")
Back to Journals » International Journal of Nanomedicine » Volume 13
Liposome delivery systems for the treatment of Alzheimer’s disease
Authors Ross C, Taylor M , Fullwood N , Allsop D
Received 24 September 2018
Accepted for publication 6 November 2018
Published 12 December 2018 Volume 2018:13 Pages 8507—8522
DOI https://doi.org/10.2147/IJN.S183117
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Callum Ross, Mark Taylor, Nigel Fullwood, David Allsop
Division of Biomedical and Life Sciences, Faculty of Health and Medicine, Lancaster University, Lancaster LA1 4YQ, UK
Abstract: Alzheimer’s disease (AD) will affect around 115 million people worldwide by the year 2050. It is associated with the accumulation of misfolded and aggregated proteins (β-amyloid and tau) in the senile plaques and neurofibrillary tangles found in the brain. Currently available drugs for AD only temporarily alleviate symptoms and do not slow the inevitable progression of this disease. New drugs are required that act on key pathologies in order to arrest or reverse cognitive decline. However, there has been a spectacular failure rate in clinical trials of conventional small molecule drugs or biological agents. Targeted nanoliposomes represent a viable and promising drug delivery system for AD that have not yet reached clinical trials. They are biocompatible, highly flexible, and have the potential to carry many different types of therapeutic molecules across the blood–brain barrier (BBB) and into brain cells. They can be tailored to extend blood circulation time and can be directed against individual or multiple pathological targets. Modifications so far have included the use of brain-penetrating peptides, together with Aβ-targeting ligands, such as phosphatidic acid, curcumin, and a retro-inverted peptide that inhibits Aβ aggregation. Combining several modifications together into multifunctional liposomes is currently a research area of great interest. This review focuses on recent liposomal approaches to AD therapy, including mechanisms involved in facilitating their passage across the BBB, and the evaluation of new therapeutic agents for blocking Aβ and/or tau aggregation.
Keywords: amyloid, blood–brain barrier, cell-penetrating peptides, neurofibrillary tangles, senile plaques, tau
Introduction
Alzheimer’s disease (AD) affects 36 million people worldwide, and this number is set to rise to over 115 million by 2050, as populations age.1 AD is a neurodegenerative disorder that severely affects the functioning of the brain, triggering widespread neuronal and synaptic loss, memory impairment, and cognitive and behavioral disturbances.2 A major hypothesis regarding the underlying cause of AD is that the progressive accumulation of protein aggregates in the brain leads to neurodegeneration and dementia3 (see Karran and de Strooper4 for a critical review). These protein aggregates include extracellular senile plaques, containing the β-amyloid (Aβ) peptide, and neurofibrillary tangles (NFTs), consisting of intra-neuronal paired helical filaments (PHFs) composed of hyper-phosphorylated tau protein.5 Aβ spontaneously and progressively aggregates to form oligomers and amyloid fibrils, with oligomers being a strong candidate for inducing synaptic damage and memory deficits in AD.6 However, AD is likely to be a multifactorial disorder, with many mechanisms, including NFT formation, contributing to neuronal cell damage.
It is well established that Aβ and tau underlie the neuropathology of AD, but many aspects of the neuropathogenesis of this disease remain unanswered. There is good evidence that aggregation and accumulation of these molecules induce membrane damage and oxidative damage through an increase in free radical production and promotion of pro-inflammatory processes.7–9 NFT formation is also likely to interfere with axonal transport systems. The drugs currently available for the treatment of AD (acetylcholinesterase inhibitors [AChEIs] and the NMDA antagonist memantine) do not target any of these underlying disease mechanisms10 and do not slow down the long-term progression of the disorder. Instead, they provide only temporarily improved cognition by counterbalancing neurotransmitter disturbances, in addition to having many limitations and side effects.11 It is therefore essential to develop alternative therapies, with minimal adverse side effects, which aim to prevent, slow or reverse the neurodegeneration responsible for AD. Unfortunately, clinical trials on >200 drugs, with different molecular targets, have so far failed to find a therapeutic approach that arrests or reverses cognitive decline in patients with AD.12–14
Any effective treatment for AD would most likely require the therapeutic agent concerned to be transported across the blood–brain barrier (BBB),15 which prevents access to the brain of around 98% of potential neuropharmaceuticals.16 Liposomes are a biocompatible and highly flexible drug delivery system, with the potential for carrying many different types of bioactive molecules across the BBB. The therapeutic potential of liposomes was recognized shortly after their development in 1961, but only recently they have been considered as a suitable vehicle for the delivery of drugs that act on the central nervous system (CNS).17,18 Their physiological composition facilitates numerous modifications, in comparison with other nano-carriers. Simple “non-targeted” liposomes can carry a drug cargo into the brain (eg, via the olfactory route, see “Intranasal delivery” section), but modified “targeted” liposomes are required for effective delivery across the BBB and can also be designed to interact with specific molecular targets relevant to the treatment or prevention of AD. Liposomes can incorporate hydrophilic or lipophilic/hydrophobic therapeutic agents, where hydrophilic drugs are entrapped in the aqueous core, and lipophilic compounds are contained in the hydrophobic region of the lipid bilayer.19 Moreover, various types of therapeutic agent can be attached to the surface of the liposomes. These various configurations can be combined to give a targeted multi-drug delivery system, which is particularly relevant for the treatment of any multifactorial disease. These attractive properties of liposomes have provoked an interest in their development as a possible new and effective therapy for AD. This narrative review presents our own perspective on recent and promising liposome approaches to AD therapy, including mechanisms involved in facilitating their passage across the BBB, and the evaluation of new therapeutic agents for blocking Aβ and/or tau aggregation.
The BBB
The BBB is a metabolic and transport barrier that protects the brain from harmful stimuli. It comprises brain capillary endothelial cells that are attached to each other through tight junctions, resulting in restricted paracellular transport.20,21 Degrading enzymes are also present at the BBB, which can destroy molecules during their attempted passage into the brain.22 However, specific mechanisms are in place for the BBB transport of molecules that are essential for brain function,23 and these can be exploited for therapeutic drug delivery.
Early strategies for BBB transportation
Passive diffusion permits the passage of some small lipophilic compounds across the BBB, such as certain amino acids, nucleosides, and small peptides. An early strategy utilized this simple mechanism by developing small lipophilic drugs that might pass through the endothelial cells. However, this excluded the vast majority of potential therapeutic molecules. A second approach was to develop small water-soluble drugs, in order to facilitate BBB transport via the paracellular hydrophilic diffusion pathway. However, most of these potential molecules could not penetrate past the tight endothelial cell junctions.24
Alternatives
Intracerebral or intracerebroventricular injection of drugs provides a strategy that avoids the BBB, but these procedures are highly invasive, and so are not widely used. Drugs administered by any peripheral route (eg, orally or by intravenous injection) will encounter the BBB.25 Alternative methods, such as bypassing the BBB by delivery through the olfactory region (eg, as a nasal spray) have some potential.26–28 Another strategy looks to utilize existing active BBB transport mechanisms, namely carrier-mediated transcytosis (also known as transporter-mediated transcytosis), receptor-mediated transcytosis, cell-mediated endocytosis, and adsorptive transcytosis (Table 1).29
 | Table 1 Transport mechanisms for BBB transit and brain delivery |
Liposomal approaches to AD therapy
Several different types of nanoparticles (eg, solid lipid, magnetic, dendrimers, and liposomes) are under development as possible vectors for AD therapy. A popular strategy is to aim for optimum brain selectivity by targeting receptors or transporters that are highly expressed at the BBB, and improving stability, for example, by using non-natural amino acids in the case of peptide drugs, so that they are resistant to proteolysis.30 A second route considers intranasal administration of the active compounds, without any BBB interruption. Recent studies have begun to focus on liposomes as a possible carrier of drugs for the treatment of AD.31–33
BBB transport
Possible mechanisms for the transport of liposomes across the BBB have been highly debated.25 An initial theory was that the phospholipid bilayer of the liposomes on its own might facilitate transportation across various biological membranes, including the BBB, but this simple mechanism proved to be ineffective. Modifications proposed for enhancing the transport of liposome carriers across the BBB utilize the existing active transport mechanisms, involving absorptive, carrier- or receptor-mediated transcytosis.34 Absorptive methods take advantage of the BBB’s negative charge. Cationic liposomal drug vehicles have been developed that can trigger cell internalization through electrostatic interactions. However, the nonspecific uptake of the cationic liposomes by peripheral tissues and their binding to serum proteins has meant that a toxic dose would often be required to reach therapeutic efficacy,35 thus limiting therapeutic potential. Carrier-mediated transcytosis utilizes nutrients capable of passing across the BBB, such as glucose and glutathione (GSH), that can be attached to the surface of liposomes and facilitate translocation.19 Ultimately, receptor-mediated transcytosis has great potential for success, since there are possibilities to target one or more of the many different receptors at the BBB by using their respective ligands. Yet there is still substantial interest in direct penetration of the BBB membrane, which has been achieved largely through the use of cell-penetrating peptides (CPPs) (Figure 1).
