")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Liver-targeted cyclosporine A-encapsulated poly (lactic-co-glycolic) acid nanoparticles inhibit hepatitis C virus replication
Authors Jyothi KR, Beloor J, Jo A, Nguyen MN, Choi TG, Kim J, Akter S, Lee S, Maeng CH, Baik HH, Kang I, Ha J, Kim SS
Received 22 September 2014
Accepted for publication 1 November 2014
Published 30 January 2015 Volume 2015:10(1) Pages 903—921
DOI https://doi.org/10.2147/IJN.S74723
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
KR Jyothi,1 Jagadish Beloor,2 Ara Jo,1 Minh Nam Nguyen,1 Tae Gyu Choi,1 Jin-Hwan Kim,1 Salima Akter,1 Sang-Kyung Lee,2 Chi Hoon Maeng,3 Hyung Hwan Baik,1 Insug Kang,1 Joohun Ha,1 Sung Soo Kim1
1Department of Biochemistry and Molecular Biology, School of Medicine, Kyung Hee University, Seoul, Republic of Korea; 2Department of Bioengineering and Institute for Bioengineering and Biopharmaceutical Research, Hanyang University, Seoul, Republic of Korea; 3Department of Medical Oncology and Hematology, Kyung Hee University Hospital, Seoul, Republic of Korea
Abstract: Therapeutic options for hepatitis C virus (HCV) infection have been limited by drug resistance and adverse side effects. Targeting the host factor cyclophilin A (CypA), which is essential for HCV replication, offers a promising strategy for antiviral therapy. However, due to its immunosuppressive activity and severe side effects, clinical application of cyclosporine A (CsA) has been limited as an antiviral agent. To overcome these drawbacks, we have successfully developed a liver-specific, sustained drug delivery system by conjugating the liver-targeting peptide (LTP) to PEGylated CsA-encapsulated poly (lactic-co-glycolic) acid (PLGA) nanoparticles. Furthermore, our delivery system exhibited high specificity to liver, thus contributing to the reduced immunosuppressive effect and toxicity profile of CsA. Finally, targeted nanoparticles were able to effectively inhibit viral replication in vitro and in an HCV mouse model. As a proof of principle, we herein show that our delivery system is able to negate the adverse effects of CsA and produce therapeutic effects in an HCV mouse model.
Keywords: HCV, liver-targeting peptide, targeted drug delivery
Introduction
Hepatitis C virus (HCV) is a leading cause of liver cancer and induces a persistent infection in more than 170 million people worldwide.1 The current triple therapy for HCV infection involves the combination of interferon-α (IFN-α) and ribavirin with a direct-acting antiviral (DAA) such as telaprevir or boceprevir.2 Despite the increase in sustained virological response rates, triple therapy has been accompanied by drug resistance and adverse side effects such as nausea, diarrhea, dysgeusia, and neutropenia.3 Since the major limitation of first-generation DAAs is their low genetic barrier of resistance, the second-generation viral protease, NS5A and NS5B inhibitors are currently clinically assessed in combination with or without IFN-α or ribavirin.4–6 Although development of second-generation DAAs has improved the antiviral effect compared to the first-generation DAAs, there might be differences in the antiviral resistance and sustained virological response between the genotypes. Despite the improved sustained virological response of IFN-α free DAA therapy against non-genotype 1 infection, emergence of resistance by the oral therapies is still a major concern.7,8 In order to address the limitations of current antiviral therapies, alternative approaches have sought to target host factors required for HCV-associated pathogenesis.9,10 IFN-α free DAA regimens with the optimal combination of host-targeting agent have been shown to have very promising antiviral efficacy with a high genetic barrier of resistance against all genotypes including genotype 2 and 3.11,12
Several lines of evidence support the notion that host factor cyclophilin A (CypA) has a major influence on the conformation of two HCV nonstructural proteins, NS5A and NS5B, and thus enhances HCV replication activity.13–15 Although cyclosporine A (CsA) can effectively inhibit HCV replication, its immunosuppressive activity and adverse side effects have caused the therapeutic use of the drug to decline.16 Also, because of a very narrow therapeutic window, excessive levels of CsA can lead to severe side effects including nephrotoxicity, hepatotoxicity, neurotoxicity, hypertension, and dyslipidemia.17,18 Alisporivir, a non-immunosuppressive analog of CsA, is the current CypA inhibitor in the clinical trials for HCV. Although the combination of alisporivir with IFN-α/ribavirin was put on partial clinical hold at Phase III, it is now under development in an IFN-α free regimen and yet to be approved by the US Food and Drug Administration (FDA).19 Since the development of new drugs is currently quite challenging, it is important to investigate the possibility that new therapeutic formulations might reduce adverse side effects.
With the recent advances in the field of nanoscience, poly (lactic-co-glycolic) acid (PLGA) has been intensively investigated as a nanocarrier of small hydrophobic and hydrophilic drugs as well as biological macromolecules.20 As a drug delivery carrier, PLGA-based nanoparticles have the ability to achieve sustained release of the therapeutic agent, which results in improved pharmacokinetic and phamacodynamic profiles. Notably, PLGA has been approved by the FDA for use in clinical applications. During the last decade, extensive research has shown that a CsA-based, PLGA nanoparticle is an effective ocular drug delivery system for treating keratoconjunctivitis and other ocular-related inflammatory diseases.21,22 Moreover, PLGA nanoparticulate formulation of CsA resulted in lower nephrotoxicity compared to the conventional CsA.23 Therefore, nanoparticulate formulation strategies provide a means to incorporate an old drug into a new delivery system, thereby minimizing related side effects and maximizing the therapeutic value.
PLGA drug-loaded nanoparticles have also been designed with a targeting moiety for selective delivery via receptor-mediated internalization.24,25 Since nontargeted nanoparticles have low cellular uptake, anticancer drugs have been engineered so as to be directly targeted to tumor cells. Targeted delivery of platinum-based drugs such as cisplatin has been shown to enhance the drug’s antitumor efficacy against lung and breast cancer.26–28 Interestingly, a new target-specific delivery system of IFN-α using hyaluronic acid-conjugated gold nanoparticles has enhanced the expression levels of key components of the innate immune response associated with the anti-HCV effect in normal BALB/c mice; this system may also reduce lung-associated toxicity.29
Based upon the encouraging results with targeted nanoparticles as compared to nontargeted nanoparticles, we developed a new formulation of CsA-encapsulated PLGA nanoparticles conjugated with a liver-targeting peptide (LTP). The LTP includes conserved region I from the circumsporozoite protein (CSP) of Plasmodium sporozoites that specifically binds to heparin sulfate proteoglycans (HSPGs) on the surface of hepatocytes.30 The feasibility of using targeted CsA encapsulated PLGA nanoparticles as a nanosized HCV replication inhibitor was assessed by measuring the size and surface charge distribution, encapsulation efficiency, and drug release profile. The liver specificity of targeted nanoparticles was analyzed using biodistribution studies. Finally, the toxicity profile and antiviral efficacy of liver-specific CsA-encapsulated PLGA nanoparticles were evaluated both in vitro and in an HCV mouse model.
Methods
Preparation and surface modification of PLGA nanoparticles
The PLGA nanoparticles were prepared by oil-in-water emulsification solvent evaporation followed by lyophilization. Briefly, PLGA (50 mg) was dissolved in 1 mL dichloromethane with or without CsA (5 mg). This organic mixture was added dropwise to 10 mL of aqueous 4% poly vinyl alcohol (PVA) solution under constant stirring. The micro-size PLGA particles were subjected to sonication (60 seconds, 30% amplitude) to achieve homogenous and nanosized particles. The resulting PLGA nanoparticles were poured into 200 mL of 2% PVA solution and stirred for 4 hours at room temperature to evaporate the organic content. To collect the dispersed PLGA nanoparticles, centrifugation was performed at 15,000 rpm for 20 minutes at 4°C; collected PLGA nanoparticles were washed three times with distilled water. The resulting nanosuspension was subsequently cooled to –20°C and lyophilized.
N-Hydroxysuccinimide (NHS)/ethyl (dimethylaminopropyl) carbodiimide (EDC) coupling was employed for the attachment of both the polyethylene glycol (PEG) spacer and the LTP to PLGA nanoparticles. First, the PEG spacer was linked by the formation of amide bonds between the primary amine group of PEG-bis (amine) (NH2-PEG-NH2) and the carboxylic group of PLGA nanoparticles. Then, the LTP was conjugated to PEGylated nanoparticles via amide bonds between the amine group of conjugated NH2-PEG-NH2 and carboxyl group of the LTP. Briefly, the carboxylates of the 1 mg PLGA nanoparticles were activated by resuspension in 500 μL of buffer containing 0.1 M 2-(N-morpholino) ethanesulfonic acid and 0.5 M NaCl, pH 6.0, and then incubation with 5 mM EDC and 10 mM sulfo-NHS for 15 minutes at room temperature on a rotator. Removal of unreacted excess EDC/NHS and buffer change (phosphate-buffered saline [PBS], pH 7.4) was done by centrifuging the particles at 15,000 rpm for 5 minutes. After activating carboxyl groups, the PLGA nanoparticles were reacted with PEG-bis (amine) (2 kDa) to form CsA nanoparticles (CsANP) at a 1:1.5 molar ratio for 45 minutes at room temperature on a rotator. CsANP were further conjugated with 500 μg of carboxyl group-activated LTP using an EDC/NHS reaction to yield the final targeted nanoparticles (CsANP-LTP).
Fluorescent nanoparticles for cellular uptake and biodistribution studies were prepared by linking Alexa-488 directly to PEG-conjugated nanoparticles (Alexa-488-CsANP) or to targeted nanoparticles (Alexa-488-LTP/CsANP) using the standard manufacturer’s protocol. DiI fluorescent dye-encapsulated nanoparticles (Alexa-488-LTP/DiINP) were prepared as described above for CsANP-LTP.
Characterization
Mean size distribution and zeta (ζ) potentials of PLGA nanoparticles were determined by dynamic light scattering (DLS) (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK). The nanoparticles were dissolved and dispersed in distilled water at a concentration of 1 mg/mL.
Scanning electron microscope measurements
Lyophilized nanoparticles were mounted on the holder and coated with gold under vacuum, and their morphology was investigated using a scanning electron microscope (SEM) (S-4160 FE-SEM; Hitachi Ltd., Tokyo, Japan).
