Back to Journals » International Journal of Nanomedicine » Volume 13
Liposomal nanoparticle-based conserved peptide influenza vaccine and monosodium urate crystal adjuvant elicit protective immune response in pigs
Authors Dhakal S ![]() , Cheng X, Salcido J, Renu S
, Cheng X, Salcido J, Renu S ![]() , Bondra K, Lakshmanappa YS
, Bondra K, Lakshmanappa YS ![]() , Misch C, Ghimire S, Feliciano-Ruiz N, Hogshead B, Krakowka S, Carson K, McDonough J, Lee CW, Renukaradhya GJ
, Misch C, Ghimire S, Feliciano-Ruiz N, Hogshead B, Krakowka S, Carson K, McDonough J, Lee CW, Renukaradhya GJ ![]()
Received 1 July 2018
Accepted for publication 18 August 2018
Published 24 October 2018 Volume 2018:13 Pages 6699—6715
DOI https://doi.org/10.2147/IJN.S178809
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Santosh Dhakal,1,2 Xingguo Cheng,3 John Salcido,3 Sankar Renu,1,2 Kathy Bondra,1,2 Yashavantha Shaan Lakshmanappa,1,2 Christina Misch,1,2 Shristi Ghimire,1,2 Ninoshkaly Feliciano-Ruiz,1,2 Bradley Hogshead,1,2 Steven Krakowka,4 Kenneth Carson,3 Joseph McDonough,3 Chang Won Lee,1,2 Gourapura J Renukaradhya1,2
1Food Animal Health Research Program, Ohio Agricultural Research and Development Center, Wooster, OH 44691, USA; 2Department of Veterinary Preventive Medicine, College of Veterinary Medicine, The Ohio State University, Columbus, OH 43210, USA; 3Pharmaceuticals and Bioengineering Department, Chemistry and Chemical Engineering Division, Southwest Research Institute, San Antonio, TX 78238-0510, USA; 4The Department of Veterinary Biosciences, College of Veterinary Medicine, The Ohio State University, Columbus, OH, USA
Background: Influenza (flu) is a constant threat to humans and animals, and vaccination is one of the most effective ways to mitigate the disease. Due to incomplete protection induced by current flu vaccines, development of novel flu vaccine candidates is warranted to achieve greater efficacy against constantly evolving flu viruses.
Methods: In the present study, we used liposome nanoparticle (<200 nm diameter)-based subunit flu vaccine containing ten encapsulated highly conserved B and T cell epitope peptides to induce protective immune response against a zoonotic swine influenza A virus (SwIAV) H1N1 challenge infection in a pig model. Furthermore, we used monosodium urate (MSU) crystals as an adjuvant and co-administered the vaccine formulation as an intranasal mist to flu-free nursery pigs, twice at 3-week intervals.
Results: Liposome peptides flu vaccine delivered with MSU adjuvant improved the hemagglutination inhibition antibody titer and mucosal IgA response against the SwIAV challenge and also against two other highly genetically variant IAVs. Liposomal vaccines also enhanced the frequency of peptides and virus-specific T-helper/memory cells and IFN-γ response. The improved specific cellular and mucosal humoral immune responses in adjuvanted liposomal peptides flu vaccine partially protected pigs from flu-induced fever and pneumonic lesions, and reduced the nasal virus shedding and viral load in the lungs.
Conclusion: Overall, our study shows great promise for using liposome and MSU adjuvant-based subunit flu vaccine through the intranasal route, and provides scope for future, pre-clinical investigations in a pig model for developing potent human intranasal subunit flu vaccines.
Keywords: influenza A virus peptides, liposome nanoparticles, monosodium urate crystal adjuvant, intranasal vaccination, swine influenza virus, pigs
Introduction
The WHO estimates that approximately three to five million people are infected with seasonal influenza (flu) annually and approximately 290,000 to 650,000 people die as a result worldwide.1,2 The highest risk of flu-associated complications occurs among children aged less than 5 years, pregnant women, elderly individuals, and people with chronic medical conditions. Influenza A virus (IAV) subtypes H1N1 and H3N2 are currently the major circulating viruses among humans. In pigs, IAV subtypes H1N1, H1N2, and H3N2 are circulating globally and cause considerable economic loss to the swine industry through morbidity, loss of body weight gain, increased time to market, medication and veterinary expenses.3 Due to the presence of receptors for both avian (α2,3-Gal) and human (α2,6-Gal) flu viruses in the respiratory tract, pigs can act as a source for novel zoonotic flu pathogens of pandemic potential.4 One such example is the 2009 pandemic IAV, where this swine origin IAV (SwIAV), H1N1, caused over 200,000 human deaths.5 Recently, a novel H3N2 SwIAV termed “H3N2 variant” with a matrix gene segment derived from 2009 pandemic H1N1 virus has caused over 370 human infections in the US.6
Vaccination is one of the most effective ways to prevent flu in humans and pigs. Current human flu vaccines are a mixture of either trivalent or quadrivalent IAV available in injectable forms (inactivated and recombinant vaccines) or as a nasal spray (live attenuated vaccines). However, these vaccines have limitations such as: i) they are most effective only when circulating viruses perfectly match the vaccine strains (homologous protection), and thus require vaccine virus updates every flu season; ii) uncertainty exists on selection of current and correct strains; iii) the production process is slow and complicated as it involves hazardous biological work; and iv) they are unable to prevent pandemics or protect against flu variants frequently generated by antigenic drift.7,8 Moreover, for bulk vaccine production all the selected virus strains must grow well in chicken eggs. It has been observed that certain virus strains of H3N2 subtype of IAV grow poorly in eggs,9 making it difficult to obtain candidate vaccine viruses in a timely manner. Similar to humans, multivalent inactivated vaccines are also being used in pigs predominantly as intramuscular injections. Swine flu vaccines also suffer from the limitations of human flu vaccines including: i) limited homologous but not heterologous protection; ii) lack of mucosal immune response in the respiratory tract; and in addition iii) infection of vaccinated pigs with a mismatched homosubtypic virus can induce more severe respiratory disease which has been attributed to the presence of non-neutralizing anti-hemagglutinin (HA) IgG.10–12 Such vaccine-enhanced respiratory disease can also occur in piglets with vaccine-induced maternal antibodies.13 All these suggest the necessity of developing an innovative flu vaccine to use in pigs, and also in humans with greater efficacy against a broad range of variants and subtypes.
Highly conserved peptides of flu virus have gained significant attention as a universal flu vaccine candidate in the last few decades. Among these peptides, the extracellular domain of M2 protein (M2e, 23 amino acids) is an attractive vaccine target for induction of broad protection, as it is highly conserved among all the avian and mammalian flu viruses.14 The other strategies involving peptide-based universal flu vaccine include HA stalk domain and T cell-based multivalent conserved epitopes of IAV internal proteins.7,15,16 Compared to the conventional inactivated or attenuated vaccines, peptide-based vaccines are safer, easier to produce, and can be synthesized in large quantities at cheaper cost when needed. However, they are less immunogenic in the absence of appropriate adjuvant/s and a suitable delivery system which protect the peptides from proteolytic degradation and facilitate controlled release of antigens to the immune cells.17
Biodegradable and biocompatible polymer-based nanoparticles are an important endeavor of the 21st century and have been utilized as a potent carrier system, and importantly they are US Food and Drug Administration (FDA)-approved for drug delivery.18–20 Synthetic liposome-based vaccine has been a great success in vaccine development. For example, the FDA-approved liposome-encapsulated vaccine (Epaxal) for hepatitis A has shown superior clinical efficacy (single dose, 100% protection with no adverse effects) in infants compared with a traditional vaccine (only 66.6% protection and requires repeated boosters).21 Encapsulation of vaccine antigens in nanoparticles has proven beneficial in activating the immune system through augmenting internalization, processing and presentation of entrapped antigens by the professional antigen presenting cells (APCs).22 Therefore, liposome-encapsulated subunit flu vaccine is a promising universal flu vaccine, as it can protect humans and swine from a wide range of variant flu strains. Additionally, for efficient mitigation of flu virus infections, induction of robust mucosal immune response is of the utmost importance, and the intranasal route of vaccine delivery can be instrumental as it produces both mucosal and systemic immune responses.23 Recently, we used poly(lactic-co-glycolic acid) polymer-based nanoparticles to encapsulate M2e and four highly conserved IAV T and B cell epitope peptides, and evaluated the candidate vaccine’s efficacy in pigs administered intranasally with no secondary adjuvant against a virulent zoonotic IAV H1N1 challenge infection. Our results showed that PLGA encapsulation of peptides elicited robust peptide-specific T cell response, which partially helped in the clearance of the challenge virus from the lungs. However, this PLGA vaccine did not improve induction of mucosal IgA and systemic antibody responses.24 In this study, our aim was to achieve induction of robust mucosal IgA, cellular and humoral immune responses against variant flu viruses in pigs. We used liposomes as an antigen carrier system incorporating ten highly conserved T and B cell epitope peptides, and co-administered the candidate vaccine intranasally as a mist with adjuvant monosodium urate (MSU). MSU is a naturally occurring crystal in humans, and it is safe and non-toxic.25 Our results suggested that the liposomal subunit flu vaccine formulation enhanced both cellular and humoral immune responses leading to an enhanced protective immune response in pigs.
