LPs. Mice in each preparation group were administered 5 mg/kg Cur and 25 mg/kg CA4P every 2 days. During the treatment course, mice were weighed and diameters of tumors measured. Tumor volumes were calculated according to the formula L × W2/2 in which L is the longest and W the shortest tumor diameter (mm). Mice were killed and tumors harvested after completion of 2 weeks of treatment. Tumor weights were measured, and tumor growth–inhibition rate45 was calculated: (1 − tumor weight of drug-treated group/tumor weight of control group) ×100%.
Histochemical staining
After the drug administration in the in vivo antitumor study, tumor tissue samples were removed, fixed in 4% formaldehyde for >24 hours, and embedded in paraffin. Paraffin-embedded tumor samples were cut into slices of 4 μm using an ultrathin slicer (KD3358; Cody Instrument Equipment). For histological evaluation, H&E was used for tumor-tissue staining.46 Images were magnified 400 times by observation and photography with positive fluorescence microscopy (IX51; Olympus, Tokyo, Japan).
Statistical analyses
All data are expressed as means ± SD. Student’s t-test was used to determine whether differences between test groups were statistically significant. P<0.05 was considered statistically significant.
Results and discussion
Synthesis and characterization of DSPE-PEG2,000-GA
The targeting material DSPE-PEG2,000-GA was synthesized by coupling aminated GA to the DSPE-PEG2,000-NHS. Figure 2 indicates that GA was first modified by ethylenediamine to form GA-N. Second, the free-amino group in GA-N was reacted with DSPE-PEG2,000-NHS to generate DSPE-PEG2,000-GA. Figure 3 shows the characteristic peaks of DSPE-PEG2,000-NHS