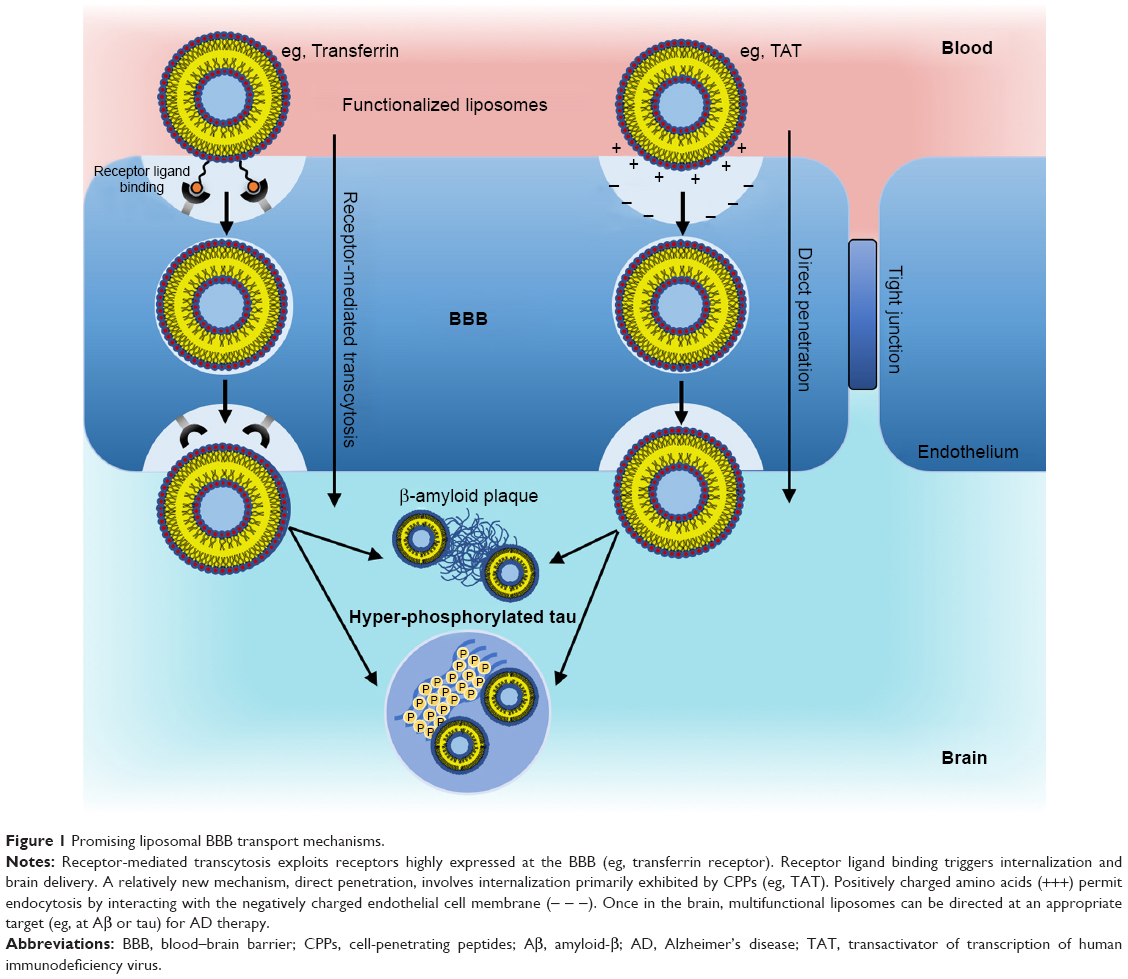 | Figure 1 Promising liposomal BBB transport mechanisms. |
Intranasal delivery
A second route involves the intranasal administration of liposomes. This strategy avoids the BBB by offering direct nose-to-brain absorption through the olfactory and trigeminal nerves. Liposomal encapsulation of compounds can result in improved penetration via this route, by protecting them from degradation and facilitating their transport across the mucosal barrier. A recent study has demonstrated effective administration of a liposome-encapsulated “β-breaker” peptide, known as H102, using this strategy.27 Results showed satisfactory drug concentrations in the brain, with limited toxicity. Thus, the intranasal route offers a promising alternative to BBB transport and delivery.
Vesicle optimization
Liposome size
Liposome size, as well as lipid composition, affects their circulation in the bloodstream and uptake into the brain.36,37 Brain delivery requires liposomes to be roughly nano- or microsized and consists of one or more lipid bilayers surrounding an aqueous core. Only certain sizes will allow passage across the BBB for neurotherapy (such as in AD) and so small vesicles (100 nm and less) are often preferred. There are studies, however, that have shown that liposomes from 100 to 140 nm have certain advantages, such as a longer half-life in blood circulation and avoidance of plasma proteins. Large nano-liposomes (250 nm in diameter) are cleared twice as fast as 100 nm liposomes.38 Yet, liposomes 100 nm and smaller have more limited storage capacity, leading to poor encapsulation efficiency.39 A consideration of size should be made based on factors such as their therapeutic application, encapsulation efficiency, and stability.
Lipid composition
Phospholipids commonly used in liposomes include synthetic lipids such as 1,2-dipalmitoyl-sn-glycero-3-phosphocholine and ethyl-phosphatidylcholine, or natural lipids such as phosphatidylcholine (PC), sphingomyelin (SP), and lecithin (LC). Natural lipids are at risk of contamination from viruses, prions, or toxins, especially if they are isolated from a mammalian source, for example, bovine brain. They are, however, cheaper in large-scale production, use fewer solvents and other chemicals in their extraction, and are more readily accepted by regulatory authorities, especially when derived from a non-animal (eg, plant) source. Cholesterol is often added to liposomes during assembly, which not only maintains the stability of membranes in vivo and in vitro but also reduces permeability and alters the structure and function of the vesicles.40
Aβ has been shown to insert preferentially into any anionic phospholipids incorporated into liposomes, which could have a protective effect, by removing toxic Aβ.41,42 The majority of treatments aimed at AD have used PEGylated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-PEG 2000 (DSPE) to improve circulation time (see “Stealth liposomes” section), although wheat germ agglutinin, chitosan, silk fibroin, and polyvinyl alcohol have also been used to achieve similar “stealth” properties.
Liposome modifications
Liposomes can be functionalized in various ways in order to realize their potential as a therapeutic carrier (Figure 2). Modification can involve the attachment of molecules to the exterior, or the encapsulation of molecules internally, either in the aqueous core or lipid bilayers.19 Figure 2 considers possible modifications to liposomes that are particularly relevant for the treatment of AD.
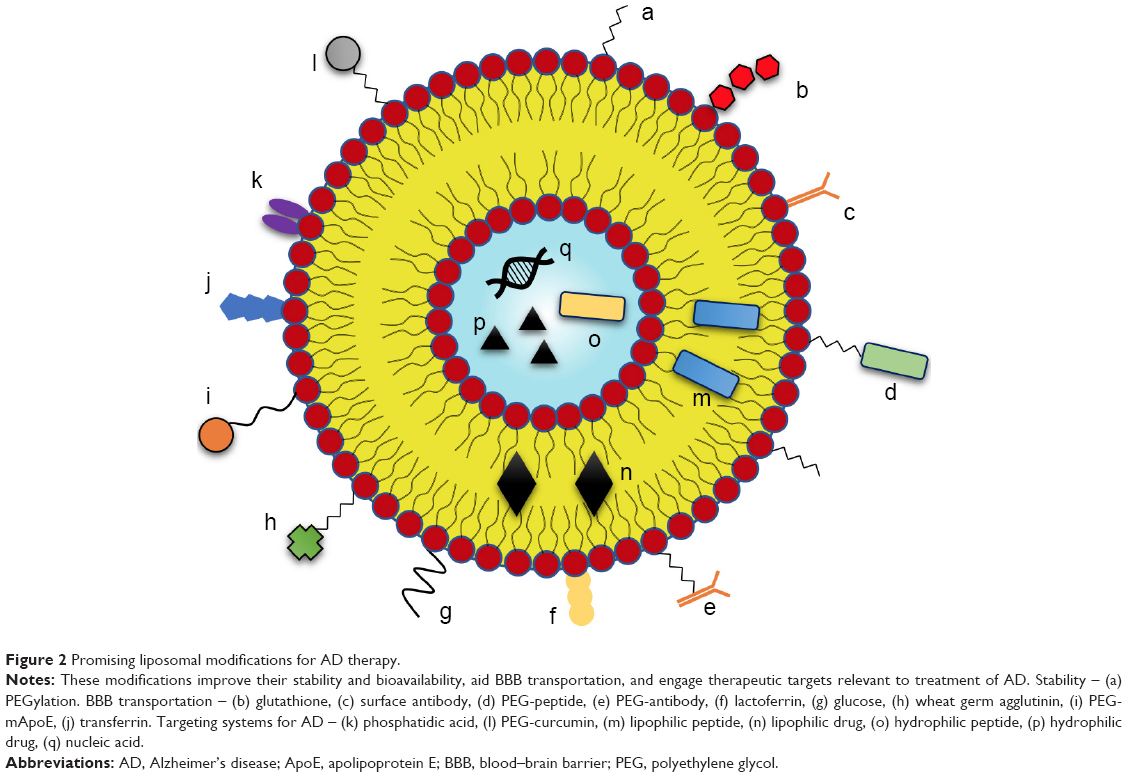 | Figure 2 Promising liposomal modifications for AD therapy. |
Stealth liposomes
Simple liposomes are covered by plasma proteins (eg, fibrinogen, immunoglobulins, and complement proteins) when in circulation, so that a “protein corona” is formed around them. This leads to the activation of phagocytic systems and removal of these liposomes from the bloodstream.43 In order to improve their pharmacokinetic profile and allow longer time-periods in circulation, so-called “stealth” liposomes were developed, with polyethylene glycol (PEG), or another polysaccharide, incorporated into one of the lipid components of the liposomal membrane. PEG has high hydration, and this increases hydrodynamic volume and allows the formation of a water cloud around the polymer. Any hydrophilic molecule bound covalently to PEG also exhibits these properties, providing increased solubility and resistance to interaction with plasma proteins. Subsequently, PEGylated liposomes in circulation are less prone to formation of a protein corona, allowing dramatically improved circulation times. However, there is no certainty that these liposomes will be transported across the BBB. To provide this, further modifications are required, often involving the use of PEGylated lipids for attachment of suitable components to the liposome surface.44
BBB transport
GSH
The transport of liposomes across the BBB can be facilitated by the attachment of appropriate molecules to the lipid surface. One of these is GSH, an endogenous tripeptide and antioxidant found in almost every cell in the human body. Due to its essential protective role in the brain, GSH is transported actively across the BBB, via a sodium-dependent GSH transporter that is highly expressed on the BBB epithelium. However, the exact mechanism of action of this transporter remains unclear.45–47 “G-Technology®” is a strategy involving PEGylated liposomes with glutathione (GSH-PEG)-mediated delivery across the BBB into the brain, and this has shown some success in double transgenic APPswe/PS1ΔE9 (APP/PS1) mice,47 but receptor saturation limits this system’s effectiveness.48 Additional research is required to enhance this strategy further.