Material and methods
Cells and viruses
Madin-Darby canine kidney (MDCK) epithelial cells (CRL-2285, American Type Culture Collection [ATCC] Manassas, VA, USA) were used to grow flu viruses used in the study. Field isolate of SwIAV A/SW/OH/24366/2007 (H1N1-OH7, γ lineage) was used for challenge infection of pigs.26 It is a virulent zoonotic virus with 100% identical genome sequence to the human virus isolated from infected individuals at an Ohio county fair. For cross-protective antibody analysis we used other field isolates of SwIAV, A/SW/OH/FAH10-1/10 (H1N2-OH10, δ1 lineage)27 and A/turkey/OH/313053/2004 (H3N2-OH4).28 The H1N2-OH10 isolate has nucleoprotein (NP) and M genes of the 2009 pandemic H1N1 IAV, and it is heterologous to the H1N1-OH7 SwIAV used in our challenge infection.29 The H3N2-OH4 is heterosubtypic to challenge virus H1N1-OH7, which was isolated from turkeys in 2004 in a farm in Ohio and shown to transmit between swine and turkeys.28
Peptides’ selection for vaccine preparation
A total of ten highly conserved and well-characterized IAV peptides were selected for this study (Table 1). All peptides were chemically synthesized. Peptide 1 (M2e) represents the extracellular domain of SwIAV M2 protein.30 Peptide 2 is a 34-mer peptide derived from HA1 domain of the human A/South Carolina/1/18 (H1N1) IAV.24,31 Peptides 3, 4, and 5 represent the IAV HA of human isolate A/New Caledonia/20/99,24,31 avian isolate A/VietNam/1203/04 (H5N1),31 and human isolate A/Catalonia/63/2009 (H1N1) IAV, respectively.31 Peptides 6–10 were from NP, RNA-dependent-RNA PB1, neuraminidase (NA), and polymerase acidic (PA) proteins. These last five peptides are nonamers of 2009 pandemic swine origin IAV A/Beijing/01/2009 (H1N1) strain, and are predicted to be potential cytotoxic T lymphocyte (CTL) epitope peptides.32 Peptides 2, 3, 9, and 10 were used earlier in flu vaccination trials in pigs.24,31
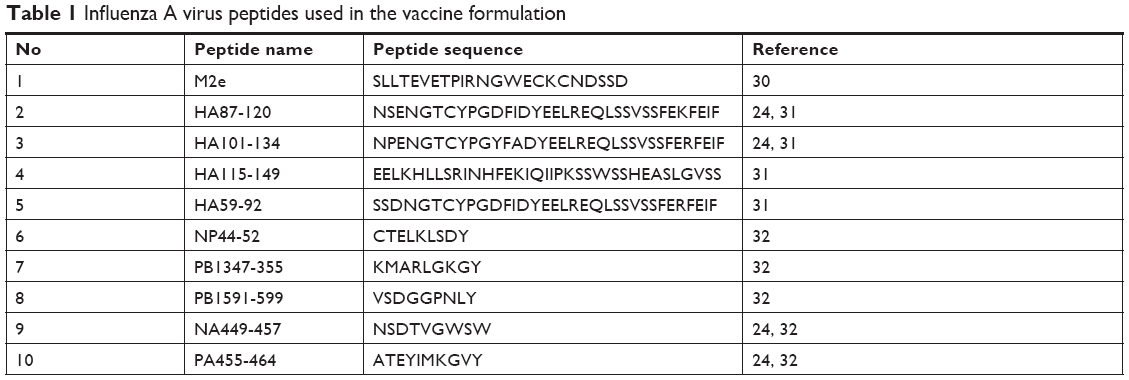 | Table 1 Influenza A virus peptides used in the vaccine formulation |
Liposomal encapsulation of peptides
M2e and peptides were encapsulated in liposomes using the method described previously.33 Briefly, 1 g soy lecithin, 125 mg cholesterol, and 24 μL alpha tocopherol were dissolved in 25 mL MeOH/CHCl3 (1:1 ratio) to form a clear solution. The 25 mL lipid solution was aliquoted (5 mL each) into glass vials (40 mL). Each vial was placed on a Rotavapor for removal of the solvent to form lipid cakes. Each vial was flushed with nitrogen and dried in vacuum for 1.5 hours to remove residual solvent. The lipid cake was hydrated with 2 mL PBS, freeze-thawed five times, added 7 mL PBS and was extruded seven times through a Lipex extruder fitted with 0.2 μm and 0.1 μm membrane filters, and the total volume was made up to 10.5 mL. This procedure resulted in formation of liposomes. One of the five vials was used as a negative control (without IAV peptides). The other four vials with lipid cakes were used to encapsulate ten peptides (Table 1) based on their solubility and charge characteristics. Acidic peptides 1, 2, 6, 8, and 9 (2 mg each dissolved in PBS pH 7.5) were used together to hydrate one vial of lipid cake. Two vials of lipid cakes were used to hydrate hydrophobic peptides 3 and 5, and basic peptides 4 and 7 separately (2 mg of each peptide dissolved in PBS pH 6). The last vial of lipid cake was used to hydrate the neutral peptide 10 (2 mg dissolved in PBS in 8 M urea). The four hydrated lipid cakes were combined, freeze-thawed five times, and 40.5 mL PBS was added to make up the total volume to 42 mL and it was extruded through 0.1 μm membrane to form a homogenous liposome preparation. Particle size, size distribution, and zeta potential of liposomal formulations were quantified using a particle sizer and Zeta potential analyzer (Brookhaven Instruments Corporation, Holtsville, NY, USA).
For determining the precise encapsulation efficiency of each of the peptides in our liposome peptide candidate vaccine, we selected a representative peptide from the acidic (#2), basic (#7), hydrophobic (#5), and neutral (#10) peptide groups (Table 2). These were encapsulated individually in liposomes as described previously. A method to separate the encapsulated peptide from non-encapsulated peptide was developed based on size exclusion column made from Sephadex G-50 beads (medium size).34 Briefly, fully PBS-hydrated Sephadex G-50 beads were loaded into a mini-column and centrifuged at 1,000× g for 3 minutes to remove excess buffer. An amount of 1 mL of liposomes was applied to the Sephadex beads and the mini-column was placed inside a 50 mL centrifuge tube and centrifuged at 100× g for 10 minutes followed by 1,000× g for 3 minutes to expel the liposomal material from the column into the test tube. An amount of 2 mL PBS was used to elute the non-encapsulated peptide and the eluate was recovered by centrifugation at 1,000× g for 3 minutes. The non-encapsulated peptide was quantified by UV-vis spectroscopy at 223 nm, the mass of peptide was calculated based on the elution volume, and eluted peptide concentration was determined by using a pre-determined calibration curve. The loading efficiency of each representative peptide was quantified using the formula: loading efficiency (%) = (mass of total peptide – mass of non-encapsulated peptide)/mass of total peptide ×100.
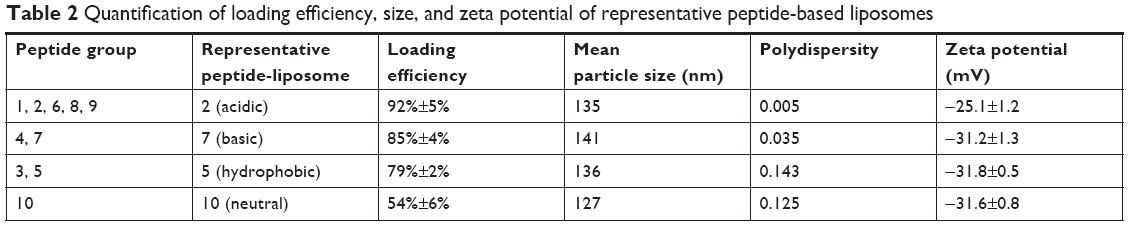 | Table 2 Quantification of loading efficiency, size, and zeta potential of representative peptide-based liposomes |
Synthesis of MSU crystal adjuvant
MSU crystals were synthesized by following the procedure described previously,35 which yielded crystals with a similar morphology and birefringence to those found in gout patients. Briefly, 1.68 g of solid uric acid was added to 400 mL sodium hydroxide solution (0.4 g of NaOH, 25 mM). The resultant opaque solution was allowed to remain overnight at 80°C and the filtrate was rinsed with cold distilled water three times and air dried in the fume hood for 2 days. The dried MSU particles were sieved to a size range of 1–5 μm in length and were nanosized in diameter. They were divided into multiple 5 mg aliquots and dispensed into individual vials and sterilized with ethylene trioxide. Imaging of MSU crystals was performed by spreading the crystals onto a carbon tape placed on an aluminum stub and sputter coated with gold/palladium for electrical conductivity. The morphology of MSU crystals was examined by a scanning electron microscope (FEI-Quanta 650; FEI company, Hillsboro, OR, USA).