at 3.6 ppm (PEG group) in the NMR spectra obtained. Peaks at 0.7–1.5 ppm were assigned to the triterpenoid protons of GA-N, and peaks of DSPE-PEG2,000-GA were observed at 3.6 and 0.7–1.3 ppm. These results showed that GA-N had been successfully introduced into DSPE-PEG2,000-NHS by the presence of characteristic peaks at 3.6 and 0.7–1.3 ppm in the DSPE-PEG2,000-GA.
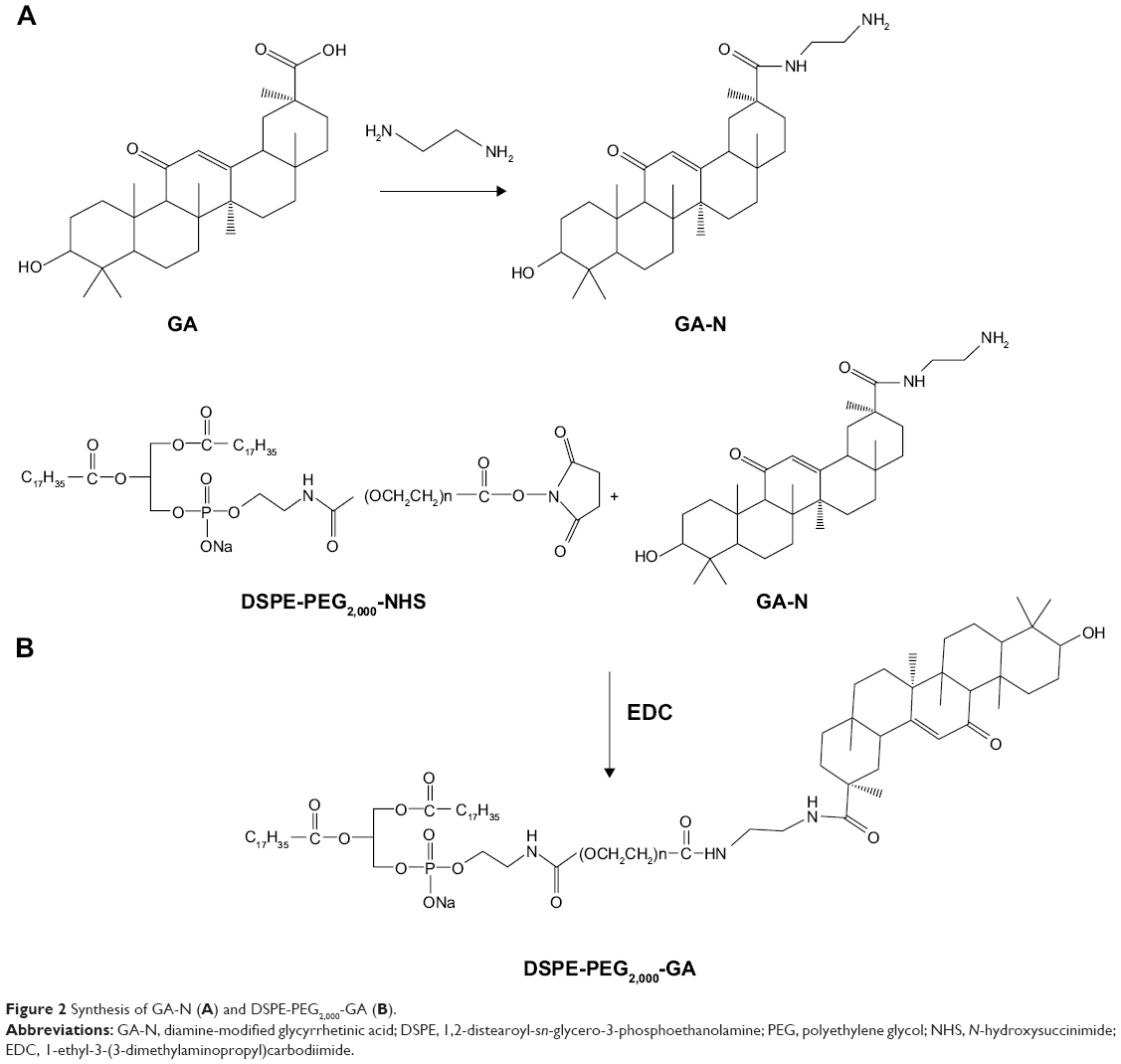 | Figure 2 Synthesis of GA-N (A) and DSPE-PEG2,000-GA (B). |
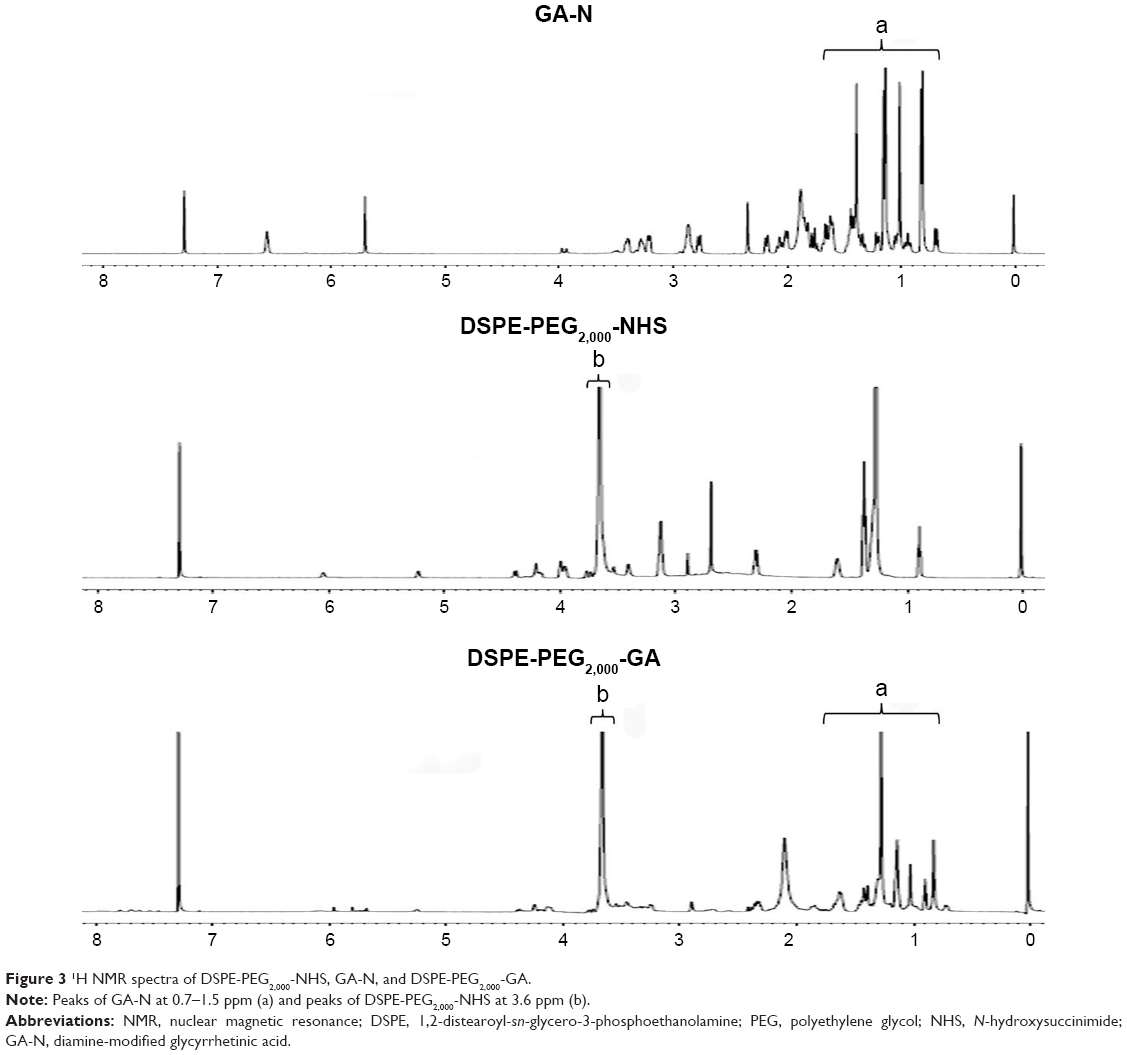 | Figure 3 1H NMR spectra of DSPE-PEG2,000-NHS, GA-N, and DSPE-PEG2,000-GA. |
Characterization of liver-targeting LPs for codelivery of Cur and CA4P
Mean particle size, ζ-potential, EE, and LC of each LP formulation are shown in Figure 4. LC and EE were measured when the ratios of Cur to CA4P ranged from 1:1 to 10:1, and the third group (Cur:CA4P 1:5) was selected as the representative candidate for further research, owing to its higher LC and EE values compared to those of other groups. Cur-CA4P/GA LPs were prepared by membrane dispersion (Figure 1). LPs were composed of a hydrophilic core and hydrophobic shell. The hydrophobic Cur was loaded into the hydrophobic membranes, whereas CA4P was encapsulated in the hydrophilic core. DSPE-PEG2,000-GA was embedded in the outer membrane. Morphological characteristics of the LPs were observed by transmission electron microscopy. Cur-CA4P/GA LPs showed a circular bubble structure with uniform size, which was similar to that of the blank LPs (Figure 5A and B). As shown in Figure 5C, Cur-CA4P/GA LPs showed an average particle size of ~166 nm with narrow size distribution (polydispersity index 0.23). LPs <200 nm in size are