Glucose
In a similar way to GSH, there are carrier-mediated transport mechanisms in place to facilitate the transport of glucose across the BBB, and the incorporation of glucose onto the liposome surface can enhance BBB delivery.30 Glucose-modified PEGylated liposomes prepared with various PEG chain lengths have been tested for BBB penetration in mice. Results show that liposomes with shorter PEG chain lengths have little evidence of brain penetration, because the glucose cannot interact effectively with the BBB surface, and therefore cannot utilize the glucose transporter. Longer PEG chain lengths are highly flexible and are able to fold back on themselves in such a way that the ability to cross the BBB is also impaired. Liposomes prepared with medium length PEG chains attached to glucose can cross the BBB more effectively.49 Thus, liposomal preparations with medium length PEGylation are likely to be the most effective method for utilizing glucose to enhance BBB transport.
Transferrin (Tf)
Tf is the most commonly targeted receptor (TfR), due to its localization on BBB endothelia. This localization is likely to be due to Tf’s essential role as an iron-binding blood plasma glycoprotein that controls the level of free iron and oxidative stress in the brain, as well as in other tissues.50 Fishman et al demonstrated that Tf could cross the BBB via receptor-mediated endocytosis,51 and more recent studies have shown that liposomes modified with Tf can also penetrate the BBB in vitro.52,53 However, TfR-mediated endocytosis of liposomes is limited due to endogenous Tf competing for binding to the TfR.54 This issue can in part be resolved by using antibodies that bind with higher affinity than Tf to TfR, thus avoiding this competition. Still, Tf is likely to be most effective as part of a highly functionalized liposome, where multiple modifications allow an additive increase in BBB transport; a view supported by recent studies.55,56
Lactoferrin (Lf)
The lactoferrin receptor (LfR), bound by Lf, is also heavily overexpressed on the BBB, which has led to the development of Lf functionalized liposomes to enhance transport into the brain via receptor-mediated endocytosis.57 This delivery system has the potential to be particularly efficient, as the expression of the LfR on microvessels and neurons is increased in AD,58 allowing more effective targeting. Chen et al57 investigated the ability of Lf procationic liposomes (Lf-PCLs) to cross the BBB of rodents, for potential treatment of AD. Lf-PCLs become positively charged upon binding to the Lf receptor on the BBB epithelium. This allows them to fuse more easily with the BBB and overcome disadvantages such as non-specific binding and poor stability in circulation.59
mApoE
The ApoE glycoprotein is responsible for the transport and delivery of cholesterol and other lipids from the plasma to the CNS.60 It contains a peptide binding domain that permits BBB transport. A peptide sequence containing this binding domain (mApoE) has been used to facilitate BBB transport in vitro.61,62 However, the success of this peptide alone to facilitate BBB transport is uncertain, as the BBB still limits the transport of ApoE, and therefore mApoE, in and out of the brain.63 It is likely, therefore, to be used in combination with other liposome modifications.
CPPs
In the last 20 years, CPPs have been identified, which are able to translocate across biological membranes, including the BBB.64 They cross in a non-toxic manner, independent of membrane receptors and energy, which could reduce the limitations of receptor saturation that have been observed with other methods. Most CPPs rely on positively charged amino acids interacting with the negatively charged membrane. Arginine and to a lesser extent lysine are particularly effective, as they form hydrogen bonds with the negatively charged phosphates, which may lead to internalization. CPPs with different properties vary in their internalization mechanisms (Table 2), but remain similar in their effective direct penetration of cell membranes.
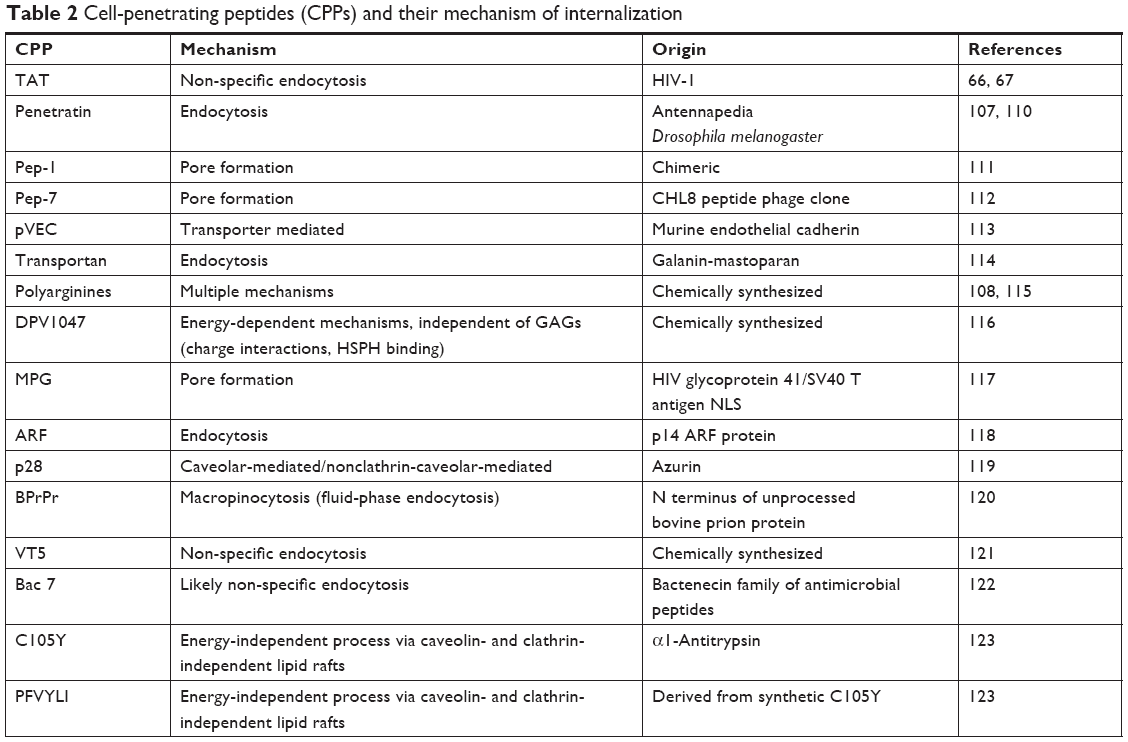 | Table 2 Cell-penetrating peptides (CPPs) and their mechanism of internalization |
Among all CPPs, the HIV-1 tat (TAT) protein has been best described and has been used successfully for delivery of liposome nanoparticles into the brain.65 TAT triggers steps for non-specific endocytotic delivery due to ionic interactions between positive charges of the peptide and negative charges of the BBB.66 Recent studies have utilized a retro-inverted version of TAT, where L-amino acids are replaced with D-amino acids, and the sequence is reversed. This reduces the potential problems with TAT, such as protease degradation and poor bioavailability in vivo, but still maintains the ability for transport across the BBB.67 Alternative CPPs, such as polyarginines (eg, octa-arginine) and penetratin have also shown potential for the delivery of therapeutics directly to the brain in SAMP8 mice.68
Multifunctionalized liposomes
Many of the modifications discussed above are likely to be more effective as part of a multifunctionalized liposomal system. This will include one or more molecules that enhance BBB transport, but most importantly, it will allow the incorporation of molecules that permit both BBB transport and targeting for AD therapy. The following are multifunctionalized liposomal systems that utilize some of the modifications discussed above and are in development as possible treatments for AD.
Aβ targeting
It is likely that the accumulation and aggregation of Aβ in the brain have an important role, directly or indirectly, in the induction of synaptic damage and memory deficits in AD.69,70 The accumulation of Aβ oligomers or fibrils may be caused by overproduction, inefficient clearance from the brain, or by a combination of both of these. Effective therapeutic molecules can have an impact on one of these mechanisms to reduce Aβ burden. Many of these molecules on their own have low uptake across the BBB, but their incorporation into a multifunctionalized liposomal system can help to resolve this problem, allowing effective targeting to Aβ.