Experimental design
Cesarean-delivered, colostrum-deprived, and bovine colostrum-fed Large White-Duroc crossbred piglets (n=26) were raised in our Biological Safety Labels 2 facility at Ohio Agricultural Research and Development Center. Piglets were confirmed seronegative for hemagglutination inhibition (HI) antibodies against IAV subtypes H1N1, H1N2, and H3N2. Piglets derived from four sows were randomly divided into five experimental groups (n=4–6 pigs/group) (Table 3).
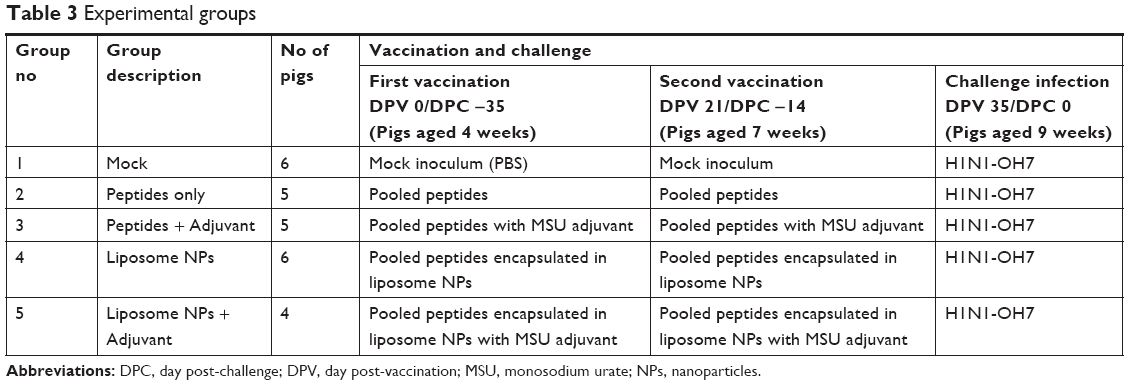 | Table 3 Experimental groups |
Vaccination, virus challenge, and collection of samples
Primary vaccination of piglets was performed at 4 weeks of age followed by booster vaccination after 3 weeks. Vaccine was delivered through the intranasal route as a mist using a multi-dose vaccine delivery device (Prima Tech USA, Kenansville, NC, USA) in the final volume of 2 mL in PBS per dose as described previously.36 Each dose of vaccine had 500 μg of the total ten peptides (50 μg of each peptide) either in soluble form or encapsulated in liposomes. A group of pigs received MSU adjuvant (5 mg/dose) co-administered with the vaccine formulation. A similar dose of 50 μg of each peptide was used in our recent PLGA NPs vaccine trial in pigs, delivered by intranasal route as a mist.24 One hour before intranasal delivery of the vaccine formulation in pigs, MSU microcrystals (Figure 1B) were mixed with the liposome suspension, and MSU crystals were not encapsulated in liposomes. MSU has not been used in pigs as an adjuvant but was tested in mice and proved to be a potent adjuvant; we used 5 mg/dose. Challenge infection was established using the virulent, zoonotic and highly infectious H1N1 SwIAV (6×106 TCID50 2 mL), 1 mL was administered intranasally as a mist and the other 1 mL intratracheally.36,37
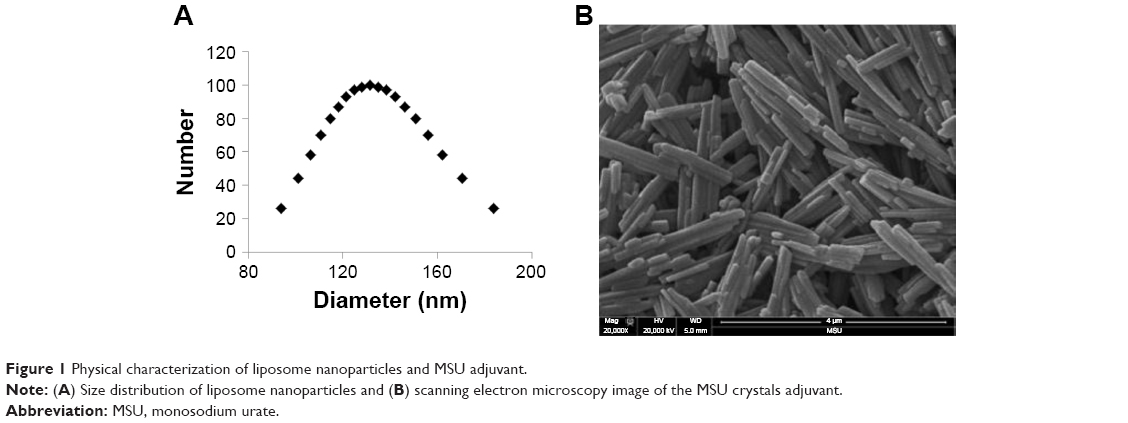 | Figure 1 Physical characterization of liposome nanoparticles and MSU adjuvant. |
Blood samples were collected at day post-vaccination (DPV) 0, 21, 35, and 41. From day post-challenge 0 (DPC 0) to euthanasia at DPC 6, rectal temperature of pigs was recorded daily. Nasal swab samples were collected at DPC 4 and 6 in 2 mL DMEM containing antibiotics and the clarified fluid aliquots were frozen and stored. Pigs were euthanized at DPC 6 and lungs were examined and scored for macroscopic pneumonic lesions. Average scores of all the lung lobe sections were considered for final grading as described previously.36 Bronchoalveolar lavage (BAL) fluid was collected using PBS.36 Aliquots of plasma, nasal swab fluid, and BAL fluid were stored at −80°C. Lung tissue samples were collected from the identical regions of the right apical, cardiac, and diaphragmatic lobes of each pig and fixed in 10% neutral buffered formalin for histopathological evaluation. Peripheral blood mononuclear cells (PBMCs) and tracheobronchial lymph nodes mononuclear cells (TBLN MNCs) were isolated at DPC 6. Cells were used in lymphocyte proliferation assay upon stimulation with live SwIAV or pooled peptides, and similarly re-stimulated cells were also subjected to flow cytometry to determine the frequency of lymphocyte subsets.36,37
Animals were maintained, cared for, fed with ad-libitum food and water, inoculated, had clinical samples collected, and were euthanized as per the approved protocol of the Institutional Animal Care and Use Committee at The Ohio State University. The liposome peptides vaccine and MSU adjuvant were prepared in Pharmaceuticals and Bioengineering Department, Southwest Research Institute, San Antonio, TX, by following the institution-approved biosafety procedures.
This study was carried out in strict accordance with the recommendations by the Public Health Service Policy, USDA Regulations, National Research Council’s Guide for the Care and Use of Laboratory Animals and the Federation of Animal Science Societies’ Guide for the Care and Use of Agricultural Animals in Agricultural Research and Teaching.
Virus titration and antibody analyses
Serial 10-fold dilutions of nasal swab fluid and BAL fluid samples were transferred to a monolayer of MDCK cells and incubated for 72 hours at 37°C in a 5% CO2 incubator. Cells were fixed using acetone and immunostained using IAV NP-specific mAb (clone MO58) followed by Alexa fluor 488 conjugated secondary antibody, and the specific signals of immunofluorescence indicating the virus infectivity were recorded as described previously.36 The virus-specific HI titers and specific IgG and IgA antibody levels in test samples against three SwIAV isolates (H1N1, H1N2, and H3N2) were determined as described previously.36 In the IgA and IgG antibody assays 1:100 dilution of BAL fluid and plasma samples, and for IgA analysis 1:2 dilution of nasal swab samples were used, and the results were compared among the pig groups.
Histopathology
Five micrometer sections of apical, cardiac, and diaphragmatic lung lobes of pigs were stained with H&E and examined microscopically for histopathological changes as described previously.36,37 The severity of inflammation was scored based on the distribution of lesions and infiltration of mononuclear cells and graded from 0–3.