Encapsulation efficiency
The content of the drug in the nanoparticles was determined using high-performance liquid chromatography (1525 dual pump; Waters, Milford, MA, USA) equipped with an ultraviolet detector and a C18 column as described previously.22 The column was equilibrated with a mobile phase (80% acetonitrile and 20% water), and samples were eluted during isocratic mobile phase conditions at a rate of 1 mL/min and detected at 210 nm. The encapsulation efficiency of CsA was calculated as reported previously.22,23
Release profile
CsA-encapsulated PLGA nanoparticles were dispersed in PBS (pH 7.4) and incubated in a shaking water bath at 200 rpm for various time intervals at 37°C. At each respective time point, nanoparticles were removed by centrifuging at 14,000 rpm for 15 minutes at 4°C. The released CsA content in the solution was analyzed by high-performance liquid chromatography, as described in encapsulation efficiency method.
In vitro studies
Luciferase assay
The luciferase activity of HCV replicon cells directly correlates with the levels of HCV RNA synthesis.34 HCV replication in Con1b replicon cells was determined by monitoring Renilla luciferase activity (Promega Corporation, Fitchburg, WI, USA), as described previously.31 In brief, HCV replicon cells were seeded in each well of a 12-well plate at a density of 1.5×105/well. The cells were treated with CsA, CsANP, or CsANP-LTP (equivalent concentration of 2.5 μg CsA each) for 48 or 72 hours. The luciferase signal was measured in triplicate. For the sustained antiviral effect, HCV replicon cells were treated with CsA, CsANP, or CsANP-LTP (equivalent concentration of 2.5 μg CsA each). After 12 hours of treatment, the cells’ media were replaced with fresh culture medium followed by culturing for different time points and analyzed for luciferase activity. The relative luciferase activity obtained from CsA-, CsANP-, or CsANP-LTP-treated cells were normalized to the corresponding values obtained with untreated cells.
In vivo studies
All animal experiments were approved by the Institutional Animal Care and Use Committee of Kyung Hee University (Seoul, Korea).
Biodistribution
Balb/c mice (n=3) were intravenously injected with Alexa-488-labeled nanoparticles. At 24 hours after administration, the distribution of Alexa-488-labeled CsANP or CsANP-LTP in Balb/c mice was analyzed by ex vivo imaging of different organs using a Kodak imaging station (4000MM; Eastman Kodak Company, Scientific Imaging Systems, New Haven, CT, USA). The mean fluorescence intensity in the region of interest (ROI) was determined and expressed as a fold change in the intensity with respect to mock-injected mice.
Toxicity profile
Male, 6-week-old Balb/c mice (Orient Bio Inc, Seongnam, Korea) were used to assess the toxicity of free CsA and different nanoparticle formulations. The animals were divided into four groups (n=6): untreated control, and groups treated with either free CsA, CsANP, or CsANP-LTP. The mice were injected intravenously every other day with CsA, CsANP, or CsANP-LTP (15 mg/kg of CsA or equivalently loaded respective nanoparticles) for a period of 21 days. Body weights were obtained and blood and organ samples were collected from control and treated groups to assess systemic toxicity in mice. To assess hepatotoxicity, the activities of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured using a GOT·GPT kit (Asan Pharm, Gyeonggi-do, Korea) according to the manufacturer’s protocol adjusted to a 96-well microplate. For nephrotoxicity, blood urea nitrogen (BUN) levels in the serum were measured using a BUN Enzymatic kit (Bioo Scientific, Austin, TX, USA).
Immunosuppressive experiments
To assess immunosuppressive effects of the nanoparticle formulations, Balb/c (male, 6-week-old) mice were injected intravenously with CsA, CsANP, or CsANP-LTP (15 mg/kg of CsA or equivalently loaded respective nanoparticles) every other day for a period of 21 days. At 24 hours after the last administration, the mice were sacrificed and their spleens were removed into a Petri dish containing Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum, 100 units/mL of penicillin, and 100 μg/mL of streptomycin. Spleens were dispersed through a nylon mesh to generate a single cell suspension followed by depletion of RBC by using an red blood cells (RBC) lysis buffer (Sigma-Aldrich Co., St Louis, MO, USA). The splenocytes were washed and cultured in RPMI complete medium.
IL-2 enzyme-linked immunosorbent assay
Isolated splenocytes (2×106/well) from different treatment groups were seeded in a 24-well plate and then maintained under unstimulated or stimulated conditions with 1 μg/mL of ionomycin (Calibiochem, La Jolla, CA, USA) and 20 ng/mL phorbol-12-myristate-13-acetate (PMA) (Sigma-Aldrich Co.) for different time intervals. After the indicated incubation time, the culture media from triplicate wells were collected and assayed for IL-2 levels by enzyme-linked immunosorbent assay (ELISA) according to the protocol from Enzo Life Sciences (Plymouth Meeting, PA, USA).
HCV mouse model
Immunodeficient NOD/SCID mice (Charles River Laboratories, Wilmington, MA, USA), aged 6 weeks, were used. Mice were engrafted with Huh7 cells containing the HCV-Con1b replicon (1×106 cells in 200 μL PBS) by intrasplenic injection, as described previously.32 Cell transplantation and surgical procedures were performed under anesthesia and prophylactic antibiotics were given. After 5 weeks of cell transplantation, free CsA or CsANP-LTP (15 mg/kg of CsA or equivalently loaded respective nanoparticles) were intravenously administered every other day for a period of 21 days to assess the antiviral efficacy. To analyze the sustained and prolonged effects, mice were divided into four groups: untreated control, and groups treated with free CsA, CsANP, or CsANP-LTP. The treatment was discontinued after three injections (every other day) and antiviral efficacy was checked after 21 days. Each treatment group consisted of three animals. After completion of treatment, the mice were sacrificed, and liver tissues were obtained for analysis.
Fluorescent immunohistochemistry
The deparaffinized and rehydrated mouse liver tissue sections were incubated overnight at 4°C with monoclonal antibody against human hepatocytes (Hep par 1) and HCV NS5A (Santa Cruz Biotechnology Inc., Dallas, TX, USA) at a 1:50 dilution. After three washes, the sections were incubated with Alexa Fluor-488 (green)- or Alexa Fluor-546 (red)-conjugated anti-rabbit or anti-mouse Immunoglobulin G (IgG) antibodies (Thermo Fisher Scientific, Waltham, MA, USA) at a dilution of 1:100 for 2 hours. Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI) for 5 minutes. Analysis was performed using confocal microscopy.
Statistical analysis
Data analysis was performed using a two-tailed Student’s t-test. Data are expressed as the mean ± standard deviation of at least three independent experiments, unless stated otherwise.
Results
Preparation and characterization of CsA-encapsulated PLGA nanoparticles
CsA-encapsulated PLGA nanoparticles were prepared by the single-emulsion method (oil-in-water).Figure 1 shows the stepwise preparation and engineering of liver-targeted PLGA nanoparticles. To avoid excessive aggregation of nanoparticles and stearic hindrance between the cell surface HSPGs and the liver-targeting moiety conjugated to the nanoparticles, as well as to enhance the in vivo circulation time, nanoparticles were PEGylated using NH2-PEG-NH2. The LTP, which specifically interacts with HSPGs, was conjugated to PEGylated PLGA nanoparticles to impart liver cell specificity.
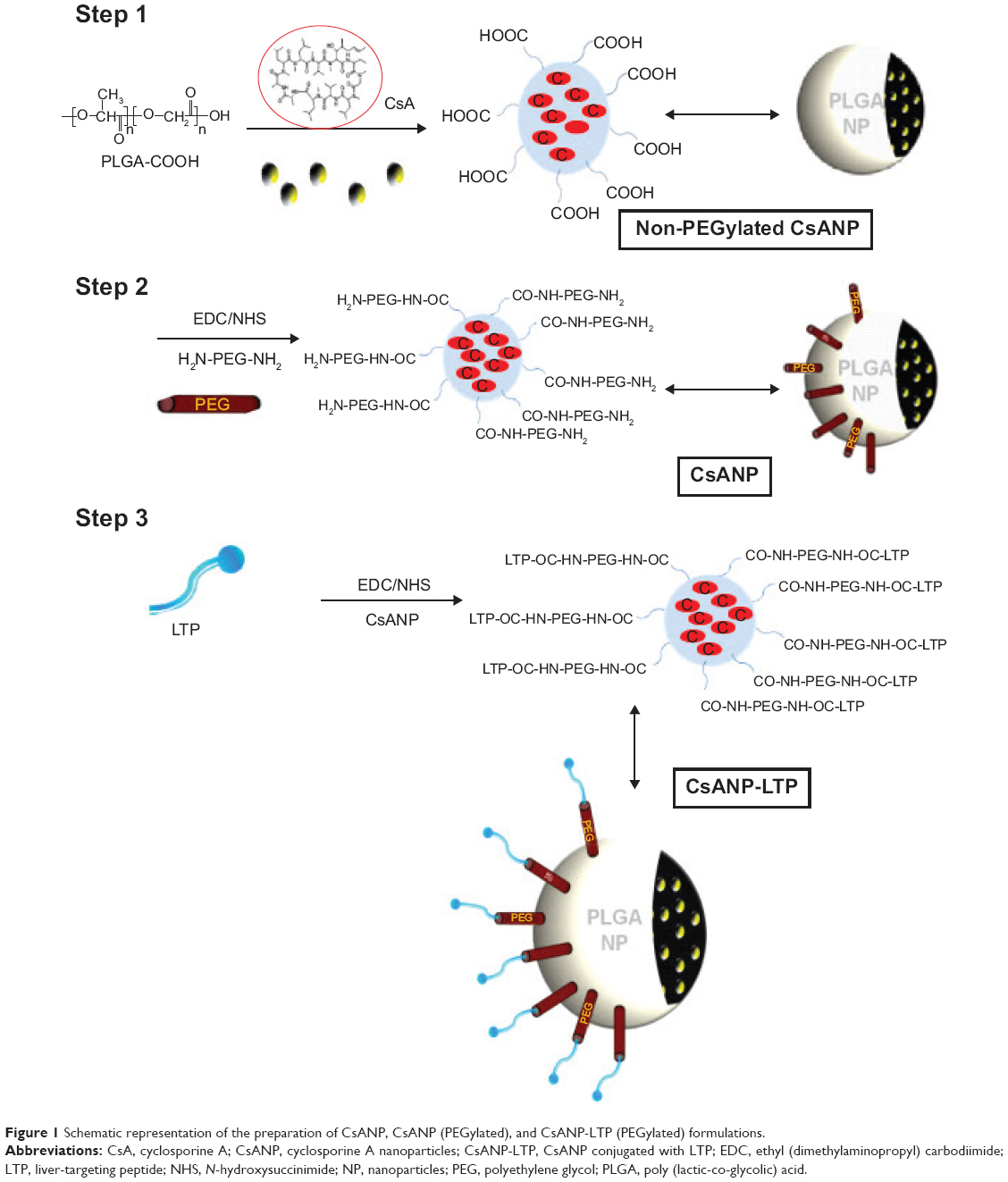 | Figure 1 Schematic representation of the preparation of CsANP, CsANP (PEGylated), and CsANP-LTP (PEGylated) formulations. |
The physicochemical properties of the non-PEGylated, PEGylated nontargeted (CsANP), and PEGylated liver-targeted CsA-encapsulated PLGA nanoparticles (CsANP-LTP) were analyzed using DLS and SEM. The size distribution of all the prepared nanoparticles was homogenous and appropriate for both in vitro and in vivo studies. Generally, the clinically applicable size of nanomedicines is less than 250 nm, as this size tends to have a similar range of properties based on physiological and anatomical consequences.33 Our non-PEGylated nanoparticles, CsANP, AND CsANP-LTP particles had an average size of 188 nm with a polydispersity index (PDI) value of 0.088, 212 nm with a PDI value of 0.125, and 229 nm with a PDI value of 0.912, respectively (Figure 2A). These results were further supported by SEM images, which showed evenly-dispersed morphology (Figure 2B). In order to evaluate the surface properties of the nanoparticles, we again utilized DLS analysis to measure the ζ potential. The ζ potential values were found to vary depending upon the surface modification of nanoparticles. The non-PEGylated nanoparticles possessed the highest negative charge, at −32.5 mV, followed by the CsANP (−17.4 mV) and CsANP-LTP (−8.2 mV) (Table 1). The reduction in the negative charge on CsANP or CsANP-LTP indirectly indicates the decreased number of carboxyl groups and addition of amine groups on the surface of the nanoparticles which contribute toward the net surface charge. Targeted nanoparticles with LTP possessed increased ζ potential values on average, owing to the additional positive charge amino acids in the peptide.