mApoE-PA liposomes
Balducci et al71 conducted a key study to look at the ability of multifunctional liposomes to target Aβ. These liposomes were bi-functionalized with mApoE to enhance crossing of the BBB, and with phosphatidic acid (PA), which is a high affinity ligand for Aβ.61,71 This bifunctional liposome (mApoE-PA-LIP) was able to disaggregate Aβ fibrils in vitro, a property that was not exhibited by liposomes mono-functionalized with either mApoE or PA alone. This synergistic effect could be due to simultaneous interaction of the negatively charged PA phosphate group with positively charged amino acid residues on Aβ and of positively charged amino acids on mApoE with negatively charged regions of Aβ.72 Results from in vivo studies, showing a reduction in amyloid plaque load only with mApoE-PA-LIP, are supportive of this idea. However, the uptake of mApoE-PA-LIP in the brain was very low, despite the mApoE modification. Mancini et al conducted a follow-up investigation on the mechanism behind this therapy, and a strategy known as the “sink effect” was proposed, which reduces Aβ burden by peripheral administration of a binding agent that draws excess Aβ out of the brain (Figure 3).73 The study showed that peripheral administration of mApoE-PA-LIP increased the level of plasma Aβ without significant amounts of mApoE-PA-LIP entering the brain. Aβ oligomers were found to be transported out of the brain, across the BBB, with mApoE-PA-LIP acting in the periphery to mediate a fivefold increase in this efflux.74
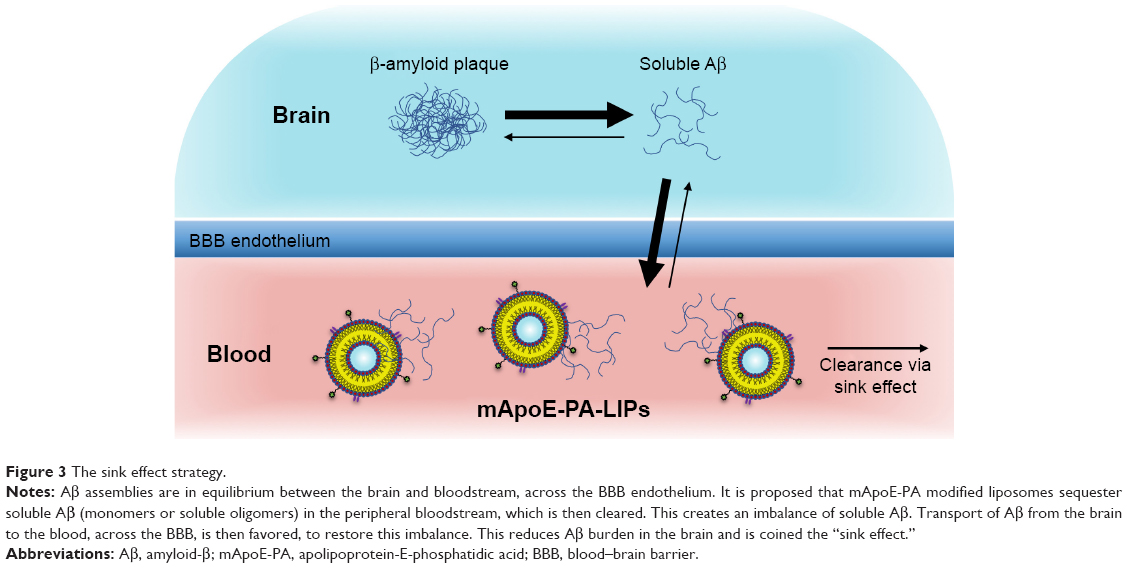 | Figure 3 The sink effect strategy. |
This provides an insight into future therapies that may only require sequestration of Aβ in the plasma, without the need to cross the BBB. It is evident, however, that this therapy might slow down neurodegeneration, but does not eliminate the cause of Aβ overproduction. Yet, this bifunctional liposome system provides a valuable insight into future research that may effectively prevent amyloid plaque formation.
Curcumin-lipid liposomes
Oxidative damage occurs very early on in course of AD,75,76 and antioxidants can protect neurons from β-amyloid toxicity,77 and so clinical trials involving antioxidants for the treatment of AD have been undertaken. So far, these have been unsuccessful,78 but the use of curcumin, a turmeric derivative, has shown some potential due to its anti-amyloid effects.79 This phenolic antioxidant binds to amyloid deposits in vitro and in vivo, and it not only disrupts the aggregation of the amyloid peptide but also disaggregates pre-formed fibrils.80,81 A curcumin analog with a substitution at the C-4 position has been reported to be more effective in this regard than curcumin itself.82
Curcumin has relatively poor bioavailability, hence, liposomal delivery systems for curcumin have been developed in an attempt to overcome this.83,84 In the curcumin-lipid modified liposome developed by Mourtas et al,84 a PEGylated lipid coating provides stealth characteristics and acts as an anchor allowing the attachment of anti-transferrin monoclonal antibodies, for mediation of transport across the BBB, in addition to a lipid-PEG-curcumin derivative. Results showed that this system has high affinity for amyloid plaques and reduces amyloid plaque formation in APPswe transgenic mice.79 These liposomes may be useful in the future for the diagnosis and treatment of AD.
Peptide inhibitor nanoparticles (PINPs)
Austen et al85 showed that a small peptide, named OR2 (RGKLVFFGR-NH2), inhibits the aggregation of Aβ into oligomers and fibrils, and blocks the toxic effects of Aβ on cultured cells. However, this peptide is sensitive to proteolysis and was not designed to cross the BBB.85 To improve its stability, a retro-inverso version (RI-OR2) was made,86 and this was enhanced further by the addition of a retro-inverted version of TAT to RI-OR2, producing RI-OR2-TAT (Ac-rGffvlkGrrrrqrrkkrGy-NH2).67 Following its peripheral injection, a fluorescein-labeled version of RI-OR2-TAT was found to cross the BBB and bind to the amyloid plaques present in the cerebral cortex of APPswe/PS1ΔE9 transgenic mice. Daily intraperitoneal (ip) injection of RI-OR2-TAT, for 3 weeks, into these mice resulted in substantial (25%–45%) reductions in brain Aβ oligomer levels, amyloid plaque counts, oxidative damage, and inflammatory processes. However, RI-OR2-TAT inhibits Aβ aggregation only at relatively high concentrations (ie, at a molar ratio of RI-OR2-TAT:Aβ of 1:5 at best) and so its therapeutic potential is likely to be limited.67
In order to improve RI-OR2-TAT further, it was attached to the surface of stealth liposomes by covalent linkage to the PEGylated lipid, via an additional cysteine residue on the peptide, to produce PINPs (Figure 4).65 The presence of the liposome carrier greatly improves the potency of RI-OR2-TAT, so that a molar ratio of 1:2,000 of liposome-linked inhibitory peptide to Aβ now gives ~50% inhibition of Aβ aggregation. This great increase in potency could be due to a multivalent effect. Many copies of the RI-OR2-TAT decorate each liposome, which should allow the simultaneous interaction of multiple inhibitory peptides with any multimeric form of Aβ, thus increasing efficiency as an aggregation inhibitor. EM studies have shown that PINPs can attach themselves to the free ends of amyloid fibrils (Figure 5),87 apparently terminating fibril growth, and possibly preventing the formation of Aβ oligomers (the free ends of fibrils act as “factories” for generation of oligomeric Aβ).88 Studies in transgenic mice have shown that peripheral injection of PINPs protects against memory loss in TG2576 mice.65 The TAT region of the inhibitory peptide facilitates brain penetration, with a brain:blood ratio of around 50% being achieved shortly after intravenous (iv) injection. However, most of the dose accumulates in peripheral tissues, such as lungs, liver, and spleen, most likely due to clearance of liposomes via the reticuloendothelial system, and so it is possible that the “sink” effect (see above) could explain some of the in vivo properties of PINPs. The effectiveness of these PINPS could be improved in future by incorporation of alternative or additional brain delivery systems.
