Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Liposomes as nanomedical devices
Authors Bozzuto G ![]() , Molinari A
, Molinari A ![]()
Received 5 June 2014
Accepted for publication 11 October 2014
Published 2 February 2015 Volume 2015:10(1) Pages 975—999
DOI https://doi.org/10.2147/IJN.S68861
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Giuseppina Bozzuto,1,2 Agnese Molinari2
1Chemical Methodology Institute, CNR, 2Department of Technology and Health, Istituto Superiore di Sanità, Rome, Italy
Abstract: Since their discovery in the 1960s, liposomes have been studied in depth, and they continue to constitute a field of intense research. Liposomes are valued for their biological and technological advantages, and are considered to be the most successful drug-carrier system known to date. Notable progress has been made, and several biomedical applications of liposomes are either in clinical trials, are about to be put on the market, or have already been approved for public use. In this review, we briefly analyze how the efficacy of liposomes depends on the nature of their components and their size, surface charge, and lipidic organization. Moreover, we discuss the influence of the physicochemical properties of liposomes on their interaction with cells, half-life, ability to enter tissues, and final fate in vivo. Finally, we describe some strategies developed to overcome limitations of the “first-generation” liposomes, and liposome-based drugs on the market and in clinical trials.
Keywords: liposomes, nanomedicine, drug delivery, ultrastructure
Introduction
Liposomes were discovered by Alec D Bangham in the 1960s at the Babraham Institute, University of Cambridge, and consist of single or multiple concentric lipid bilayers encapsulating an aqueous compartment (Figure 1).1,2 The first formulations were composed solely of natural lipids; at present they can include natural and/or synthetic lipids and surfactants. They have the capability of entrapping both lipophilic and hydrophilic agents, in the lipid membrane and in the aqueous core, respectively. The size of these nearly spherical lipid vesicles can range from a few nanometers to several micrometers. However, liposomes applied to medical use range between 50 and 450 nm.3
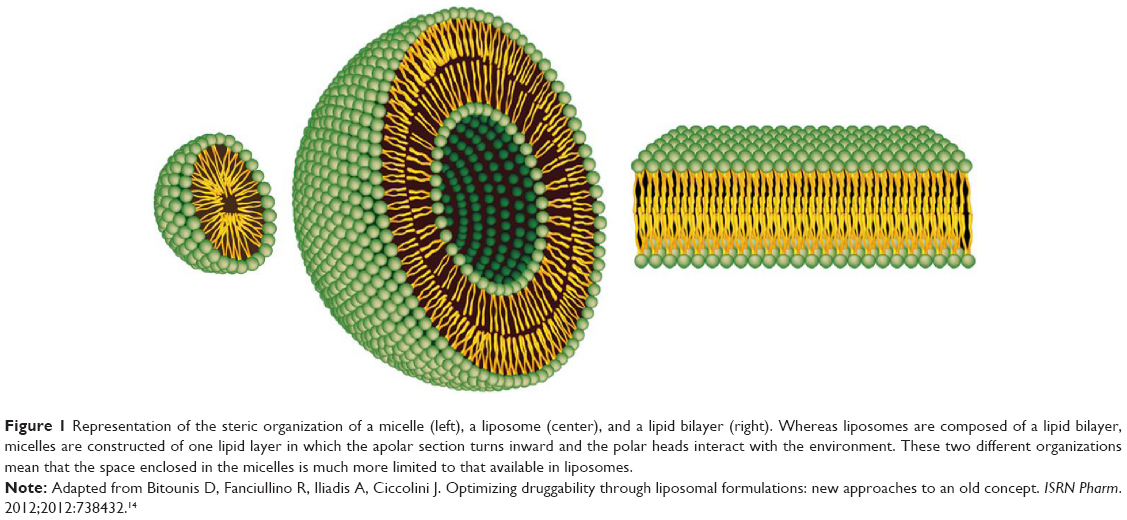 | Figure 1 Representation of the steric organization of a micelle (left), a liposome (center), and a lipid bilayer (right). Whereas liposomes are composed of a lipid bilayer, micelles are constructed of one lipid layer in which the apolar section turns inward and the polar heads interact with the environment. These two different organizations mean that the space enclosed in the micelles is much more limited to that available in liposomes. |
Liposomes seem to be an almost ideal drug-carrier system, since their morphology is similar to that of cellular membranes and because of their ability to incorporate various substances. Therefore, for the last 50 years liposomes have been widely investigated and they continue to be the subject of intense research. They are valued for their biological and technological advantages as optimal delivery systems for biologically active substances, both in vitro and in vivo, and are considered to be the most successful drug-carrier system known to date.4 During the two last decades, notable progress has been made, and several biomedical applications of liposomes are either in clinical trials or are about to be put on the market, while others have already been approved for public use.5
Therefore, the goal of this review is not to undertake an exhaustive report on the plethora of data published on liposomes since their first synthesis in the 1960s, but to focus on some points that play a key role in the development of liposomal formulation for therapy.
We briefly analyze how the efficacy of liposomes depends on the physicochemical properties of their membranes, on the nature of their components, and on their size, surface charge, and lipid organization. In addition, we discuss how the physicochemical properties of liposomes influence their stability in the bloodstream, their ability to enter various tissues, their interaction with cells, and their final fate in vivo. We also describe some strategies developed to overcome the limitations of the “first-generation” liposomes, and how this has been crucial in opening the way from the laboratory bench to clinical trials or to the market.
The physicochemistry of liposomes
The adequacy of liposomes as a carrier system for drugs strictly depends on the physicochemical properties of their membranes, on the nature of their components, on their size, surface charge, and lipid organization.6
Liposomes are mainly composed of phospholipids, amphiphilic molecules that have a hydrophilic head and two apolar hydrophobic chains. When phospholipids are dispersed in aqueous solutions, due to their amphipathic nature they have a strong tendency to form membranes.7 On the one hand, their polar heads prefer to interact with the aqueous environment; on the other, their long apolar aliphatic chains promote interaction with one another. In aqueous solution, these dual properties favor the formation of two lipid layers. The hydrophobic chains of each layer face each other and constitute a lipophilic inner compartment that acts as a permeability barrier, both inward and outward. Hydrophobic interactions are behind the formation of these lipid bilayers, and van der Waals forces keep the long hydrocarbon tails together, thus strengthening this architecture. Lastly, hydrogen bonds and polar interactions between the water molecules of the aqueous environment and the polar heads of lipids stabilize this organization. The final organization of lipids depends on their nature, concentration, temperature, and geometric form.8 If ions or molecules are present during the formulation process, they can be encapsulated inside these membranes.
Liposomes can be classified on the basis of the preparation method (reverse-phase evaporation vesicles or vesicle extruded technique), size (small, intermediate, or large), and lamellarity (uni-, oligo-, and multilamellar vesicles). The formation of unilamellar vesicles (ULVs) or multilamellar vesicles (MLVs) depends on the synthesis methods and postformation processing used for their preparation (refer to the “Methods for the preparation of liposomes” section for more details). Since ULVs (one lipid bilayer, 50–250 nm) enclose a large aqueous core, they are ideally suited for the encapsulation of hydrophilic drugs. On the other hand, MLVs (two or more concentric lipid bilayers organized like an onion-skin, 1–5 μm) preferentially entrap lipid-soluble drugs.9 In addition to the ability to entrap drugs with different solubility characteristics, it has been hypothesized that ULVs and MLVs have different release kinetics. In general, MLVs are formed more easily at larger hydrodynamic diameters, and thus have greater entrapped volume than ULVs. As a result, unilamellar liposomes with a hydrodynamic diameter of 130 nm exhibit a much faster release rate than MLVs with two to three lamellar bilayers and a hydrodynamic diameter of 250 nm.10,11 The difference in the release rate is due overall to the number of phospholipid bilayer that it have to cross before being released.
The ongoing interest of researchers in liposome characteristics, such as stability, pharmacokinetic properties, and therapeutic efficacy, has led to second-generation liposomes by the modulation of lipid composition, size, and the charge of the vesicle. The addition of cholesterol to the lipid bilayer of liposomes reduces their permeability and increases their in vivo and in vitro stability, because the presence of cholesterol induces a dense packing of phospholipids and inhibits their transfer to high-density lipoprotein (HDL) and low-density lipoprotein (LDL). In fact, cholesterol is a hydrophobic molecule and preferentially interacts with the core of the membrane, thus stabilizing it. Further, cholesterol can be used to anchor other molecules, such as polyethylene glycol (PEG) or deoxyribonucleic acid (DNA), to the liposomes for their application in biosensing or as “stealth” drug carriers12 (reviewed in Hosta-Rigau et al).13 Finally, the use of phosphatidylcholine with saturated fatty acyl chains and materials that stretch the transition temperature beyond 37°C offered even greater stabilization.14 For prolongation of the in vivo liposome circulation time, a milestone is the inclusion of hydrophilic carbohydrates or polymers, such as monosialoganglioside (GM1) and PEG in liposome composition. GM1 decreases the blood proteins absorbed on the liposomal surface and improves the half-life of liposomes in the blood.15,16 Similarly, the PEGylation of the liposomal carrier proved to extend the blood-circulation time while diminishing the uptake by the reticuloendothelial system (RES). Further, by modifying the PEG-molecule terminus, liposomes can be actively addressed with specific ligands or monoclonal antibodies (more details are reported in the section “Pharmacokinetics of liposomes”).17
Methods for the preparation of liposomes
There are many different methods for the preparation of liposomes. The choice of the appropriate method depends on several factors: 1) the physicochemical characteristics of the liposome components and those of the drug to be loaded; 2) the toxicity and the concentration of the loaded substance; 3) the type of the medium in which the liposomes are dispersed; 4) the additional processes during the application/delivery of the liposomes; 5) the size and the half-life desired for the successful application; and 6) the costs, reproducibility, and applicability regarding large-scale production for clinical purpose and good manufacturing practice-relevant issues.18–20
One of the most widely used techniques for liposome-manufacture preparation is the thin-film hydration or Bangham method.21,22 Briefly, this method involves dissolution of the lipid in an organic solvent, evaporation of the solvent, and the dispersion of the obtained lipid film in aqueous media. The drug to be entrapped can be included in the aqueous media (for hydrophilic drugs) or in the lipid film (for lipophilic drugs). However, the encapsulation efficiency of water-soluble drugs is low (5%–15%). Moreover, this method produces large and nonhomogeneous MLVs that require sonication or extrusion processes to be produced in homogeneous small ULVs.
The reverse-phase evaporation and solvent-injection methods provide hydration of the lipids directly from an organic solvent, and achieve an aqueous suspension of MLVs and ULVs, respectively.23–25 The use of these methods is affected by the solubility of lipids in the organic solvent and the elimination of the latter from the products. Nevertheless, these procedures guarantee higher encapsulation efficiency than thin-film hydration.
The detergent-depletion method involves the hydration of a lipid film with a detergent solution and leads to the formation of large MLVs.22,26,27 This method is rarely used, because it needs long preparation time and poor trapping efficiency. On the contrary, the dehydration–rehydration technique described by Kirby and Gregoriadis provides high drug-encapsulation efficiency and is widely used in nanomedicine. This procedure induces the fusion of preformed vesicles by means of dehydration and controlled rehydration.28
To ensure the desired size, lamellarity, and homogeneity properties of liposomes manufactured with the aforementioned techniques, postformation processing is required. The most common methods for postformation processing are sonication, extrusion, and high-pressure homogenization. Sonication is used to reduce the size of the vesicles and give energy to lipid suspension. This can be obtained by applying an ultrasonic irradiation to the suspensions of MLVs.29 The resulting small ULVs are purified by ultracentrifugation. The membrane-extrusion method reduces the size of the liposomes (large ULVs or MLVs) by passing them through a membrane filter with a defined pore size.30–32 The high-pressure homogenization is a fluid mechanical process that involves the subdivision of vesicles into smaller sizes and occurs in a special homogenizing valve.33
Since industrial-scale production of liposomes has become a reality, the range of liposome-preparation methods has been extended by a number of techniques. Among these are the heating method, spray-drying, freeze-drying, supercritical reverse-phase evaporation, and several modified ethanol-injection techniques that are increasingly attractive, as extensively reviewed in Laouini et al.34
Finally, the microfluidic-based method is an emerging technology for liposome synthesis that allows strict control of the lipid-hydration process. Microfluidics is a method to manipulate liquid flows in channels with dimensions of tens to hundreds of micrometers.35,36 The characteristic of laminar flow and rapid and tunable mixing has some advantages in liposome formation over traditional methods, such as thin-film hydration and reverse-phase evaporation. In continuous microfluidic flow systems, precise control of liposome size and size distribution can be implemented by controlling flow and mixing conditions. However, the problem of scaling up liposome production needs to be addressed during the implementation of microfluidic technology for practical application. Additionally, the ultrafine structure of liposomes produced with the microfluidic method needs to be further investigated.37
Microfluidic remote loading for rapid single-step liposomal drug preparation has been recently reported. In this method, microfluidic-directed formation of liposomes is combined with in-line sample purification and remote drug loading for single-step, continuous-flow synthesis of nanoscale vesicles containing high concentrations of stably loaded drug compounds. Using an on-chip microdialysis element, the system enables rapid formation of large transmembrane pH and ion gradients, followed by immediate introduction of amphipathic drugs for real-time remote loading into the liposomes. The microfluidic process enables in-line formation of drug-laden liposomes with drug:lipid molar ratios of up to 1.3, and a total on-chip residence time of approximately 3 minutes, representing a significant improvement over conventional bulk-scale methods, which require hours to days for combined liposome synthesis and remote drug loading. The microfluidic platform may be further optimized to support real-time generation of purified liposomal drug formulations with high concentrations of drugs and minimal reagent waste for effective liposomal drug preparation at or near the point of care.38
Real-time, highly sensitive, and low-cost monitoring of drug-loading and delivery dynamics in different pharmaceutical and biomedical environments could be very useful both for pharmaceutical manufacturing and for quality-assurance assays applied to liposomal formulations. Currently, the techniques used to investigate liposomal structures and their stability in different environments, as well as drug-loading and -delivery mechanisms, operate basically off-line and/or with prepared sampling. Organic electrochemical transistors (OECTs), promising devices for applications in bioelectronics and nanomedicine, have been recently proposed as ideally suitable for sensing and real-time monitoring of liposome-based structures. These systems seem to be particularly suited for real-time monitoring of liposomes in solution. OECTs are sensitive devices for detecting liposomes on a wide dynamic range down to 10−5 mg/mL (with a lowest detection limit, assessed in real-time monitoring, of 10−7 mg/mL), thus matching the needs of typical drug-loading/drug-delivery conditions. Furthermore, OECTs proved to be able to sense and discriminate successive injections of different liposomes, and so could be good candidates in quality-control assays or in the pharmaceutical industry.39
Parameters including shape, size, surface features, and lamellarity strongly influence the biological behavior of liposomes. Therefore, they have to be extensively characterized prior to their use in order to ensure in vitro and in vivo performance. Liposome shape can be assessed using electron microscopy techniques. Lamellarity of liposomes can be determined by using negative staining and/or freeze-fracturing for transmission electron microscopy (TEM) and P-31 nuclear magnetic resonance analysis.40
Several techniques are available for the determination of size and size distribution, among which the most widely applied include dynamic light scattering (DLS), size-exclusion chromatography, and field-flow fractionation (FFF).41–44 Various microscopy techniques, including cryo-TEM, have also been found useful for the characterization of liposome size.45,46
DLS measures the time-dependent fluctuations in the intensity of scattered light, which occur because particles (liposomes) in a suspension undergo random Brownian motion due to collisions between suspended particles and solvent molecules. An analysis of the intensity fluctuations allows the determination of the distribution of the diffusion coefficients of the liposomes, which are converted into a size distribution using established theories. DLS is a simple and rapid method, but it provides an average property of liposome bulk.