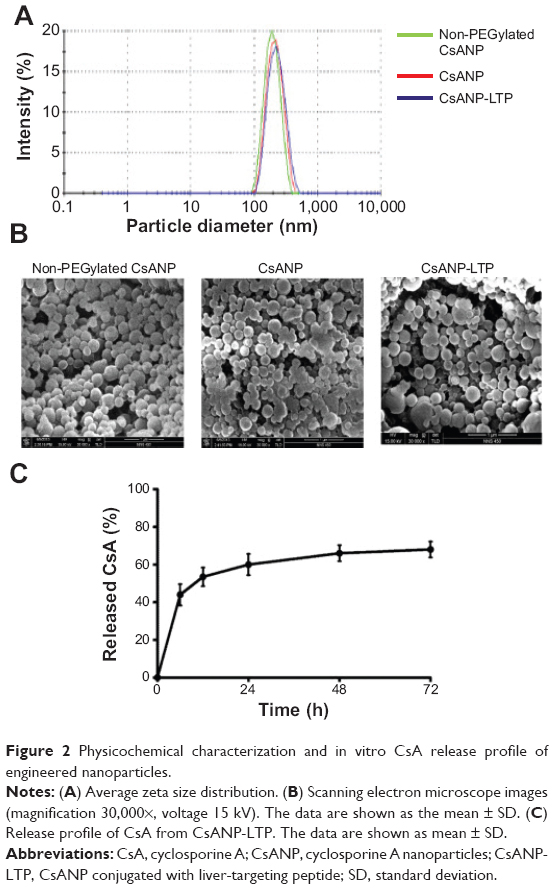 | Figure 2 Physicochemical characterization and in vitro CsA release profile of engineered nanoparticles. |
 | Table 1 Characterization of nanoparticles: representative polydispersity index and zeta potential of non-PEGylated CsANP, CsANP (PEGylated), and CsANP-LTP (PEGylated) |
The encapsulation efficiency and the release profile of the encapsulated drug are crucial for the overall functionality of the nanoparticles. In addition, the stability of the drug should be retained after the process of nanoparticle preparation. Our results also demonstrated good encapsulation efficiency of pre- and post-surface modifications of the nanoparticles (Table 2). The in vitro release profile of the CsA from the PLGA nanoparticles at physiological pH and room temperature was biphasic in nature, composed of an initial phase of rapid release followed by a latter slow and sustained release. As shown in Figure 2C, 67% of the CsA was released from CsANP-LTP within 72 hours in PBS, suggesting the sustained release of CsA from CsANP-LTP. The initial phase of the CsA release and the sustained release of the second phase occurs in cells in which uptake of CsANP-LTP occurs via specific receptor-mediated endocytosis.
 | Table 2 EE of non-PEGylated CsANP, CsANP (PEGylated) and CsANP-LTP (PEGylated) |
Antiviral effect of liver-targeted CsA encapsulated PLGA nanoparticles
To evaluate the anti-HCV activity of the formulated nanoparticles, HCV replicon cells expressing luciferase were used. The luciferase activity of HCV replicon cells directly correlates with the level of HCV RNA synthesis.34 CsA is known to effectively reduce HCV RNA levels by blocking the interaction of CypA with viral proteins.15,35 Both the luciferase assay (Figure 3A) and Western blot analysis (Figure 3B) showed that CsANP or CsANP-LTP can decrease HCV replication levels as efficiently as CsA for up to 72 hours. CsA treatment resulted in significantly higher levels of toxicity in HCV replicon cells, while CsANP or CsANP-LTP treatment did not affect the cell viability in time- and concentration-dependent experiments (Figure S1). Next, the prolonged antiviral effect of CsANP-LTP under sustained release condition was investigated via treatment-withdrawal experiments. After 12 hours’ incubation with CsA or nanoparticle formulations, the treatment was withdrawn and the cells were replaced with new media followed by culturing for the different time periods. Despite the withdrawal of treatment, CsANP-LTP showed significant sustained reduction in the replication of HCV replicon RNA and NS5A protein levels even up to 5 days (Figure 3C and D). CsANP-LTP decreased the NS5A levels more efficiently than free CsA or CsANP. This enhanced antiviral effect of CsANP-LTP was significant compared with free CsA. Cellular uptake of targeted PLGA nanoparticles showed that CsANP-LTP is specifically targeted into liver cells (Figure S2). Furthermore, we could not detect significant uptake of targeted PLGA nanoparticles in other cell lines such as A549 cells (human lung cancer) and mouse splenocytes (Figure S3 and S4). Moreover, attachment of a PEG moiety between the nanoparticle and LTP improved the anti-HCV effect of CsANP-LTP (Figure S5). In summary, these results suggest that CsANP-LTP effectively enhances the inhibition of HCV replication.
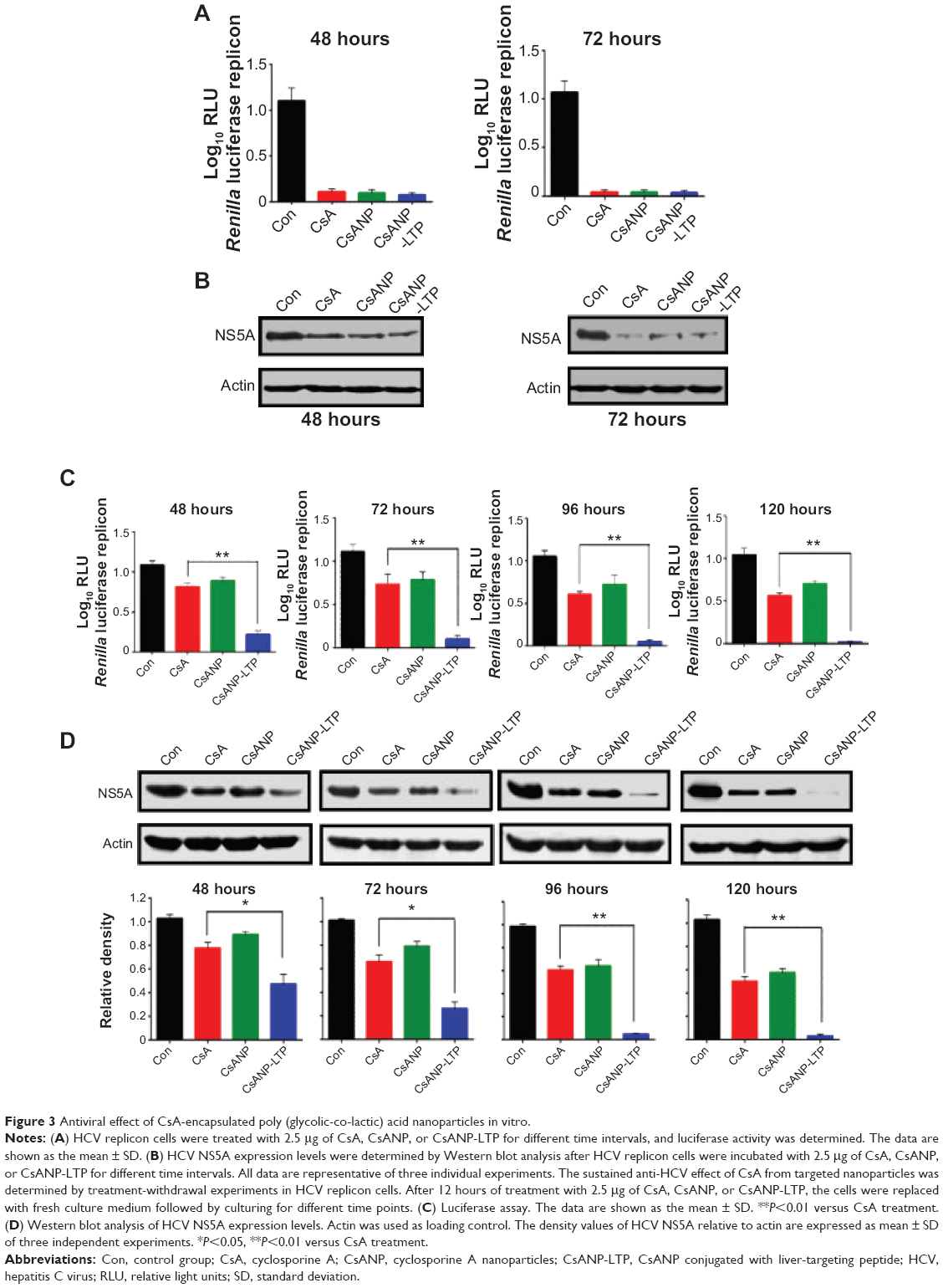 | Figure 3 Antiviral effect of CsA-encapsulated poly (glycolic-co-lactic) acid nanoparticles in vitro. |
Biodistribution and in vivo toxicity profile of engineered nanoparticles
We investigated the biodistribution of Alexa-488-labeled CsANP or CsANP-LTP 24 hours after intravenous injection in Balb/c mice. Use of LTP-CsANP resulted in an alteration in the location of accumulation, mainly to the liver, in contrast to CsANP, which were found to be deposited in the lung in addition to the liver. Imaging results showed significantly higher accumulation of the targeted nanoparticles in the liver (1.8- to 2.0-fold) and also a slight deposition in kidney in comparison to nontargeted nanoparticles, even 24 hours after intravenous administration (Figure 4A). These results indicate that LTP-conjugated nanoparticles specifically target liver and deliver therapeutic agents.