effectively easily accumulated in tumor via the EPR effect.47 The surface charge of LPs is an indication of their stability and interaction with cells.48 Table 1 shows that the ζ-potential of Cur-CA4P LPs and Cur-CA4P/GA LPs was ~−25 mV and exhibited good stability. The stability of Cur-CA4P/GA LPs was investigated for 2 weeks, and no significant changes in particle size, ζ-potential, EE, and LC were observed (P>0.05, Figure 6).
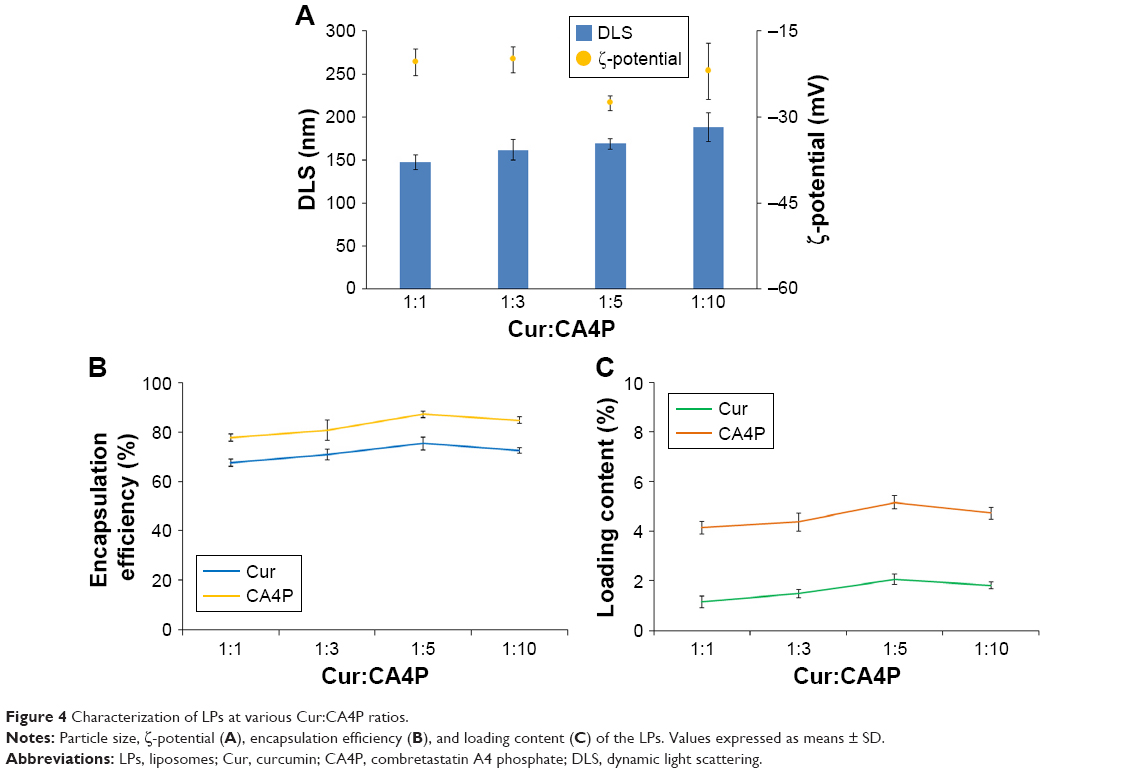 | Figure 4 Characterization of LPs at various Cur:CA4P ratios. |
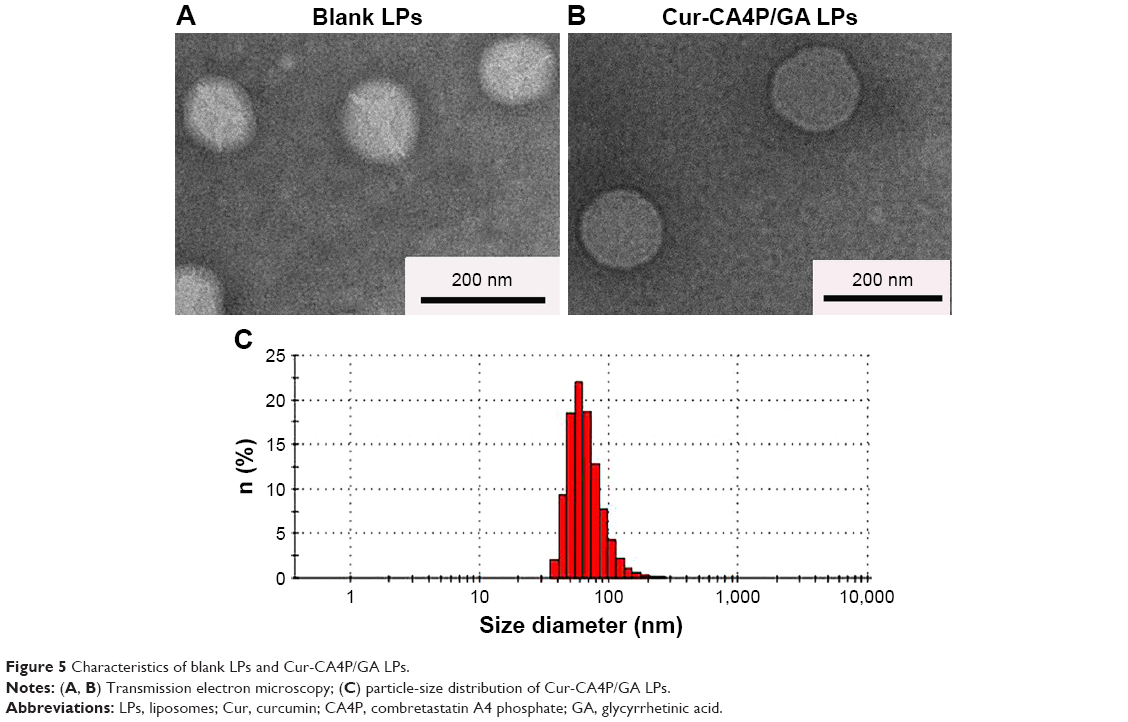 | Figure 5 Characteristics of blank LPs and Cur-CA4P/GA LPs. |
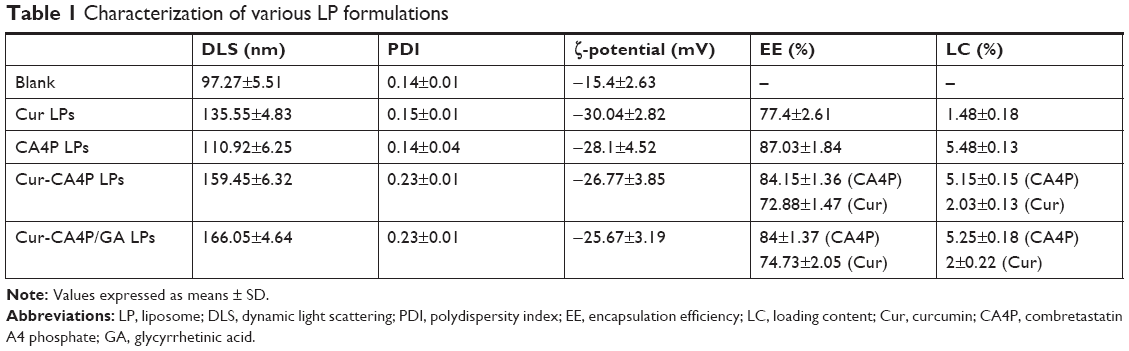 | Table 1 Characterization of various LP formulations |
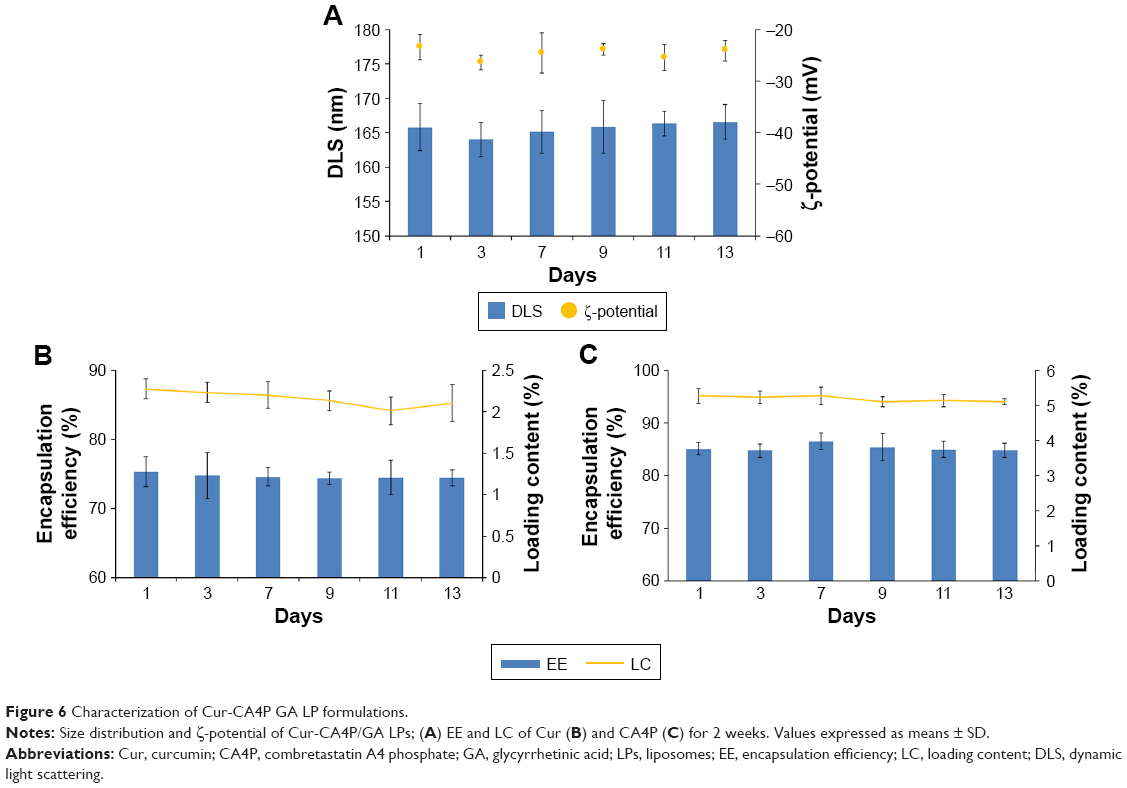 | Figure 6 Characterization of Cur-CA4P GA LP formulations. |
Cur and CA4P release from LPs in vitro
In vitro drug release of Cur and CA4P from Cur-CA4P/GA LPs was evaluated in PBS (pH 7.4) containing 1% Tween 80 and 20% anhydrous ethanol at 37°C. As shown in Figure 7, no obvious burst release of either drug was observed. Both drugs showed sustained release within 48 hours. The results showed certain sustained-release effects of agents encapsulated in the LP system. Cumulative release rates for both drugs were almost the same during the 10 hours of the experiment, whereas CA4P released more quickly than Cur after 10 hours. Approximately 60% of the CA4P was released within 24 hours, whereas <50% of Cur was released during the same time. At 48 hours, cumulative release was ~65% for CA4P and 50% for Cur, which might have been due to the faster release of hydrophilic CA4P than hydrophobic Cur in water-soluble PBS.