On the contrary, electron microscopy techniques, such as cryo-TEM and TEM using freeze-fracturing, provide a precise determination of liposome size, since they allow for the visualization of single liposomes and can resolve particles of varying size. The result is exact information about the profile of the liposome population over the whole range of sizes. Unfortunately, these techniques are very expensive and require specific equipment.45 Another microscopic technique utilized to analyze liposome morphology and size is atomic force microscopy, which provides information with high resolution on the three-dimensional profile of liposomes without removing them from their native environment.47,48
Size-exclusion chromatography is a technique that can separate and quantify liposome populations by exploiting the time-based resolution of hydrodynamic size. The process of separation is based on large-particle elution before that of smaller particles. The large particles are left out from the internal pore volume of the porous substrate used in this technique, and are eluted more quickly from the column.43,49
FFF is a separation technique based on the laminar flow of particles in a solution, and uses a semipermeable membrane that allows only the carrier fluid to pass through the membrane. FFF separates liposomes based on size, and can separate materials over a wide colloidal size range while maintaining high resolution.50
Liposomes as nanocarriers for drug delivery
Despite considerable progress in recent years, the diagnosis and treatment of various diseases, especially cancer, continue to present constraints, such as low sensitivity or specificity, drug toxicity, and severe side effects. In particular, many therapeutic agents have a very narrow window, ie, the therapeutic dose is not much lower than a toxic one. In many cases, the employment of an appropriate drug carrier can reduce the toxicity by changing the temporal and spatial distribution of a drug.
Since they were first described in the 1960s, liposomes have long been recognized as drug-delivery vehicles. They are very appropriate for this aim, due to their biocompatibility and biodegradability.9 Due to their nature, liposomes are in fact considered safe nanocarriers. However, the addition of nonphysiological additives can induce chemical modifications that are useful to improve efficacy in drug delivery but potentially toxigenic.
In addition to being biocompatible, all liposomes have in common a structure that gives them the ability to contain both hydrophilic and hydrophobic drugs. The encapsulation of the active form of a drug into the lipid bilayer protects it against naturally occurring phenomena, such as enzymatic degradation and immunologic and chemical inactivation. Therefore, liposomes prevent a drug from being metabolized prior to reaching target tissues, and simultaneously they minimize exposure of healthy tissue to the encapsulated drug during its circulation in the blood. Both of these effects contribute to increase the therapeutic index. In fact, high levels of the active form of a drug are delivered to the tumor site so that the expected cytotoxic effect can be achieved. Meanwhile, any undesirable side effects of the encapsulated drug are substantially reduced when compared to the free form.
The delivery of the encapsulated drug depends on the nature of the lipid bilayer, the size of the drug molecules, their partition coefficient in oil/water, and their interactions with the lipid membrane. The mechanism and extent of liposome-delivery is also strongly influenced by the nature and density of the charge (ζ-potential) of the liposome surface (refer to the “Charged liposomes for drug delivery” section for more details).
The encapsulation efficiency of a molecule in a liposome depends on its polarity and partition coefficient, which also determines its localization in the liposomal membrane. If a drug is hydrophobic in nature, it resides in the acyl hydrocarbon chain of the liposome, and hence encapsulation is dependent on the properties of the acyl chains of the liposome, such as length and packing density. It is also expected that the encapsulation efficiency of hydrophobic molecules is influenced by changes in the drug-to-lipid ratio. On the other hand, if a drug is polar/hydrophilic, it tends to localize in the aqueous core or adjacent to the water–lipid interface, near the polar head groups of the liposome. Therefore, its encapsulation efficiency does not exhibit a strong dependence on the drug-to-lipid ratio. On the contrary, the introduction of hydrophilic chains in the liposome surface favors greater entrapment of hydrophilic molecules when compared with hydrophobic molecules.51
Drugs loaded in liposomes are not bioavailable; they only become bioavailable when they are released. Therefore, optimizing the release rate of a liposome-vehicle drug is strategic to reach a level within its therapeutic window and at a sufficient rate for a sufficient period to have optimal therapeutic activity. On the other hand, premature drug release should be avoided. To overcome this inconvenience, several experimental approaches have been pursued, either by modifying the lipid bilayer or entrapping drugs suitable for the purpose.
Switching from a fluid-phase phospholipid bilayer to a solid-phase bilayer, eg, by incorporating cholesterol (bilayer-tightening effect) or sphingomyelin into liposomes, the retaining of cargo into liposomes is increased.52–54 Another approach to control the release rate of entrapped substances is to choose drugs with physical characteristics favoring retention in the lipid nanovector. Similarly to biological membranes, liposomes have high permeability to hydrophobic drugs and low permeability to hydrophilic drugs. Anticancer drugs of high hydrophilicity retained in the aqueous internal compartment of liposomes (albeit with low trapping efficiency) are slowly released from the liposomes over several hours to several days.55,56 Drugs of high hydrophobicity efficiently inserted into the fatty acyl chain region of the lipid bilayer can be easily released. Retention of highly hydrophobic drugs, such as paclitaxel, in liposomes is problematic, and many formulations and their pharmacokinetics have been studied to increase drug–liposome associations.57
Many anticancer drugs are of intermediate solubility and readily partition between the liposome bilayer and the exterior or interior aqueous phase, resulting in their rapid release from liposomes. However, manipulation of the interior pH of the liposomes or the formation of molecular complexes within the liposomes can result in excellent retention of weak bases, such as doxorubicin (Dox) or daunorubicin, in liposomes.58,59 Drug retention can be improved by loading drugs to achieve high intraliposomal drug concentrations above their solubility limits, thus enhancing precipitation, or by encapsulating polyanions, such as dextran sulfate.60,61 Drugs that are not weak bases, such as docetaxel, can be converted to weak-base prodrugs, thus allowing encapsulation and liposomal retention.62
Liposomes should store, protect, and transfer substantial quantities of drugs while being well tolerated in patients receiving the cure. These unique characteristics could provide for an improved biopharmaceutical profile through reduced toxicity, favorable pharmacokinetic behavior, and an enhanced therapeutic index in comparison to the free-form drug. Liposomes as drug-delivery systems could show several advantages over conventional dosage forms, particularly for parenteral (ie, local or systemic injection or infusion), topical, and pulmonary routes of administration.
Several clinical studies have demonstrated that liposomal encapsulation of drugs typically leads to a change in toxicity profiles. When used in clinical settings, liposomal treatments proved to improve patient outcome dramatically, reducing some of the side effects associated with chemotherapy, such as cardiotoxicity, nausea, and vomiting, when compared to unencapsulated drugs.63,64 Therapeutic activity of vincristine, widely used in the treatment of a number of human carcinomas, significantly increased after its encapsulation in appropriately designed liposomal systems. In fact, vincristine sulfate-liposome injection improved the therapeutic index by facilitating increased dose intensification, while maintaining a predictable and manageable safety profile. This effect is a consequence of a lower clearance and a higher area under the curve compared with conventional free vincristine sulfate.65 In the field of antimicrobial agents, liposomal amphotericin B showed better tolerance and higher efficacy than the antibiotic amphotericin B deoxycholate. At present, liposomal amphotericin B is the drug of choice for the treatment of patients with disseminated histoplasmosis and acquired immunodeficiency syndrome (AIDS).66 Nystatin entrapped in pH-sensitive liposomes enhanced anticryptococcal efficacy in a murine model.67
Antitumoral anthracyclines, such as Dox, daunorubicin, and epirubicin, achieve highly efficient encapsulation. Liposomal anthracyclines proved to be effective and showed reduced cardiotoxicity when compared to free agents, either as a single agent or in combination with other drugs.68,69 A meta-analysis study compared the safety and toxicity of liposomal Dox versus conventional anthracyclines. Both liposomal Dox and PEGylated liposomal Dox (PLD) showed favorable toxicity profiles, with better cardiac safety and less myelosuppression, alopecia, nausea, and vomiting compared with conventional anthracyclines, making them a favorable choice over conventional anthracyclines in elderly patients, patients with risk factors for cardiac disease, and patients with prior use of anthracyclines.70
Among the pharmaceutical options available for treatment of ovarian cancer, increasing attention has been progressively focused on PLD, whose unique formulation prolongs the persistence of the drug in the circulation and potentiates intratumor accumulation. PLD has become a major component in the routine management of epithelial ovarian cancer (extensively reviewed in Pisano et al).71 Nonrandomized Phase II trials of PLD in platinum-resistant ovarian cancer patients documented the biological activity of this agent in this clinical setting, with objective response rates of approximately 10%–20% being reported in several trials.72–74 Data indicated that palmar–plantar erythrodysesthesia (hand–foot syndrome), toxic acral erythema, and mucositis were the most common toxicities of PLD, reported in up to 50% of treated patients. Although not life-threatening, palmar–plantar erythrodysesthesia can negatively impact quality of life, and it is a major cause of both dose reduction and treatment discontinuation.75,76 As regards cardiac toxicity, in several trials PLD formulation has been related to a better safety profile compared to conventional Dox.77 Compared to the 7.5% incidence of irreversible cardiotoxicity at cumulative doses of 400–550 mg/m2 reported with Dox, most of the studies of PLD showed a lower incidence of cardiac failure even at doses higher than 500 mg/m2.78–80 In a prospective trial performed on patients with advanced gynecological malignancies treated with PLD, cardiac safety was further assessed at histology (endomyocardial biopsies), showing no myocardial damage in patients treated with PLD (median PLD dose of 708 mg/m2).81 Therefore, the optimal cardiac safety profile of PLD may allow prolonged treatment. Encouraging results from a Phase II trial in AIDS-related Kaposi’s sarcoma patients treated with PLD up to a 2,360 mg/m2 cumulative dose have been reported.82 In metastatic breast cancer patients also, doses greater than 450 mg/m2 were not associated with a significant decrease in left ventricular ejection fraction from baseline compared to conventional Dox.80 In a relapsed ovarian cancer patient responding to second-line chemotherapy, maintenance therapy with PLD for more than 1 year was reported to be safe by Andreopoulou et al with no cardiac events reported.83
Effect of size on liposome fate
Pathological tissues, such as inflammatory or solid tumor tissues, are characterized by increased vascular permeability.84 The enhanced permeation and retention (EPR) effect is the phenomenon characterizing malignant tissues by which nanocarriers of an appropriate size can pass through tumor-vessel walls and enter the neoplastic lesion (Figure 2). More particularly, solid tumors that undergo angiogenesis develop a discontinuous endothelium, with large fenestrations allowing molecules of up to approximately 4,000 kDa, or 500 nm, to enter the interstitial space.85,86 Liposomes can satisfy the size conditions needed to pass through tumor vessels and concentrate in the target site. This mechanism represents the major targeting principle for intravenously administered long-circulating liposomes. However, because no specific targeting ligands are used to interact with the tumor target site, this tumor-localization process is referred to as “passive targeting.”
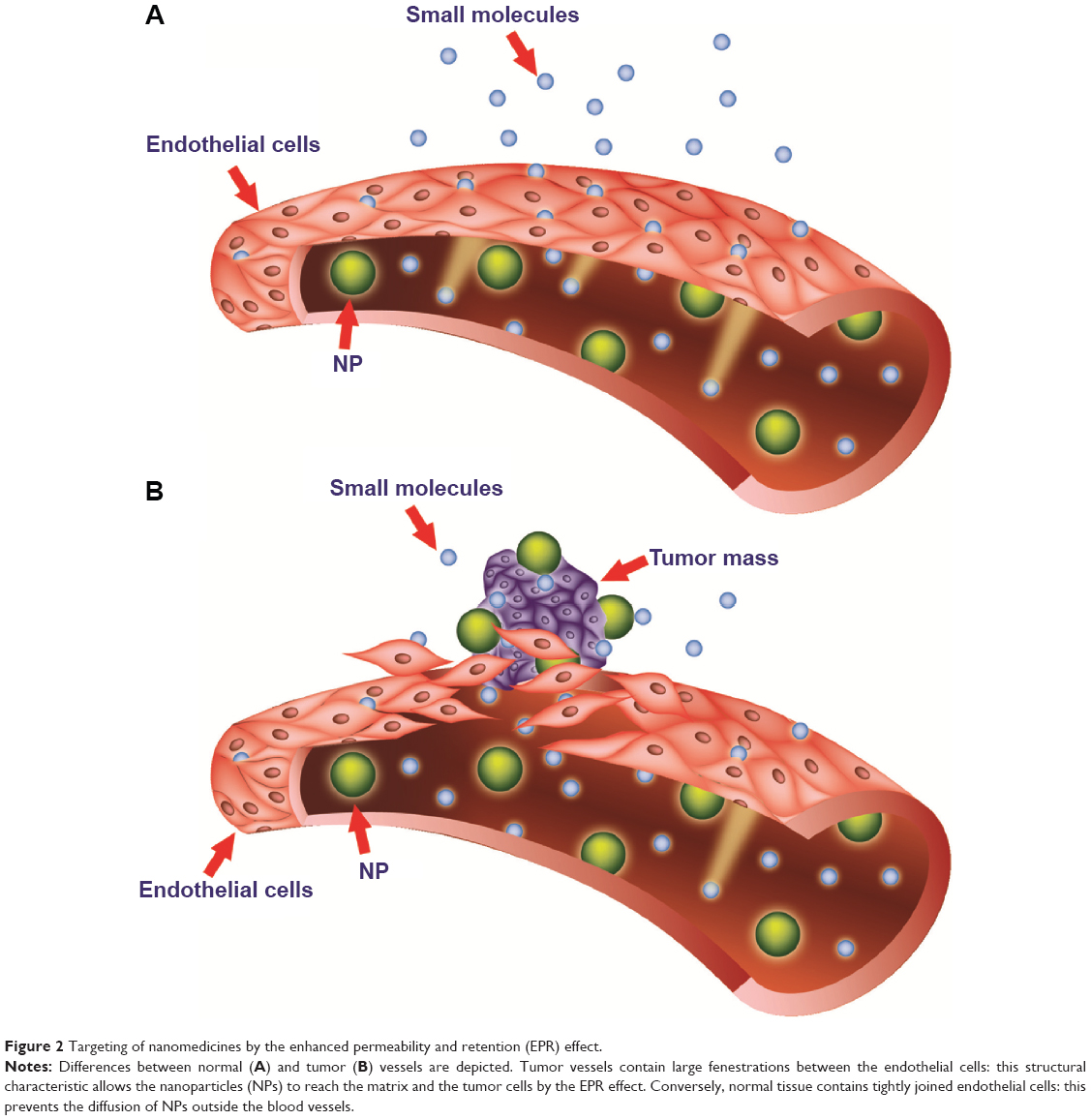 | Figure 2 Targeting of nanomedicines by the enhanced permeability and retention (EPR) effect. |
This passive targeting depends on the mechanical and physical properties of liposomes. More importantly, once liposomes have entered the tumoral tissue, they are retained from the malfunctioning lymphatic system. Therefore, after its release, the drug encapsulated into liposomal carriers can exert its therapeutic effect.9
The overall size of the liposome-based drugs is an essential physical aspect that determines the clinical successes of the nanocarriers. Experimenting with variations in liposomal size, researchers observed that liposomes smaller than 100 nm in diameter interacted less with plasma proteins, evaded capture by the RES, had a longer half-life in the blood, and accumulated passively at the tumoral site.5 Conversely, it was found that larger liposomes were eliminated more rapidly from blood circulation and did not escape RES uptake. On the other hand, it was observed that small liposomes had reduced drug-storage capacities.17
Even if the size of these drug-delivery systems can be easily modulated, theoretically an ideal liposome designed for the delivery of chemotherapeutics should be of 50–100 nm in diameter. The lower size limit (50 nm) should prevent intravenous-based nanocarriers from randomly penetrating normal vessel walls while in circulation.87 However, an upper size limit to these systems could also exist. In order to gain access to tumor tissue, nanocarriers should retain the ability to extravasate from vessels through the large vascular fenestrations (250 nm or larger) that are present in and around tumor sites and are attributed to ongoing angiogenesis.84,87,88 At the tissue level, many nanomedicine products attempt to target sites passively through the EPR effect, with feature sizes typically in the 100–200 nm range, but particles up to 400 nm have demonstrated extravasation and accumulation in tumors (although this is an extreme case). However, when size is increased, capture by the RES also increases.68,89 For example, a previous study reported that PEGylated liposomes 250 nm in diameter were removed from circulation more than twice as fast as liposomes 100 nm in diameter with similar lipid composition.90 This is particularly problematic, since it is imperative that liposomes loaded, eg, with an antitumoral agent, will remain in circulation until they accumulate within tumor tissue and release the active molecule at a concentration sufficient to exert a therapeutic effect.