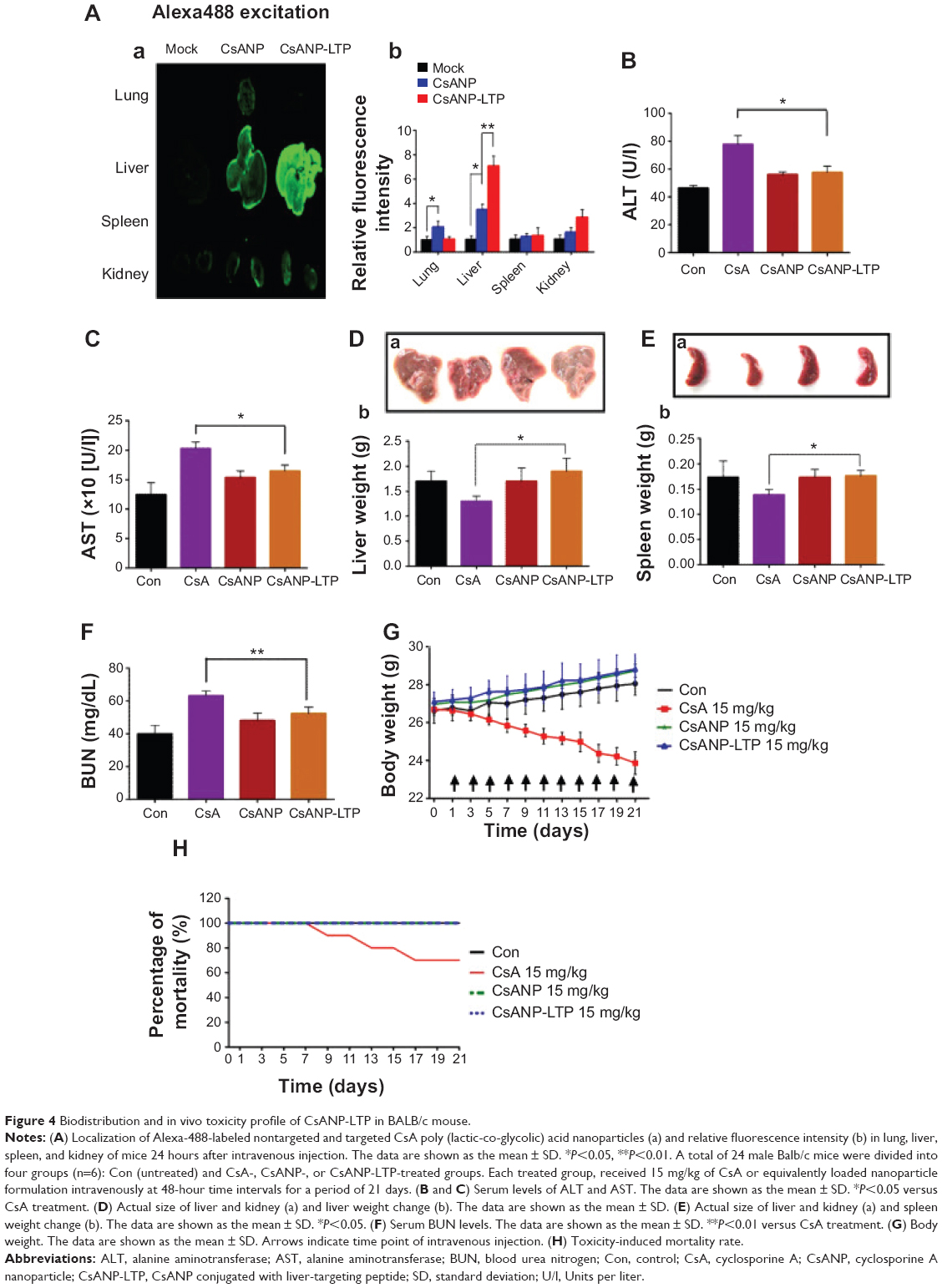 | Figure 4 Biodistribution and in vivo toxicity profile of CsANP-LTP in BALB/c mouse. |
Despite its remarkable biological activity, CsA is associated with severe hepatotoxicity, nephrotoxicity, and significant body weight reduction. Hence, we tested whether sustained release of CsA from PLGA nanoparticles resolved CsA-associated toxicities. Normal Balb/c mice were administered free CsA, CsANP, or CsANP-LTP (CsA 15 mg/kg equivalents, eleven intravenous doses, every two days). Serum ALT and AST levels were quantified to evaluate hepatic function. Free CsA treatment resulted in an approximately two fold increase in serum ALT and AST levels in comparison to the control group; however, encapsulated CsA in CsANP or CsANP-LTP resulted in ~70%–80% (P<0.05) lower AST and ALT levels than those observed after free CsA treatment (Figure 4B and C). In addition, shrinkage of the liver was found in mice treated with free CsA (the liver weight was ~20%–26% lower than that of the control group), while liver weight in the CsANP or CsANP-LTP group did not show any significant change (Figure 4D). Spleen toxicity was characterized by loss of spleen weight in the CsA-treated mice (~13%), while no spleen weight loss was observed in the CsANP or CsANP-LTP groups (Figure 4E). Serum BUN levels were measured to assess renal function. Mice in the free CsA-treated group showed a 1.8-fold increase in BUN levels as compared to the control group, while CsANP or CsANP-LTP treatment resulted in approximately normal BUN levels (Figure 4F). Although the biodistribution results showed higher levels of CsANP-LTP accumulation in kidney compared to CsANP, there was no significant difference in BUN levels. Free CsA treatment resulted in ~12%–17% reduction in average body weight, yet neither the CsANP nor CsANP-LTP treated group experienced an adverse effect on body weight (Figure 4G). We also found that intravenous administration of free CsA resulted in mortality for three out of ten injected mice; however, all mice survived until the end of the experiment when administered with the same amount and number of doses of nanoparticle formulations (Figure 4H). Therefore, we conclude that CsANP or CsANP-LTP formulations are well tolerated and abolish acute toxicities of free CsA with prolonged circulation time and sustained drug release.
Effect of targeted CsA encapsulated PLGA nanoparticles on immunosuppression
We speculated that a nanoparticle-based drug delivery strategy might be beneficial to overcome the drawbacks of CsA. After treatment with free CsA, CsANP, or CsANP-LTP (CsA 15 mg/kg equivalents, eleven intravenous doses, every other day for 21 days), mice were sacrificed, and isolated splenocytes were further cultured under unstimulated or stimulated conditions (Figure 5A). Interestingly, encapsulation of CsA in both the nontargeted and targeted nanoparticles strongly reduced the immunosuppressive activity (Figure 5B). These results were also supported by both the biodistribution (Figure 4A) and histological analysis (Figure S4A), which showed no deposition of formulated nanoparticles in the spleen. The observed low levels of IL-2 inhibition in CsANP or CsANP-LTP treatments may be due to the slow release of CsA from nanoparticles during its circulation in the blood. We further confirmed the specificity of targeted nanoparticles by testing their ability to be internalized by splenocytes. Alexa-488-labeled CsANP or CsANP-LTP showed no internalization by splenocytes (Figure S4B), thus implying that live-targeted nanoparticles do not exhibit nonspecific delivery to spleen. Taken together, target-specific CsANP might be more advantageous than conventional CsA as an improved nanomedicine for the treatment of HCV.
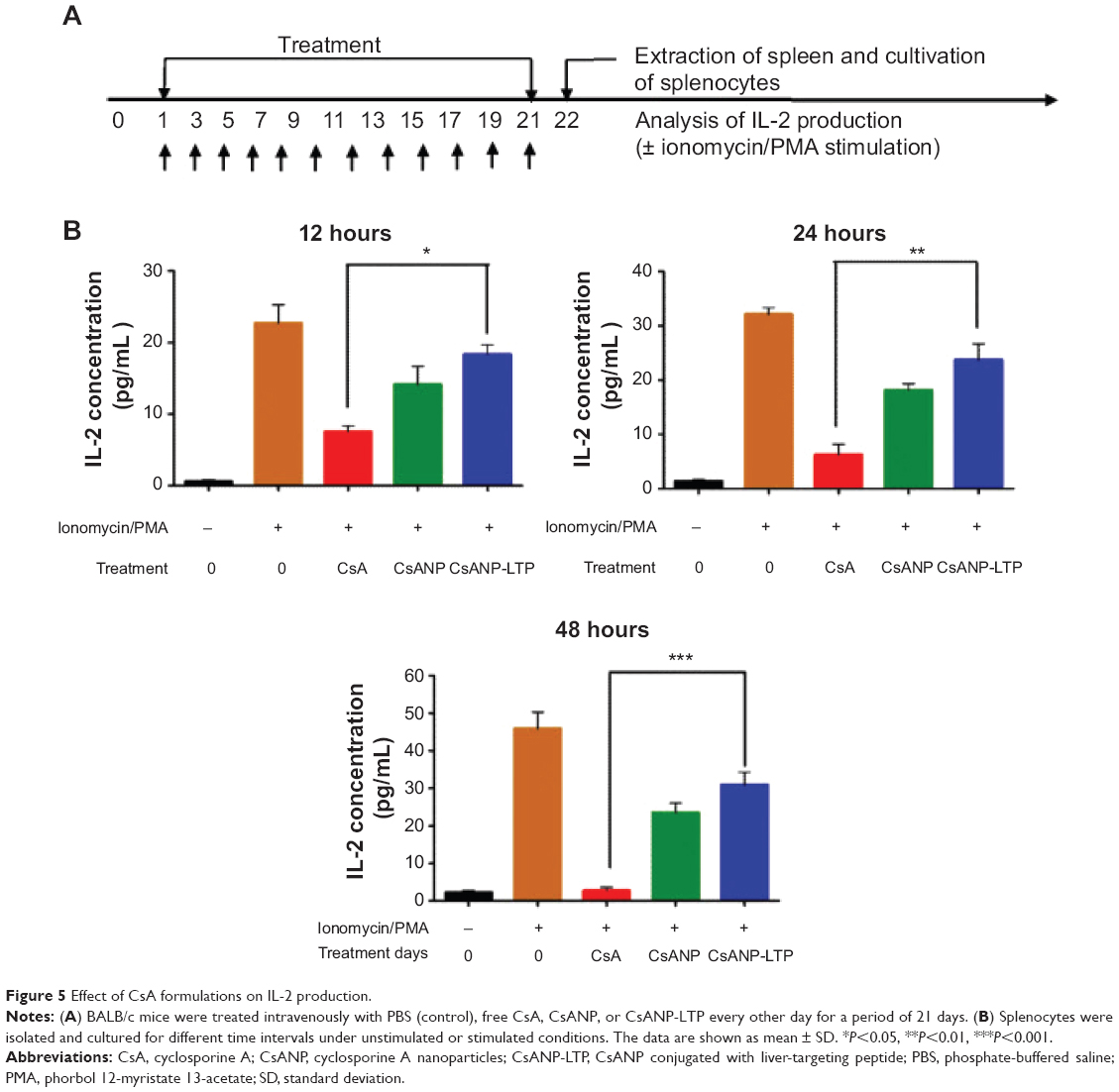 | Figure 5 Effect of CsA formulations on IL-2 production. |
Antiviral effect of liver-targeted CsA encapsulated PLGA nanoparticles in an HCV mouse model
We next determined the inhibition of HCV replication after systemic delivery of targeted CsA encapsulated PLGA nanoparticles in an HCV mouse model. Five weeks after transplantation of HCV replicon cells, mice were randomly divided into three groups. One group was maintained as control, and the other two groups were injected intravenously with either CsA or CsANP-LTP (CsA 15 mg/kg equivalents, eleven intravenous doses, every other day).In contrast to free CsA, no significant loss of body weight was observed in the CsANP-LTP group (data not shown). Inhibition of HCV replication was confirmed by measuring HCV RNA and NS5A protein levels after continuous treatment. The CsANP-LTP-treated group inhibited HCV replication as effectively as those treated with free CsA (Figure 6A and B). In addition, confocal microscopic analysis detected NS5A in control human hepatocytes, while hepatocytes from the free CsA- and CsANP-LTP-treated groups showed faint detection of NS5A, thus confirming the reduction of viral replication (Figure 6C). Furthermore, the prolonged antiviral effect was also investigated in the HCV mouse model. Mice were divided into four groups (n=3 mice per group); one group was maintained as untreated control. Free CsA and nanoparticulate formulations (CsANP or CsANP-LTP) were administered intravenously on days 1, 3, and 5 (CsA 15 mg/kg equivalents). Treatment was discontinued and antiviral efficacy was determined after 21 days. In contrast to free CsA or CsANP, CsANP-LTP resulted in a significant decrease in HCV RNA and NS5A protein levels even after withdrawal of treatment (Figure 6D and E). These data are consistent with a prolonged and sustained anti-HCV effect of CsANP-LTP in vitro (Figure 3C and D). Our results indicate that CsANP-LTP treatment results in enhanced anti-HCV activity while reducing the frequency of doses required and severe toxicities associated with free CsA treatment.