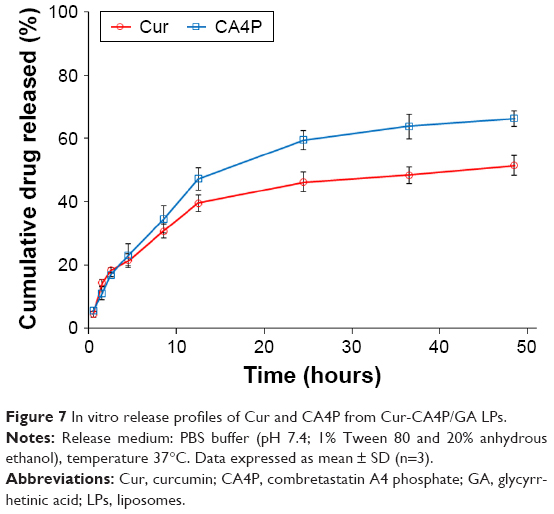 | Figure 7 In vitro release profiles of Cur and CA4P from Cur-CA4P/GA LPs. |
Cellular uptake analysis
The cellular uptake of FITC LPs and FITC LPs-GA was evaluated by confocal laser scanning microscopy. As shown in Figure 8, DAPI (blue) was regarded as a fluorescence marker for BEL7402 cell nuclei. The green fluorescence in the cytoplasm indicates FITC-labeled LPs. In the merged image, colocations of blue and green fluorescence indicate that the LPs had been ingested into the cells. As shown in Figure 8, FITC-GA LPs showed stronger green fluorescence than FITC LPs. The possible explanation is that the GA LPs were taken up by the cancer cells via GA-receptor-mediated endocytosis.49 The results suggest that the GA molecule may promote the uptake of LPs, resulting in additional fluorescence signal accumulated in the cytoplasm.
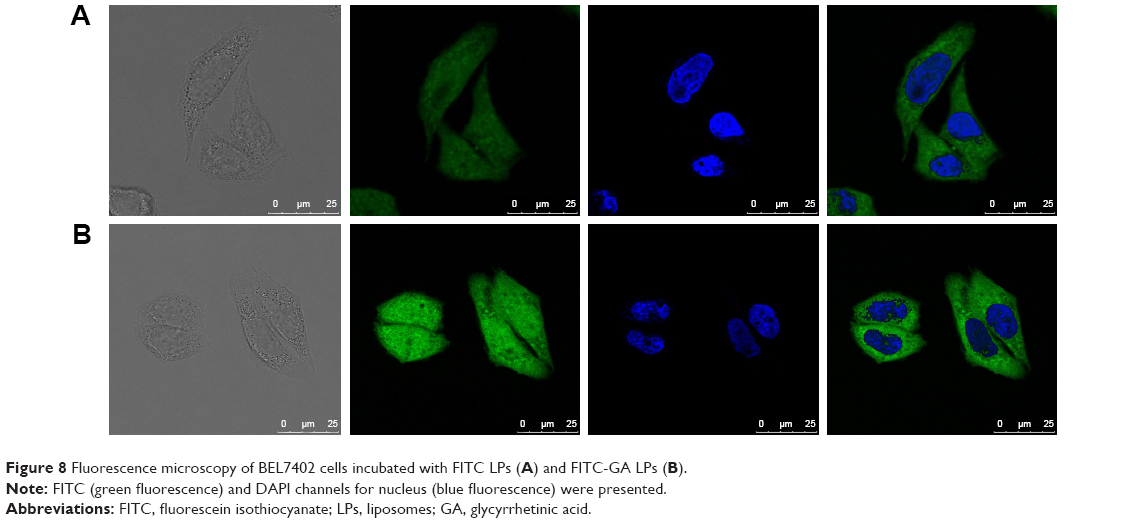 | Figure 8 Fluorescence microscopy of BEL7402 cells incubated with FITC LPs (A) and FITC-GA LPs (B). |
In vitro cytotoxicity assays
First, MTT was used to investigate the cytotoxicity of blank GA LPs against BEL7402 and B16 cells. Figure 9A and B show that cellular viability was >86% after blank GA-LP treatment for 24 hours. This result suggested that blank GA LPs exhibited no obvious toxicity at experimental conditions and can be used as drug-delivery carriers.
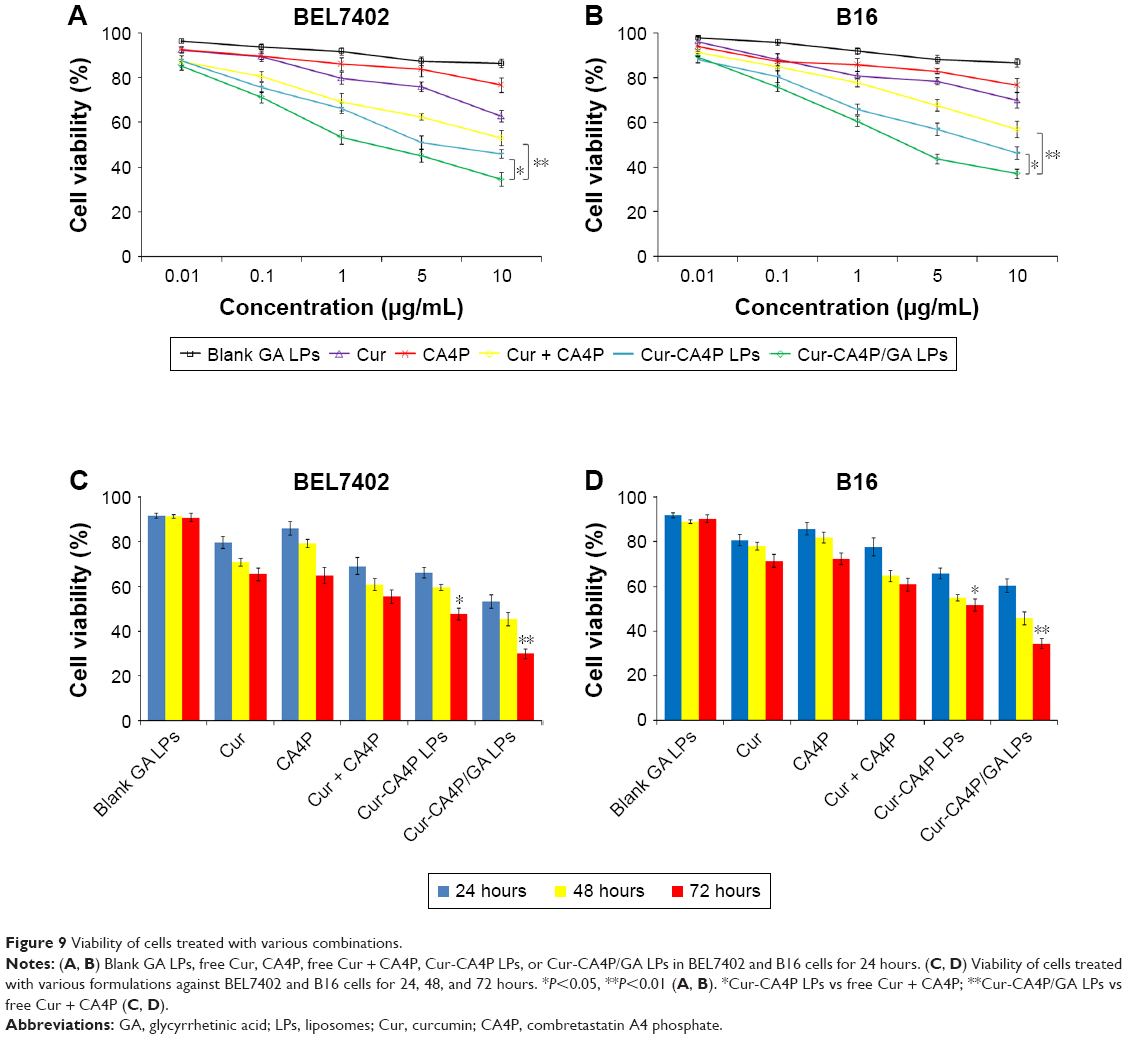 | Figure 9 Viability of cells treated with various combinations. |
In vitro cytotoxicity testing of drug-loaded LPs was conducted against BEL7402 and B16 cells. As shown in Figure 9A, free Cur, CA4P, and Cur + CA4P, Cur-CA4P LPs, and Cur-CA4P-GA LPs expressed dose-dependent cytotoxic effects in BEL7402 cells after incubation for 24 hours, which was similar to that in B16 cells (Figure 9B). Similar to the free-drug combination, codelivery of LP formulations exhibited time-dependent cytotoxicity against the two cell lines for 24, 48, and 72 hours (Figure 9C and D). Compared with free Cur and CA4P, the combination of Cur and CA4P showed higher cytotoxicity against BEL7402 and B16 cells, as shown in Figure 9A and B. The result suggested that the combination of chemotherapy drugs with antiangiogenic drugs exhibited cytotoxicity. Moreover, as shown in Figure 9C and D, Cur-CA4P LPs displayed stronger cytotoxicity than the free mixture of Cur and CA4P. Interestingly, Cur-CA4P/GA LPs showed higher cytotoxicity compared with Cur-CA4P LPs (P<0.05). This was because the introduction of GA increased cellular uptake of drug-loaded LPs, leading to additional accumulation of drugs in tumor cells.