Pharmacokinetics of liposomes
The pharmacokinetics of liposomes focuses on their distribution throughout the body fluids and tissues and their metabolism. The latter mainly includes liposome chemical degradation and excretion, which is achieved through their uptake and clearance by the RES.
As mentioned previously, one important goal of the design of a new liposomal carrier is the modulation of the pharmacokinetic profiles of a drug. The advantages of liposomal-based drugs should be greater solubility of the cargo, increased half-life, selective delivery to the site of action, improved therapeutic index, and the ability to overcome resistance against chemotherapeutics. When a therapeutic agent is loaded into liposomes, it adopts the carrier’s pharmacokinetics until it is delivered. As a result, liposomes modify both the tissue distribution and the rate of clearance of the loaded drug.91 The pharmacokinetics of liposomal-based drugs depends on the physicochemical characteristics of the lipid vehicle, such as lipid composition, size, membrane lipid packing, steric stabilization, surface charge, dose, and route of administration. It has been reported that the primary sites of accumulation of carrier-mediated agents are the tumor, liver, and spleen, compared with noncarrier formulations. Factors affecting the pharmacokinetic and pharmacodynamic variability of these agents remain unclear, but most likely include the RES.63
Once inside the organism, liposomes’ stability in the bloodstream, as well as their capacity to enter target tissues, determines the fate of liposomes. During their circulation in the blood, liposomes meet plasma proteins, such as opsonins and HDLs and LDLs. Opsonins include various protein types, like immunoglobulins and fibronectin, which help RES recognize and eliminate liposomes. Blood carrying HDL and LDL interacts with liposomes and reduces their stability. The interaction with lipoproteins causes lipid transfers and rearrangements on the surface of liposomes. This frequently induces lipid depletion, liposome breakdown, and rapid release of the cargo to the plasma.5 By modulation of lipid composition, this effect can be avoided (see later in this paper for more details).
After interaction with target cells, the delivery of the encapsulated drug depends on the nature of the lipid bilayer, the size of the drug molecules, their partition coefficient in oil/water, and their interactions with the lipid membrane. The final fate of the drug, ie, the extracellular fluid or the cytoplasm of the target cell, depends on the molecular architecture, mechanism of release, and the composition of the carrier.63
Elimination of liposomes takes place in different ways. The first involves absorption of plasma proteins on the surface of liposomes and then their recognition by the RES. This event results in the excretion of the cargo at the hepatic level and its subsequent metabolism by Kupffer cells. In the second way, liposomes are metabolized by splenic macrophages. Finally, after their accumulation, they are metabolized and eliminated by the target tissues.5,92,93
Despite all the hopes invested in conventional liposomes, they have presented various problems and pharmacological implications over the years. A major drawback of conventional liposomes is their quick capture by the RES.94 Liposomes are mainly accumulated in the liver and the spleen, due to their generous blood irroration and the abundance of tissue-resident phagocytic cells.63,92,93 This is extremely advantageous in the case of local infections: the high concentration of antimicrobial agents in the RES can help treat infective pathogens. However, during chemotherapy, it may lead to partial depletion of the macrophages and interfere with the important host-defense functions of this cell type.95 On the other hand, the marked increase in retention and accumulation of liposomal drugs in such organs as the spleen and the liver may lead to the delayed removal of lipophilic anticancer drugs from the circulation.96
A number of different strategies were then tested in the following years in order to overcome the aforementioned limitations, giving rise to a “second generation” of liposomes. The best strategy was described in the early 1990s, when experiments carried out by several groups of scientists demonstrated that PEGylation of the liposome surface was able to improve the stability and circulation time of liposomes dramatically after intravenous administration, by rendering the liposomes invisible to macrophages.50 These “long-circulating liposomes” were then named Stealth liposomes because of their ability to evade the immune system; this results in a significant increase in blood-circulation time in vivo.97–99
PEG is a linear or branched polyether diol with many useful properties, such as biocompatibility, solubility in aqueous and organic media, lack of toxicity, very low immunogenicity and antigenicity, and good excretion kinetics. These properties permit the employment of PEG in a variety of applications, including the biomedical field, after US Food and Drug Administration (FDA) approval for internal administration (http://www.accessdata.fda.gov/scripts/cder/drugsatfda). PEG chains protect liposomes from mononuclear phagocytic system cells by building a protective, hydrophilic film on the liposomal surface. Their presence prevents the interaction of liposomes with other molecules, such as various serum components. One possible explanation for the impaired interaction is the PEG-induced “steric hindrance.” The mechanism of steric hindrance by the PEG-modified surface has been thoroughly examined.100 The water molecules form a structured shell through hydrogen bonding to the ether oxygen molecules of PEG. The tightly bound water forms a hydrated film around the particle and repels the protein interactions.101 In addition, the presence of PEG on the surface may also increase the hydrodynamic size of the particle, decreasing its clearance, a process that is dependent on molecular size as well as particle volume.102
PEG-bearing liposomes are not opsonized or affected by complement components, and consequently evade capture by mononuclear phagocytic system cells.103,104 Finally, the presence of PEG in liposome formulations prevents aggregation, favors the formation of small, monodisperse particles, and increases the EPR effect, due to the extended circulation time and escape from the RES.105
The practical consequences of these phenomena are evident when the biopharmaceutical profiles of Stealth liposomes and conventional liposomes are contemplated. Stealth liposomes have a longer half-life (which leads to longer blood-circulation times), low systemic plasma clearance, and low volume of distribution (minimal interaction with nondiseased tissue). This results in multiple-fold greater area-under-the-curve values (drug concentration–time profile) and improved tissue distribution (targeting of target sites).90
The advantage of PEGylation is credible when the relative half-lives of non-PEGylated and PEGylated liposomes are compared. In fact, the half-life of liposomes after PEGylation increases from a few hours to 45 hours.106 Therefore, it is not surprising that the clinically approved antitumoral drug Doxil® is PEGylated in order to improve tumor-site accumulation of the drug.107 However, while PEG coating increases liposome-circulation times, it could also negatively influence the uptake by target cells due to PEG-induced steric hindrance.97
Although clinically useful activity has been demonstrated, currently available liposome-based therapies do not exhibit active targeting at the cellular level. Existing FDA-approved liposome technologies against cancer rely on passive accumulation through the EPR effect.108 In other words, these nanomedicines possess no functionality to actuate release other than passive efflux from the liposome at the tumor site. However, this uncontrolled, passive release in some cases results in suboptimal pharmacokinetics or reduced efficacy, as observed with cisplatin-loaded liposomes.109–111 An additional level of sophistication and specificity for the target cell can be achieved through ligand-mediated targeting, which is defined as active targeting. The goal is to develop platforms with improved biodistribution, pharmacokinetic properties, and active targeting. The properties of such targeted liposomes can be modulated and adapted to different needs. Peptides, carbohydrates, glycoproteins, receptor ligands, monoclonal antibodies, and growth factors have been applied as ligands. Ligand-targeted liposomes can selectively recognize the antigens or the receptors located on the surface of target cells. Due to this high selectivity toward cancer cells, almost all of the administered liposomal drug would accumulate at the tumor site, leaving uninjured healthy bystander cells. That way, the required dose for the expected cytotoxic effect will be significantly smaller when compared to nontargeted therapies: this contributes to a better therapeutic index, with higher drug efficacy and fewer side effects.112–115
As extensively reviewed in Noble et al116 ligand-targeted liposomes have demonstrated improved efficacy over passively targeted equivalents through enhanced targeting and intracellular uptake, but they have raised new challenges, such as hindered diffusion and penetration through the target tissue, immune recognition, and deactivation of targeting through the nonspecific binding of serum proteins. As a result, ligand-targeted liposome systems have not demonstrated consistently successful outcomes in preclinical settings, and other studies are necessary to address issues related to their efficiency.116
Charged liposomes
When a liposome interacts with a cell, the delivery of the drug and its distribution in the target cell can occur in several ways. Liposomes can adsorb into the membrane of cells, where the lipid bilayer of the carrier is degraded by enzymes, such as lipases, or by mechanical strain. This leads to the release of the active ingredients into the extracellular fluid, where they can diffuse through the cell membrane and cytoplasm. However, the latter process cannot easily occur when the loaded molecules are hydrophilic. A second way requires the fusion of the liposomal membrane with the plasma membrane of the target cell: this phenomenon causes the release of liposomal content directly into the cytoplasm. The third and most frequent way is receptor-mediated endocytosis. This process only regards vesicles of a maximum diameter of 150 nm and active ingredients that can endure the acidic environment of lysosomes, where liposomes are enzymatically processed. Phagocytosis can also occur, but involves liposomes of a diameter larger than 150 nm. These large liposomes are phagocytosed by specialized cells of the immune system, such as macrophages, monocytes, and Kupffer cells (Figure 3).5,84,117
 | Figure 3 Liposome–cell interaction. |
The mechanism and extent of liposome–cell interaction is strongly influenced by the nature and density of the charge of the liposomes surface. By changing the lipid composition, both of these parameters can be modified. The liposomes can include charged components that confer them an overall neutral, positive, or negative charge. Lack of surface charge (neutral liposomes) increases the aggregation of liposomes, reducing their physical stability. Moreover, neutral liposomes do not interact significantly with cells, and this causes drug release from the liposomes in the extracellular space.118,119 On the other hand, charged liposomes present numerous advantages compared with neutral liposomes. For example, the presence of a charge on the surface induces electrostatic repulsion among liposomes by creating a ζ-potential, positive or negative, that prevents their aggregation and flocculation. Moreover, a high electrostatic surface charge could promote the interaction of liposomes with cells.
The literature includes several studies carried out on the potential use of charged liposomes for biomedical applications. However, the great majority of these studies focused on positively charged liposomes, due to the encouraging results obtained in in vitro and in vivo experimentations.
Negatively charged liposomes are generally constituted by anionic lipids, such as dimyristoyl phosphatidylglycerol and dipalmitoyl phosphatidylglycerol. Literature data show that the negative liposomes, due to their electrostatic properties, are less stable than neutral and positive liposomes when injected into the blood circulation. In fact, anionic liposomes rapidly interact with the biological system subsequently to their opsonization with complement and other circulating proteins.120–122 Such an interaction has at least two acute consequences: a rapid uptake by the RES, and toxic effects, such as pseudoallergy that is manifested as vasoconstriction, pulmonary hypertension, dyspnea, and drop in circulating platelets and leukocytes.123 For this reason, anionic liposomes have not been widely used as drug-delivery systems for intravenous administration. Moreover, several studies examining the effect of negative and positive charge on the adjuvant activities of liposomes have been carried out, but the results obtained were rather conflicting and inconclusive.124–127 However, in recent years, there has been increased interest in developing charged liposomes as carriers for transdermal drug delivery, due to their enhanced penetration properties through the skin.128 To clarify the effect of the surface charge on percutaneous absorption, histological studies revealed that the negatively charged liposomes diffused to the dermis and the lower portion of hair follicles through the stratum corneum and the follicles much more quickly than the positive vesicles. Therefore, the rapid penetration of negatively charged liposomes would contribute to increased permeation of drugs through the skin.129
Cationic liposomes (CLPs), first described in 1987 by Felgner et al are typically used for gene delivery, based on the electrostatics between positively charged lipids and negatively charged nucleic acids.130 They consist of natural neutral phospholipids and positively charged lipids. Commonly used neutral phospholipids include dioleoyl phosphatidylethanolamine (DOPE) or dioleoyl phosphatidylcholine, and are in most cases required for the stabilization of the liposome/DNA complex. A variety of lipid formulations with a positive charge are already on the market, such as DC-cholesterol HCl, (3β-[N-(N′, N′-dimethylaminoethane)-carbamyl]cholesterol hydrochloride), DOTAP (1,2-dioleoyl-3-trimethylammonium-propane [chloride salt]), DOBAQ (N-[4-carboxybenzyl]-N, N-dimethyl-2,3-bis(oleoyloxy)propan-1-aminium), DDAB (dimethyldioctadecylammonium [bromide salt]), and MLV5 (N1-[2-((1S)-1-[(3-aminopropyl)amino]-4-[di(3-amino-propyl)amino]butylcarboxamido)ethyl]-3,4-di[oleyloxy]-benzamide), and many others are under development.
In the case of CLP–cell interaction, the endocytic pathway seems to be the preferential route of internalization. The pathway of internalization is of the utmost importance in the final fate of the drug loaded in the lipid carriers, as, eg, in the case of drugs sensitive to an acidic environment. Both the interaction with the cell membrane and the pathway of internalization are strictly dependent on the physicochemistry of liposomes. Morphological and ultrastructural studies are invaluable tools to study the mechanisms of liposome–cell interaction and analyze at nanoresolution the temporal and spatial parameters involved. As an example, morphological and ultrastructural studies by the freeze-fracturing technique on the interaction of CLPs with tumor cells are reported in Figures 4 and 5. In these experiments, CLPs formulated as described in Bombelli et al were used.131 Freeze-fracturing is an election technique used to study plasma-membrane architecture and its modifications occurring upon the interaction with xenobiotics, microbial agents, nanomaterials, and so on. As shown by the replica performed on cryofixed and cross-fractured samples, before being internalized, CLPs appear clustered on the plasma membrane of the glioblastoma cell. The capping phenomenon strongly suggests the involvement of an endocytic pathway (Figure 4). The replica of cross-fractured tumor cells allows the visualization of liposomes interacting with cytoplasmic organelles, very likely endosomes (Figure 5).