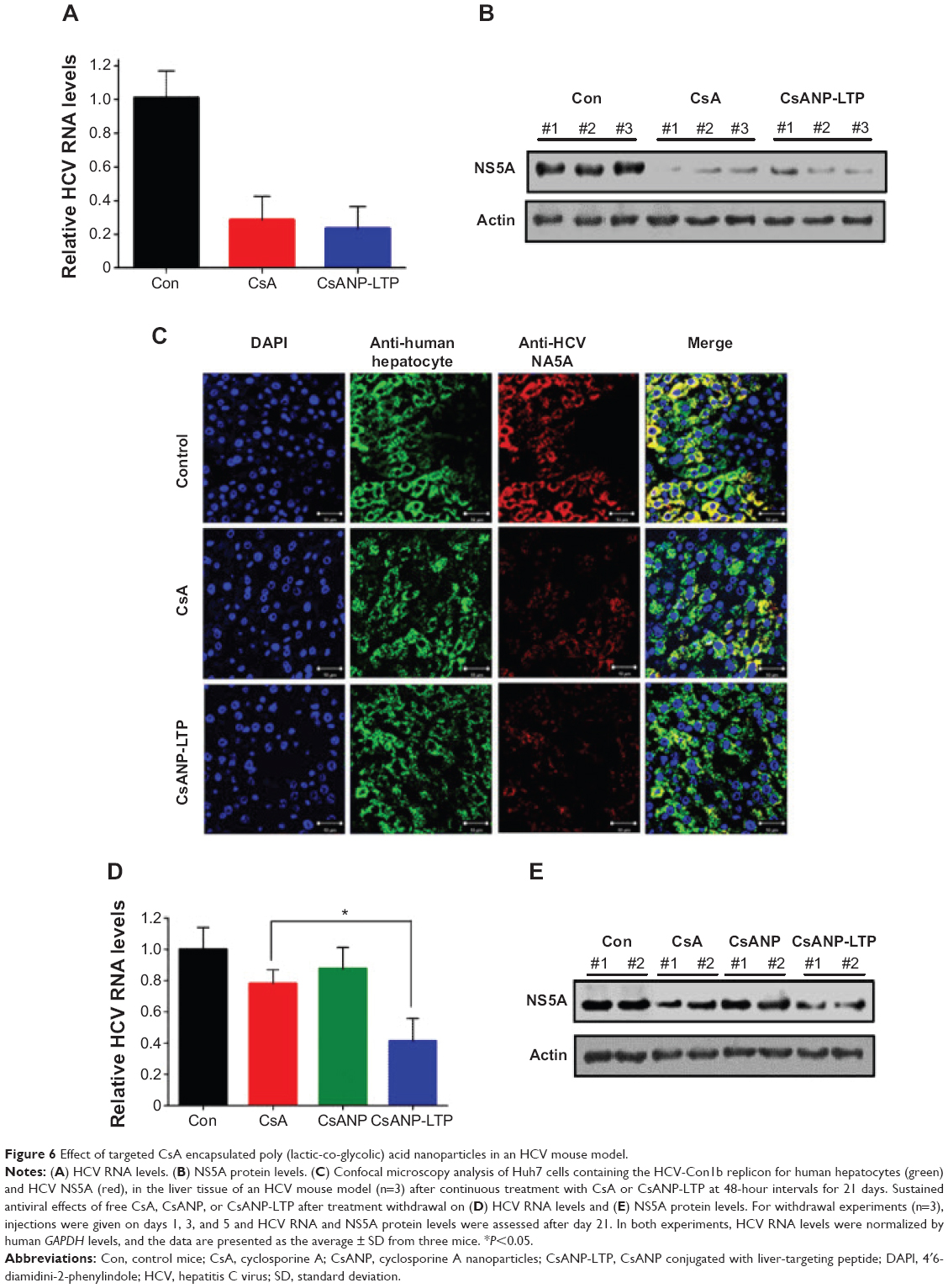 | Figure 6 Effect of targeted CsA encapsulated poly (lactic-co-glycolic) acid nanoparticles in an HCV mouse model. |
Discussion
HCV remains a global health challenge, causing chronic liver disease worldwide, even after a decade of intense research.1 The current therapy for HCV involves the combination of standard of care (SOC) with drugs targeting viral proteins or host factors.9,36 Small molecules, due to their size and molecular weight, exhibit disorderly pharmacokinetic profiles such as rapid clearance, indiscriminate distribution and localization in undesired tissue and organs, causing dose-limiting toxicity. Also, as small molecule drugs have poor specificity, administering high doses can become crucial, which in turn increases the toxicity burden to patients. Therefore, there is a need for target-specific delivery of drugs in order to reduce the side effects to nontargeted tissues and organs, and increase the effectiveness of therapy. In this regard, the use of targeted nanoparticle formulations seems to open new paths for HCV treatment with increased efficacies.
CsA is a potent host-targeted HCV inhibitor, but its immunosuppressive activity, associated severe toxicities, rapid elimination from the human body, and narrow therapeutic window have been limiting factors in its clinical success. Herein, we have demonstrated the application of liver-targeted CsA-encapsulated PLGA nanoparticles for enhanced anti-HCV therapy. The uniqueness of this delivery system is the conjugation of nanoparticles with conjugation of nanoparticle with the targeting moiety that binds to the HSPGs on the surface of hepatocytes (Figure 1). A variety of techniques have been developed for PEGylation of PLGA nanoparticles, such as incorporation of polymer conjugates (eg, poly[lactic acid]–PEG)37 or covalent attachment via amino or carboxyl-terminated PLGA.38 In this report, we used NH2-PEG-NH2 for surface modification of drug-loaded PLGA particles that can help to overcome the excessive aggregation of nanoparticles, avoid stearic hindrance for the interaction between the cell surface receptor and liver targeting moiety conjugated on the nanoparticles, and enhance the in vivo circulation time of nanoparticles. The overall charge on all prepared nanoparticles was negative, which is critical to achieve a low rate of nonspecific cellular uptake; thus the negative charges eventually contribute toward the development of long-circulating nanoparticles. Our results establish a strategy by which to achieve favorable results utilizing specific uptake into hepatocytes, which is otherwise unattainable with nontargeted nanoparticles (Figure S2). The observed superior activity of the targeted nanoparticles can be attributed to rapid internalization of nanoparticles via specific interaction of LTP with HSPGs on hepatocytes followed by sustained release of CsA over the period of time. In addition, CsA-encapsulated PLGA nanoparticles showed no cytotoxicity, unlike free CsA. Our results also showed the benefits of sustained release of CsA from CsANP-LTP, with greater anti-HCV efficacy over nontargeted nanoparticles or free CsA in HCV replicon cells.
Since HCV primarily infects hepatocytes, delivery of drug to the infected site at a high dose is a prerequisite for nanoparticles to attain anti-HCV efficacy. To achieve high-dose drug delivery to the liver and to reduce nonspecific cellular uptake in the nontarget tissues, we employed a nanoparticle-targeting strategy by conjugating LTP. Biodistribution of targeted nanoparticles yielded significant results regarding the accumulation of nanoparticles in the liver (Figure 4A). The rapid internalization of CsANP-LTP into hepatocytes facilitated the retention in the liver, providing an advantage over the CsANP. Due to the negative surface charge, nontargeted nanoparticles were not taken up by hepatocytes and thus entered the circulation over the experimental time period and were easily excreted via the kidneys. Localization of targeted nanoparticles in the kidneys might be due to the slow rate of circulation of targeted nanoparticles which leached out from the liver. Thus, biodistribution data indicate that high antiviral efficacy can be facilitated by nanoparticle-based targeting strategies.