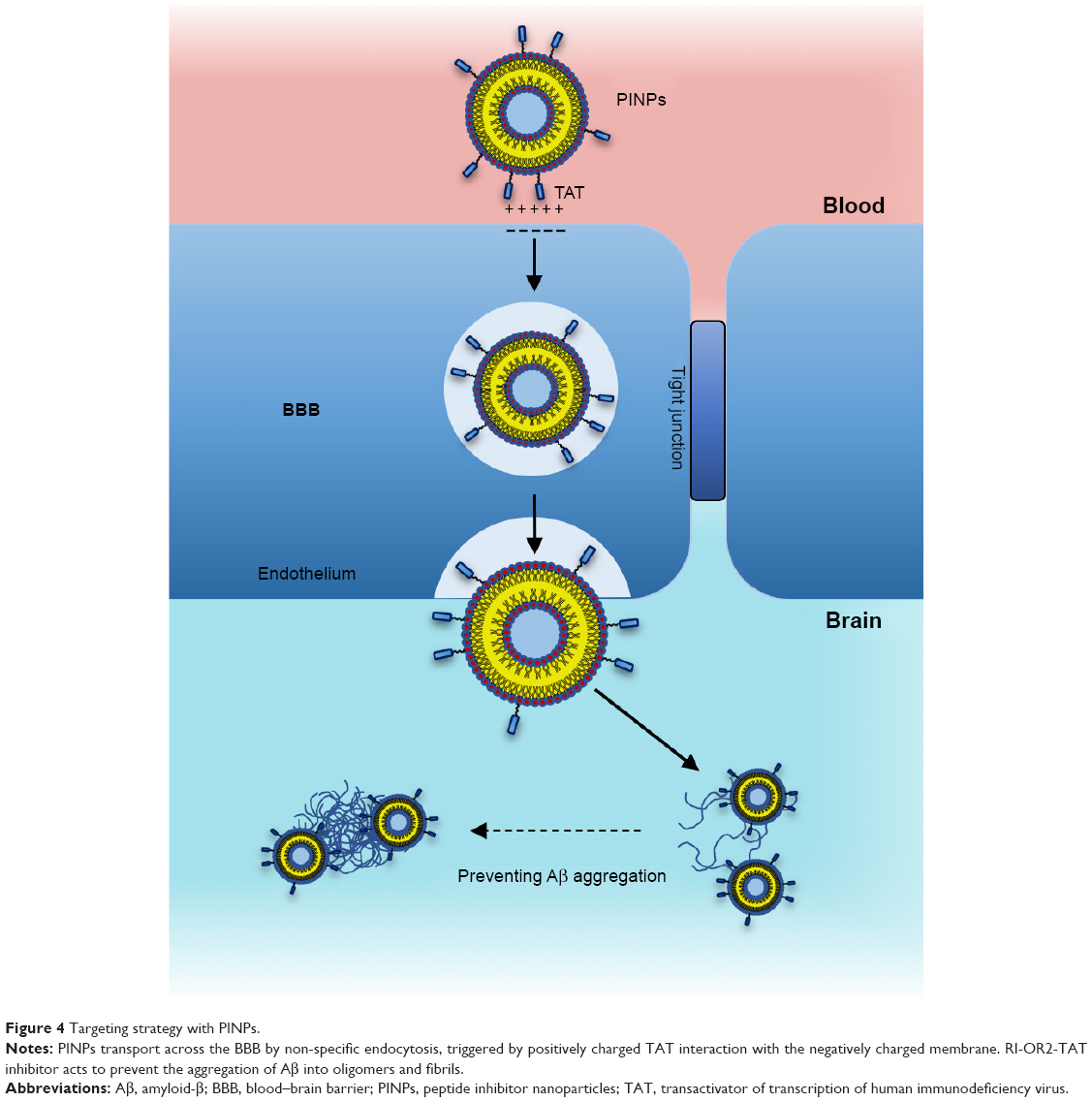 | Figure 4 Targeting strategy with PINPs. |
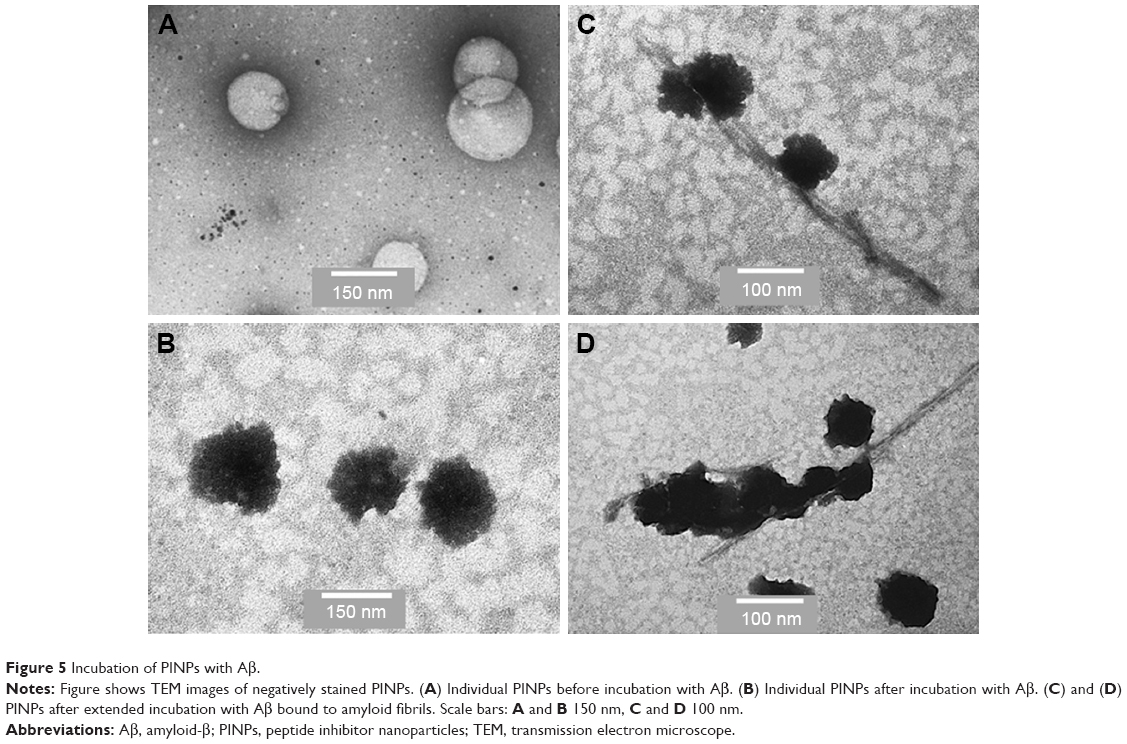 | Figure 5 Incubation of PINPs with Aβ. |
Neurotransmission targeting
Synaptic loss and degeneration in AD include the loss of brain cholinergic neurons, which results in decreased acetylcholine (ACh) levels, reduced ACh receptor (AChR) density, and an overall decrease in cholinergic neurotransmission. Many limitations and side effects are associated with the existing AChEIs that attempt to rebalance cholinergic neurotransmission in AD. They have poor stability in circulation, unpredictable uptake and bioavailability, and can cause gastrointestinal complications and, occasionally, even esophageal tears.89 They also provide only temporarily improved cognition. Yet, it may be worthwhile to improve these existing therapies, especially since viable alternatives have failed to emerge. Subsequently, studies have investigated the potential use of multifunctionalized liposomes, incorporating existing drugs, to treat AD.
Rivastigmine-loaded liposomes
Rivastigmine is a reversible, noncompetitive inhibitor of brain acetylcholinesterase (AChE).90 As stated previously, oral rivastigmine administration has many limitations, such as poor stability and low ability to pass through the BBB. An alternative intranasal delivery route has shown some potential as an alternative for rivastigmine administration.91 A study from Yang et al92 has considered whether intranasal delivery of liposomes loaded with rivastigmine can enhance brain delivery and improve therapeutic effect. These liposomes were modified with a PEGylated derivative of a poly-arginine CPP (DSPE-PEG-CPP) to increase stability and enhance BBB delivery, respectively, in an attempt to improve the limitations exhibited by the administration of rivastigmine alone. It was found that liposomes improved the therapeutic effect of rivastigmine, due to enhanced transcytosis across both the BBB and cellular membranes.92
El-Helaly et al94 have reported an alternative modification strategy for the intranasal administration of rivastigmine-loaded liposomes. In order to maintain their stability, a positively charged inducer called didecyldimethyl ammonium bromide was added to these liposomes, leading to electrostatic repulsion and reduced interactions between them. This, coupled with the use of a PEGylated lipid, resulted in a highly stabile “electrosteric stealth liposome.”93 Results showed a fourfold increase in both plasma and brain drug levels, compared to rivastigmine itself. This effect was increased further with liposomes bound to Tween 80, a nonionic surfactant that has been found to enhance BBB transport.94 Tween 80 is likely to act as an anchor for apolipoproteins (APO-E and APO-B), enhancing BBB transport via receptor-mediated endocytosis.95 Further preclinical studies on rivastigmine-loaded liposomes are required, such as toxicity and dose–response studies, before they can be considered as suitable for therapeutic use.
Liposomes with multiple therapeutic targets
Liposomes have great potential for modification, including the development of multifunctional liposomes directed against more than one therapeutic target. In the case of AD, one of the legitimate goals would be to target the formation of both Aβ plaques and tau tangles. Theoretically, it is possible to conceive of a multifunctional liposome that also incorporates other therapeutics, such as those directed at the ACh system, neuroprotectants, autophagy, anti-inflammatory agents, or antioxidants.
Wheat germ agglutinin-conjugated liposomes
The multifunctional liposomes produced by Kuo et al aim to improve the overall neuronal survival in AD by inhibiting the phosphorylation of p38 and c-Jun N-terminal kinase (JNK), two key members of the mitogen-activated protein kinase (MAPK) family, in neurons damaged by the accumulation of Aβ (Figure 6).42 Study of this kinase cascade is essential for the understanding of AD progression, since MAPK-catalyzed phosphorylation events in the brain have a key role in the downstream activity of cellular components (eg, phosphatases, kinases) that impact on apoptosis and overall neuronal survival. It would also provide valuable insight into possible future therapies.