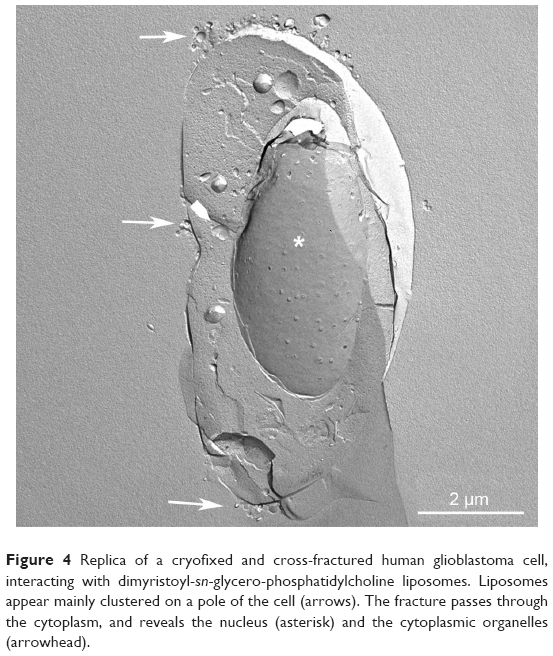 | Figure 4 Replica of a cryofixed and cross-fractured human glioblastoma cell, interacting with dimyristoyl-sn-glycero-phosphatidylcholine liposomes. Liposomes appear mainly clustered on a pole of the cell (arrows). The fracture passes through the cytoplasm, and reveals the nucleus (asterisk) and the cytoplasmic organelles (arrowhead). |
 | Figure 5 Replica of a cryofixed and cross-fractured human glioblastoma cell, interacting with dimyristoyl-sn-glycero-phosphatidylcholine liposomes. Liposomes (arrows) appear in the extracellular space and the cytoplasm of the cell. Some of them are interacting with cytoplasmic organelles (arrowhead). |
Since the overall positive charge of CLPs enhances the transfection of anionic animal target cells, liposome/DNA complexes are commonly used for delivering genes. However, preclinical and clinical results encourage the use of CLPs for the delivery of antitumoral agents to the tumor vasculature. In fact, it has been demonstrated that the presence of anionic sites in angiogenic endothelial cells (such as anionic phospholipids, proteoglycans, hyperglycosylated and hypersialylated membrane proteins, and so on) makes newly formed tumor vessels selective targeting sites for CLPs. It has been reported that such drugs as paclitaxel, Dox, and oxaliplatin carried by CLPs showed enhanced antitumor efficacy associated with a reduced functionality of the tumor microvasculature.132,133 Campbell et al demonstrated that the presence of a positive charge on liposomes is needed to improve their interactions with the glycoproteins of the endothelial cell membranes.134 These data support the use of CLPs to target cytotoxic drugs preferentially to the tumor vascular endothelium, and to achieve long circulation half-lives.135
An important goal of CLPs is to cross the blood–brain barrier (BBB) and achieve brain drug concentration. The BBB is composed of specific structures formed by brain-capillary endothelial cells and basement membrane, sheathed with astrocytic end-feet. BBB impedes drug penetration into the central nervous system, and for this reason many brain drug-delivery strategies have focused on crossing it. The mechanism by which CLPs have been shown to cross the BBB easily is either absorptive-mediated transcytosis or receptor-mediated transcytosis. Both mechanisms are triggered by the electrostatic interaction between liposome cationic components and membrane anionic microdomains of the brain’s capillary endothelial cells.136
Even if an increased amount of cationic lipids in liposomes may enhance affinity for angiogenic endothelial cells and the subsequent anticancer effect, some points need to be considered. The increased positive charge on the unshielded CLP surface could result in their aggregation in the bloodstream through electrostatic interactions with anionic species in the blood, in an enhanced uptake by the RES, and in reduced accumulation in the tumor.122,133,137–141 These effects negatively influence the fate and therapeutic efficacy of CLPs in vivo.
Even in the case of CLPs, PEG protects from circulating proteins, improving their plasma clearance and enhancing their anticancer effects. PEG-modified CLPs have also been shown to improve oligonucleotide loading and delivery and the solubility of various therapeutic agents.142,143
The use of CLPs may improve the efficacy and intrinsic safety of photodynamic therapy, a protocol that involves the administration of a photosensitizer and its irradiation in light of the appropriate wavelength (near infrared radiation) to excite and activate it and to induce the formation of cytotoxic species. In fact, liposomes can efficiently solubilize hydrophobic photosensitizers, thus increasing the photoactive population. Further, they can improve the accumulation in tumors and the pharmacokinetics of the photosensitizer, thus reducing the side effect of skin photosensitization due to poor target specificity.144 In our studies, CLPs formulated with dimyristoyl-sn-glycero-phosphatidylcholine (DMPC) and the cationic gemini surfactant (S, S)-2,3-dimethoxy-1,4-bis(N-hexadecyl-N, N-dimethylammonio)butane bromide 1a (DMPC/1a) was developed to deliver the photosensitizer m-tetrahydroxyphenylchlorin (m-THPC) to human colon adenocarcinoma and glioblastoma cells.131,145 The presence of gemini surfactant strongly influenced the interaction of liposomes with the cell membrane and the delivery efficacy of CLPs. It significantly increased the cell uptake of m-THPC and the cytotoxic effect of the photosensitizer after the irradiation, when compared with the related pharmaceutical formulation Foscan.145 A subsequent study demonstrated that the stereochemistry of the gemini spacer strongly influenced the physicochemistry of liposomal formulations and the final intracellular fate of the loaded drug.146
Stimuli-responsive liposomes
Conventional and long-circulating liposomes may present a slow release of the loaded drug or may be unable to fuse with the endosome after internalization. To overcome these problems, stimuli-responsive liposomes have been developed. Lipids in these liposomes generally include a triggerable component that is responsible for gating the stability and/or permeability of the lipid bilayer. Stimuli-responsive liposomes are capable of reacting when triggered by stimuli from target tissues (pH, redox potential) or applied from outside the organism (hyperthermia, ultrasound, and [electro]magnetic field).147,148 pH-sensitive, redox potential-sensitive, temperature-sensitive, magnetic field-sensitive, and ultrasound-sensitive liposomes have been produced.
The development of pH-sensitive liposomes was planned after the consideration that some pathological tissues, including tumors or areas of inflammation and infection, display an acidic environment compared with normal tissues.149 A pH-sensitive liposome is generally stable at physiological pH, but can be subjected to destabilization and acquire fusogenic properties under acid conditions, thus leading to the release of its aqueous contents.150 To achieve the pH-sensitive release of liposome content, liposomes are formulated with pH-sensitive components. After being endocytosed in an intact form, these fuse with the endovacuolar membrane as a result of the lower pH inside the endosome, and release their contents into the cytoplasm. Long-circulating PEGylated pH-sensitive liposomes, despite having decreased pH sensitivity, still effectively deliver their contents into the cytoplasm.151
Redox potential-sensitive liposomes take advantage of the high redox-potential difference that exists between the reducing intracellular space and the oxidizing extracellular space. Redox potential-sensitive liposomes release their content inside cells when disulfide bonds present in lipids or other components are reduced by glutathione to thiol groups. Consequently, the integrity of the liposome structure, which is maintained under normal conditions by disulfide bonds, is compromised, and the entrapped cargo can be released. Endogenous triggering of liposomal payload release by overexpressed enzyme activity in affected tissues offers the possibility of active and site-specific release. Redox-triggered content release from liposomes was reported when applying liposomes made from quinone-DOPE (Q-DOPE) lipids. Complete payload release occurs upon their redox activation when the quinone head group possesses a “trimethyl-locked” quinone redox switch, attached to the N-terminus of DOPE lipids, that undergoes a cleavage event upon two-electron reduction.152 The authors expect that Q-DOPE liposomes and their variants will be important in treating diseases with associated tissues that overexpress quinone reductases, such as cancers and inflammatory diseases, because the quinone redox switch is a known substrate for this group of reductase.152
Thermosensitive liposomes have been widely investigated since 1978.153 In the last decade, there has been increased interest in delivery mediated by temperature-sensitive liposomes, in part due to advances in image-guided hyperthermia applicators. Temperature-sensitive liposomes in combination with heating the target region can selectively enhance the bioavailability of the drug locally while minimizing systemic exposure. Temperature-sensitive liposomes promptly discharge their cargo upon heating (within seconds to minutes), while at body temperature the payload is somewhat stably encapsulated.154–156 Temperature-sensitive liposomes release their encapsulated drugs at the melting-phase transition temperature (Tm) of the lipid bilayer. At this Tm, the lipid membrane changes its permeability because of the transition from the gel to the liquid crystalline phase.157,158 Temperature-sensitive liposomes frequently include dipalmitoylphosphatidylcholine as the key component, because the gel-to-liquid crystalline phase transition occurs for these lipids at 41°C, as extensively reviewed in Kono.159
Temperature-sensitive liposomes have been successfully applied in both preclinical and clinical studies in combination with heat-based thermal therapies, including radiofrequency ablation, ultrasound hyperthermia, and microwave hyperthermia.160–163
A formulation based on these thermosensitive liposomes took the brand name ThermoDox® and was further developed by Celsion Corporation. ThermoDox liposomes can be triggered to release their payload by any heat-based treatment, such as radiofrequency thermal ablation, microwave hyperthermia, and high-intensity-focused ultrasound.157
A complete regression of local cancer using temperature-sensitive liposomes combined with ultrasound-mediated hyperthermia was recently reported by Kheirolomoom et al.164 These authors employed temperature-sensitive liposomes containing lysolipid, loaded with a pH-sensitive complex formed by Dox and copper, ie, CuDox. The complex remains associated at neutral pH, but dissociates to give free Dox in lower-pH environments. The resulting liposomes were injected intravenously into a syngeneic murine breast cancer model. Successively, the intravascular release of the drug was triggered by ultrasound. The entire tumor was insonified for 5 minutes prior to drug administration and 20 minutes after drug injection. A single-dose administration of CuDox lysolipid-containing temperature-sensitive liposomes (LTSLs) combined with insonation suppressed tumor growth. Moreover, after twice-weekly treatment over a period of 28 days, a complete response was achieved in which the NDL tumor cells and the tumor interstitium could no longer be detected. All mice treated with ultrasound combined with CuDox LTSLs survived, and the tumor was undetectable at 8 months posttreatment. Iron- and copper-laden macrophages were observed at early time points following treatment with this temperature-sensitive formulation. Systemic toxicity indicators, such as cardiac hypertrophy, leukopenia, and weight and hair loss, were not detected with CuDox LTSLs after the 28-day therapy.164
Liposomes in theranostics
Nanotechnology gives the opportunity to assemble therapeutic and diagnostic agents as a single theranostic platform, ie, a molecular platform that simultaneously integrates diagnosis and therapy.165–168 The main goal is to diagnose and treat the diseases at their earliest stage. A theranostic platform is multifunctional in nature, able to detect and specifically deliver therapeutic agents to the diseased cells with the help of targeting ligands and biomarkers.168–171
Liposomes are a valid platform for theranostic nanomedicine, owing to their size, hydrophobic and hydrophilic character, biocompatibility, biodegradability, low toxicity, and immunogenicity. In 2007, the concept of liposome–nanoparticle hybrids was presented by Al-Jamal and Kostarelos as a general methodology to be used as a platform for the delivery of novel nanoparticles. Such hybrid constructs present great opportunities to engineer theranostic nanoscale delivery systems.172 Liposome–nanoparticle hybrids can be designed by embedding, encapsulation, or conjugation of nanoparticles onto various types of liposomes. The theranostic potential of such hybrids is illustrated in Al-Jamal and Kostarelos.173 The authors described, in particular, Dox-loaded, lipid bilayer-embedded quantum-dot vesicle hybrids capable of chemotherapy (cytotoxic activity of Dox) and optical imaging (embedded quantum dots).
For imaging purposes, nanosize diagnostic agents can be entrapped within the theranostic liposomes, and the therapeutic agent can be either encapsulated in the core or embedded in the lipophilic bilayer shell.173–178 For example, for magnetic resonance imaging, superparamagnetic iron oxides can either be coated with a lipid layer (small magnetoliposomes [MLs]) or several superparamagnetic iron oxides or gadolinium(III) chelates can be entrapped into the aqueous core of liposomes (large MLs). These large MLs provide additional cargo space for drug encapsulation, into either the core or the lipid membrane, rendering MLs theranostic agents.179,180 Multimodal imaging properties have been obtained by loading quantum dots or fluorescent dyes, such as calcein, into the lipid membrane of liposomes. For example, recently, Li et al constructed a multifunctional liposome containing gadolinium-DOTA lipids for magnetic resonance imaging, a lipidized near-infrared dye for near-infrared fluorescence imaging, Dox loading for therapeutic activity, and radiolabeling with 99mTc and 64Cu for single-photon emission computed tomography and positron-emission tomography imaging. These liposomes were applied in vivo to a squamous cell carcinoma of head and neck tumor xenograft in nude rats after intratumoral injection.181
Muthu et al prepared tocopheryl polyethylene glycol 1000 succinate-coated theranostic liposomes containing Dox and quantum dots with and without targeting moieties.182 Folic acid was used as the targeting probe to target folate receptors overexpressing MCF-7 breast cancer cell lines. Along similar lines, Wen et al developed quantum dots and apomorphine-incorporated theranostic liposomes to eliminate uptake by the liver and to enhance brain targeting.183 In their most recent work, Wen et al prepared theranostic liposomes loaded with quantum dots, camptothecin, and irinotecan for simultaneous bioimaging and drug delivery.184