Cell-migration assays
To confirm the cytotoxic effect of various formulations further, we performed wound-healing assays in BEL7402 and B16 cells. Cell movement and migration to the denuded area was measured after 24 hours. Photos were taken at specific time points, as shown in Figure 10. Tumor cells showed differences in migration and invasion abilities after treatment with the four different drug formulations. The scratched gap had almost disappeared in control group by 24 hours’ incubation. Free Cur slightly inhibited the migration of cells compared with the control group, with migration rates of 45.68% and 58.9% in BEL7402 and B16, respectively. Free CA4P also played a minimal role in inhibiting tumor-cell migration. However, Cur + CA4P showed improved migration inhibition, with rates of 38.29% and 47.28% in BEL7402 and B16, respectively (Figure 10C and D). This indicated that combination treatment of Cur and CA4P can increase migration-inhibitory ability over single-Cur therapy. The combination-treatment therapy was superior to the single drug. Obviously, Cur-CA4P LPs and Cur-CA4P/GA LPs showed stronger inhibition efficacy on cell migration than the free mixture of Cur and CA4P. The possible cause was that LPs, as a carrier, increased cell internalization of drugs. Compared with Cur-CA4P LPs, cell migration was remarkably inhibited for Cur-CA4P/GA LPs, with migration rates of only 15.74% and 15.69%, respectively. The results of the cell-migration test were consistent with cytotoxicity test results, where the Cur-CA4P/GA LPs group showed highest cell killing ability (P<0.01). GA molecules increased the active targeting of LPs and increased the cellular uptake of drugs. Therefore, Cur-CA4P/GA LPs inhibited the migration of cancer cells significantly.
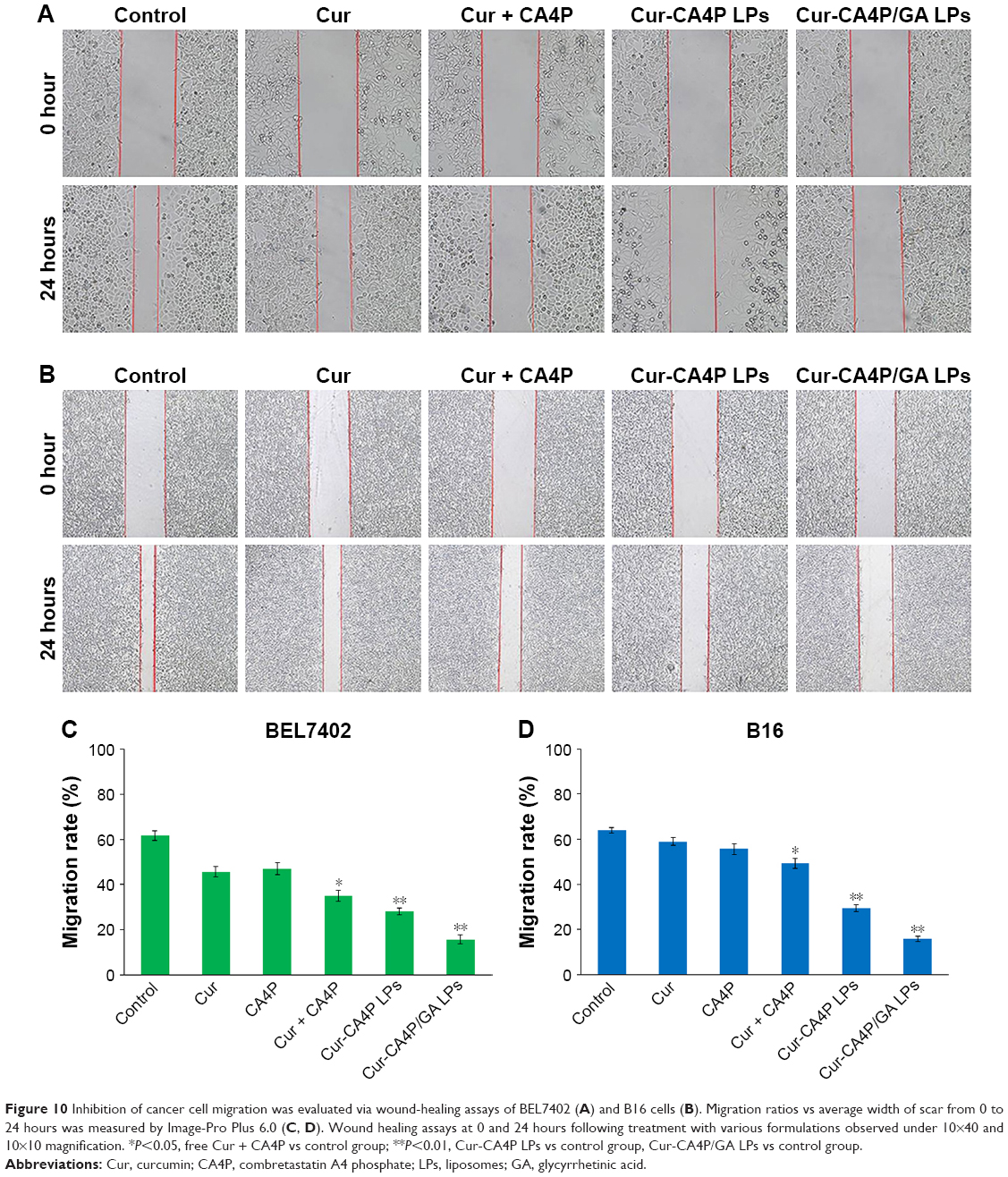 | Figure 10 Inhibition of cancer cell migration was evaluated via wound-healing assays of BEL7402 (A) and B16 cells (B). Migration ratios vs average width of scar from 0 to 24 hours was measured by Image-Pro Plus 6.0 (C, D). Wound healing assays at 0 and 24 hours following treatment with various formulations observed under 10×40 and 10×10 magnification. *P<0.05, free Cur + CA4P vs control group; **P<0.01, Cur-CA4P LPs vs control group, Cur-CA4P/GA LPs vs control group. |
In vivo real-time imaging of GA LPs
To investigate liver-targeting properties of GA LPs, we injected free DiR and DiR-GA LPs into H22 tumor-bearing mice. Results showed that NIRF signals of the free-DiR group were lower than the DiR-GA LPs group from 1 to 48 hours (Figure 11). No fluorescence signals in the tumor regions were detected for the free-DiR group, except at 2 hours. By contrast, DiR-GA LPs increased in accumulation in tumor regions and signals were sustained at 48 hours after injection. The GA LP drug loading system increased the accumulation of DiR in tumors. We concluded that the retention time of LPs were significantly prolonged in blood circulation. Moreover, GA improved the targeting of LPs and increased the accumulation of DiR in tumors. Therefore, the preferable active targeting ability of LPs to tumor sites will provide a key prerequisite for the superior combination therapy of Cur and CA4P.