Cell proliferation assay
PBMCs and TBLN MNCs were isolated at DPC 6 as described previously.36,38 Antigen-specific lymphocyte proliferation was carried out using cell titer 96 aqueous non-radioactive proliferation assay kit as per the manufacturer’s instructions (Promega Corporation, Madison, WI, USA).36,37 Briefly, 1 million cells were stimulated either with pooled peptides (2 μg/peptide/mL) or SwIAV H1N1-OH7 (at multiplicity of infection 0.1) and incubated at 37°C in a 5% CO2 incubator. After 72 hours of incubation [3-(4,5-dimethylthiazol-2-yl)-5-(3- carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium + phenazine methosulfate solution was added and it was incubated for an additional 4 hours to determine the optical density at 490 nm.
Re-stimulation of MNCs for flow cytometry and supernatant for IFN-γ ELISA
PBMCs and TBLN MNCs isolated at DPC 6 were re-stimulated with: i) each of the peptides used in the vaccine formulation separately at 2 μg/mL concentration; ii) pooled peptides (each 2 μg/mL); or iii) live challenge virus SwIAV H1N1-OH7 (at 0.1 multiplicity of infection). One million cells per well were cultured in 200 μL E-RPMI medium for 72 hours at 37°C. After 72 hours of incubation the cells were subjected to flow cytometry to determine the frequency of T-helper/memory cells (CD3+ CD4+ CD8α+) as described previously.36 Briefly, 1×106 cells were plated in 96 well-plates, blocked with 2% pig serum, and surface-labeled with swine lymphocyte-specific purified, fluorochrome- or biotin-conjugated monoclonal antibody followed by fluorochrome labeled anti-mouse isotype-specific antibody or streptavidin. The surface immunostained cells were fixed with 1% paraformaldehyde, washed, and acquired using the flow cytometer BD Aria II (BD Biosciences, San Jose, CA, USA) and analyzed using the FlowJo software (Tree Star, Ashland, OR, USA). Antibodies used in flow cytometry were: anti-porcine CD3, CD4α, and CD8α, procured from Southernbiotech, Birmingham, AL, USA.36 Culture supernatants harvested from re-stimulated PBMCs were harvested at 72 hours and subjected to IFN-γ cytokine ELISA as described previously.36,37
Cytokine ELISA
IL-6, IL-10, IL-12, and IFN-γ cytokine secretion in the BAL fluid samples were analyzed by ELISA as described previously.39
Statistical analysis
Data were presented as mean and standard error of mean (SEM) of 4–6 pigs. HI titers were presented as geometric mean with 95% CI. Virus titers were log-transformed and analyzed. All the data were first subjected to Shapiro-Wilk normality test and it was found that the data were not normally distributed, and hence statistical analysis was carried out by non-parametric Kruskal–Wallis test followed by Dunn’s post hoc test using GraphPad Prism (version 5; GraphPad Software, Inc., La Jolla, CA, USA). Pig body temperature data were analyzed by repeated measure ANOVA using Friedman test followed by Dunn’s pairwise comparison. A P-value <0.05 was considered statistically significant.
Results
Physical characterization of peptide-loaded liposomes and MSU adjuvant
The ten pooled peptide-loaded liposomes had a concentration of total peptide loading as 0.47 mg/mL. The size and charge of peptide-loaded liposomes were characterized by ZetaPas particle sizer (Brookhaven Instruments Corporation). The mean diameter of liposome peptide NPs was 134 nm, the median was 131 nm (Figure 1A), and the charge/zeta potential was −20.3±2.4 mV. Zeta potential is the potential difference existing between the surface of a nanoparticle dispersed in a conducting liquid (eg, water) and the bulk of the liquid. It is a measure of surface charge (positive, negative or neutral), normally in terms of millivolt. The phospholipid used to make liposome NPs was highly negatively charged. There was a slight effect of type of peptides used on the zeta potential of liposomes. In general, our liposome peptide NPs were highly negatively charged (<−20 mV) and were very stable as a liquid suspension. Encapsulation efficiency of individual peptides in liposomes was determined by using a representative peptide from each of the four peptide categories belonging to acidic, basic, hydrophobic, and neutral peptide groups. In general, the loading efficiency of each peptide was very high (80%–90%), a finding consistent with the reported peptide-loading efficiency in similar liposome formulations.40 However, only the neutral peptide (#10) had lower (54%) encapsulation efficiency (Table 3). The peptide encapsulated liposomes were used in pigs without separating the non-encapsulated peptides from encapsulated peptides. Therefore, the peptide vaccine groups received 500 μg of non-encapsulated peptides while liposome peptide groups received the same amount of peptides with 70%–80% encapsulated in liposome NPs and the rest in soluble form. Scanning electron microscopy was used to image MSU crystals. The crystals were 1–5 μm in diameter and nanosize width (Figure 1B). Fourier-transform infrared spectroscopy images indicated that ethylene trioxide gas sterilization did not affect the chemical signature of MSU crystals (data not shown).
Liposomal vaccine protected pigs from clinical flu and reduced nasal viral shedding and viral load
Virulent flu infection in pigs often induces fever that persists during the first few days.36 In our study, mock vaccinated and peptides only vaccinates had fever until DPC 3. In contrast, both the liposomal vaccine groups (with or without adjuvant) showed fever only at DPC 1 and recovered to normal body temperature subsequently (Figure 2A). The body temperature in both the liposomal vaccine groups (group 4 and 5) was significantly reduced (P<0.05) compared to group 2 (peptide only vaccinated animals) after challenge. The flu virus replicates in the respiratory tract epithelial cells and is transmitted through nasal shedding. In pigs vaccinated with liposome peptide nanovaccine with MSU adjuvant, nasal virus shedding was significantly reduced (P<0.05) compared to mock-challenge group, and 8–16-fold reduced compared to other vaccine groups (Figure 2B). Similarly, the challenge virus titer in BAL fluid was significantly reduced (P<0.05) (approximately 2-log) in both liposomal vaccine groups compared to mock-challenged pigs (Figure 2C). Though not statistically significant (P>0.05), the virus titer in BAL fluid of pigs vaccinated with liposome peptide nanovaccine without adjuvant (group 4) was five times lower than peptide only vaccinated animals (group 2). Similarly, virus titer in pigs vaccinated with liposome peptide nanovaccine with MSU adjuvant (group 5) was 14 times lower (P>0.05) than pigs receiving peptides with MSU adjuvant (group 3) (Figure 2C).
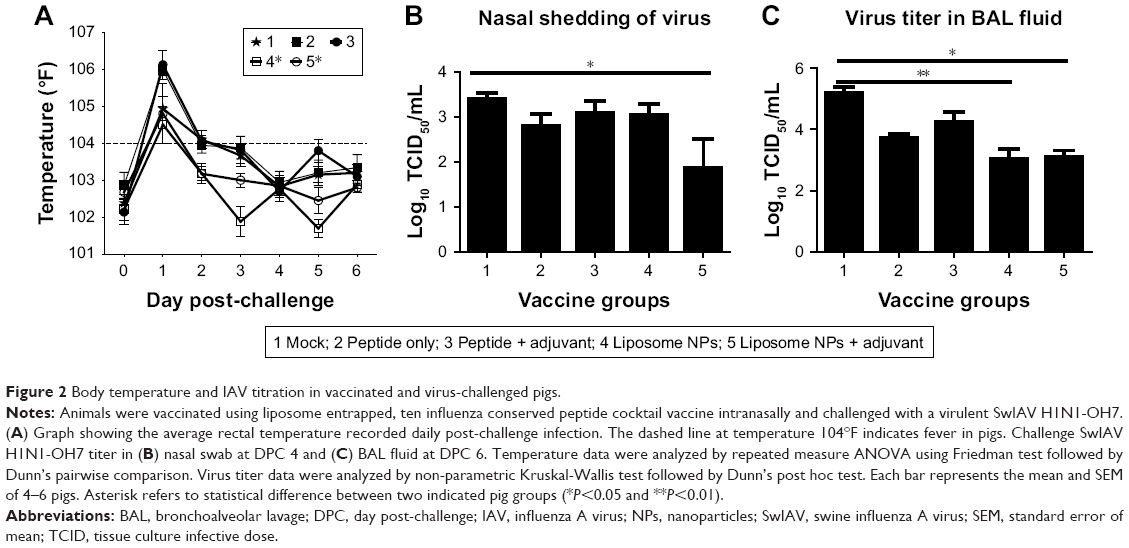 | Figure 2 Body temperature and IAV titration in vaccinated and virus-challenged pigs. |
Liposomal peptide vaccines reduced pulmonary lesions and favored cytokine production
On the day of necropsy at DPC 6 the lungs were examined and scored for gross pneumonic lesions. We noticed, in both liposomal vaccine pig groups, a significant reduction (P<0.05) in gross pneumonic lesions compared to mock-challenged pigs (Figure 3A). Microscopic analysis of the H&E stained sections of the lungs of liposome peptide with adjuvant vaccinated pigs showed lower lung inflammatory changes compared to other vaccine groups, but the data were not statistically significant (Figure 3B). We analyzed secretion of cytokines in BAL fluid of pigs collected at DPC 6 (Figure 3C–F). It is worthwhile to note that the mock-challenged animals had higher levels of all the detected cytokines (Figure 3C–F) compared to all the vaccinated groups, supporting the robust, acute inflammatory response observed in the lungs of those animals (Figure 3A and B). Specifically, the liposomal vaccines induced lower amounts of inflammatory cytokine IL-6 than mock-infected group (Figure 3C). Production of IL-10 (Figure 3D) and IL-12 (Figure 3E), which are secreted mainly by activated dendritic cells and macrophages, and the cytokine IFN-γ (Figure 3F), secreted specifically by activated lymphocytes, were increased in pigs vaccinated with liposome peptide nanovaccine with adjuvant compared to other vaccine groups. Cytokines IL-12 and IFN-γ promote the cell-mediated immunity required for induction of immune response against viral pathogens,41 while the cytokine IL-10 initiates B cell differentiation and growth and induces IgG, IgM, and IgA production.42 Overall, the cytokine response and reduced lung pathology data in liposome peptide vaccinated pigs correlated with dampened clinical flu symptoms and reduced viral load in the respiratory tract.