Smith et al used heat-sensitive liposomes with modified HER2 affisomes (HER2+ affisomes).185 Affibody® affinity ligands are innovative protein-engineering technologies. The liposomes were either loaded with rhodamine phosphatidylethanolamine and calcein or with Dox. HER2+ cells and HER2– cells were both incubated with the liposomes.186
As stated by Svenson, even if liposomes are an ideal platform for theranostics, several issues remain to be addressed. Nanocarrier polydispersity is an issue for clinical translation and regulatory approval of nanocarriers. Random entrapment and surface conjugation of diagnostic and therapeutic agents into liposomes or other nanocarriers is a tempting and easily achievable approach. However, the polydispersity of the resulting nanocarriers and questionable reproducibility of the approach will create high hurdles for clinical translation and regulatory approval. Adding active targeting ligands to a nanocarrier not only adds at least another step to its production but also adds to polydispersity, complicates regulatory evaluation and approval, increases the costs of goods, and can have negative biological outcomes because of multivalency binding and enhanced recognition by the RES with reduced circulation time.187
Liposomes on the market or in clinical trials
Doxil (100 nm) was the first pharmaceutical product in a PEGylated liposomal formulation that received FDA approval (1995) for the treatment of chemotherapy-refractory Kaposi’s sarcoma in AIDS patients, and more recently for recurrent epithelial ovarian cancer.188 Currently, several liposome-based drugs are approved for clinical practice; many others are still in the various stages of clinical trials (Tables 1 and 2).189 Most liposomal drug formulations, such as Doxil and Myocet® (190 nm), have been approved for intravenous application.190 Generally, liposome products have a longer circulating half-life when compared with respective unencapsulated drugs. The time of circulation in the blood depends on size, charge density, fluidity of the lipid bilayer, or coating by PEG. For the delivery of surface antigens derived from the influenza virus (Inflexal® V) or hepatitis A (Epaxal®), intramuscular delivery has been approved. Inflexal V and Epaxal are both vaccine products: it has been reported that cell-mediated and humoral immune response is potentiated when viral membrane proteins or peptide antigens are incorporated into liposomes.191,192 For the administration of liposomal vaccines, oral delivery has also been considered; however, this is more problematic, due to the potential for liposome breakdown following exposure to bile salts. Therefore, injections remain the best route of administration for therapeutic peptides.193 Since the first liposomal pharmaceutical product, Doxil, liposomes have been widely utilized as carriers for various therapeutic agents in clinical trials (extensively reviewed in Chang and Yeh).189 Until now, virosomes (Epaxal and Inflexal V), CLPs (EndoTAG1-1®), PEGylated liposomes (Doxil and Lipodox), and temperature-sensitive liposomes (ThermoDox) have been considered for clinical use. In contrast with the liposome-based drugs on the market, liposome-based drugs in clinical trials display a large variety of loaded drugs (eg, cisplatin, BLP25 lipopeptide, Grb2 antisense oligodeoxynucleotide, bacteriophage T4 endonuclease 5) for several therapeutic applications (from topical delivery systems to portable aerosol delivery systems). As previously described, PEGylation may extend the blood-circulation time of liposomes, modify drug distribution in the body, and hence reduce related adverse effects (eg, cardiotoxicity). However, a significant incidence of stomatitis in clinical trials contemplating the use of PEGylated liposomes (Doxil and Lipodox) has been reported.189
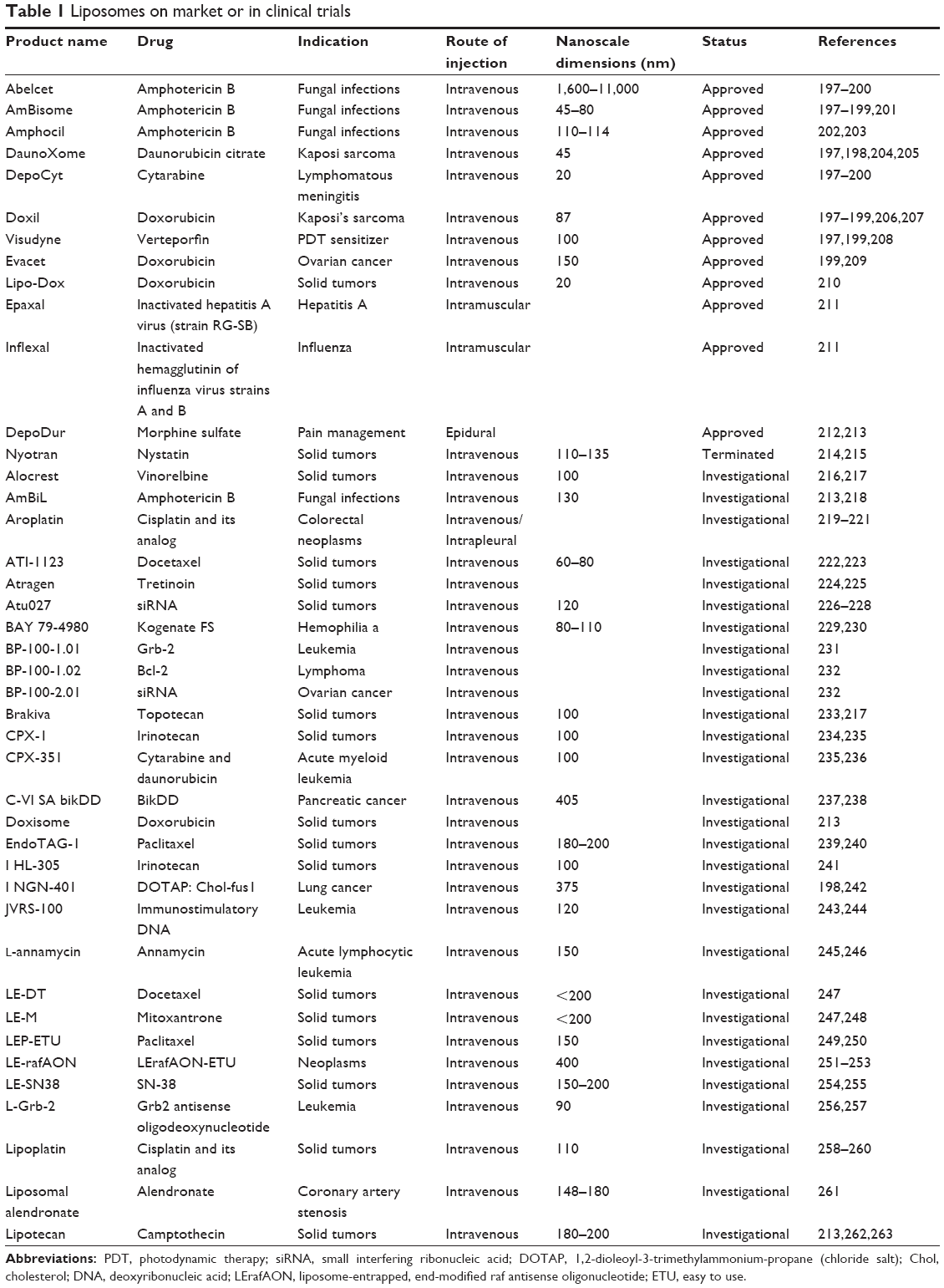 | Table 1 Liposomes on market or in clinical trials |
 | Table 2 Liposomes on market or in clinical trials |
In addition, it has been observed that some of the new-generation liposomes demonstrated only comparable or even poor therapeutic efficiency when they were compared with relative free drugs or conventional vesicles in clinical trials. In comparison with Doxil, ThermoDox showed a significant decrease of Dox accumulation in mouse tumors at 24 hours after administration.162,189 EndoTAG-1 plus gemcitabine showed beneficial survival and efficacy in a randomized controlled Phase II clinical trial in advanced pancreatic cancer and triple receptor-negative breast cancer.194 In addition, a positive efficacy trend of the EndoTAG-1 combination therapy for triple receptor-negative breast cancer was reported by MediGene (http://www.medigene.com). SPI-77, the first liposomes loaded with cisplatin, showed limited clinical efficacy in a Phase II clinical trial of advanced non-small-cell lung cancer. On the other hand, the same formulation induced increased cisplatin tumor accumulation in preclinical models.195 Similarly to SPI-77, a Phase II study demonstrated that liposomal annamycin had no detectable antitumor activity in the treatment of Dox-resistant breast cancer.196
Conclusion
Due to their biocompatibility and biodegradability, liposomes were the first drug-delivery system approved for clinical purposes. Despite their long history in the field of research and development, there are still unresolved problems that limit their ultimate therapeutic outcome.
The advantages of liposomal-based drugs should be greater solubility of the cargo, increased half-life, selective delivery to the site of action, and the ability to overcome resistance against chemotherapeutics. The consequential pharmacokinetic changes could result in a reduction of adverse effects and an improvement in the therapeutic index of the encapsulated drugs. To reach these therapeutic outcomes, liposomes were firstly modified (PEGylated) in order to solve pharmacological challenges, such as destabilization by blood lipoproteins, uptake by RES, and rapid clearance from blood circulation.
PEGylated liposomes have been approved and are on the market, but their clinical success is hampered by some limitations, such as a lack of specificity. To increase their target-specificity and the amount of released therapeutic agent at the site of disease, stimuli-sensitive liposomes and multifunctional carriers for theranostics have been designed.
However, the transfer to large-scale production and to the clinic of these liposomal formulations suffers drawbacks, such as instability, polydispersity, toxicity at repeated administration, and capability of inducing immunostimulation and complement activation. The precise control of liposome size and distribution can be optimized by new preparation methods, such us microfluidic-based methods and microfluidic remote loading (rapid single-step liposomal drug preparation). For pharmaceutical manufacturing and for quality-assurance assays, we can foresee progress in automation and control of processes of conventional liposomes as well as multifunctional liposomes. OECTs have been recently proposed for real-time monitoring of liposome-based structures.
The increase of complexity of liposomal formulation even more needs accurate in vitro and in vivo preclinical studies before transfer to the clinic. The analysis of physicochemical characteristics, toxicity, hematocompatibility, delivery, and therapeutic efficiency is mandatory. Dialogue between scientists, clinicians, and industry is indispensable in the design phase of new liposomal formulations to increase the success rate for liposomes as nanomedical formulations.
Acknowledgments
We would like to thank Giuseppe Formisano for his technical assistance with the freeze-fracturing technique and Cosimo Curianò for the graphics (Figures 1–3).
Disclosure
The authors report no conflicts of interest in this work.
References
Bangham AD, Horne RW. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J Mol Biol. 1964;8:660–668. | ||
Bangham AD, Hill MW, Miller NG. Preparation and use of liposomes as models of biological membranes. In: Korn ED, editor. Methods in Membrane Biology. Vol 1. New York: Plenum; 1974:1–68. | ||
Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. The big picture on nanomedicine: the state of investigational and approved nanomedicine products. Nanomedicine. 2013;9:1–14. | ||
Felice B, Prabhakaran MP, Rodríguez AP, Ramakrishna S. Drug delivery vehicles on a nano-engineering perspective. Mater Sci Eng C Mater Biol Appl. 2014;41:178–195. | ||
Fanciullino R, Ciccolini J. Liposome-encapsulated anticancer drugs: still waiting for the magic bullet? Curr Med Chem. 2009;16:4361–4373. | ||
Euliss LE, DuPont JA, Gratton S, DeSimone J. Imparting size, shape, and composition control of materials for nanomedicine. Chem Soc Rev. 2006;35:1095–1104. | ||
Papahadjopoulos D, Kimelberg HK. Phospholipid vesicles (liposomes) as models for biological membranes: their properties and interactions with cholesterol and proteins. In: Progress in Surface Science. Vol. Oxford: Pergamon; 1973:141–149. | ||
Frolov VA, Shnyrova AV, Zimmerberg J. Lipid polymorphisms and membrane shape. Cold Spring Harb Perspect Biol. 2011;3:a004747. | ||
Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1:297–315. | ||
Betageri GV, Parsons DL. Drug encapsulation and release from multilamellar and unilamellar liposomes. Int J Pharm. 1992; 81:235–241. | ||
Niven RW, Speer M, Schreier H. Nebulization of liposomes. II. The effects of size and modeling of solute release profiles. Pharm Res. 1991; 8:217–221. | ||
Schechter E. Aspects structuraux et fonctionnels. In: Schechter E, Rossignol B, editors. Biochimie et Biophysique des Membranes. Paris: Dunod; 2002. | ||
Hosta-Rigau L, Zhang Y, Teo BM, Postma A, Städler B. Cholesterol – a biological compound as a building block in bionanotechnology. Nanoscale. 2013;5:89–109. | ||
Bitounis D, Fanciullino R, Iliadis A, Ciccolini J. Optimizing druggability through liposomal formulations: new approaches to an old concept. ISRN Pharm. 2012;2012:738432. | ||
Milla P, Dosio F, Cattel L. PEGylation of proteins and liposomes: a powerful and flexible strategy to improve the drug delivery. Curr Drug Metab. 2012;13:105–109. | ||
Gabizon A, Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumors. Proc Natl Acad Sci U S A. 1988;85:6949–6953. | ||
Allen TM. Long-circulating (Stealth) liposomes: therapeutic applications. In: Puisieux F, Couvreur P, Delattre J, Devissaguet JP, editors. Liposomes: New Systems and New Trends in Their Applications. Paris: Editions de Santé; 1995:125–155. | ||
Gómez-Hens A, Fernández-Romero JM. Analytical methods for the control of liposomal delivery systems. Trends Analyt Chem. 2006;25: 167–178. | ||
Mozafari MR, Johnson C, Hatziantoniou S, Demetzos C. Nanoliposomes and their applications in food nanotechnology. J Liposome Res. 2008;18:309–327. | ||
Wagner A, Vorauer-Uhl K. Liposome technology for industrial purposes. J Drug Deliv. 2011;2011:591325. | ||
Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965; 13:238–252. | ||
New RC. Preparation of liposomes. In: New RC, editor. Liposomes: A Practical Approach. New York: Oxford University Press; 1990. | ||
Szoka F, Papahadjopoulos D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci U S A. 1978;75:4194–4198. | ||
Batzri S, Korn ED. Single bilayer liposomes prepared without sonication. Biochim Biophys Acta. 1973;298:1015–1019. | ||
Deamer D, Bangham AD. Large volume liposomes by an ether vaporization method. Biochim Biophys Acta. 1976;443:629–634. | ||
Lasch J, Weissig V, Brandl M. Preparation of liposomes. In: Torchilin V, Weissig V, editors. Liposomes: A Practical Approach. New York: Oxford University Press; 2003. | ||
Brunner J, Skrabal P, Hausser H. Single bilayer vesicles prepared without sonication physicochemical properties. Biochim Biophys Acta. 1976;455:322–331. | ||
Kirby C, Gregoriadis G. Dehydration-rehydration vesicles: a simple method for high-yield drug entrapment in liposomes. Nat Biotechnol. 1984;2:979–984. | ||
Woodbury DJ, Richardson ES, Grigg AW, Welling RD, Knudson BH. Reducing liposome size with ultrasound: bimodal size distributions. J Liposome Res. 2006;16:57–80. | ||
Hope MJ, Bally MB, Webb G, Cullis PR. Production of large unilamellar vesicles by a rapid extrusion procedure – characterization of size distribution, trapped volume and ability to maintain a membrane-potential. Biochim Biophys Acta. 1985;812:55–65. | ||
Berger N, Sachse A, Bender J, Schubert R, Brandl M. Filter extrusion of liposomes using different devices: comparison of liposome size, encapsulation efficiency, and process characteristics. Int J Pharm. 2001;223:55–68. | ||
MacDonald RC, MacDonald RI, Menco BP, Takeshita K, Subbarao NK, Hu LR. Small volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim Biophys Acta. 1991;1061:297–303. | ||
Bachmann D, Brandl M, Gregoriadis G. Preparation of liposomes using a mini-lab 8.30 H high-pressure homogenizer. Int J Pharm. 1993;91: 69–74. | ||
Laouini A, Jaafar-Maalej C, Limayem-Blouza I, Sfar S, Charcosset C, Fessi H. Preparation, characterization and applications of liposomes: state of the art. J Colloid Sci Biotechnol. 2012;1:147–168. | ||
Stone HA, Stroock AD, Adjari A. Engineering flows in small devices: microfluidics toward a lab-on-a-chip. Annu Rev Fluid Mech. 2004;36: 381–411. | ||
Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368–373. | ||
Yu B, Lee RJ, Lee LJ. Microfluidic methods for production of liposomes. Methods Enzymol. 2009;465:129–141. | ||
Hood RR, Vreeland WN, DeVoe DL. Microfluidic remote loading for rapid single-step liposomal drug preparation. Lab Chip. 2014;14: 3359–3367. | ||
Tarabella G, Balducci AG, Coppedè N, et al. Liposomes sensing and monitoring by organic electrochemical transistors integrated in microfluidics. Biochim Biophys Acta. 2013;1830:4374–4380. | ||
Lasic DD. Magnetic resonance methods in the studies of liposomes. Bull Magn Reson. 1991;13:3–13. | ||
Ostrovsky N. Liposome size measurements by photon correlation spectroscopy. Chem Phys Lipids. 1993;64:45–56. | ||
Palmer AF, Wingert P, Nickels J. Atomic force microscopy and light scattering of small unilamellar actin-containing liposomes. Biophys J. 2003;85:1233–1247. | ||