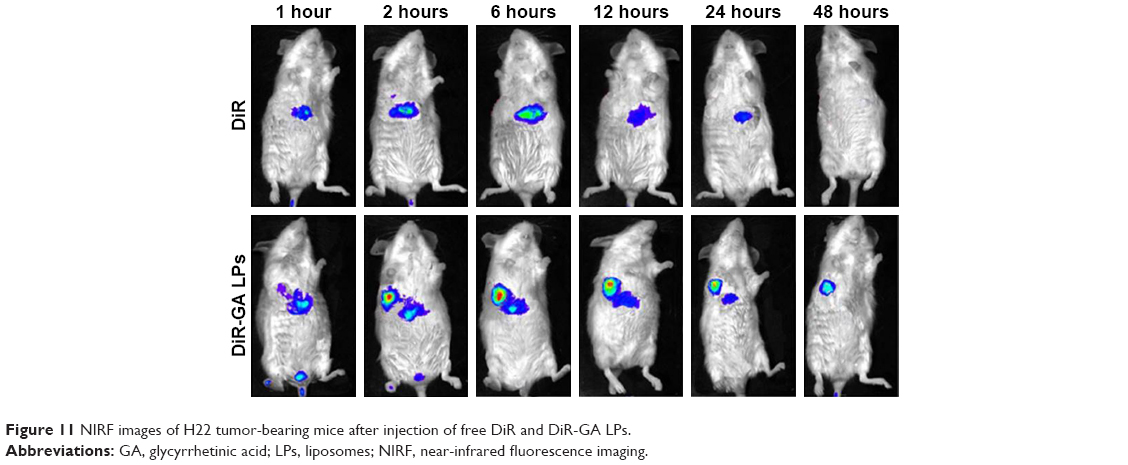 | Figure 11 NIRF images of H22 tumor-bearing mice after injection of free DiR and DiR-GA LPs. |
In vivo therapeutic efficacy
In vivo antitumor efficacy of Cur-CA4P/GA LPs for H22 tumor-bearing mice was tested for 2 weeks. As demonstrated in Figure 12A, body weight in the free-Cur group showed an evident decreasing trend compared with the control group, indicating the systemic toxicity of free Cur. Interestingly, no evident weight difference between LP groups and the control was observed, suggesting that LP formulations can reduce systemic toxicity. In Figure 12B, mice treated with physiological saline displayed a rapid increase in tumor size during the experimental period, and those treated with the four drug formulations showed a slight increase, indicating that these drug formulations inhibited the development of tumors. Moreover, compared with single-drug treatment, the combination of Cur and CA4P was more effective in antitumor therapy, where the two drugs achieved an additive effect by proapoptotic and antiangiogenic activities.50 Compared with free Cur + CA4P, the two LP formulations showed stronger antitumor efficacy (Figure 12B–D), which was consistent with MTT assays. The result was due to the fact that LPs improved the accumulation of Cur and CA4P in tumor regions via the EPR effect, resulting in high inhibition efficiency. Furthermore, as shown in Figure 12D, the tumor-inhibition rate of Cur-CA4P/GA LPs was 90.5%, which was higher than the Cur-CA4P LPs. A possible explanation was that GA LPs were taken up by tumor cells via GA-receptor-mediated endocytosis, which increased the antitumor effect by active liver-targeted delivery.
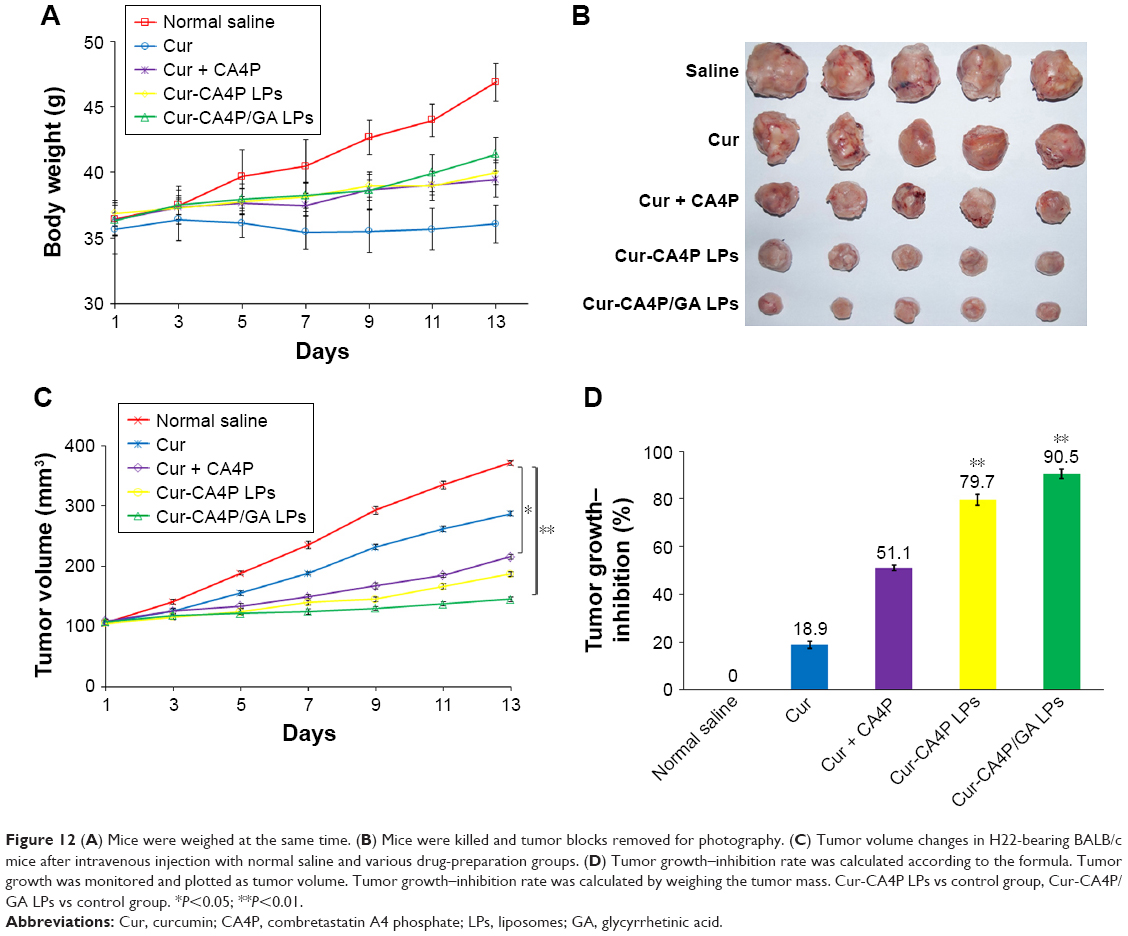 | Figure 12 (A) Mice were weighed at the same time. (B) Mice were killed and tumor blocks removed for photography. (C) Tumor volume changes in H22-bearing BALB/c mice after intravenous injection with normal saline and various drug-preparation groups. (D) Tumor growth–inhibition rate was calculated according to the formula. Tumor growth was monitored and plotted as tumor volume. Tumor growth–inhibition rate was calculated by weighing the tumor mass. Cur-CA4P LPs vs control group, Cur-CA4P/GA LPs vs control group. *P<0.05; **P<0.01. |
Histochemical staining
H&E staining was conducted to evaluate the antitumor effect of Cur-CA4P/GA LPs further. As shown in Figure 13, cancer cells showed no significant necrosis in the saline control group, in which nuclei were maintained in their full state. By contrast, tumor cells treated with drug formulations exhibited evident morphologic changes, with shrunken nuclei and low cellular density, suggesting inhibition in the development of tumor cells. Compared with the free-Cur group, the combination group produced more nuclear shrinkage, suggesting that the combination of Cur and CA4P achieved enhanced antitumor effects by proapoptotic and antiangiogenic activities.51 Moreover, tumor cells treated with Cur-CA4PLP and Cur-CA4P GALP formulations showed lower cellular density and more nuclear shrinkage in comparison with the Cur + CA4P group, indicating more drugs accumulated in tumor regions under treatment with the LP formulation. Among all the formulations, Cur-CA4P/GA LPs showed the most potent efficacy. This was probably due to the fact that GA LPs effectively delivered drugs to the tumor microenvironment via the EPR effect and increase drug uptake by tumor cells via GA-receptor-mediated endocytosis.