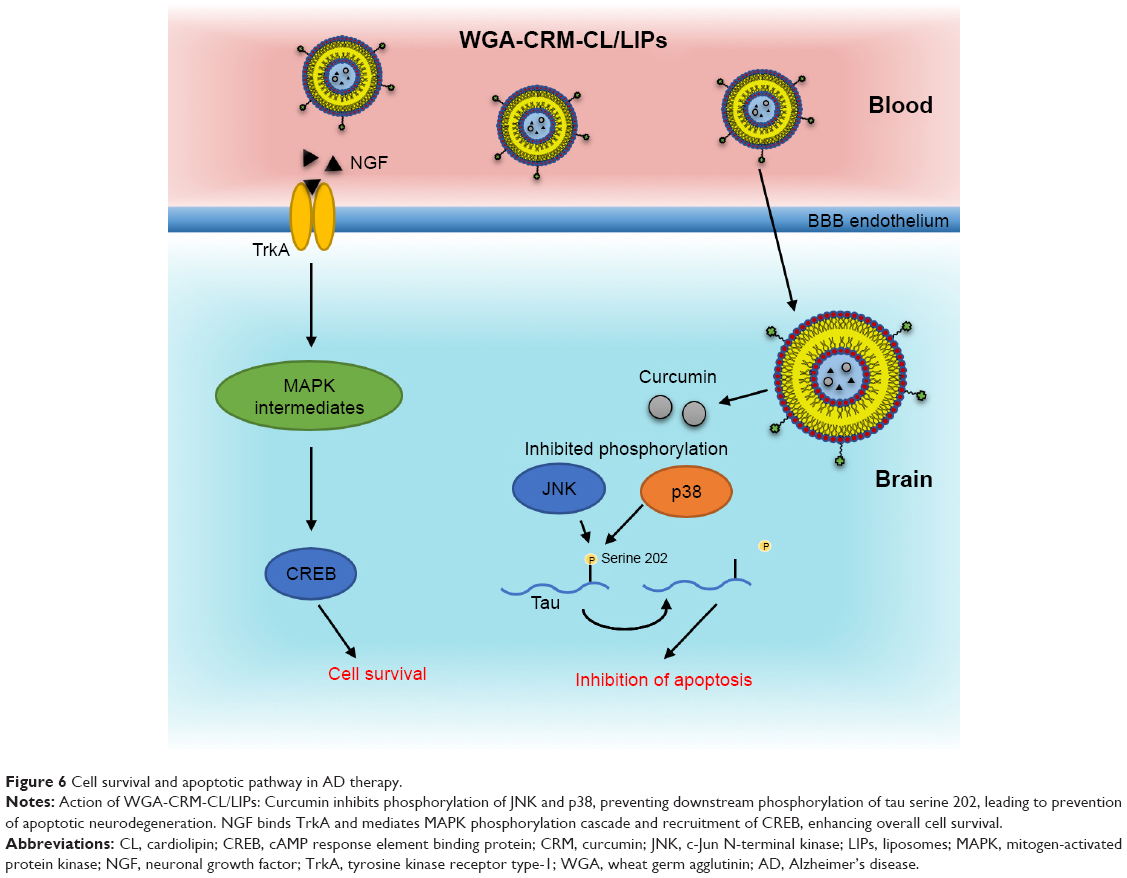 | Figure 6 Cell survival and apoptotic pathway in AD therapy. |
The key constituents of this multifunctional liposome (coined WGA-CRM-CL/LIP) are curcumin (CRM), nerve growth factor (NGF), cardiolipin (CL), and wheat germ agglutinin (WGA). In addition to acting as an Aβ aggregation inhibitor, curcumin alters the expression of phosphorylated p38 (p-p38) and phosphorylated c-Jun N-terminal kinase (p-JNK), key players in the apoptotic pathway. NGF promotes the activity of tyrosine kinase receptor type 1 (TrkA), which is involved in slowing down neuronal apoptosis. In order to enhance the targeting of apoptotic neurons surrounding Aβ, CL was included, as it has a strong affinity for Aβ. In this case, both PEGylation and WGA provided an additional novel dual functionalization, with the aim of improving delivery across the BBB.42
WGA-CRM-CL/LIPs reduced Aβ “plaque” deposition in AD model SK-N-MC cells and increased the percentage of healthy neurons with cholinergic activity in a Wistar rat AD model (established by injection of Aβ into CA1 neurons). Additionally, curcumin mediates downstream inhibition of tau phosphorylation at serine 202, which is a key site for axonogenesis (Figure 6).96 While the therapeutic potential of this inhibition is unclear, this is an illustrative method of targeting and altering tau phosphorylation, while also targeting and reducing Aβ burden. The authors suggest that WGA-CRM-CL/LIP should have little effect on a healthy brain, since it is not in an inflammatory state and that this type of AD therapy would most likely be given to patients in the later stages of AD.42
Aβ targeting nanosweepers
Luo et al97 have developed “nanosweepers” (M3) that not only capture extracellular Aβ and direct it into cells but also upregulate autophagy and digestion of Aβ. The nanosweepers are composed of a cationic chitosan core with PEGylated GKLVFF (that can co-assemble with Aβ) and Becln-1 (that induces autophagy to degrade Aβ). They work by capturing and co-assembling with extracellular Aβ, inhibiting toxic Aβ formation, then specifically directing Aβ into cells and activating autophagy. This strategy has been shown to degrade insoluble Aβ from a level of 1,539 down to 914 ng/mg, and soluble Aβ from 585 to 190 ng/mg, in APPswe/PS1dE9 transgenic mice, and also increase neuronal cell viability. M3 is a multifunctional peptide-polymer that could provide a novel therapeutic approach for the treatment of AD, by clearance of Aβ.97 By design, it is not a good aggregation inhibitor and could be useful in combination with additional therapies.
Future prospects
The majority of studies employing liposomal carrier systems to target the CNS are still restricted to early experimental work on cell and animal models, or follow on preclinical development, with none so far entering human clinical trials. Further investment and research is required to develop and optimize these carrier systems for AD therapy, since many of the studies considered in this article report promising findings, with a drive toward early clinical testing. A reoccurring problem is that some of the modifications on their own do not allow sufficient penetration through the BBB, or adequate targeting for AD therapy. Since liposomes have great potential for heavy functionalization, future steps should incorporate several different modifications into the same liposomes that can work together in synergy.
Furthermore, current systems in development have tended to target only Aβ, due to its fundamental role in AD, yet liposomes have the potential to be adapted to incorporate a therapeutic cocktail of molecules that aid BBB transport and are able to treat multiple areas of AD pathology. Further studies could prevent the formation of tau tangles, by using an anti-tau antibody, or tau-directed peptides, or small molecule inhibitors, in addition to preventing formation or dissolution of amyloid plaques, and targeting of phosphorylation cascades. A therapeutic system that can target Aβ, tau, neuronal survival, and apoptosis, and cholinergic neurotransmission, for example, could become the gold standard treatment for AD; liposomes are certainly a good candidate for the development of such a therapy. CPPs have great potential for AD therapy, yet their non-specific uptake by peripheral tissue limits their use. TAT peptide, which is derived from HIV, is an example of utilizing aspects of pathogens for therapeutic purposes. The future of CPPs for brain delivery could involve using trans-membrane peptide domains derived from trypanosomes,98 or the Zika virus,99 both of which contain peptides that target enhanced uptake into the brain.
Drawbacks and barriers
There remain some issues for liposomes as a drug delivery system. There is an increased cost when they are used, but this is ameliorated by the fact that they may allow a drug to reach its target with a liposome, but not without, or the overall dose may be decreased as a greater proportion of the drug reaches its target. They are not suitable for oral delivery because they will rupture due to osmotic shock when entering the hypotonic environment of the stomach (depending on the osmolarity of the internal aqueous filling of the liposome) and the lipids will be digested in the small intestine. Once in the bloodstream, the majority of liposomes are taken up non-specifically by the mononuclear phagocyte system, and this includes those with a stealth coating. This reduces the number available to be delivered to the brain, although this can be overcome by administering a larger dose in a single bolus, rather than multiple smaller doses.100 There are two main strategies for crossing the BBB (see “BBB transport” section) by either targeting a transporter system or by use of a CPP. The former approach may lead to saturation of the transporter system, whereas this is less of a limiting factor in the latter as it is non-specific. Finally, as this is a relatively new technology, there are still some technical issues regarding the supply of liposome products to the consumer in terms of sterility and product shelf life. Liposomes are not suitable for heat sterilization, but bacterial contamination can be avoided by filtration due to the small size of the liposomes.101 In terms of stability, liposomes in suspension will eventually start to fuse together, changing the size to the point at which they become ineffective as a delivery system. To avoid this, the liposomes can be lyophilized or spray dried in the presence of trehalose or sucrose and will retain their size on rehydration.102,103
Conclusion
Despite these challenges, the use of liposomal carriers is proving to be a potentially good strategy for future therapy of AD. They have a unique ability for modification to cross the BBB, while also showing some evidence of effectiveness without brain delivery, via the peripheral sink effect. Multiple molecules are able to enhance BBB transport, and many others allow targeting toward key molecular systems involved in AD pathogenesis. It is likely that future development will focus mainly on early intervention to slow down the progression of this disease, rather than attempting to reverse it in more advanced cases. Current study is moving toward targeting more than just one aspect of brain pathology, using multifunctional liposomes, and this holds great promise for the future, but is currently in the very early stages of development. Further study and substantial funding will be required to achieve success, so that this type of strategy can be driven into human clinical trials and beyond.
Acknowledgments
Research in our own laboratory on peptide-liposome-based therapy has received funding from the European Community’s Seventh Framework Program (FP7/2007–2013) under grant agreement no 212043, Alzheimer’s Research UK, The Alzheimer’s Society UK, The Sir John Fisher Foundation, and Lancaster University’s “Defying Dementia” charity.
Disclosure
Lancaster University has a granted patent on intellectual property related to this area of research based on the inventions of DA and MT. The other authors (CR and NF) report no conflicts of interest in this work.