Grabielle-Madelmont C, Lesieur S, Ollivon M. Characterization of loaded liposomes by size exclusion chromatography. J Biochem Biophys Methods. 2003;56:189–217. | ||
Moon MH, Giddings JC, Pharm J. Size distribution of liposomes by flow field-flow fractionation. J Pharm Biomed Anal. 1993;11:911–920. | ||
Ruozi B, Belletti D, Tombesi A, et al. AFM, ESEM, TEM, and CLSM in liposomal characterization: a comparative study. Int J Nanomedicine. 2011;6:557–563. | ||
Frederik PM, Hubert DH. Cryoelectron microscopy of liposomes. Methods Enzymol. 2005;391:431–448. | ||
Jass J, Tjärnhage T, Puu G. Atomic force microscopy imaging of liposomes. Methods Enzymol. 1993;367:199–213. | ||
Ruozi B, Tosi G, Forni F, et al. Atomic force microscopy and photon correlation spectroscopy: two techniques for rapid characterization of liposomes. Eur J Pharm Sci. 2005;25:81–89. | ||
Ruysschaert T, Marque A, Duteyrat JL, Lesieur S, Winterhalter M, Fournier D. Liposome retention in size exclusion chromatography. BMC Biotechnol. 2005;10:5–11. | ||
Korgel BA, Van Zanten JH, Monbouquette HG. Vesicle size distributions measured by flow field-flow fractionation coupled with multiangle light scattering. Biophys J. 1998;74:3264–3272. | ||
Mohan A, Narayanan S, Sethuraman S, Krishnan UM. Novel resveratrol and 5-fluorouracil coencapsulated in pegylated nanoliposomes improve chemotherapeutic efficacy of combination against head and neck squamous cell carcinoma. Biomed Res Int. 2014;2014: 424239. | ||
Storm G, Roerdink FH, Steerenberg PA, de Jong WH, Crommelin DJ. Influence of lipid composition on the antitumor activity exerted by doxorubicin-containing liposomes in a rat solid tumor model. Cancer Res. 1987;47:3366–3372. | ||
McIntosh TJ. The effect of cholesterol on the structure of phosphatidylcholine bilayers. Biochim Biophys Acta. 1978;513:43–58. | ||
Cullis PR, Hope MJ. The bilayer stabilizing role of sphingomyelin in the presence of cholesterol: a 31P NMR study. Biochim Biophys Acta. 1980;597:533–542. | ||
Defrise-Quertain F, Chatelain P, Delmelle M, et al. Model studies for drug entrapment and liposome stability. In: Gregoriadis G, editor. Liposome Technology. Boca Raton (FL): CRC; 1984:1–17. | ||
Allen TM, Mehra T, Hansen C, Chin YC. Stealth liposomes: an improved sustained release system for 1-beta-D-arabinofuranosylcytosine. Cancer Res. 1992;52:2431–2439. | ||
Fahr A, van Hoogevest P, May S, Bergstrand N, Leigh ML. Transfer of lipophilic drugs between liposomal membranes and biological interfaces: consequences for drug delivery. Eur J Pharm Sci. 2005;26: 251–265. | ||
Mayer LD, Bally MB, Cullis PR. Uptake of adriamycin into large unilamellar vesicles in response to a pH gradient. Biochim Biophys Acta. 1986;857:123–126. | ||
Forssen EA, Malé-Brune R, Adler-Moore JP, et al. Fluorescence imaging studies for the disposition of daunorubicin liposomes (DaunoXome) within tumor tissue. Cancer Res. 1996;56:2066–2075. | ||
Johnston MJ, Edwards K, Karlsson G, et al. Influence of drug-to-lipid ratio on drug release properties and liposome integrity in liposomal doxorubicin formulations. J Liposome Res. 2008;18:145–157. | ||
Drummond DC, Noble CO, Guo Z, Hong K, Park JW, Kirpotin DB. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66:3271–3277. | ||
Zhigaltsev IV, Winters G, Srinivasulu M, et al. Development of a weak-base docetaxel derivative that can be loaded into lipid nanoparticles. J Control Release. 2010;144:332–340. | ||
Zamboni WC. Concept and clinical evaluation of carrier mediated anticancer agents. Oncologist. 2008;13:248–260. | ||
Lasic DD. Novel applications of liposomes. Trends Biotechnol. 1998;16:307–321. | ||
Silverman JA, Reynolds L, Deitcher SR. Pharmacokinetics and pharmacodynamics of vincristine sulfate liposome injection (VSLI) in adults with acute lymphoblastic leukemia. J Clin Pharmacol. 2013;53:1139–1145. | ||
Moen MD, Lyseng-Williamson KA, Scott LJ. Liposomal amphotericin B: a review of its use as empirical therapy in febrile neutropenia and in the treatment of invasive fungal infections. Drugs. 2009;69:361–392. | ||
Nasti TH, Khan MA, Owais M. Enhanced efficacy of pH sensitive nystatin liposomes against Cryptococcus neoformans in murine model. J Antimicrob Chemother. 2006;57:349–352. | ||
Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51:691–743. | ||
Lao J, Madani J, Puértolas T, et al. Liposomal doxorubicin in the treatment of breast cancer patients: a review. J Drug Deliv. 2013;2013:456409. | ||
Rafiyath SM, Rasul M, Lee B, Wei G, Lamba G, Liu D. Comparison of safety and toxicity of liposomal doxorubicin vs. conventional anthracyclines: a meta-analysis. Exp Hematol Oncol. 2012;1:10. | ||
Pisano C, Cecere SC, Di Napoli M, et al. Clinical trials with pegylated liposomal doxorubicin in the treatment of ovarian cancer. J Drug Deliv. 2013;2013:898146. | ||
Gordon AN, Granai CO, Rose PG, et al. Phase II study of liposomal doxorubicin in platinum- and paclitaxel-refractory epithelial ovarian cancer. J Clin Oncol. 2000;18:3093–3100. | ||
Katsumata N, Fujiwara Y, Kamura T, et al. Phase II clinical trial of pegylated liposomal doxorubicin (JNS002) in Japanese patients with mullerian carcinoma (epithelial ovarian carcinoma, primary carcinoma of fallopian tube, peritoneal carcinoma) having a therapeutic history of platinum-based chemotherapy: a phase II study of the Japanese Gynecologic Oncology Group. Jpn J Clin Oncol. 2008;38:777–785. | ||
Markman M, Kennedy A, Webster K, Peterson G, Kulp B, Belinson J. Phase 2 trial of liposomal doxorubicin (40 mg/m2) in platinum/paclitaxel-refractory ovarian and fallopian tube cancers and primary carcinoma of the peritoneum. Gynecol Oncol. 2000;78:369–372. | ||
Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475–1480. | ||
Alberts DS. Muggia FM, Carmichael J, et al. Efficacy and safety of liposomal anthracyclines in phase I/II clinical trials. Semin Oncol. 2004;31:53–90. | ||
Gabizon AA. Liposomal anthracyclines. Hematol Oncol Clin North Am. 1994;8:431–450. | ||
Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–717. | ||
Batist G, Ramakrishnan G, Rao CS, et al. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J Clin Oncol. 2001;19:1444–1454. | ||
O’Brien ME, Wigler N, Inbar M, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol. 2004;15:440–449. | ||
Gabizon AA, Lyass O, Berry GJ, Wildgust M. Cardiac safety of pegylated liposomal doxorubicin (Doxil/Caelyx) demonstrated by endomyocardial biopsy in patients with advanced malignancies. Cancer Invest. 2004;22:663–669. | ||
Mustafa MH. Decreased risk of cardiotoxicity with long-term use of Doxil/Caelyx at high lifetime cumulative doses in patients with AIDS-related Kaposi/Es sarcoma (KS). Poster presented at: ASCO Annual Meeting 2001; May 12–15, 2001; San Francisco, CA. | ||
Andreopoulou E, Gaiotti D, Kim E, et al. Pegylated liposomal doxorubicin HCL (PLD; Caelyx/Doxil): experience with long-term maintenance in responding patients with recurrent epithelial ovarian cancer. Ann Oncol. 2007;18:716–721. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131–135. | ||
Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–1380. | ||
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. | ||
Brown S, Khan DR. The treatment of breast cancer using liposome technology. J Drug Deliv. 2012;2012:212965. | ||
Roberts WG, Delaat J, Nagane M, Huang S, Cavenee WK, Palade GE. Host microvasculature influence on tumor vascular morphology and endothelial gene expression. Am J Pathol. 1998;153:1239–1248. | ||
O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209:171–176. | ||
Woodle MC, Matthay KK, Newman MS, et al. Versatility in lipid compositions showing prolonged circulation with sterically stabilized liposomes. Biochim Biophys Acta. 1992;1105:193–200. | ||
Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of PEGylated liposomal doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42:419–436. | ||
Ishida T, Harashima H, Kiwada H. Liposome clearance. Biosci Rep. 2002;22:197–224. | ||
Semple SC, Harasym TO, Clow KA, Ansell SM, Klimuk SK, Hope MJ. Immunogenicity and rapid blood clearance of liposomes containing polyethylene glycol-lipid conjugates and nucleic acid. J Pharmacol Exp Ther. 2005;312:1020–1026. | ||
Gabizon AA, Shmeeda H, Zalipsky S. Pros and cons of the liposome platform in cancer drug targeting. J Liposome Res. 2006;16:175–183. | ||
Daemen T, Hofstede G, Ten Kate MT, Bakker-Woudenberg IA, Scherphof GL. Liposomal doxorubicin-induced toxicity: depletion and impairment of phagocytic activity of liver macrophages. Int J Cancer. 1995;61:716–721. | ||
Juliano RL, Stamp D. Pharmacokinetics of liposome-encapsulated anti-tumor drugs. Studies with vinblastine, actinomycin D, cytosine arabinoside, and daunomycin. Biochem Pharmacol. 1978;27: 21–27. | ||
Gabizon AA. Stealth liposomes and tumor targeting: one step further in the quest for the magic bullet. Clin Cancer Res. 2001;7:223–225. | ||
Bedu-Addo FK, Tang P, Xu Y, Huang L. Effects of polyethyleneglycol chain length and phospholipid acyl chain composition on the interaction of polyethyleneglycol-phospholipid conjugates with phospholipid: implications in liposomal drug delivery. Pharm Res. 1996;13:710–717. | ||
Photos PJ, Bacakova L, Discher B, Bates FS, Discher DE. Polymer vesicles in vivo: correlations with PEG molecular weight. J Control Release. 2003;90:323–334. | ||
Harris JM, editor. Poly(Ethylene Glycol) Chemistry: Biotechnical and Biomedical Applications. New York: Plenum;1992. | ||
Gref R, Domb A, Quellec P, et al. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv Drug Deliv Rev. 1995;16:215–233. | ||
Moghimi SM, Hedeman H, Muir IS, Illum L, Davis SS. An investigation of the filtration capacity and the fate of large filtered sterically-stabilized microspheres in rat spleen. Biochim Biophys Acta. 1993;1157:233–240. | ||
Harris JM, Martin NE, Modi M. PEGylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet. 2001;40:539–551. | ||
Wu H, Ramanathan RK, Zamboni BA, et al. Population pharmacokinetics of PEGylated liposomal CKD-602 (S-CKD602) in patients with advanced malignancies. J Clin Pharmacol. 2011;52:180–194. | ||
Heyes J, Hall K, Tailor V, Lenz R, MacLachlan I. Synthesis and characterization of novel poly(ethylene glycol)-lipid conjugates suitable for use in drug delivery. J Control Release. 2006;112:280–290. | ||
Allen TM. Liposomes. Opportunities in drug delivery. Drugs. 1997;54:8–14. | ||
Gabizon AA. PEGylated liposomal doxorubicin: metamorphosis of an old drug into a new form of chemotherapy. Cancer Invest. 2001;19:424–436. | ||
Dawidczyk CM, Kim C, Park JH, et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–144. | ||
Andresen TL, Jensen SS, Jørgensen K. Advanced strategies in liposomal cancer therapy: problems and prospects of active and tumor specific drug release. Prog Lipid Res. 2005;44:68–97. | ||
Barenholz Y. Liposome application: problems and prospects. Curr Opin Colloid Interface Sci. 2011;6:66–77. | ||
Bandak S, Goren D, Horowitz A, Tzemach D, Gabizon A. Pharmacological studies of cisplatin encapsulated in long-circulating liposomes in mouse tumor models. Anticancer Drugs. 1999;10:911–920. | ||
Asai T, Oku N. Angiogenic vessel-targeting DDS by liposomalized oligopeptides. Methods Mol Biol. 2010;605:335–347. | ||
Gabizon A, Tzemach D, Gorin J, et al. Improved therapeutic activity of folate-targeted liposomal doxorubicin in folate receptor-expressing tumor models. Cancer Chemother Pharmacol. 2010;66:43–52. | ||
Garg A, Tisdale AW, Haidari E, Kokkoli E. Targeting colon cancer cells using PEGylated liposomes modified with a fibronectin-mimetic peptide. Int J Pharm. 2009;366:201–210. | ||
Vingerhoeds MH1, Haisma HJ, van Muijen M, van de Rijt RB, Crommelin DJ, Storm G. A new application for liposomes in cancer therapy. Immunoliposomes bearing enzymes (immuno-enzymosomes) for site-specific activation of prodrugs. FEBS Lett. 1993;336:485–490. | ||
Noble GT, Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol. 2014;32:32–45. | ||
Fanciullino R, Giacometti S, Aubert C, et al. Development of stealth liposome formulation of 2′-deoxyinosine as 5-fluorouracil modulator: in vitro and in vivo study. Pharm Res. 2005;22:2051–2057. | ||
Senior JH, Trimble KR, Maskievicz R. Interaction of positively charged liposomes with blood: implications for their application in vivo. Biochim Biophys Acta. 1991;1070:173–179. | ||
Zhao W, Zhuang S, Qi XR. Comparative study of the in vitro and in vivo characteristics of cationic and neutral liposomes. Int J Nanomedicine. 2011;6:3087–3098. | ||
Harashima H, Matsuo H, Kiwada H. Identification of proteins mediating clearance of liposomes using a liver perfusion system. Adv Drug Delivery Rev. 1998;32:61–79. | ||
Miller CR, Bondurant B, McLean SD, McGovern KA, O’Brien DF. Liposome-cell interactions in vitro: effect of liposome surface charge on the binding and endocytosis of conventional and sterically stabilized liposomes. Biochemistry. 1998;37:12875–12883. | ||
Cullis PR, Chonn A, Semple SC. Interactions of liposome and lipid-based carrier systems with blood proteins: relation to clearance behavior in vivo. Adv Drug Delivery Rev. 1998;32:3–17. | ||
Campbell PI. Toxicity of some charged lipids used in liposome preparations. Cytobios. 1983;37:21–26. | ||
Allison AG, Gregoriadis G. Liposomes as immunological adjuvants. Nature. 1974;252:252. | ||
Nakanishi T, Kunisawa J, Hayashi A, et al. Positively charged liposome functions as an efficient immunoadjuvant in inducing cell-mediated immune response to soluble proteins. J Control Release. 1999;61: 233–240. | ||
Afrin F, Rajesh R, Anam K, Gopinath M, Pal S, Ali N. Characterization of Leishmania donovani antigens encapsulated in liposomes that induce protective immunity in BALB/c mice. Infect Immun. 2002;70:6697–6706. | ||
Badiee A, Jaafari MR, Khamesipour A, et al. The role of liposome charge on immune response generated in BALB/c mice immunized with recombinant major surface glycoprotein of Leishmania (rgp63). Exp Parasitol. 2009;121:362–369. | ||
González-Rodríguez ML, Rabasco AM. Charged liposomes as carriers to enhance the permeation through the skin. Expert Opin Drug Deliv. 2011;8:857–871. | ||
Ogiso T, Yamaguchi T, Iwaki M, Tanino T, Miyake Y. Effect of positively and negatively charged liposomes on skin permeation of drugs. Drug Target. 2001;9:49–59. | ||