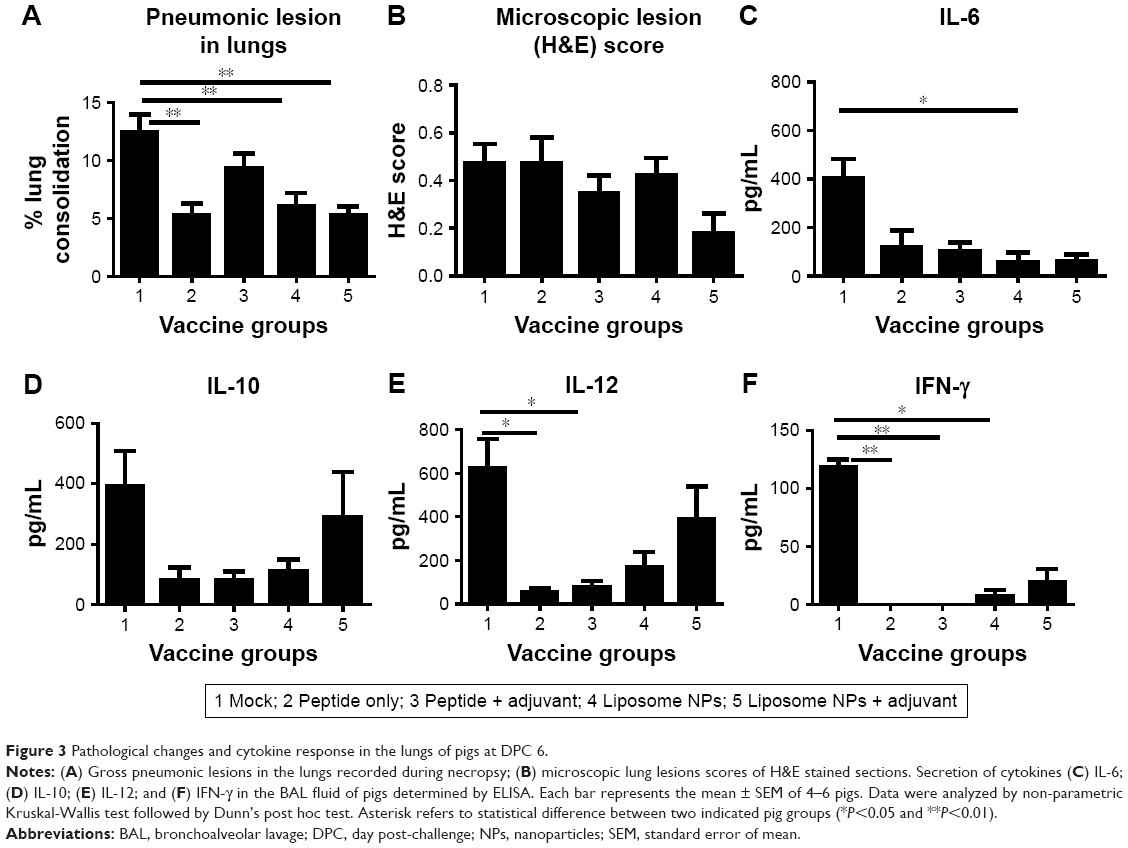 | Figure 3 Pathological changes and cytokine response in the lungs of pigs at DPC 6. |
Liposomal vaccine with adjuvant resulted in improved mucosal antibody response
Immunologically, the liposome peptide vaccine with adjuvant (group 5) induced significantly higher (P<0.05) HI antibody titer in BAL fluid against the challenge virus H1N1-OH7 at DPC 6 compared to mock-challenged pigs (Figure 4A). Importantly, a similar increase in HI titers was also observed against SwIAV H1N2-OH10 (heterologous to H1N1-OH7) (Figure 4B) and H3N2-OH4 (heterosubtypic to H1N1-OH7) (Figure 4C) in group 5 pigs. Similarly, the virus-specific mucosal IgA antibody response in nasal swab samples at DPC 6 was remarkably higher in group 5 pigs against all three SwIAV subtypes compared to all other vaccine groups, though statistically significant differences were observed only with group 4 animals (Figure 4D–F). The higher IgA response was consistent with the reduced nasal virus shedding in group 5 pigs (Figure 2B). Similarly, IgA response in BAL fluid at DPC 6 in group 5 pigs was also increased against all three IAVs namely H1N1-OH7, H1N2-OH10, and H3N2-OH4, but the data were not statistically significant (data not shown). The overall humoral response data indicated that the liposome peptide vaccine with adjuvant increased the production of specific functional cross-reactive mucosal antibodies in the respiratory tracts of pigs.
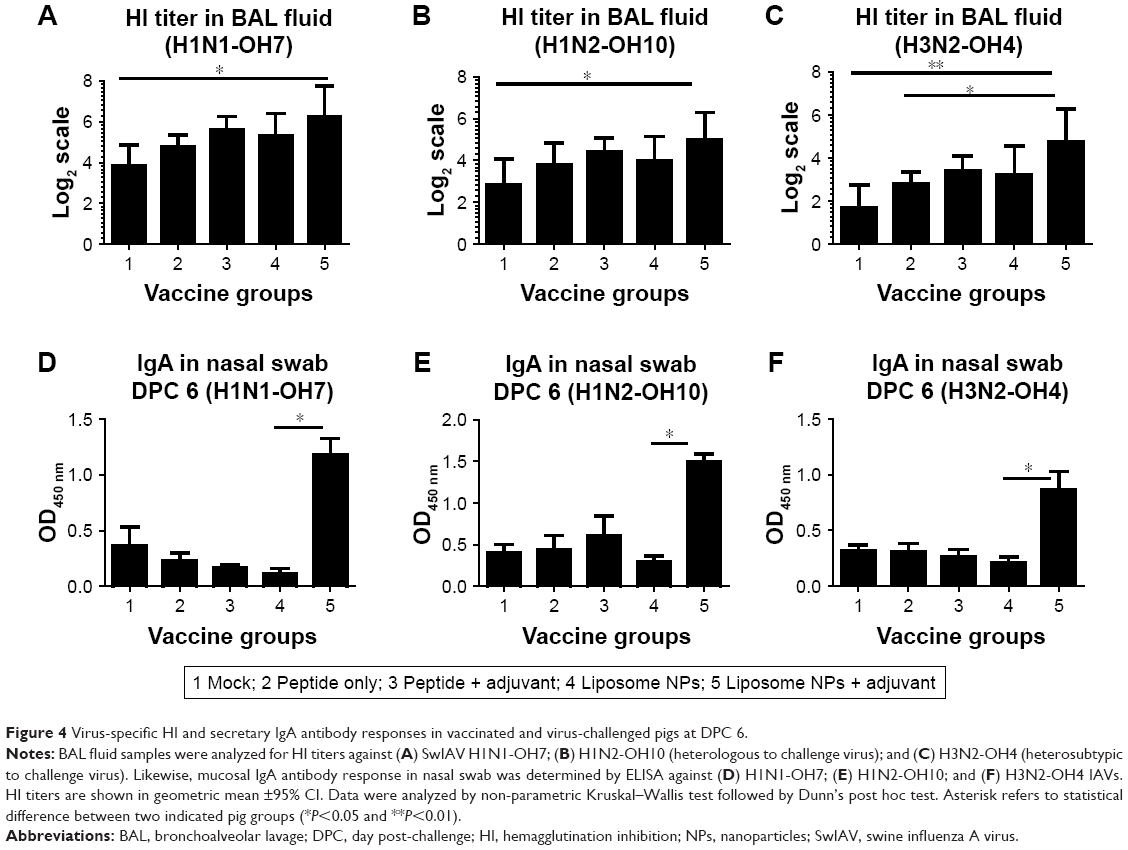 | Figure 4 Virus-specific HI and secretary IgA antibody responses in vaccinated and virus-challenged pigs at DPC 6. |
Liposome peptide vaccine boosted the frequency of virus-specific T-helper/memory cells
PBMCs isolated at DPC 6 were re-stimulated with both individual and ten pooled peptides as well as the challenge virus (H1N1-OH7) for 3 days and then subjected to flow cytometry to determine the frequency of T-helper/memory cells (CD3+ CD4+ CD8α+). In pigs, increased frequency of T-helper/memory cell population is indicative of a protective response against viral infections.43,44 Our data indicated that the frequency of virus-specific T-helper/memory cells was significantly higher (P<0.05) in both the liposomal vaccine with or without adjuvant groups compared to mock-challenged pigs re-stimulated with pooled peptides (Figure 5A), H1N1-OH7 virus (Figure 5B), and individual peptides 1, 2, 6, 7, 8, 9, and 10 (Figure 5C, D and H–L). A similar increase in the frequency of T-helper/memory cells was also observed in PBMCs re-stimulated with peptides 3, 4, and 5 in both liposomal vaccine groups, but statistical significance was observed only in group 4 pigs (Figure 5E–G).