References
World Health Organization. Neurological Disorders: Public Health Challenges. Geneva: WHO; 2006. | ||
Prince M, Ali G-C, Guerchet M, Prina AM, Albanese E, Wu Y-T. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimers Res Ther. 2016;8(1):13. | ||
Krstic D, Knuesel I. Deciphering the mechanism underlying late-onset Alzheimer disease. Nat Rev Neurol. 2013;9(1):25–34. | ||
Karran E, de Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem. 2016;139 (Suppl 2):237–252. | ||
Murphy MP, Levine H. Alzheimer’s disease and the amyloid-β peptide. J Alzheimers Dis. 2010;19(1):311–323. | ||
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. | ||
Lipton SA, Gu Z, Nakamura T. Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders. Int Rev Neurobiol. 2007;82:1–27. | ||
Moreira PI, Siedlak SL, Aliev G, et al. Oxidative stress mechanisms and potential therapeutics in Alzheimer disease. J Neural Transm. 2005;112(7):921–932. | ||
Oddo S, Laferla FM. The role of nicotinic acetylcholine receptors in Alzheimer’s disease. J Physiol Paris. 2006;99(2–3):172–179. | ||
Hansen RA, Gartlehner G, Webb AP, Morgan LC, Moore CG, Jonas DE. Efficacy and safety of donepezil, galantamine, and rivastigmine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. Clin Interv Aging. 2008;3(2):211–225. | ||
Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2013;6(1):19–33. | ||
Brown D, Superti-Furga G. Rediscovering the sweet spot in drug discovery. Drug Discov Today. 2003;8(23):1067–1077. | ||
Szuromi P, Vinson V, Marshall E. Rethinking drug discovery – Introduction. Science. 2004;303(5665):1795. | ||
Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5(12):993–996. | ||
Gregori M, Masserini M, Mancini S. Nanomedicine for the treatment of Alzheimer’s disease. Nanomedicine. 2015;10(7):1203–1218. | ||
Wolburg H, Noell S, Mack A, Wolburg-Buchholz K, Fallier-Becker P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009;335(1):75–96. | ||
Lai F, Fadda AM, Sinico C. Liposomes for brain delivery. Expert Opin Drug Deliv. 2013;10(7):1003–1022. | ||
Vieira DB, Gamarra LF. Getting into the brain: liposome-based strategies for effective drug delivery across the blood-brain barrier. Int J Nanomedicine. 2016;11:5381–5414. | ||
Noble GT, Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. Ligand-targeted liposome design: challenges and fundamental considerations. Trends Biotechnol. 2014;32(1):32–45. | ||
Bhowmik A, Khan R, Ghosh MK. Blood brain barrier: a challenge for effectual therapy of brain tumors. Biomed Res Int. 2015;2015:1–20. | ||
Sánchez-Navarro M, Giralt E, Teixidó M. Blood-brain barrier peptide shuttles. Curr Opin Chem Biol. 2017;38:134–140. | ||
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. | ||
Chen Y, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv Drug Deliv Rev. 2012;64(7):640–665. | ||
Hersh DS, Wadajkar AS, Roberts N, et al. Evolving drug delivery strategies to overcome the blood brain barrier. Curr Pharm Des. 2016;22(9):1177–1193. | ||
Agrawal M, Ajazuddin, Tripathi DK, et al. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J Control Release. 2017;260:61–77. | ||
Li W, Zhou Y, Zhao N, Hao B, Wang X, Kong P. Pharmacokinetic behavior and efficiency of acetylcholinesterase inhibition in rat brain after intranasal administration of galanthamine hydrobromide loaded flexible liposomes. Environ Toxicol Pharmacol. 2012;34(2):272–279. | ||
Zheng X, Shao X, Zhang C, et al. Intranasal H102 peptide-loaded liposomes for brain delivery to treat Alzheimer’s disease. Pharm Res. 2015;32(12):3837–3849. | ||
Illum L. Nasal drug delivery – recent developments and future prospects. J Control Release. 2012;161(2):254–263. | ||
Gao H. Progress and perspectives on targeting nanoparticles for brain drug delivery. Acta Pharm Sin B. 2016;6(4):268–286. | ||
Patching SG. Glucose transporters at the blood-brain barrier: Function, regulation and gateways for drug delivery. Mol Neurobiol. 2017;54(2):1046–1077. | ||
Arumugam K, Subramanian GS, Mallayasamy SR, Averineni RK, Reddy MS, Udupa N. A study of rivastigmine liposomes for delivery into the brain through intranasal route. Acta Pharm. 2008;58(3):287–297. | ||
Mutlu NB, Değim Z, Yilmaz Ş, Eşsiz D, Nacar A. New perspective for the treatment of Alzheimer diseases: liposomal rivastigmine formulations. Drug Dev Ind Pharm. 2011;37(7):775–789. | ||
Confaloni A, Tosto G, Tata AM. Promising therapies for Alzheimer’s disease. Curr Pharm Des. 2016;22(14):2050–2056. | ||
Spuch C, Navarro C. Liposomes for targeted delivery of active agents against neurodegenerative diseases (Alzheimer’s disease and Parkinson’s disease). J Drug Deliv. 2011;469679. | ||
Joshi S, Singh-Moon RP, Ellis JA, et al. Cerebral hypoperfusion-assisted intra-arterial deposition of liposomes in normal and glioma-bearing rats. Neurosurgery. 2015;76(1):92–100. | ||
Abra RM, Hunt CA. Liposome disposition in vivo. III. Dose and vesicle-size effects. Biochim Biophys Acta. 1981;666(3):493–503. | ||
Harashima H, Kiwada H. Liposomal targeting and drug delivery: kinetic consideration. Adv Drug Deliv Rev. 1996;19(3):425–444. | ||
Fanciullino R, Ciccolini J. Liposome-encapsulated anticancer drugs: still waiting for the magic bullet? Curr Med Chem. 2009;16(33):4361–4373. | ||
Allen TM. Liposomes. Opportunities in drug delivery. Drugs. 1997;54(Suppl 4):8–14. | ||
Briuglia ML, Rotella C, McFarlane A, Lamprou DA. Influence of cholesterol on liposome stability and on in vitro drug release. Drug Deliv Transl Res. 2015;5(3):231–242. | ||
Alarcón JM, Brito JA, Hermosilla T, Atwater I, Mears D, Rojas E. Ion channel formation by Alzheimer’s disease amyloid β-peptide (Aβ40) in unilamellar liposomes is determined by anionic phospholipids. Peptides. 2006;27(1):95–104. | ||
Kuo YC, Lin CY, Li JS, Lou YI. Wheat germ agglutinin-conjugated liposomes incorporated with cardiolipin to improve neuronal survival in Alzheimer’s disease treatment. Int J Nanomedicine. 2017;12:1757–1774. | ||
Palchetti S, Colapicchioni V, Digiacomo L, et al. The protein corona of circulating PEGylated liposomes. Biochim Biophys Acta. 2016;1858(2):189–196. | ||
Veronese FM, Mero A. The impact of PEGylation on biological therapies. BioDrugs. 2008;22(5):315–329. | ||
Ballatori N, Krance SM, Marchan R, Hammond CL. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol Aspects Med. 2009;30(1–2):13–28. | ||
Gaillard PJ, Appeldoorn CC, Dorland R, et al. Pharmacokinetics, brain delivery, and efficacy in brain tumor-bearing mice of glutathione pegylated liposomal doxorubicin (2B3-101). PLoS One. 2014;9(1):10. | ||
Rotman M, Welling MM, Bunschoten A, et al. Enhanced glutathione PEGylated liposomal brain delivery of an anti-amyloid single domain antibody fragment in a mouse model for Alzheimer’s disease. J Control Release. 2015;203:40–50. | ||
Zhang CX, Zhao WY, Liu L, et al. A nanostructure of functional targeting epirubicin liposomes dually modified with aminophenyl glucose and cyclic pentapeptide used for brain glioblastoma treatment. Oncotarget. 2015;6(32):32681–32700. | ||
Xie F, Yao N, Qin Y, et al. Investigation of glucose-modified liposomes using polyethylene glycols with different chain lengths as the linkers for brain targeting. Int J Nanomedicine. 2012;7:163–175. | ||
Li H, Qian ZM. Transferrin/transferrin receptor-mediated drug delivery. Med Res Rev. 2002;22(3):225–250. | ||
Fishman JB, Rubin JB, Handrahan JV, Connor JR, Fine RE. Receptor-mediated transcytosis of transferrin across the blood-brain barrier. J Neurosci Res. 1987;18(2):299–304. | ||
Gao JQ, Lv Q, Li LM, et al. Glioma targeting and blood-brain barrier penetration by dual-targeting doxorubincin liposomes. Biomaterials. 2013;34(22):5628–5639. | ||
Chen ZL, Huang M, Wang XR, et al. Transferrin-modified liposome promotes α-mangostin to penetrate the blood-brain barrier. Nanomedicine. 2016;12(2):421–430. | ||
Chen YC, Chiang CF, Chen LF, Liang PC, Hsieh WY, Lin WL. Polymersomes conjugated with des-octanoyl ghrelin and folate as a BBB-penetrating cancer cell-targeting delivery system. Biomaterials. 2014;35(13):4066–4081. | ||
Papadia K, Markoutsa E, Mourtas S, et al. Multifunctional LUV liposomes decorated for BBB and amyloid targeting. A. In vitro proof-of-concept. Eur J Pharm Sci. 2017;101:140–148. | ||
Song XL, Liu S, Jiang Y, et al. Targeting vincristine plus tetrandrine liposomes modified with DSPE-PEG2000-transferrin in treatment of brain glioma. Eur J Pharm Sci. 2017;96:129–140. | ||
Chen H, Tang L, Qin Y, et al. Lactoferrin-modified procationic liposomes as a novel drug carrier for brain delivery. Eur J Pharm Sci. 2010;40(2):94–102. | ||
Kawamata T, Tooyama I, Yamada T, Walker DG, McGeer PL. Lactotransferrin immunocytochemistry in Alzheimer and normal human brain. Am J Pathol. 1993;142(5):1574–1585. | ||
Zhong Z-R, Liu J, Deng Y, et al. Preparation and characterization of a novel nonviral gene transfer system: procationic-liposome-protamine-DNA complexes. Drug Deliv. 2007;14(3):177–183. | ||
Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240(4852):622–630. | ||
Gobbi M, Re F, Canovi M, et al. Lipid-based nanoparticles with high binding affinity for amyloid-β1-42 peptide. Biomaterials. 2010;31(25):6519–6529. | ||
Re F, Cambianica I, Sesana S, et al. Functionalization with ApoE-derived peptides enhances the interaction with brain capillary endothelial cells of nanoliposomes binding amyloid-β peptide. J Biotechnol. 2011;156(4):341–346. | ||
Lane-Donovan C, Wong WM, Durakoglugil MS, et al. Genetic restoration of plasma ApoE improves cognition and partially restores synaptic defects in ApoE-deficient mice. J Neurosci. 2016;36(39):10141–10150. | ||