Felgner PL, Gadek TR, Holm M, et al. Lipofection: a highly efficient lipid-mediated DNA transfection procedure. Proc Natl Acad Sci U S A. 1987;84:7413–7417. | ||
Bombelli C, Caracciolo G, Di Profio P, et al. Inclusion of a photosensitizer in liposomes formed by DMPC/gemini surfactant: correlation between physicochemical and biological features of the complexes. J Med Chem. 2005;48:4882–4891. | ||
Strieth S, Eichhorn ME, Sauer B, et al. Neovascular targeting chemotherapy: encapsulation of paclitaxel in cationic liposomes impairs functional tumor microvasculature. Int J Cancer. 2004;110:117–124. | ||
Wu J, Lee A, Lu Y, Lee RJ. Vascular targeting of doxorubicin using cationic liposomes. Int J Pharma. 2007;337:329–335. | ||
Campbell RB, Fukumura D, Brown EB, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62:6831–6836. | ||
Campbell RB, Ying B, Kuesters GM, Hemphill R. Fighting cancer: from the bench to bedside using second generation cationic liposomal therapeutics. J Pharm Sci. 2009;98:411–429. | ||
Schnyder A, Huwyler J. Drug transport to brain with targeted liposomes. NeuroRx. 2005;2:99–107. | ||
Krasnici S, Werner A, Eichhorn ME, et al. Effect of the surface charge of liposomes on their uptake by angiogenic tumor vessels. Int J Cancer. 2003;105:561–567. | ||
Stamatatos L, Leventis R, Zuckermann MJ, Silvius JR. Interactions of cationic lipid vesicles with negatively charged phospholipid vesicles and biological membranes. Biochemistry. 1988;27:3917–3925. | ||
Litzinger DC, Brown JM, Wala I, et al. Fate of cationic liposomes and their complex with oligonucleotide in vivo. Biochim Biophys Acta. 1996;1281:139–149. | ||
McLean JW, Fox EA, Baluk P, et al. Organ-specific endothelial cell uptake of cationic liposome-DNA complexes in mice. Am J Physiol. 1997;273:H387–H404. | ||
Dass CR. Improving anti-angiogenic therapy via selective delivery of cationic liposomes to tumour vasculature. Int J Pharm. 2003;267:1–12. | ||
Meyer O, Kirpotin D, Hong K, et al. Cationic liposomes coated with polyethylene glycol as carriers for oligonucleotides. J Biol Chem. 1998;273:15621–15627. | ||
Gursel I, Gursel M, Ishii KJ, Klinman DM. Sterically stabilized cationic liposomes improve the uptake and immunostimulatory activity of CpG oligonucleotides. J Immunol. 2001;167:3324–3328. | ||
Skupin-Mrugalska P, Piskorz J, Goslinski T, Mielcarek J, Konopka K, Düzgüneş N. Current status of liposomal porphyrinoid photosensitizers. Drug Discov Today. 2013;18:776–784. | ||
Molinari A, Bombelli C, Mannino S, et al. m-THPC-mediated photodynamic therapy of malignant gliomas: assessment of a new transfection strategy. Int J Cancer. 2007;121:1149–1155. | ||
Bombelli C, Stringaro A, Borocci S, et al. Efficiency of liposomes in the delivery of a photosensitizer controlled by the stereochemistry of a gemini surfactant component. Mol Pharm. 2010;7:130–137. | ||
Torchilin V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur J Pharm Biopharm. 2009;71:431–444. | ||
Perche F, Torchilin VP. Recent trends in multifunctional liposomal nanocarriers for enhanced tumor targeting. J Drug Deliv. 2013;2013:705265. | ||
Gullino PM, Grantham FH, Smith SH, Haggerty AC. Modifications of the acid-base status of the internal milieu of tumors. J Natl Cancer Inst. 1965;34:857–869. | ||
Simões S, Moreira JN, Fonseca C, Düzgüneş N, de Lima MC. On the formulation of pH-sensitive liposomes with long circulation times. Adv Drug Deliv Rev. 2004;56:947–965. | ||
Caldeira de Araújo Lopes S, Vinícius Melo Novais M, Salviano Teixeira C, et al. Preparation, physicochemical characterization, and cell viability evaluation of long-circulating and pH-sensitive liposomes containing ursolic acid. Biomed Res Int. 2013;2013:467147. | ||
Ong W, Yang Y, Cruciano AC, McCarley RL. Redox-triggered contents release from liposomes. J Am Chem Soc. 2008;130:14739–14744. | ||
Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Design of liposomes for enhanced local release of drugs by hyperthermia. Science. 1978;202:1290–1293. | ||
Needham D, Dewhirst MW. The development and testing of a new temperature-sensitive drug delivery system for the treatment of solid tumors. Adv Drug Deliv Rev. 2001;53:285–305. | ||
Li L, ten Hagen TL, Schipper D, et al. Triggered content release from optimized stealth thermosensitive liposomes using mild hyperthermia. J Control Release. 2010;143:274–279. | ||
Kong G, Anyarambhatla G, Petros WP, et al. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: importance of triggered drug release. Cancer Res. 2000;60:6950–6957. | ||
Thanou M, Gedroyc W. MRI-guided focused ultrasound as a new method of drug delivery. J Drug Deliv. 2013;2013:616197. | ||
Evans E, Needham D. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion, and colloidal interactions. J Phys Chem. 1987;91:4219–4228. | ||
Kono K. Thermosensitive polymer-modified liposomes. Adv Drug Deliv Rev. 2001;53:307–319. | ||
Gasselhuber A, Dreher MR, Negussie A, Wood BJ, Rattay F, Haemmerich D. Mathematical spatio-temporal model of drug delivery from low temperature sensitive liposomes during radiofrequency tumour ablation. Int J Hyperthermia. 2010;26:499–513. | ||
Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: a novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin Pharmacother. 2009;10:333–343. | ||
Dromi S, Frenkel V, Luk A, et al. Pulsed-high intensity focused ultrasound and low temperature-sensitive liposomes for enhanced targeted drug delivery and antitumor effect. Clin Cancer Res. 2007;13:2722–2727. | ||
Hauck ML, LaRue SM, Petros WP, et al. Phase I trial of doxorubicin-containing low temperature sensitive liposomes in spontaneous canine tumors. Clin Cancer Res. 2006;12:4004–4010. | ||
Kheirolomoom A, Lai CY, Tam SM, et al. Complete regression of local cancer using temperature-sensitive liposomes combined with ultrasound-mediated hyperthermia. J Control Release. 2013;172:266–273. | ||
Wang LS, Chuang MC, Ho JA. Nanotheranostics – a review of recent publications. Int J Nanomedicine. 2012;7:4679–4695. | ||
Sumer B, Gao J. Theranostic nanomedicine for cancer. Nanomedicine (Lond). 2008;3:37–140. | ||
Deveza L, Choi J, Yang F. Therapeutic angiogenesis for treating cardiovascular diseases. Theranostics. 2012;2:801–814. | ||
Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Adv Drug Deliv Rev. 2010;2:1052–1063. | ||
Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2:3–44. | ||
Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064–1079. | ||
Ye Y, Chen X. Integrin targeting for tumor optical imaging. Theranostics. 2011;1:102–126. | ||
Al-Jamal WT, Kostarelos K. Liposome-nanoparticle hybrids for multimodal diagnostic and therapeutic applications. Nanomedicine (Lond). 2007;2:85–98. | ||
Al-Jamal WT, Kostarelos K. Liposomes: from a clinically established drug delivery system to a nanoparticle platform for theranostic nanomedicine. Acc Chem Res. 2011;44:1094–1104. | ||
Nie Y, Ji L, Ding H, et al. Cholesterol derivatives based charged liposomes for doxorubicin delivery: preparation, in vitro and in vivo characterization. Theranostics. 2012;2:1092–1103. | ||
Leung SJ, Romanowski M. Light-activated content release from liposomes. Theranostics. 2012;2:1020–1036. | ||
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4:145–160. | ||
Papahadjopoulos D, Allen TM, Gabizon A, et al. Sterically stabilized liposomes – improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88:11460–11464. | ||
Al-Jamal WT, Al-Jamal KT, Tian B, et al. Lipid-quantum dot bilayer vesicles enhance tumor cell uptake and retention in vitro and in vivo. ACS Nano. 2008;2:408–418. | ||
Soenen SJ, Vande Velde G, Ketkar-Atre A, Himmelreich U, De Cuyper M. Magnetoliposomes as magnetic resonance imaging contrast agents. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2011;3:197-211. | ||
Kamaly N, Miller AD. Paramagnetic liposome nanoparticles for cellular and tumour imaging. Int J Mol Sci. 2010;11:1759-1776. | ||
Li S, Goins B, Zhang L, Bao A. Novel multifunctional theranostic liposome drug delivery system: construction, characterization, and multimodality MR, near-infrared fluorescent, and nuclear imaging. Bioconjug Chem. 2012;23:1322-1332. | ||
Muthu MS, Kulkarni SA, Raju A, Feng SS. Theranostic liposomes of TPGS coating for targeted co-delivery of docetaxel and quantum dots. Biomaterials. 2012;33:3494–3501. | ||
Wen CJ, Zhang LW, Al-Suwayeh SA, Yen TC, Fang JY. Theranostic liposomes loaded with quantum dots and apomorphine for brain targeting and bioimaging. Int J Nanomedicine. 2012;7:1599–1611. | ||
Wen CJ, Sung CT, Aljuffali IA, Huang YJ, Fang JY. Nanocomposite liposomes containing quantum dots and anticancer drugs for bioimaging and therapeutic delivery: a comparison of cationic, PEGylated and deformable liposomes. Nanotechnology. 2013;24:325101. | ||
Smith B, Lyakhov I, Loomis K, et al. Hyperthermia-triggered intracellular delivery of anticancer agent to HER2(+) cells by HER2-specific affibody (ZHER2-GS-Cys)-conjugated thermosensitive liposomes (HER2(+) affisomes). J Control Release. 2011;153:187–194. | ||
Löfblom J, Feldwisch J, Tolmachev V, Carlsson J, Ståhl S, Frejd FY. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584:2670–1680. | ||
Svenson S. Theranostics: are we there yet? Mol Pharm. 2013;10:848–856. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160:117–134. | ||
Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. | ||
Mross K, Niemann B, Massing U, et al. Pharmacokinetics of liposomal doxorubicin (TLC-D99; Myocet) in patients with solid tumors: an open-label, single-dose study. Cancer Chemother Pharmacol. 2004;54:514–524. | ||
Stegmann T, Morselt HW, Booy FP, van Breemen JF, Scherphof G, Wilschut J. Functional reconstitution of influenza virus envelopes. EMBO J. 1987;6:2651–2659. | ||
Glück R, Mischler R, Brantschen S, Just M, Althaus B, Cryz SJ Jr. Immunopotentiating reconstituted influenza virus virosome vaccine delivery system for immunization against hepatitis A. J Clin Invest. 1992;90:2491–2495. | ||
Shaji J, Patole V. Protein and peptide drug delivery: oral approaches. Indian J Pharm Sci. 2008;70:269–277. | ||
Löhr JM, Haas SL, Bechstein WO, et al. Cationic liposomal paclitaxel plus gemcitabine or gemcitabine alone in patients with advanced pancreatic cancer: a randomized controlled phase II trial. Ann Oncol. 2012;23:1214–1222. | ||
White SC, Lorigan P, Margison GP, et al. Phase II study of SPI-77 (sterically stabilised liposomal cisplatin) in advanced non-small-cell lung cancer. Br J Cancer. 2006;95:822–828. | ||
Booser DJ, Esteva FJ, Rivera E, et al. Phase II study of liposomal annamycin in the treatment of doxorubicin-resistant breast cancer. Cancer Chemother Pharmacol. 2002;50:6–8. | ||
Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211–1218. | ||
Bawa R. Nanoparticle-based therapeutics in humans: a survey. NLB. 2008;5:135–155. | ||
Faraji AH, Wipf P. Nanoparticles in cellular drug delivery. Bioorg Med Chem. 2009;17:2950–2962. | ||
Sigma-Tau Pharmaceuticals, Inc. – Producs (Website). Available from: http://www.sigmatau.com/products/abelcet_rx.asp. Accessed Sep 04, 2013. | ||
Ambisome (amphotericin B) liposome for injection (Website). Available from: http://www.ambisome.com/. Accessed Sep 03, 2013. | ||
Tollemar J, Ringden O. Lipid formulations of amphotericin B. Less toxicity but at what economic cost? Drug Saf. 1995;13:207–218. | ||
Samaritan Pharmaceuticals (Website). Available from: http://www.samaritanpharma.com/amphocil/. Accessed Sep 03, 2013. | ||
Griese N, Blaschke G, Boos J, Hempel G. Determination of free and liposome-associated daunorubicin and daunorubicinol in plasma by capillary electrophoresis. J Chromatogr A. 2002;979:379–388. | ||
DaunoXome (Website). Available from: http://daunoxome.com/. Accessed Sep 05, 2013. | ||
Bao A, Goins B, Klipper R, Negrete G, Phillips WT. Direct 99mTc labeling of pegylated liposomal doxorubicin (Doxil) for pharmacokinetic and non-invasive imaging studies. J Pharmacol Exp Ther. 2004;308:419–425. | ||
Doxil (Website). Available from: http://www.doxil.com/. Accessed Sep 05, 2013. | ||
Visudyne (Website). Available from: http://www.visudyne.com/. Accessed Sep 05, 2013. | ||
Muggia FM. Liposomal encapsulated anthracyclines: new therapeutic horizons. Curr Oncol Rep. 2001;3:156–162. | ||
TLC – Product (Website). Cited: 2011 Mar 31. Available from: http://www.tlcbio.com/en/product.html | ||
Crucell (Website). Available from: http://www.crucell.com/Products. Accessed Oct 2, 2013. | ||
Patil SD, Burgess DJ. Liposomes, design and manufacturing. In: Burgess DJ, editor. Injectable Dispersed Systems: Formulation, Processing and Performance (Drugs and The Pharmaceutical Sciences Series). New York: Marcel Dekker; 2005:249–303. | ||
Pacira Pharmaceuticals, INC – Produtcs (Website). Cited: 2013 Sep 3. Aivable from: http://www.pacira.com/products/depodur.php | ||
NCT00002742. Antifungal therapy for fever and neutropenia in patients receiving treatment for hematologic cancer (Clinical Trial). ClinicalTrials.gov. | ||
Moribe K, Maruyama K. Pharmaceutical design of the liposomal antimicrobial agents for infectious disease. Curr Pharm Des. 2002;8:441–454. | ||
NCT00364676. Study of Vinorelbine liposomes injection for advanced solid tumors, non Hodgkin’s lymphoma or Hodgkin’s disease (Clinical Trial). ClinicalTrials.gov. | ||
Talon Therapeutics, Inc. (Website). Talon product portfolio. Available from: http://www.talontx.com/pipeline.php. Accessed Sep 03, 2013. | ||
NCT01652859. An bioequivalence study to compare two 2 mg/mL liposomal Amphotericin B injections in healthy subjects (Clinical Trial). ClinicalTrials.gov. | ||
NCT00043199. A safety and effectiveness study of aroplatin in patients with advanced colorectal cancer resistant to standard therapies (Clinical Trial). ClinicalTrials.gov. | ||
Lu C, Perez-Soler R, Piperdi B, et al. Phase II study of a liposome-entrapped cisplatin analog (L-NDDP) administered intrapleurally and pathologic response rates in patients with malignant pleural mesothelioma. J Clin Oncol. 2005;23:3495–3501. | ||
Dragovich T, Mendelson D, Kurtin S, Richardson K, Von Hoff D, Hoos A. A Phase 2 trial of the liposomal DACH platinum L-NDDP in patients with therapy-refractory advanced colorectal cancer. Cancer Chemother Pharmacol. 2006;58:759–764. | ||