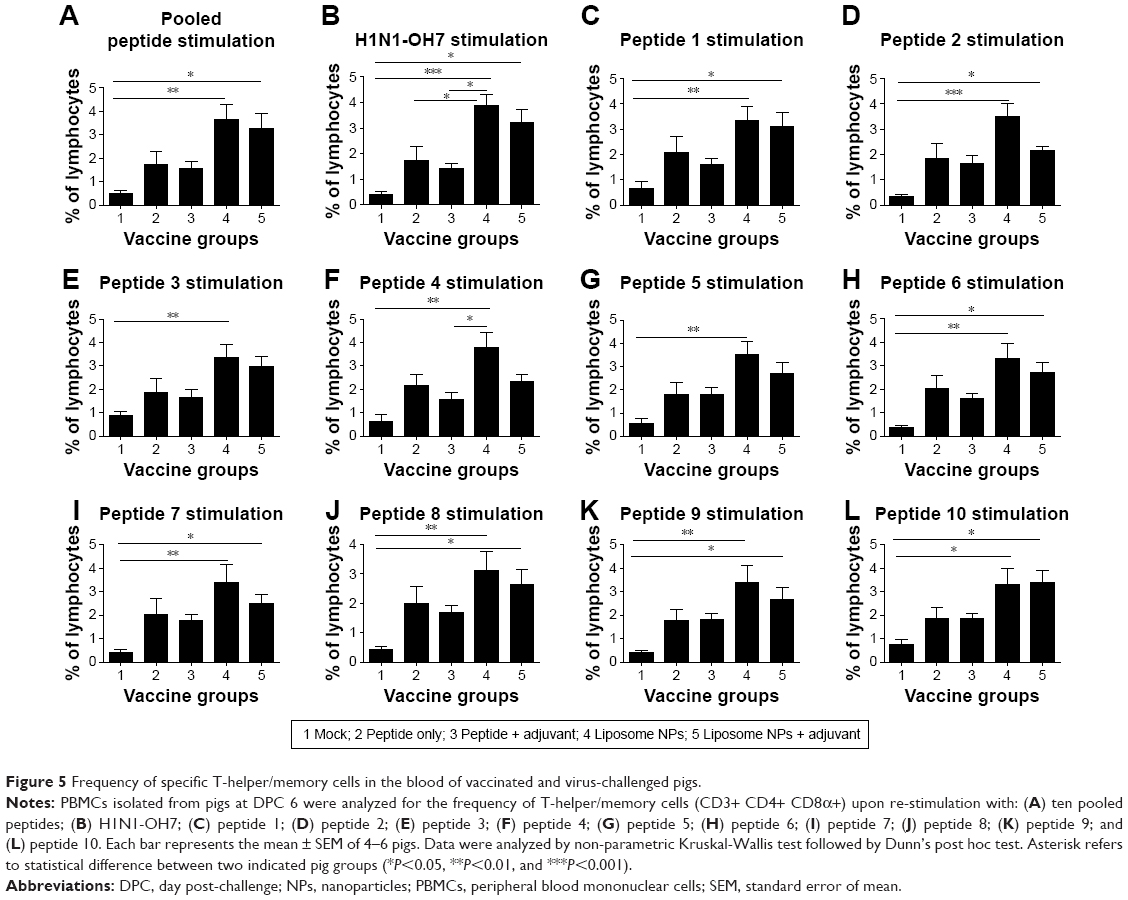 | Figure 5 Frequency of specific T-helper/memory cells in the blood of vaccinated and virus-challenged pigs. |
A similar mucosal cellular immune response analysis was performed in TBLN MNCs. On stimulation with pooled peptides, the frequency of T-helper/memory cells was significantly higher (P<0.05) in pigs vaccinated with liposome peptide without adjuvant (Figure 6A), while increased T-helper/memory cell population in TBLN MNCs was observed after re-stimulation with SwIAV H1N1-OH7 in both the liposome peptide vaccine groups with or without adjuvant compared to mock-challenged pigs (Figure 6B).
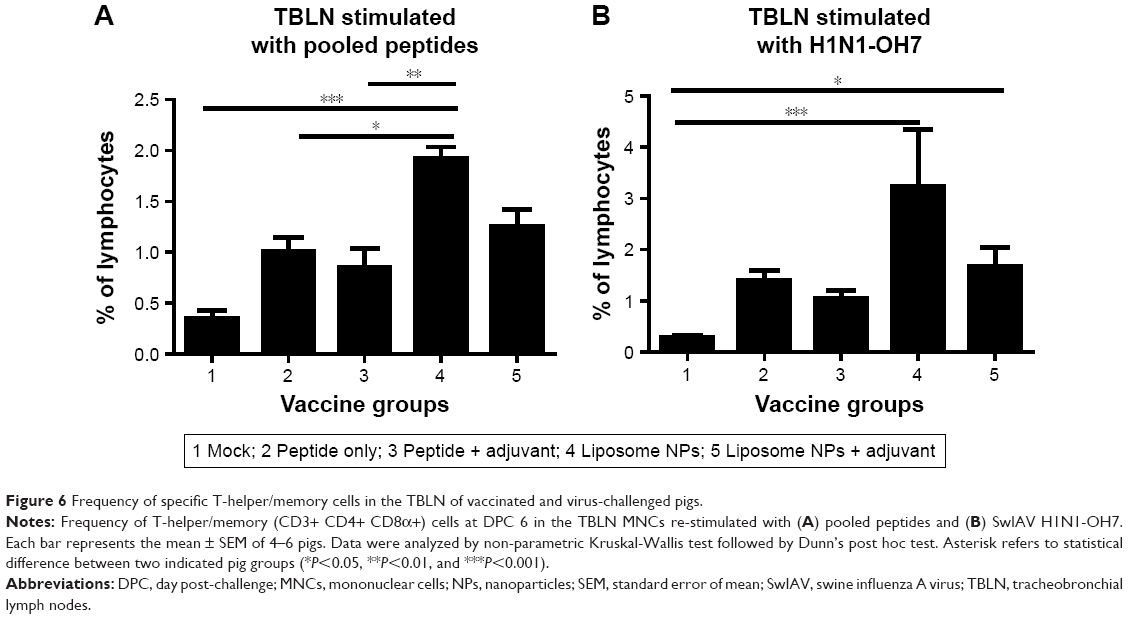 | Figure 6 Frequency of specific T-helper/memory cells in the TBLN of vaccinated and virus-challenged pigs. |
Secretion of IFN-γ and proliferation of lymphocytes in liposome peptide vaccinated pigs
Culture supernatants harvested from PBMCs re-stimulated with peptides and challenge virus were analyzed for secretion of IFN-γ (Th1 cytokine). To determine the antigen-specific recall cytokine response in vaccinated pig groups, the average cytokine production of control mock-challenged pig group was subtracted from the experimental animals (groups 2–5). Our data showed that only the PBMCs of liposome peptide vaccine groups (group 4 and 5) produced IFN-γ in excess over the mock-challenged pigs (Figure 7A–L). Interestingly, cells of liposome peptide with adjuvant pigs (group 5) secreted relatively lower levels of IFN-γ compared to group 4 animals following re-stimulation with peptides 1–7 (Figure 7A–I), and absence of any IFN-γ secretion following peptides 8–10 re-stimulation (Figure 7J–L).