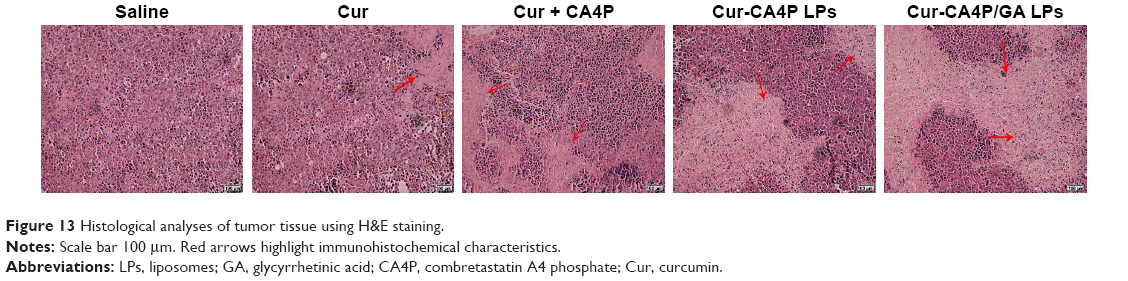 | Figure 13 Histological analyses of tumor tissue using H&E staining. |
Conclusion
In this work, novel Cur-CA4P/GA LPs were developed for effective antihepatoma therapy. GA LPs promoted cellular uptake and achieved more accumulation in tumor regions compared with GA-free LPs. Moreover, Cur-CA4P/GA LPs exhibited enhanced in vitro cytotoxicity and in vivo antitumor effects. In summary, GA-modified LPs for codelivery of proapoptotic and antiangiogenic drugs is a promising strategy for cancer therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (8180346), Natural Science Foundation of Shandong Province (ZR2017LC002), the Education Science and Technology Project of Shandong Province (J17KA141), Medical and Health Technology Development Program in Shandong Province (2016WS0673), and Project of Traditional Chinese Medicine Technology Development Program in Shandong Province (2017–212).
Disclosure
The authors report no conflicts of interest in this work.
References
Heo YA, Syed YY. Regorafenib: a review in hepatocellular carcinoma. Drugs. 2018;78(9):951–958. | ||
Xu T, Zhang J, Chen W, et al. ARK5 promotes doxorubicin resistance in hepatocellular carcinoma via epithelial-mesenchymal transition. Cancer Lett. 2016;377(2):140–148. | ||
Hanaoka H, Nakajima T, Sato K, et al. Photoimmunotherapy of hepatocellular carcinoma-targeting glypican-3 combined with nanosized albumin-bound paclitaxel. Nanomedicine. 2015;10(7):1139–1147. | ||
Zhu D, Wu S, Hu C, et al. Folate-targeted polymersomes loaded with both paclitaxel and doxorubicin for the combination chemotherapy of hepatocellular carcinoma. Acta Biomater. 2017;58:399–412. | ||
Sabra SA, Elzoghby AO, Sheweita SA, et al. Self-assembled amphiphilic zein-lactoferrin micelles for tumor targeted co-delivery of rapamycin and wogonin to breast cancer. Eur J Pharm Biopharm. 2018;128:156–169. | ||
Li J, Guo C, Feng F, et al. Co-delivery of docetaxel and palmitoyl ascorbate by liposome for enhanced synergistic antitumor efficacy. Sci Rep. 2016;6(1):38787. | ||
Yang H, Wang J, Fan JH, et al. Ilexgenin A exerts anti-inflammation and anti-angiogenesis effects through inhibition of STAT3 and PI3K pathways and exhibits synergistic effects with sorafenib on hepatoma growth. Toxicol Appl Pharmacol. 2017;315:90–101. | ||
Zhang C, An T, Wang D, et al. Stepwise pH-responsive nanoparticles containing charge-reversible pullulan-based shells and poly(β-amino ester)/poly(lactic-co-glycolic acid) cores as carriers of anticancer drugs for combination therapy on hepatocellular carcinoma. J Control Release. 2016;226:193–204. | ||
Zhu J, Hu M, Qiu L. Drug resistance reversal by combretastatin-A4 phosphate loaded with doxorubicin in polymersomes independent of angiogenesis effect. J Pharm Pharmacol. 2017;69(7):844–855. | ||
di Martino RM, Bisi A, Rampa A, Gobbi S, Belluti F. Recent progress on curcumin-based therapeutics: a patent review (2012–2016). Part II: curcumin derivatives in cancer and neurodegeneration. Expert Opin Ther Pat. 2017;27(8):953–965. | ||
Cheng Y, Zhao P, Wu S, et al. Cisplatin and curcumin co-loaded nano-liposomes for the treatment of hepatocellular carcinoma. Int J Pharm. 2018;545(1–2):261–273. | ||
Marquardt JU, Gomez-Quiroz L, Camacho AL, et al. Curcumin effectively inhibits oncogenic NF-κB signaling and restrains stemness features in liver cancer. J Hepatol. 2015;63(3):661–669. | ||
Liu Y, de Keyzer F, Wang Y, et al. The first study on therapeutic efficacies of a vascular disrupting agent CA4P among primary hepatocellular carcinomas with a full spectrum of differentiation and vascularity: correlation of MRI-microangiography-histopathology in rats. Int J Cancer. 2018. | ||
Zhu J, Xu X, Hu M, Qiu L. Co-encapsulation of combretastatin-A4 phosphate and doxorubicin in polymersomes for synergistic therapy of nasopharyngeal epidermal carcinoma. J Biomed Nanotechnol. 2015;11(6):997–1006. | ||
Yang WJ, Zhou P, Liang L, et al. Nanogel-incorporated injectable hydrogel for synergistic therapy based on sequential local delivery of combretastatin-A4 phosphate (CA4P) and doxorubicin (Dox). ACS Appl Mater Interfaces. 2018;10(22):18560–18573. | ||
Batra H, Pawar S, Bahl D. Curcumin in combination with anti-cancer drugs: a nanomedicine review. Pharmacol Res. 2019;139:91–105. | ||
Gao C, Tang F, Gong G, et al. pH-responsive prodrug nanoparticles based on a sodium alginate derivative for selective co-release of doxorubicin and curcumin into tumor cells. Nanoscale. 2017;9(34):12533–12542. | ||
Xiao B, Si X, Han MK, et al. Co-delivery of camptothecin and curcumin by cationic polymeric nanoparticles for synergistic colon cancer combination chemotherapy. J Mater Chem B. 2015;3(39):7724–7733. | ||
Pinheiro A, Silva AM, Teixeira JH, et al. Extracellular vesicles: intelligent delivery strategies for therapeutic applications. J Control Release. 2018;289:56–69. | ||
Seidi K, Neubauer HA, Moriggl R, Jahanban-Esfahlan R, Javaheri T. Tumor target amplification: implications for nano drug delivery systems. J Control Release. 2018;275:142–161. | ||
Zununi Vahed S, Salehi R, Davaran S, Sharifi S. Liposome-based drug co-delivery systems in cancer cells. Mat Sci Eng. 2017;71:1327–1341. | ||
Saleem J, Wang L, Chen C. Carbon-Based nanomaterials for cancer therapy via targeting tumor microenvironment. Adv Healthc Mater. 2018;7(20):1800525. | ||
di Bucchianico S, Gliga AR, Åkerlund E, et al. Calcium-dependent cyto- and genotoxicity of nickel metal and nickel oxide nanoparticles in human lung cells. Part Fibre Toxicol. 2018;15(1):32. | ||