NCT01041235. Safety study of a liposomal docetaxel formulation in patients with solid tumors who have failed previous therapies (Clinical Trial). ClinicalTrials.gov. | ||
Azaya Therapeutics Products (Website). Available from: http://www.azayatherapeutics.com/about-us/products. Accessed Sep 06, 2013. | ||
NCT00005969. Liposomal tretinoin in treating patients with recurrent or refractory Hodgkin’s disease (Clinical Trial). ClinicalTrials.gov. | ||
Manconi M, Sinico C, Valenti D, Loy G, Fadda AM. Niosomes as carriers for tretinoin-Preparation and properties. Int J Pharm. 2002;34:237–248. | ||
Aleku M, Schulz P, Keil O, et al. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008;68:9788. | ||
Li L, Shen Y. Overcoming obstacles to develop effective and safe siRNA therapeutics. Expert Opin Biol Ther. 2009;9:609–619. | ||
Silence Therapeutics (Website). Available from: http://silence-therapeutics.com/pipeline/preclinical-development/. Accessed Sep 06, 2013. | ||
NCT00245297. Study of the efficacy of human recombinant factor VIII (Kogenate FS) reconstituted in pegylated liposomes (Clinical Trial). ClinicalTrials.gov. | ||
Powell J, Nugent D, Harrison J, et al. Safety and pharmacokinetics of a recombinant factor VIII with pegylated liposomes in severe hemophilia A. J Thromb Haemost. 2008;6:277–283. | ||
NCT01159028. Clinical trial of L-Grb-2 antisense oligonucleotide in CML, AML, ALL and MDS (Clinical Trial). ClinicalTrials.gov. | ||
Bio-Path Holdings, Inc (Website). Available from: http://www.biopathholdings.com/pdf/factsheetv1.pdf. Accessed Sep 06, 2013. | ||
NCT00765973. Topotecan liposomes injection for small cell lung cancer (SCLC), ovarian cancer and other advanced solid tumors (Clinical Trial). ClinicalTrials.gov. | ||
NCT00361842. Multicenter study Of CPX-1 (Irinotecan HCl: Floxuridine) liposome injection in patients with advanced colorectal cancer (Clinical Trial). ClinicalTrials.gov. | ||
Mayer LD, Janoff AS. Optimizing combination chemotherapy by controlling drug ratios. Mol Interv. 2007;7:216–223. | ||
NCT00875693. A novel sequential treatment of salvage and reduced intensity conditioning (RIC) chemotherapy for allogeneic stem-cell transplantation (SCT) for primary refractory and relapsed acute myelogenous leukemia (AML) (Clinical Trial). ClinicalTrials.gov. | ||
NCT00968604. C-VISA BikDD: liposome in advanced pancreatic cancer (Clinical Trial). ClinicalTrials.gov. | ||
Xie X, Xia W, Li Z, et al. Targeted expression of BikDD eradicates pancreatic tumors in noninvasive imaging models. Cancer Cell. 2007;12:52. | ||
Matsumura Y. Preclinical and clinical studies of anticancer drug-incorporated polymeric micelles. J Drug Target. 2007;15:507–517. | ||
Thurston G, McLean JW, Rizen M, Baluk P, Haskell A, Murphy TJ, et al. Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice. J Clin Invest. 1998;101:1401–1413. | ||
NCT00364143. Safety study of IHL-305 (Irinotecan Liposome Injection) to treat advanced solid tumors (ClinicalTrial). ClinicalTrials.gov. | ||
NCT00059605. Phase I study of IV DOTAP: cholesterol-Fus1 in non-small-cell lung cancer. (ClinicalTrial). ClinicalTrials.gov. | ||
NCT00860522. JVRS-100 for the treatment of patients with relapsed or refractory leukemia (ClinicalTrial). ClinicalTrials.gov. | ||
Chang S, Warner J, Liang L, Fairman J. A novel vaccine adjuvant for recombinant flu antigens. Biologicals. 2009;37:141–147. | ||
NCT00271063. Study of Liposomal Annamycin in Patients With Refractory or Relapsed Acute Lymphocytic Leukemia (ClinicalTrial). ClinicalTrials.gov. | ||
Booser DJ, Esteva FJ, Rivera E, et al. Phase II study of liposomal annamycin in the treatment of doxorubicin-resistant breast cancer. Cancer Chemother Pharmacol. 2002;50:6–8. | ||
Neopharm, Inc. Products – Neopharm (Website). Available from: www.neopharm.com/products/. Accessed Sep 06, 2013. | ||
NCT00024492. Study of Liposome Encapsulated Mitoxantrone (LEM) in Patients With Advanced Cancer (Clinical Trial). ClinicalTrials.gov. | ||
NCT01190982. Efficacy and safety study of LEP-ETU to treat metastatic breast cancer (Clinical Trial). ClinicalTrials.gov. | ||
Zhang JA, Anyarambhatla G, Ma L, et al. Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm. 2005;59:177–187. | ||
NCT00024648. Study to determine maximum tolerated dose of LErafAON combined with radiotherapy in patients with advanced malignancies (Clinical Trial). ClinicalTrials.gov. | ||
NCT00035867. Study of TLK199 HCl liposomes for injection in myelodysplastic syndrome (Clinical Trial). ClinicalTrials.gov. | ||
Rudin CM, Marshall JL, Huang CH, et al. Delivery of a liposomal c-raf-1 antisense oligonucleotide by weekly bolus dosing in patients with advanced solid tumors. Clin Cancer Res. 2004;10:72447–77251. | ||
NCT00104754. Liposomal SN-38 in treating patients with small cell lung cancer (Clinical Trial). ClinicalTrials.gov. | ||
Zhang JA, Xuan T, Parmar M, et al. Development and characterization of a novel liposome-based formulation of SN-38. Int J Pharm. 2004;270:93–107. | ||
NCT01159028. Clinical trial of L-Grb-2 antisense oligonucleotide in CML, AML, ALL and MDS (Clinical Trial). ClinicalTrials.gov. | ||
Tari AM, Gutiérrez-Puente Y, Monaco G, et al. Liposome-incorporated Grb2 antisense oligodeoxynucleotide increases the survival of mice bearing bcr-abl-positive leukemia xenografts. Int J Oncol. 2007;31:1243–1250. | ||
Fantini M, Gianni L, Santelmo C, et al. Lipoplatin treatment in lung and breast cancer. Chemother Res Pract. 2011;2011:125192. | ||
Stathopoulos GP, Boulikas T, Vougiouka M, et al. Pharmacokinetics and adverse reactions of a new liposomal cisplatin (Lipoplatin): phase I study. Oncol Rep. 2005;13:589–595. | ||
Farhat FS, Temraz S, Kattan J, et al. A phase II study of lipoplatin (liposomal cisplatin)/vinorelbine combination in HER-2/neu-negative metastatic breast cancer. Clin Breast Cancer. 2011;11:384–389. | ||
NCT00739466. Biorest Liposomal Alendronate With Stenting sTudy (BLAST) (Clinical Trial). ClinicalTrials.gov. | ||
NCT00747474. Phase i study of intravenous LipotecanR (TLC388 HCl for Injection) in patients with advanced solid tumors (Clinical Trial). ClinicalTrials.gov. | ||
Huang ZR, Hua SC, Yang YL, Fang JY. Development and evaluation of lipid nanoparticles for camptothecin delivery: a comparison of solid lipid nanoparticles, nanostructured lipid carriers, and lipid emulsion. Acta Pharmacol Sin. 2008;29:1094–1102. | ||
NCT01052142. Safety study of a liposomal vaccine to treat malignant melanoma (Clinical Trial). ClinicalTrials.gov. | ||
Herringson TP, Altin JG. Convenient targeting of stealth siRNA-lipoplexes to cells with chelator lipid-anchored molecules. J Control Release. 2009;139:229–238. | ||
NCT00053716. Prostaglandin E1 (Liprostin) treatment with lower limb angioplasty for peripheral arterial occlusive disease (Clinical Trial). ClinicalTrials.gov. | ||
NCT00631631. L-MTP-PE for high-risk osteosarcoma (Clinical Trial). ClinicalTrials.gov. | ||
Jain V, Vyas SP, Kohli DV. Well-defined and potent liposomal hepatitis B vaccines adjuvanted with lipophilic MDP derivatives. Nanomedicine. 2009;5:334–344. | ||
NCT00144963. Liposomal Vincristine plus Dexamethasone in patients with relapsed or refractory acute lymphoblastic leukemia (Clinical Trial). ClinicalTrials.gov. | ||
NCT00964080. Study of MBP-426 in Patients with second line gastric, gastroesophageal, or esophageal adenocarcinoma (Clinical Trial). ClinicalTrials.gov. | ||
Mebiopharm – Product and Technologies (Website). Available from: http://www.mebiopharm.com/english/pro.html. Accessed Sep 06, 2013. | ||
Hamaguchi T, Matsumura Y, Nakanishi Y, Muro K, Yamada Y, Shimada Y, et al. Antitumor effect of MCC-465, pegylated liposomal doxorubicin tagged with newly developed monoclonal antibody GAH, in colorectal cancer xenografts. Cancer Sci. 2004;95:608–613. | ||
NCT00294996. Trial of Myocet in Metastatic Breast Cancer (Clinical Trial). ClinicalTrials.gov. | ||
NCT01039103. Nanocort in acute exacerbation of relapsing-remitting Multiple Sclerosis (MS). (Clinical Trial). ClinicalTrials.gov. | ||
Chow T-H, Lin Y-Y, Hwang J-J, Wang H-E, Tseng Y-L, Pang VF, et al. Therapeutic efficacy evaluation of 111 in-labeled PEGylated liposomal vinorelbine in murine colon carcinoma with multimodalities of molecular imaging. J Nucl Med. 2009;50:2073–2081. | ||
NCT00038207. Liposomal Vincristine for pediatric and adolescent patients with relapsed malignancies (Clinical Trial). ClinicalTrials.gov. | ||
Gelmon KA, Tolcher A, Diab AR, et al. Phase I study of liposomal vincristine. J Clin Oncol. 1999;17:697–705. | ||
NCT00046787. Efficacy and safety study of OSI-211 (Liposomal Lurtotecan) to treat recurrent small cell lung cancer (Clinical Trial). ClinicalTrials.gov. | ||
NCT00940758. Phase I and pharmacokinetic study of biweekly PEP02 in mCRC refractory to 1st-line oxaliplatin base therapy (Clinical Trial). ClinicalTrials.gov. | ||
NCT01191775. A Study of PNT2258 in patients with advanced solid tumors (Clinical Trial). ClinicalTrials.gov. | ||
NCT01054547. Topical formulations of liposomal local anesthetics (Clinical Trial). ClinicalTrials.gov. | ||
Bexion Pharmaceuticals (Website). Available from: http://www.bexionpharma.com/. Accessed Sep 06, 2013. | ||
Qi X, Chu Z, Mahller YY, Stringer KF, Witte DP, Cripe TP. Cancer-Selective targeting and cytotoxicity by liposomal-coupled lysosomal saposin C Protein. Clin Cancer Res. 2009;15:5840–5851. | ||
Wojton J, Chu Z, Mathsyaraja H, et al. Systemic Delivery of SapC-DOPS Has Antiangiogenic and Antitumor Effects Against Glioblastoma. Mol Ther. 2013;21:1517–1525. | ||
NCT00177281. Safety study of S-CKD602 in patients with advanced malignancies. (Clinical Trial). ClinicalTrials.gov. | ||
NCT00157209. Phase IIb randomized controlled study of blp25 liposome vaccine for immunotherapy of non-small cell lung cancer (Clinical Trial). ClinicalTrials.gov. | ||
NCT00617981. Phase 3 Study of ThermoDox with Radiofrequency Ablation (RFA) in treatment of Hepatocellular Carcinoma (HCC) (Clinical Trial). ClinicalTrials.gov. | ||
Celsion Corporation (Website). Available from: http://celsion.com/docs/pipeline_overview. Accessed Sep 06, 2013. | ||
NCT00927459. Study to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of liposomal siRNA in subjects with high cholesterol (Clinical Trial). ClinicalTrials.gov. | ||
Rossi JJ. RNAi therapeutics: SNALPing siRNAs in vivo. Gene Ther. 2006;13:583–584. | ||
Tekmira Pharmaceuticals Corporation-Products (Website). Available from: http://www.tekmirapharm.com/Programs/Products.asp. Accessed Sep 06, 2013. | ||
NCT00089180. T4N5 liposomal lotion in preventing the recurrence of nonmelanoma skin cancer in patients who have undergone a kidney transplant (Clinical Trial). ClinicalTrials.gov. | ||
Cafardi JA, Elmets CA. T4 endonuclease V: review and application to dermatology. Expert Opin Biol Ther. 2008;8:829–838. | ||
NCT01050777. Efficacy of topical liposomal form of drugs in cutaneous leishmaniasis (Clinical Trial). ClinicalTrials.gov. | ||
NCT00764361. Safety Study of Topical Doxycycline Gel for Adult Diabetic Lower Extremity Ulcers (Clinical Trial). ClinicalTrials.gov. | ||
Nanotherapeutics – Products (Website). Available from: http://www.nanotherapeutics.com/?q=products_nanodox. Accessed Sep 06, 2013. | ||
NCT00089180. T4N5 liposomal lotion in preventing the recurrence of nonmelanoma skin cancer in patients who have undergone a kidney transplant (Clinical Trial). ClinicalTrials.gov. | ||
Yarosh D, Klein J, O’Connor A, Hawk J, Rafal E, Wolf P. Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xenoderma pigmentosum study group. Lancet. 2001;357:926–929. | ||
NCT00709254. Study of single and multiple doses of inhaled AeroLEF (Liposome-Encapsulated Fentanyl) in healthy subjects (Clinical Trial). ClinicalTrials.gov. | ||
NCT00775138. A Study to Determine the Safety and Tolerability of Arikace™ Versus Placebo in Patients Who Have Bronchiectasis. | ||
NCT00492141. Aerosol L9-NC and temozolomide in ewing’s sarcoma (Clinical Trial). ClinicalTrials.gov. | ||
NCT00915187. Safety and immunogenicity study of intramuscular CCS/C-adjuvanted influenza vaccine in elderly (Clinical Trial). ClinicalTrials.gov. | ||
NCT00922363. Trial on the safety of a new liposomal adjuvant system, CAF01, when given with the tuberculosis subunit vaccine Ag85B-ESAT-6 as two injections with two months interval to healthy adult volunteers (Clinical Trial). ClinicalTrials.gov. | ||
NCT00460525. Phase II AMA-1 malaria vaccine FMP2.1/AS02A trial in mali (Clinical Trial). ClinicalTrials.gov. | ||
NCT00004806. Phase I Study of liposome-mediated gene transfer in patients with cystic fibrosis (Clinical Trial). ClinicalTrials.gov. | ||
NCT00533637. Taste and local tolerance study of NLA nasal spray in patients with allergic rhinitis (Clinical Trial). ClinicalTrials.gov. | ||
Orexo AB – OX-NLA (Website). Available from: http://www.orexo.com/en/Portfolio/OX-NLA/. Accessed Sep 05, 2013. | ||
NCT00004471. Phase I pilot study of gene therapy for cystic fibrosis using cationic liposome mediated gene transfer (Clinical Trial). ClinicalTrials.gov. | ||
NCT01095848. A phase I safety study of a cancer vaccine to treat HLA-a2 positive advanced stage ovarian, breast and prostate cancer (Clinical Trial). ClinicalTrials.gov. | ||
NCT00004104. Vaccine therapy plus interleukin-2 with or without interferon alfa-2b in treating patients with stage III melanoma (Clinical Trial). ClinicalTrials.gov. | ||
NCT01073371. Anesthetic efficacy of liposomal prilocaine in maxillary infiltration anesthesia (Clinical Trial). ClinicalTrials.gov. | ||
Saia Cereda CM, Tofoli GR, de Brito Junior RB, et al. Stability and local toxicity evaluation of a liposomal prilocaine formulation. J Liposome Res. 2008;18:329–339. | ||
NCT01334892. L-CsA in the prevention of bronchiolitis obliterans syndrome (BOS) in lung transplant (LT) patients (Clinical Trial). ClinicalTrials.gov. | ||
Behr J, Zimmermann G, Baumgartner R, et al. Lung deposition of a liposomal cyclosporine A inhalation solution in patients after lung transplantation. J Aerosol Med Pulm Drug Deliv. 2009;22:121–130. | ||
NCT00035867. Study of TLK199 HCl liposomes for injection in myelodysplastic syndrome (Clinical Trial). ClinicalTrials.gov. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.