Clinical application of immunosuppressant CsA against HCV is limited, mainly due to CsA’s immunosuppressive activity and its severe side effects.16,39 Considering that adaptive immune response is essential for the eradication of HCV, we assume that high accumulation of CsANP-LTP in the liver would be an additional advantage because non-splenic distribution of the nanoparticles protects from the immunosuppressive effect of CsA. Previous ex vivo studies40,41 have demonstrated that encapsulation of CsA in PLGA nanoparticles results in a greater sustained immunosuppressive effect, which is contradictory to our results. This discrepancy seems to be due to the experimental design of the studies. We tested the production of IL-2 in cultured splenocytes from mice administered with free CsA or nanoparticle formulations. In contrast, the other research groups shows CsA encapsulated nanoparticles suppressed IL-2 production under ex vivo conditions by continuous release of CsA into the media or by nonspecific uptake of nanoparticles by the cells. Moreover, the physicochemical properties of encapsulated nanoparticles, including a comparatively larger size and net negative surface charge, resulted in less inhibition of IL-2 production (Figure 5). Thus, specific targeting of CsANP-LTP to the liver reduced the immunosuppressive effect of free CsA in our experiments.
Several studies have reported the antiviral efficacy of CsA against HCV.15,42,43 However, side effects such as nephrotoxicity and hepatotoxicity have limited its use in the clinic. Herein, we report that use of target-specific CsA- encapsulated nanoparticles reduces CsA induced toxicity significantly (Figure 4). Encapsulation of CsA within PLGA nanoparticles seemed to minimize the liver and kidney toxicity without any significant change in the body weight and mortality, as compared to free CsA treatment. Moreover, the CsANP-LTP-treated HCV mouse model showed a sustained anti-HCV effect for a short-term treatment after intrasplenic surgery (Figure 6). Therefore, it is likely that the sustained release of CsA from targeted nanoparticles enhances antiviral efficacy without significant toxic effects.
Conclusion
In this study, we developed liver-targeted PLGA nanoparticles for CsA delivery against HCV. Both in vitro and in vivo examination of nanoparticles showed the advantage of reducing the toxic effects associated with free CsA. Additionally, compared to conventional treatment with CsA, the engineered CsA nanoparticle-based treatment resulted in decreased immunosuppressive effects. Although both formulations of nanoparticles displayed improved toxicity profiles over free CsA, only liver-specific CsA-encapsulated PLGA nanoparticles yielded a sustained and prolonged anti-HCV effect, even after treatment withdrawal. Targeted nanoparticles have the ability to reduce the amount and frequency of doses that might be important in achieving toxicity-free therapeutics. All these positive attributes make targeted CsA nanoparticles a promising drug delivery system for the safe and effective treatment of HCV infection.
Disclosure
The authors report no conflicts of interest in this work.
References
Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333–1342. | ||
Dabbouseh NM, Jensen DM. Future therapies for chronic hepatitis C. Nat Rev Gastroenterol Hepatol. 2013;10(5):268–76. | ||
Zeuzem S, Andreone P, Pol S, et al; REALIZE Study Team. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364(25):2417–2428. | ||
Kim do Y, Ahn SH, Han KH. Emerging therapies for hepatitis C. Gut Liver. 2014;8(5):471–479. | ||
Schinazi R, Halfon P, Marcellin P, Asselah T. HCV direct-acting antiviral agents: the best interferon-free combinations. Liver Int. 2014;34 Suppl 1:69–78. | ||
Lenz O, Vijgen L, Berke JM, et al. Virologic response and characterisation of HCV genotype 2-6 in patients receiving TMC435 monotherapy (study TMC435-C202). J Hepatol. 2013;58(3):445–451. | ||
Poveda E, Wyles DL, Mena A, Pedreira JD, Castro-Iglesias A, Cachay E. Update on hepatitis C virus resistance to direct-acting antiviral agents. Antiviral Res. 2014;108:181–191. | ||
Xiao F, Fofana I, Heydmann L, et al. Hepatitis C virus cell-cell transmission and resistance to direct-acting antiviral agents. PLoS Pathog. 2014;10(5):e1004128. | ||
Georgel P, Schuster C, Zeisel MB, et al. Virus-host interactions in hepatitis C virus infection: implications for molecular pathogenesis and antiviral strategies. Trends Mol Med. 2010;16(6):277–286. | ||
Zeisel MB, Lupberger J, Fofana I, Baumert TF. Host-targeting agents for prevention and treatment of chronic hepatitis C – perspectives and challenges. J Hepatol. 2013;58(2):375–384. | ||
Guedj J, Yu J, Levi M, et al. Modeling viral kinetics and treatment outcome during alisporivir interferon-free treatment in hepatitis C virus genotype 2 and 3 patients. Hepatology. 2014;59(5):1706–1714. | ||
Chatterji U, Garcia-Rivera JA, Baugh J, et al. The combination of alisporivir plus an NS5A inhibitor provides additive to synergistic anti-hepatitis C virus activity without detectable cross-resistance. Antimicrob Agents Chemother. 2014;58(6):3327–3334. | ||
Kaul A, Stauffer S, Berger C, et al. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009;5(8):e1000546. | ||
Liu Z, Yang F, Robotham JM, Tang H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J Virol. 2009;83(13):6554–6565. | ||
Yang F, Robotham JM, Grise H, et al. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog. 2010;6(9):e1001118. | ||
Garcia-Saenz-de-Sicilia M, Mukherjee S. The adverse pharmacology of calcineurin inhibitors and their impact on hepatitis C recurrence after liver transplantation: implications for clinical practice. Expert Rev Clin Pharmacol. 2012;5(5):587–593. | ||
Faulds D, Goa KL, Benfield P. Cyclosporin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs. 45(6):953–1040. | ||
Bobadilla NA, Gamba G. New insights into the pathophysiology of cyclosporine nephrotoxicity: a role of aldosterone. Am J Physiol Renal Physiol. 2007;293(1):F2–F9. | ||
Wendt A, Adhoute X, Castellani P, et al. Chronic hepatitis C: future treatment. Clin Pharmacol. 2014;6:1–17. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Jaiswal J, Gupta SK, Kreuter J. Preparation of biodegradable cyclosporine nanoparticles by high-pressure emulsification-solvent evaporation process. J Control Release. 2004;96(1):169–178. | ||
Aksungur P, Demirbilek M, Denkbaş EB, Vandervoort J, Ludwig A, Unlü N. Development and characterization of cyclosporine A loaded nanoparticles for ocular drug delivery: cellular toxicity, uptake, and kinetic studies. J Control Release. 2011;151(3):286–294. | ||
Italia JL, Bhatt DK, Bhardwaj V, Tikoo K, Kumar MN. PLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J Control Release. 2007;119(2):197–206. | ||
Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev. 2007;59(8):718–728. | ||
Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–1822. | ||
Johnstone TC, Kulak N, Pridgen EM, Farokhzad OC, Langer R, Lippard SJ. Nanoparticle encapsulation of mitaplatin and the effect thereof on in vivo properties. ACS Nano. 2013;7(7):5675–5683. | ||
Graf N, Bielenberg DR, Kolishetti N, et al. α(V)β(3) integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt(IV) prodrug. ACS Nano. 2012;6(5):4530–4539. | ||
Peng XH, Wang Y, Huang D, et al. Targeted delivery of cisplatin to lung cancer using ScFvEGFR-heparin-cisplatin nanoparticles. ACS Nano. 2011;5(12):9480–9493. | ||
Lee MY, Yang JA, Jung HS, et al. Hyaluronic acid-gold nanoparticle/interferon α complex for targeted treatment of hepatitis C virus infection. ACS Nano. 2012;6(11):9522–9531. | ||
Robertson RT, Baratta JL, Haynes SM, Longmuir KJ. Liposomes incorporating a Plasmodium amino acid sequence target heparan sulfate binding sites in liver. J Pharm Sci. 2008;97(8):3257–3273. | ||
Koutsoudakis G, Kaul A, Steinmann E, et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol. 2006;80(11):5308–5320. | ||
Kumthip K, Chusri P, Jilg N, et al. Hepatitis C virus NS5A disrupts STAT1 phosphorylation and suppresses type I interferon signaling. J Virol. 2012;86(16):8581–8591. | ||
Spivak MY, Bubnov RV, Yemets IM, Lazarenko LM, Tymoshok NO, Ulberg ZR. Gold nanoparticles – the theranostic challenge for PPPM: nanocardiology application. EPMA J. 2013;4(1):18. | ||
Krieger N, Lohmann V, Bartenschlager R. Enhancement of hepatitis C virus RNA replication by cell culture-adaptive mutations. J Virol. 2001;75(10):4614–4624. | ||
Chatterji U, Lim P, Bobardt MD, et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J Hepatol. 2010;53(1):50–56. | ||
Sarrazin C, Hézode C, Zeuzem S, Pawlotsky JM. Antiviral strategies in hepatitis C virus infection. J Hepatol. 2012;56 Suppl 1:S88–S100. | ||
Gref R, Luck M, Quellec P, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18(3–4):301–313. | ||
Li YP, Zhou ZH, Pei YY, Zhang XY, Gu ZH, Yuan WF. PEGylated polycyanoacrylate nanoparticles as salvicine carriers: synthesis, preparation, and in vitro characterization. Acta Pharmacol Sin. 2001;22(7):645–650. | ||
Fischer G, Gallay P, Hopkins S. Cyclophilin inhibitors for the treatment of HCV infection. Curr Opin Investig Drugs. 2010;11(8):911–918. | ||
Azzi J, Tang L, Moore R, et al. Polylactide-cyclosporin A nanoparticles for targeted immunosuppression. FASEB J. 2010;24(10):3927–3938. | ||
Tang L, Azzi J, Kwon M, et al. Immunosuppressive activity of size-controlled PEG-PLGA nanoparticles containing encapsulated cyclosporine A. J Transplant. 2012;2012:896141. | ||
Hopkins S, Gallay P. Cyclophilin inhibitors: an emerging class of therapeutics for the treatment of chronic hepatitis C infection. Viruses. 2012;4(11):2558–2577. | ||
Ishii N, Watashi K, Hishiki T, et al. Diverse effects of cyclosporine on hepatitis C virus strain replication. J Virol. 2006;80(9):4510–4520. |
Supplementary materials
Poly (lactic-co-glycolic) acid (PLGA (50:50)), Poly(vinyl) alcohol (PVA), N-hydroxysulfosuccinimide sodium salt (Sulfo-NHS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), MES hydrate, poly(ethylene glycol) (PEG), NH2-PEG-NH2, 1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate (DiI) dye, and cyclosporine A (CsA) were all obtained from Sigma Aldrich (St Louis, MO). Alexa-488 fluorescent dye was obtained from Invitrogen (Carlsbad, CA). Liver-targeting peptide (LTP) CKNEKKNKIERNNKLKQPP was synthesized and obtained from Peptron (Daejeon, South Korea).