Mcclements DJ. Advances in nanoparticle and microparticle delivery systems for increasing the dispersibility, stability, and bioactivity of phytochemicals. Biotechnol Adv. 2018;S0734–9750(18):30136–30138. | ||
Rezaee M, Oskuee RK, Nassirli H, Malaekeh-Nikouei B. Progress in the development of lipopolyplexes as efficient non-viral gene delivery systems. J Control Release. 2016;236:1–14. | ||
Dai Y, Xu C, Sun X, et al. Nanoparticle design strategies for enhanced anticancer therapy by exploiting the tumour microenvironment. Chem Soc Rev. 2017;46(12):3830–3852. | ||
Sun Y, Lu J, Yan D, et al. Cellular uptake mechanism and clearance kinetics of fluorescence-labeled glycyrrhetinic acid and glycyrrhetinic acid-modified liposome in hepatocellular carcinoma cells. Environ Toxicol Pharmacol. 2017;53:46–56. | ||
Zhang J, Zheng Y, Xie X, et al. Cleavable multifunctional targeting mixed micelles with sequential pH-triggered Tat peptide activation for improved antihepatocellular carcinoma efficacy. Mol Pharm. 2017;14(11):3644–3659. | ||
Han X, Wang Z, Wang M, et al. Liver-targeting self-assembled hyaluronic acid-glycyrrhetinic acid micelles enhance hepato-protective effect of silybin after oral administration. Drug Deliv. 2016;23(5):1818–1829. | ||
Sun Y, Dai C, Yin M, et al. Hepatocellular carcinoma-targeted effect of configurations and groups of glycyrrhetinic acid by evaluation of its derivative-modified liposomes. Int J Nanomedicine. 2018;13:1621–1632. | ||
Wu JL, Tian GX, Yu WJ, et al. pH-responsive hyaluronic acid-based mixed micelles for the Hepatoma-Targeting delivery of doxorubicin. Int J Mol Sci. 2016;17(4):364. | ||
D’Este M, Eglin D, Alini M. A systematic analysis of DMTMM vs EDC/NHS for ligation of amines to hyaluronan in water. Carbohydr Polym. 2014;108:239–246. | ||
Li ZL, Peng SF, Chen X, et al. Pluronics modified liposomes for curcumin encapsulation: sustained release, stability and bioaccessibility. Food Res Int. 2018;108:246–253. | ||
Sun D, Zhou JK, Zhao L, et al. Novel curcumin liposome modified with hyaluronan targeting CD44 plays an anti-leukemic role in acute myeloid leukemia in vitro and in vivo. ACS Appl Mater Interfaces. 2017;9(20):16857–16868. | ||
Juanjuan M, Jianming C. Nanoformulations of the vascular inhibitors: combretastatin A4 and combretastatin A4 phosphate. Pharm Sci-us. 2016;43(5):893–898. | ||
Hong SS, Thapa RK, Kim JH, et al. Role of zein incorporation on hydrophobic drug-loading capacity and colloidal stability of phospholipid nanoparticles. Colloids Surf B Biointerfaces. 2018;171:514–521. | ||
Su T, Long Y, Deng C, et al. Construction of a two-in-one liposomal system (TWOLips) for tumor-targeted combination therapy. Int J Pharm. 2014;476(1–2):241–252. | ||
Baghbani F, Moztarzadeh F. Bypassing multidrug resistant ovarian cancer using ultrasound responsive doxorubicin/curcumin co-deliver alginate nanodroplets. Colloids Surf B Biointerfaces. 2017;153:132–140. | ||
Li R, Deng L, Cai Z, et al. Liposomes coated with thiolated chitosan as drug carriers of curcumin. Mater Sci Eng C. 2017;80:156–164. | ||
Ju L, Cailin F, Wenlan W, et al. Preparation and properties evaluation of a novel pH-sensitive liposomes based on imidazole-modified cholesterol derivatives. Int J Pharm. 2017;518(1–2):213–219. | ||
Alemi A, Reza JZ, Haghiralsadat F, et al. Paclitaxel and curcumin coadministration in novel cationic PEGylated niosomal formulations exhibit enhanced synergistic antitumor efficacy. J Nanobiotechnology. 2018;16(1):28. | ||
Dash TK, Konkimalla VB. Selection of P-glycoprotein inhibitor and formulation of combinational Nanoformulation containing selected agent curcumin and Dox for reversal of resistance in K562 cells. Pharm Res. 2017;34(8):1741–1750. | ||
Zhang P, Li J, Ghazwani M, et al. Effective co-delivery of doxorubicin and dasatinib using a PEG-Fmoc nanocarrier for combination cancer chemotherapy. Biomaterials. 2015;67:104–114. | ||
Lv L, Qiu K, Yu X, et al. Amphiphilic copolymeric micelles for doxorubicin and curcumin co-delivery to reverse multidrug resistance in breast cancer. J Biomed Nanotechnol. 2016;12(5):973–985. | ||
Zhang J, Li J, Shi Z, et al. pH-sensitive polymeric nanoparticles for co-delivery of doxorubicin and curcumin to treat cancer via enhanced pro-apoptotic and anti-angiogenic activities. Acta Biomaterialia. 2017;58:349–364. | ||
Yang Z, Sun N, Cheng R, et al. pH multistage responsive micellar system with charge-switch and PEG layer detachment for co-delivery of paclitaxel and curcumin to synergistically eliminate breast cancer stem cells. Biomaterials. 2017;147:53–67. | ||
Deshantri AK, Moreira AV, Ecker V, et al. Nanomedicines for the treatment of hematological malignancies. J Control Release. 2018;287:194–215. | ||
Ren T, Lin X, Zhang Q, et al. Encapsulation of azithromycin ion pair in liposome for enhancing ocular delivery and therapeutic efficacy on dry eye. Mol Pharm. 2018;15(11):4862–4871. | ||
Tan J, Shen W, Shi W, et al. ONTD induces growth arrest and apoptosis of human hepatoma BEL-7402 cells though a peroxisome proliferator-activated receptor γ-dependent pathway. Toxicol In Vitro. 2017;45(Pt 1):44–53. | ||
Feng T, Wei Y, Lee RJ, Zhao L. Liposomal curcumin and its application in cancer. Int J Nanomedicine. 2017;12:6027–6044. | ||
Huang D, Zhang S, Zhong T, et al. Multi-targeting NGR-modified liposomes recognizing glioma tumor cells and vasculogenic mimicry for improving anti-glioma therapy. Oncotarget. 2016;7(28):43616–43628. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.