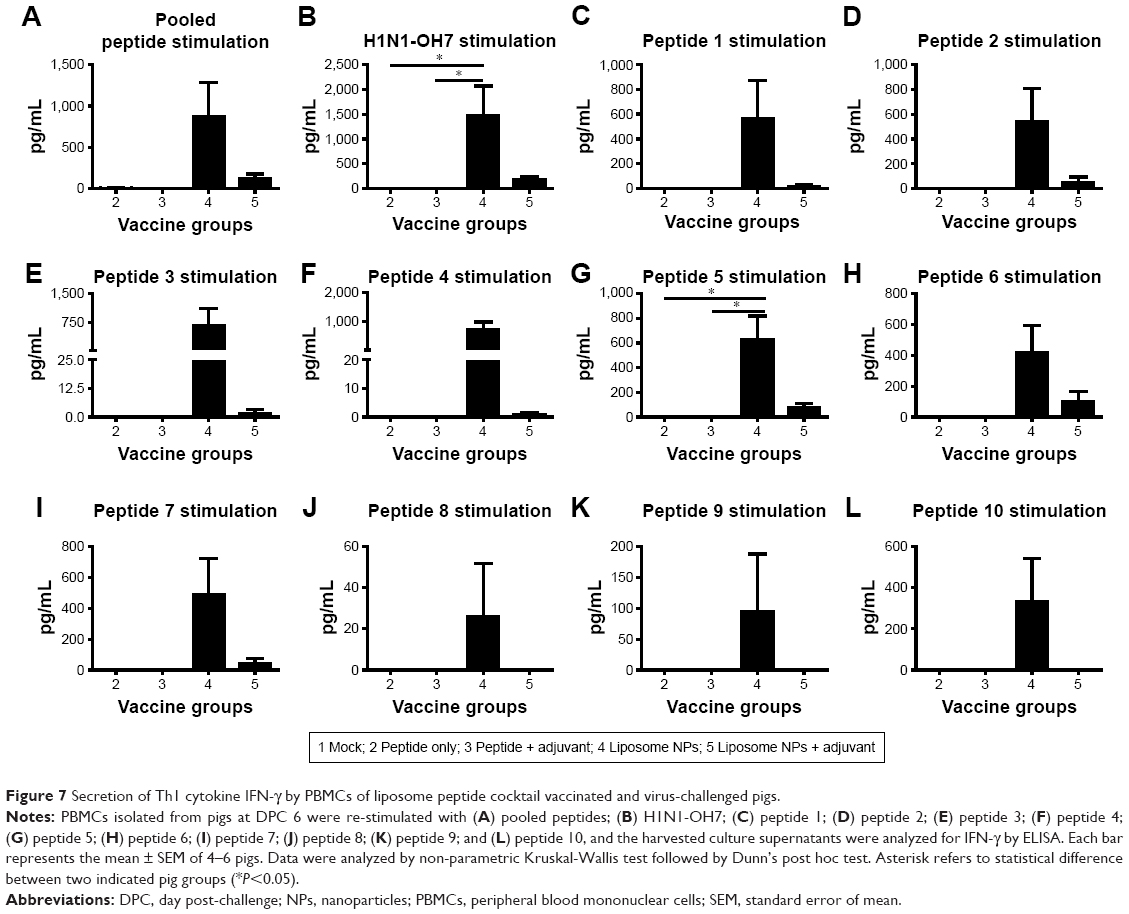 | Figure 7 Secretion of Th1 cytokine IFN-γ by PBMCs of liposome peptide cocktail vaccinated and virus-challenged pigs. |
We analyzed antigen-specific proliferation of lymphocytes in both PBMCs and TBLN MNCs of pigs re-stimulated with pooled peptides and H1N1-OH7 virus at DPC 6. In PBMCs, we observed a significantly higher (P<0.05) proliferative response in group 5 compared to group 3 pigs re-stimulated with pooled peptides (Figure 8A), but not with H1N1-OH7 virus (Figure 8B). In contrast, lymphocytes in TBLN MNCs re-stimulated with H1N1-OH7 virus (Figure 8D), but not pooled peptides (Figure 8C), had significantly higher (P<0.05) proliferative response in pig groups 4 and 5 compared to soluble peptide administered animals. Overall, specific Th1 cytokine and lymphocyte proliferation data suggested that liposome peptide vaccine improved the antigen-specific lymphocyte activation associated with improved cell-mediated immune response.
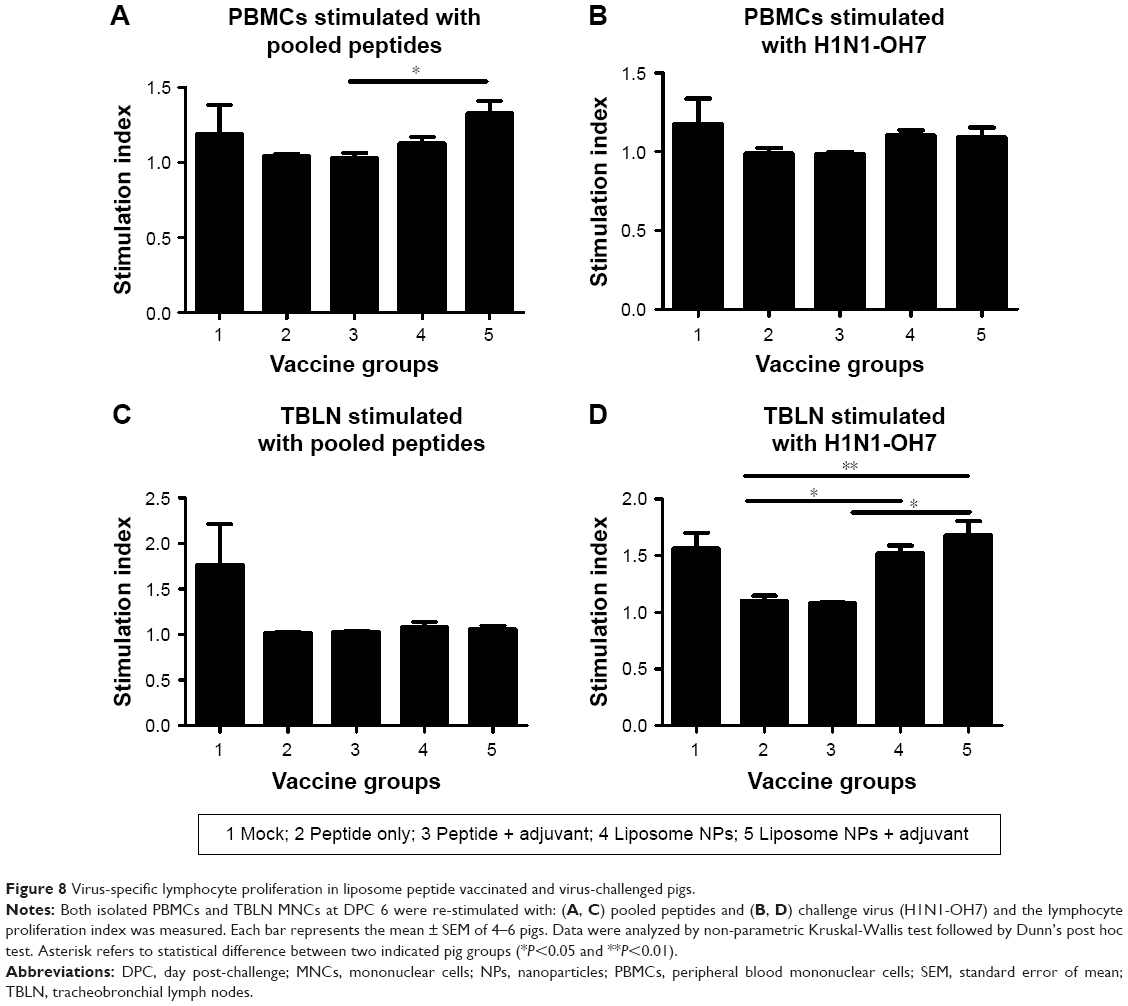 | Figure 8 Virus-specific lymphocyte proliferation in liposome peptide vaccinated and virus-challenged pigs. |
Discussion
In this study, we tested a rational design of flu subunit vaccine formulation that would induce heightened cross-protective immune response and could be readily scaled-up for bulk production. Our candidate flu vaccine was found to be effective in a pig model. Our approach included use of: i) ten highly conserved T and B cells targeting epitope peptides of different IAV strains and subtypes; ii) liposome nanoparticle encapsulation of the peptides for optimal delivery of viral antigens to the immune system; iii) novel adjuvant, MSU crystals, a safe and non-toxic agent to enhance the immunogenicity of peptide antigens; and finally iv) intranasal mist delivery of vaccine to reach the mucosa-associated lymphoid tissues in the respiratory tract. Our candidate liposome-based subunit flu nanovaccine co-administered with the MSU adjuvant partially rescued pigs from flu-induced fever, reduced the pneumonic lesions, nasal virus shedding, and lung virus load of challenge infectious virus. The observed protection was mediated through both mucosal and systemic cellular and humoral immune responses.