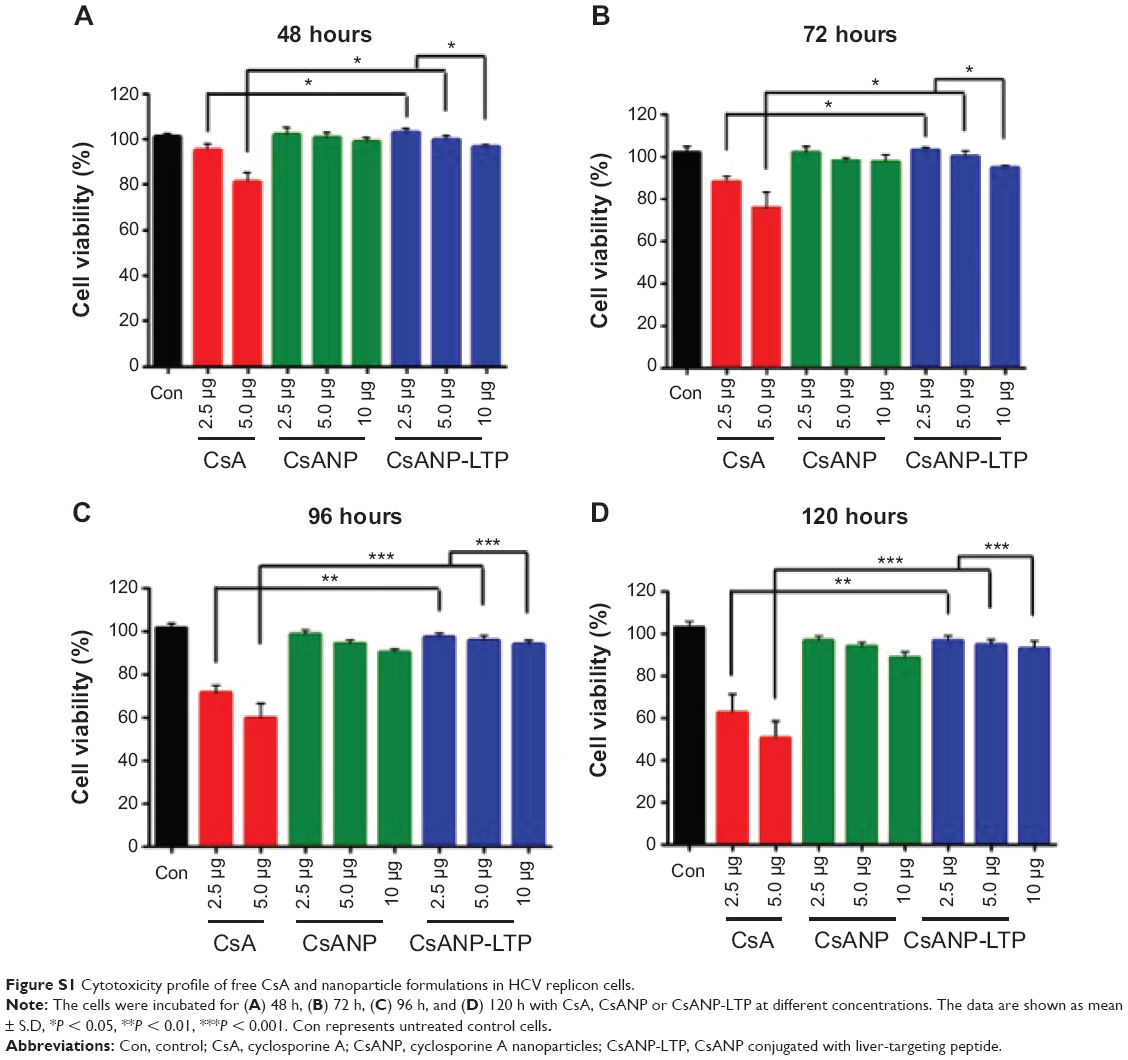 | Figure S1 Cytotoxicity profile of free CsA and nanoparticle formulations in HCV replicon cells. |
In vitro studies
Cell culture
Huh7 cells harboring subgenomic HCV genotype 1b (Con1/SG-Neo (I) hRluc FMDV2aUb) were provided by Prof. Charles Rice (The Rockefeller University and Apath, LLC, St. Louis, MO). Huh7-derived replicon cells were propagated in DMEM with 10% FBS, 100 units/mL of penicillin, 100 μg/ml of streptomycin, and 1× non-essential amino acids (NEAA) mixture and selected using 0.5 mg/ml G418 from Duchefa Biochemie (Amsterdam, The Netherlands).
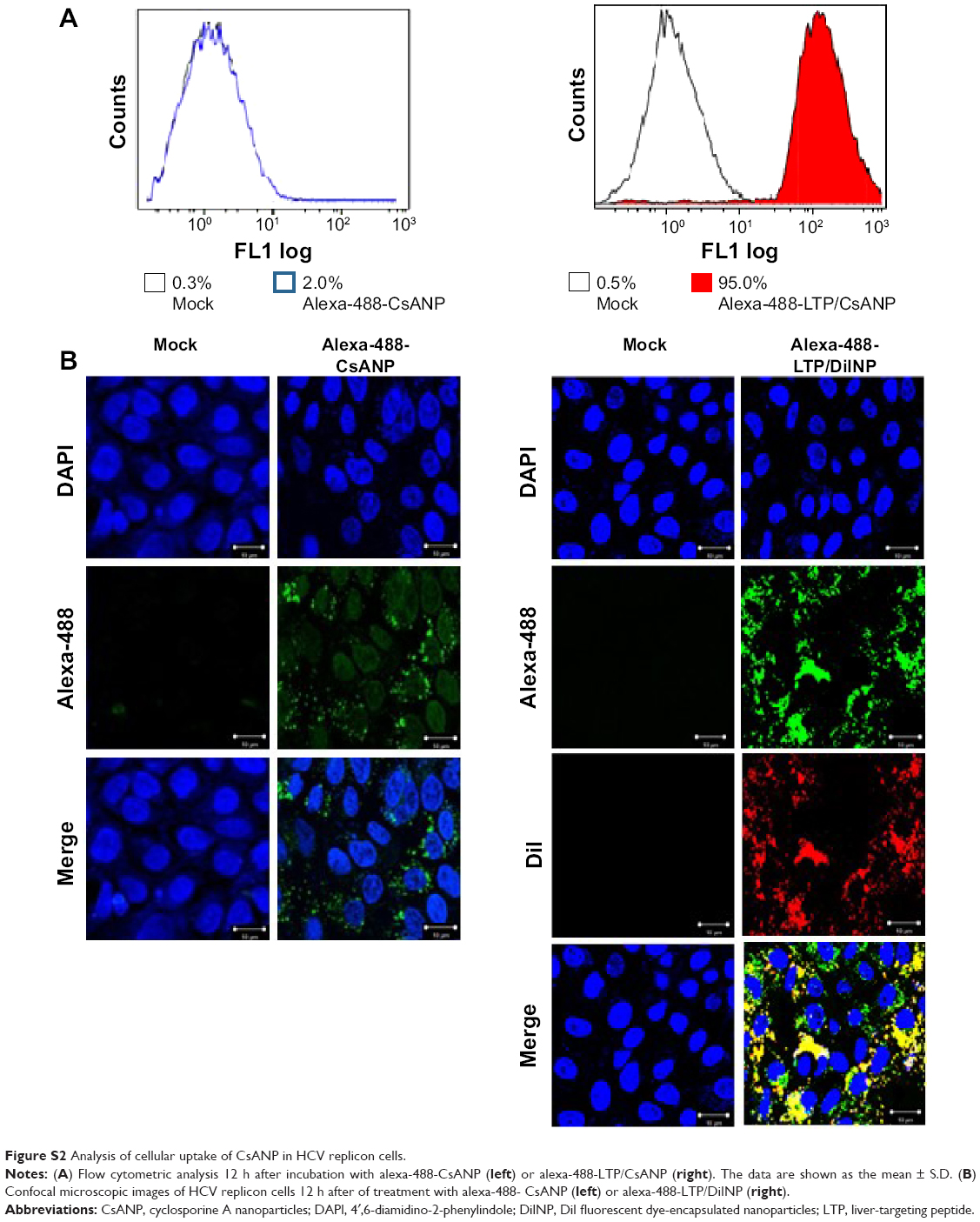 | Figure S2 Analysis of cellular uptake of CsANP in HCV replicon cells. |
Cytotoxicity assay
The HCV replicon cells (1.5×105/well) were incubated with various concentrations of free CsA, CsANP or CsANP-LTP for different time periods. Viability of treated cells was determined using 3-(4,5′dimethylthiasol-2-yl)-2,5′diphenyltetrazolium bromide (MTT) assay. MTT (1 mg/mL in PBS) was added to each well at 1/10 of the volume of the media and incubated for 2 hours to form formazan crystals, which are soluble in cell culture medium. After 4 hours, the absorbance of formazan products was measured at 570 nm using a spectrophotometer (Molecular Devices, Sunnyvale). Cell viability was expressed as the percentage of absorbance relative to untreated cells.
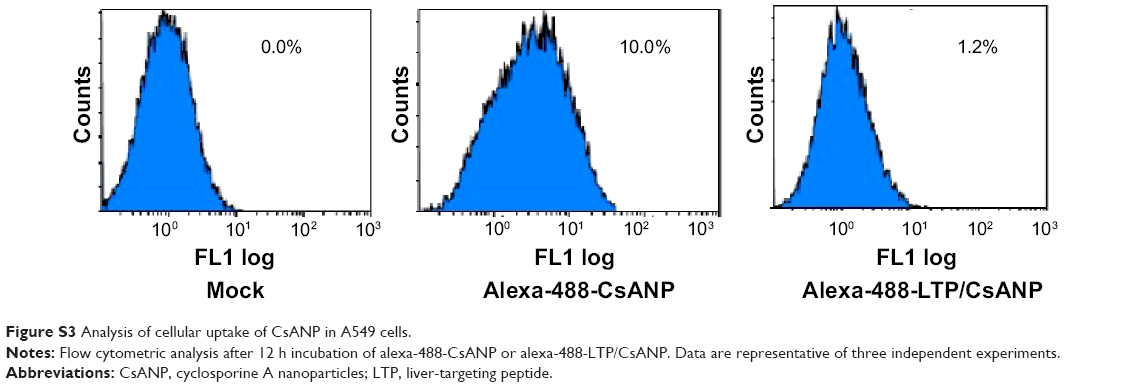 | Figure S3 Analysis of cellular uptake of CsANP in A549 cells. |
Cellular uptake
To quantify the efficiency of specific cellular uptake, HCV replicon cells (1.5×105/well) were seeded onto 12 well plates and incubated for 24 h. After 24 h, cells were treated with alexa-488-labeled CsANP or CsANP-LTP nanoparticles at 50 μg/mL final concentration for the period of 12 h. After the incubation period, the cells were trypsinized, washed two times with PBS, resuspended in fixing solution (5% formaldehyde), and evaluated using a Beckman Coulter flow cytometer. Data were analyzed using the Cell Quest Software (Beckman Coulter Inc, Brea, CA).