Lindgren M, Hällbrink M, Prochiantz A, Langel U. Cell-penetrating peptides. Trends Pharmacol Sci. 2000;21(3):99–103. | ||
Gregori M, Taylor M, Salvati E, et al. Retro-inverso peptide inhibitor nanoparticles as potent inhibitors of aggregation of the Alzheimer’s Aβ peptide. Nanomedicine. 2017;13(2):723–732. | ||
Vivès E, Richard JP, Rispal C, Lebleu B. TAT peptide internalization: seeking the mechanism of entry. Curr Protein Pept Sci. 2003;4(2):125–132. | ||
Parthsarathy V, McClean PL, Hölscher C, et al. A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. PLoS One. 2013;8(9):11. | ||
Kamei N, Tanaka M, Choi H, et al. Effect of an enhanced nose-to-brain delivery of insulin on mild and progressive memory loss in the senescence-accelerated mouse. Mol Pharm. 2017;14(3):916–927. | ||
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. | ||
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | ||
Balducci C, Mancini S, Minniti S, et al. Multifunctional liposomes reduce brain β-amyloid burden and ameliorate memory impairment in Alzheimer’s disease mouse models. J Neurosci. 2014;34(42):14022–14031. | ||
Bana L, Minniti S, Salvati E, et al. Liposomes bi-functionalized with phosphatidic acid and an ApoE-derived peptide affect Aβ aggregation features and cross the blood-brain-barrier: implications for therapy of Alzheimer disease. Nanomedicine. 2014;10(7):1583–1590. | ||
Matsuoka Y, Saito M, Lafrancois J, et al. Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to β-amyloid. J Neurosci. 2003;23(1):29–33. | ||
Mancini S, Minniti S, Gregori M, et al. The hunt for brain Aβ oligomers by peripherally circulating multi-functional nanoparticles: Potential therapeutic approach for Alzheimer disease. Nanomedicine. 2016;12(1):43–52. | ||
Friedlich AL, Butcher LL. Involvement of free oxygen radicals in β-amyloidosis: an hypothesis. Neurobiol Aging. 1994;15(4):443–455. | ||
Tabner BJ, El-Agnaf OM, Turnbull S, et al. Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia. J Biol Chem. 2005;280(43):35789–35792. | ||
Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77(6):817–827. | ||
Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, α-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med Overseas Ed. 1997;336(17):1216–1222. | ||
Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21(21):8370–8377. | ||
Yanagisawa D, Amatsubo T, Morikawa S, et al. In vivo detection of amyloid β deposition using 19F magnetic resonance imaging with a 19F-containing curcumin derivative in a mouse model of Alzheimer’s disease. Neuroscience. 2011;184:120–127. | ||
Lee I, Yang J, Lee JH, Choe YS. Synthesis and evaluation of 1-(4-[18F]fluoroethyl)-7-(4′-methyl)curcumin with improved brain permeability for β-amyloid plaque imaging. Bioorg Med Chem Lett. 2011;21(19):5765–5769. | ||
Yanagisawa D, Ibrahim NF, Taguchi H, et al. Curcumin derivative with the substitution at C-4 position, but not curcumin, is effective against amyloid pathology in APP/PS1 mice. Neurobiol Aging. 2015;36(1):201–210. | ||
Taylor M, Moore S, Mourtas S, et al. Effect of curcumin-associated and lipid ligand-functionalized nanoliposomes on aggregation of the Alzheimer’s Aβ peptide. Nanomedicine. 2011;7(5):541–550. | ||
Mourtas S, Lazar AN, Markoutsa E, Duyckaerts C, Antimisiaris SG. Multifunctional nanoliposomes with curcumin-lipid derivative and brain targeting functionality with potential applications for Alzheimer disease. Eur J Med Chem. 2014;80:175–183. | ||
Austen BM, Paleologou KE, Ali SA, Qureshi MM, Allsop D, El-Agnaf OM. Designing peptide inhibitors for oligomerization and toxicity of Alzheimer’s β-amyloid peptide. Biochemistry. 2008;47(7):1984–1992. | ||
Taylor M, Moore S, Mayes J, et al. Development of a proteolytically stable retro-inverso peptide inhibitor of β-amyloid oligomerization as a potential novel treatment for Alzheimer’s disease. Biochemistry. 2010;49(15):3261–3272. | ||
Sherer M, Fullwood NJ, Taylor M, Allsop D. A preliminary electron microscopic investigation into the interaction between Aβ1-42 peptide and a novel nanoliposome-coupled retro-inverso peptide inhibitor, developed as a potential treatment for Alzheimer’s disease. J Phys Conference Series. 2015;644:012040. | ||
Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148(6):1188–1203. | ||
Tenovuo O. Central acetylcholinesterase inhibitors in the treatment of chronic traumatic brain injury-clinical experience in 111 patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(1):61–67. | ||
Spencer CM, Noble S. Rivastigmine. A review of its use in Alzheimer’s disease. Drugs Aging. 1998;13(5):391–411. | ||
Illum L. Nasal drug delivery – possibilities, problems and solutions. J Control Release. 2003;87(1–3):187–198. | ||
Yang ZZ, Zhang YQ, Wang ZZ, Wu K, Lou JN, Qi XR. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int J Pharm. 2013;452(1–2):344–354. | ||
Yadav AV, Murthy MS, Shete AS, Sakhare S. Stability aspects of liposomes. Indian J Pharm Educ Res. 2011;45(4):402–413. | ||
Nageeb El-Helaly S, Abd Elbary A, Kassem MA, El-Nabarawi MA. Electrosteric stealth Rivastigmine loaded liposomes for brain targeting: preparation, characterization, ex vivo, bio-distribution and in vivo pharmacokinetic studies. Drug Deliv. 2017;24(1):692–700. | ||
Kreuter J, Alyautdin RN, Kharkevich DA, Ivanov AA. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). Brain Res. 1995;674(1):171–174. | ||
Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16(18):5727–5740. | ||
Luo Q, Lin Y-X, Yang P-P, et al. A self-destructive nanosweeper that captures and clears amyloid β-peptides. Nat Commun. 2018;9(1):12. | ||
Goto Y, Duthie MS, Kawazu S, Inoue N, Carter D. Biased cellular locations of tandem repeat antigens in African trypanosomes. Biochem Biophys Res Commun. 2011;405(3):434–438. | ||
Jun S-R, Wassenaar TM, Wanchai V, Patumcharoenpol P, Nookaew I, Ussery DW. Suggested mechanisms for Zika virus causing microcephaly: what do the genomes tell us? BMC Bioinformatics. 2017;18(S14):12. | ||
van Etten EW, Ten Kate MT, Snijders SV, Bakker-Woudenberg IA. Administration of liposomal agents and blood clearance capacity of the mononuclear phagocyte system. Antimicrob Agents Chemother. 1998;42(7):1677–1681. | ||
Toh M-R, Chiu GNC. Liposomes as sterile preparations and limitations of sterilisation techniques in liposomal manufacturing. Asian J Pharm Sci. 2013;8(2):88–95. | ||
Payton NM, Wempe MF, Xu Y, Anchordoquy TJ. Long-term storage of lyophilized liposomal formulations. J Pharm Sci. 2014;103(12):3869–3878. | ||
Chen C, Han D, Cai C, Tang X. An overview of liposome lyophilization and its future potential. J Control Release. 2010;142(3):299–311. | ||
Qian ZM, Li H, Sun H, Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol Rev. 2002;54(4):561–587. | ||
Guo L, Ren J, Jiang X. Perspectives on brain-targeting drug delivery systems. Curr Pharm Biotechnol. 2012;13(12):2310–2318. | ||
Fillebeen C, Descamps L, Dehouck MP, et al. Receptor-mediated transcytosis of lactoferrin through the blood-brain barrier. J Biol Chem. 1999;274(11):7011–7017. | ||
Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269(14):10444–10450. | ||
Futaki S, Ohashi W, Suzuki T, et al. Stearylated arginine-rich peptides: a new class of transfection systems. Bioconjug Chem. 2001;12(6):1005–1011. | ||
Lu W, Zhang Y, Tan YZ, Hu KL, Jiang XG, Fu SK. Cationic albumin-conjugated pegylated nanoparticles as novel drug carrier for brain delivery. J Control Release. 2005;107(3):428–448. | ||
Tseng YL, Liu JJ, Hong RL. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and tat: a kinetic and efficacy study. Mol Pharmacol. 2002;62(4):864–872. | ||
Henriques ST, Castanho MA. Translocation or membrane disintegration? Implication of peptide-membrane interactions in pep-1 activity. J Pept Sci. 2008;14(4):482–487. | ||
Gao C, Mao S, Ditzel HJ, et al. A cell-penetrating peptide from a novel pVII-pIX phage-displayed random peptide library. Bioorg Med Chem. 2002;10(12):4057–4065. | ||
Koren E, Torchilin VP. Cell-penetrating peptides: breaking through to the other side. Trends Mol Med. 2012;18(7):385–393. | ||
Pooga M, Hällbrink M, Zorko M, Langel U. Cell penetration by transportan. FASEB J. 1998;12(1):67–77. | ||
Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97(24):13003–13008. | ||
de Coupade C, Fittipaldi A, Chagnas V, et al. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem J. 2005;390(Pt 2):407–418. | ||
Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell. 2008;100(4):201–217. | ||
Johansson HJ, El-Andaloussi S, Holm T, et al. Characterization of a novel cytotoxic cell-penetrating peptide derived from p14ARF protein. Molecular Therapy. 2008;16(1):115–123. | ||
Taylor BN, Mehta RR, Yamada T, et al. Noncationic peptides obtained from azurin preferentially enter cancer cells. Cancer Res. 2009;69(2):537–546. | ||
Magzoub M, Sandgren S, Lundberg P, et al. N-terminal peptides from unprocessed prion proteins enter cells by macropinocytosis. Biochem Biophys Res Commun. 2006;348(2):379–385. | ||
Oehlke J, Krause E, Wiesner B, Beyermann M, Bienert M. Extensive cellular uptake into endothelial cells of an amphipathic β-sheet forming peptide. FEBS Lett. 1997;415(2):196–199. | ||
Sadler K, Eom KD, Yang JL, Dimitrova Y, Tam JP. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry. 2002;41(48):14150–14157. | ||
Rhee M, Davis P. Mechanism of uptake of C105Y, a novel cell-penetrating peptide. J Biol Chem. 2006;281(2):1233–1240. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.