For confocal microscopic analysis of cellular uptake, HCV replicon cells were grown on cover slips. Cells were treated with alexa-488-CsANP or alexa-488-LTP/DiINP at a final concentration of 50 μg/mL for 12 h. The cells were then rinsed with PBS three times. The cells were counterstained with DAPI (4′,6′-diamidino-2-phenylindole) and mounted on glass slides (Vector Mount, CA). Photographs of prepared samples were taken using a confocal microscope (Carl Zeiss Jena GmbH, Germany).
Western blot analysis
The HCV replicon cells were treated as described above. Cell lysates were prepared from treated samples with protein extraction solution (iNtRON Biotechnology, Chinju, Korea). Proteins were then separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto a nitrocellulose membrane, and blocked with 3% bovine serum albumin (BSA) in 1× Tris-buffered saline. Membranes were incubated in mouse anti-HCV NS5A antibody (Santa Cruz Biotechnology, Santa Cruz, CA) as a primary antibody, followed by incubation with appropriate secondary antibody. Samples were detected with enhanced chemiluminescence reagents (Santa Cruz Biotechnology, Santa Cruz, CA). Quantification of Western blots was performed by densitometry using Image J software. Actin as used as a loading control.
Quantification of HCV RNA levels by real-time PCR
Total RNA was isolated from mouse liver tissue using TRIzol reagent (Invitrogen, Carlsbad, CA) and cDNA product was obtained using an iScript cDNA synthesis kit (Bio-Rad). HCV RNA levels were analyzed using real-time PCR (qRT-PCR). The qRT-PCR was performed by using the SYBR Green PCR Master Mix (Invitrogen/Applied Biosystem, Carlsbad, CA) and the ABI Prism 7300 real-time PCR system (Applied Biosystems, Carlsbad, CA), according to the manufacturer’s instructions. Calculations, based on the 2−ΔΔCT method, 27 were performed by using the following equation: R (ratio) = 2 –[ΔCT (sample) –ΔCT (control)].Data are expressed as the fold change of the treatment groups relative to control and normalized to the levels of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences were designed by Primer 3 and UCSC In-Silico PCR, and were as follows:
HCV RNA forward 5′-ACTCCCCGGACGCT GACCTC-3′;
HCV RNA reverse 5′-GCAGGATCTCCGCCGGA ACG-3′;
GAPDH forward 5′-CAACTGGTCGTGGACAACCAT-3′;
GAPDH reverse 5′-GCACGGACACTCACAATGTTC-3′.
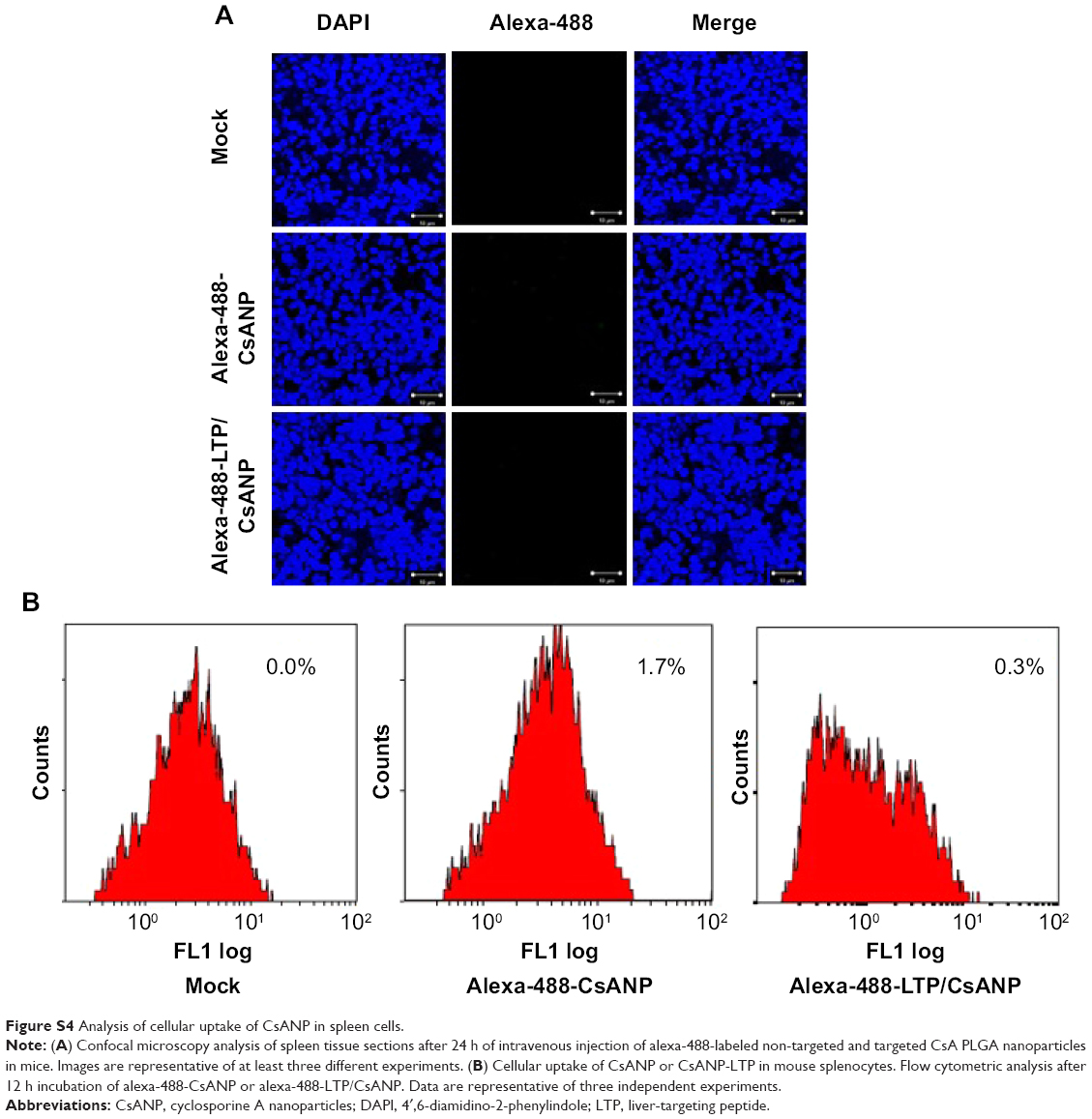 | Figure S4 Analysis of cellular uptake of CsANP in spleen cells. |
Supplementary results
Cellular uptake of CsA-encapsulated PLGA nanoparticles
Specific targeting of the CsANP-LTP was investigated using a cellular uptake assay conducted in HCV replicon cells. Alexa-488-labeled CsANP-LTP were used to evaluate the internalization of LTP-conjugated PLGA nanoparticles by flow cytometry. Only approximately 2% of the cells were positive after 12 h of treatment with alexa-488-CsANP (Figure 4A, left). By contrast, alexa-488-labeled CsANP-LTP treated cells were 95% particle positive, indicating specific binding of targeted nanoparticles on the surface of liver cells compared to non-targeted nanoparticles (Supplementary Figure 2A, right). The internalization of targeted nanoparticles was further confirmed by confocal microscopy. Alexa-488-labeled CsANP were very faintly detected in the cytoplasm of cells (Supplementary Figure 2B, left). In order to rule out the possibility that only the LTP peptide was internalized leaving the nanoparticle in the extracellular region, we loaded DiI dye into the PLGA nanoparticles and conjugated them with the alexa-488-labeled LTP and examined for the dual fluorescence in the cytoplasm. Our data clearly show the co-localization of both the DiI dye-encapsulated nanoparticles and the alexa-488 labeled LTP in the cytoplasm, thereby confirming that the LTP did facilitate the delivery of the nanoparticles into the intracellular compartment via receptor-mediated endocytosis (Supplementary Figure 2B, right). The confocal microscopy results were in agreement with those from flow cytometric analysis. Failure of non-targeted PLGA nanoparticles to enter the cells appears to be due to the overall negative surface charge which is not conducive to non-specific uptake. These data also indicate that PEGylation of nanoparticles does not aid non-specific delivery to cells. Furthermore, we could not detect significant uptake of targeted PLGA nanoparticles in other cell lines such as A549 cells (human lung cancer) and mouse splenocytes (Supplementary Figures 2, 3), strongly suggesting that cellular uptake of targeted PLGA nanoparticles was mainly due to specific binding of LTP peptide to HSPGs on the surface of liver cells and delivery into cells via receptor-mediated endocytosis. Overall, our data suggest that CsANP-LTP is specifically targeted into liver cells.
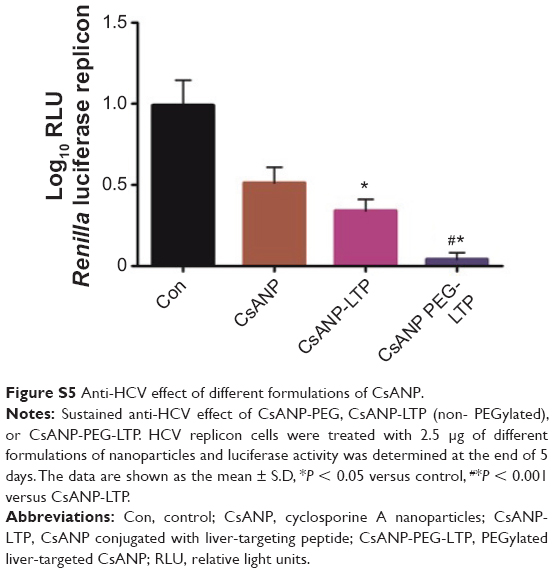 | Figure S5 Anti-HCV effect of different formulations of CsANP. |
In vitro cytotoxicity of targeted CsA encapsulated PLGA nanoparticles
After demonstrating that the targeted PLGA nanoparticles were able to be specifically internalized by hepatocytes, we evaluated the cytotoxicity of CsANP-LTP. One of the most important aspects of our anti-HCV strategy was to deliver a therapeutically sufficient amount of CsA without compromising the viability of the host cells. CsA treatment exhibited significantly higher levels of toxicity in HCV replicon cells, while CsANP or CsANP-LTP treatment did not affect the cell viability in time- and concentration-dependent experiments (Supplementary Figure 4). The non-cytotoxic effect of the nanoparticles may be attributed to the slow and sustained release of CsA from the nanoparticles.
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.