Different strategies are being explored to develop nanoparticle-based, universal flu subunit vaccines. For example, immobilized flu M2e protein on gold nanoparticles was co-delivered with CpG oligodeoxynucleotide adjuvant, and an induction of protective immunity against different IAV subtypes in mice was detected.45 Similarly, self-assembling nanoparticles were prepared through fusion of HA antigen with ferritin and showed better antibody response than commercial trivalent vaccine, and also enhanced cross-neutralizing antibodies against a wide range of unrelated flu viruses in mice.46 Smith et al used recombinant HA nanoparticle flu vaccine formulated with saponin-based adjuvant in a ferret model and showed its ability to induce better HI antibodies, neutralizing antibodies, and protective efficacy against homologous and drift viral strains.47 A recent study highlighted the use of double-layered protein nanoparticles using M2e as well as HA antigens in inducing robust and long-lasting immunity and providing protective efficacy against divergent IAV challenge in mice.48 In our current experiment, we used biodegradable liposome nanoparticles for vaccine delivery. Liposomes are artificially prepared, spherical vesicles that can entrap both water and lipid soluble molecules in their aqueous and lipid phases, respectively. This property makes them an attractive carrier system in drug delivery.49,50 Liposomes have been tested since the 1970s and have been shown to possess adjuvanticity using bacterial, viral, protozoan, tumor, and other vaccine antigens.49–51 Liposomes protect antigens from degradation, facilitate slow antigen release, and also enhance antigen uptake by APCs.52 Liposome-encapsulated antigens are processed in lysosomes, recycled, and presented by APCs to naïve T cells, leading to enhanced cellular and humoral immune responses.53 The immunogenicity of liposome nanoparticles is determined by many factors including physicochemical characteristics such as size, surface charge, type of lipid molecules used to formulate liposomes, as well as other factors such as route of immunization, animal model, and antigen/immunogen used.54 Nanoparticles of <500 nm diameter are taken-up readily and processed efficiently by APCs.55 Consistent with that requirement our subunit flu liposomal particle vaccine was <200 nm. Liposomes created in this study had high encapsulation efficiency of peptides (80%–90%). We did not remove the residual soluble peptides (~10%–20%), as the soluble peptides, with the help of adjuvant, may reach the immune cells readily while the liposome-encapsulated peptides are protected from proteolytic degradation and facilitate organized uptake and processing by APCs.
A previous study in mice has shown that intranasal immunization with liposome-encapsulated plasmid DNA encoding flu HA protein induced better mucosal and systemic antibody and cellular immune response, leading to enhanced protection.56 Similarly, liposome-encapsulated flu HA and NA proteins-based vaccine in mice showed up to 50-fold stronger and 3–5 times longer duration of humoral immune response compared to control non-liposomal proteins.57 To date, liposome-based subunit flu vaccine studies have been performed in rodent models. We first did intranasal immunization study with liposomal flu vaccine in pigs. In this study, we used liposomes to encapsulate highly conserved peptides of M2e, HA, NA, NP, PB1, and PA proteins targeting both T and B cell epitopes of different flu virus isolates and performed the immunogenicity and protective efficacy trial in pigs. Like humans, pigs are a natural host for flu with vast similarities in viral pathogenesis, clinical signs of disease, and inflammatory response.29,58–65 Pigs are quite similar to humans in terms of anatomy, physiology, and immunology.66–69 Thus, vaccine trials conducted in pigs have dual benefits, with development of better flu vaccines to use in pigs, and pre-clinical findings in pigs, resulting in better translational value for developing better human flu vaccines than studies in rodents.
Previous trials using flu subunit and peptide vaccine in pigs have shown variable results. An intradermal DNA flu vaccine containing fusion proteins of IAV M2 and NP, despite showing M2e-specific antibody and lymphocyte proliferative response, exacerbated the disease caused by SwIAV H1N1 infection in pigs.70 In another study, the recombinant IAV matrix protein injected intramuscularly reduced SwIAV-associated disease, but the nasal viral shedding was not reduced.71 Similarly, intramuscular administration of HA-based peptide vaccine showed broadly reactive antibody response in pigs, but failed to reduce nasal virus shedding.31 In contrast, the liposomal nanovaccine administered intranasally with MSU adjuvant used in this study, induced virus- and peptide-specific T-helper/memory cell population, mucosal IgA response, and reduced the nasal viral shedding and fever in vaccinated pigs.
Importantly, virus-specific lymphocyte responses in pigs vaccinated with liposome peptide vaccine were augmented both in TBLN and blood, indicating that our vaccine formulation and the delivery platform primed virus-specific T cells both in the respiratory tract and systemically. Consistent with our findings, CTL peptides conjugated with liposomes elicited a cross-protective response against heterosubtypic IAV in mice by improving the antigen-specific memory T cell pool.72,73 In our study, liposome peptide vaccines induced specific lymphocytes’ proliferation both in the TBLN and blood, and also facilitated secretion of IFN-γ, an important anti-viral Th1 cytokine.74 Likely, both the specifically activated lymphocytes and secreted mucosal antibodies have played a significant role in clearance of the challenge virus from the respiratory tracts of pigs. Particularly, peptides 6–10 in our vaccine formulation are predicted to be CTL epitopes based on their ability to form a stable complex with swine histocompatibility molecule (SLA) – SLA1*0401 binding motif.32 Peptides 6 (NP 44-52), 7 (PB1 347-355), and 8 (PB1 591-599) are also identical to the human CTL epitope sequence, indicating their potential to be cross-species flu virus CTL epitopes.32 Inclusion of these peptides in the vaccine formulation might have helped in enhancing the cellular immune response which translated into increased protective efficacy in pigs.
Flu virus primarily infects epithelial cells lining the respiratory tract mucosa, and nasal virus shedding forms the means of viral transmission. Hence, induction of strong mucosal antibody response is beneficial for efficient protection during flu epidemics and pandemics. Intranasal delivery of vaccine can enhance mucosal immunity, and intranasal but not parenteral flu liposomal vaccine encapsulating NP peptides coadministered with anti-CD40 antibody induces mucosal immunity and reduces replication of virus in the lungs of mice.75 In our earlier study in pigs, SwIAV M2e and two each of human IAV T (peptides 9 and 10) and B (peptides 2 and 3) cell epitope peptides were encapsulated in PLGA NPs and administered intranasally as a mist without any secondary adjuvant, and its efficacy was tested against SwIAV H1N1-07 challenge infection, it enhanced epitope-specific cytotoxic T cell responses but did not stimulate a B cell response, particularly on mucosal surfaces.24 Strong mucosal IgA response is essential to reduce the nasal IAV shedding and the present study also demonstrates that.36,37
Uric acid crystals activate innate host defense mechanisms and trigger robust inflammation and immune activation through NLRP3 inflammasome pathway.76 The innate immune activation by MSU crystals potentiates the adaptive immune response, particularly the antibody response.77 MSU crystals have been shown to be safe after intradermal injection (2–2,000 μg) in humans.78 They can induce maturation of dendritic cells in intraperitoneally injected mice with ovalbumin resulting in efficient priming of CD8+ T cells.79 MSU crystals stimulate production of ROS and phagolysosome maturation. In mice, subcutaneous injection of MSU crystals as an adjuvant, promotes innate anti-mycobacterial immunity and improves the efficacy of tuberculosis vaccine.80 Adjuvant effects of MSU crystals were compared with a traditional incomplete Freud’s adjuvant (IFA) in a mice, and this experiment revealed that MSU adjuvant was comparable to IFA (unpublished data). Microparticles with a size less than 5–6 μm (preferably less than 2 μm) accumulate in lungs after inhalation or intranasal administration and interact with pulmonary mucosal immune cells within 24–48 hours.81,82 In this study in pigs, we hypothesized that MSU adjuvant liposome peptide IAV vaccine is better than the candidate vaccine administered without the adjuvant intranasally. Our data showed that the liposome-based peptide vaccine formulation with or without adjuvant was superior to soluble antigens. MSU adjuvant liposome NP vaccine further potentiated the immune responses, as indicated by reduced lung pneumonic lesions, improved cytokine responses, and enhanced mucosal IgA response resulting in 15-fold reduced nasal viral shedding compared to the candidate liposome peptide vaccine delivered without the MSU adjuvant. However, such an adjuvant effect of MSU with soluble (eg, non-liposome-bound) peptides was not observed. These data indicate that improved immune response and protective efficacy in pigs vaccinated with liposome peptide vaccine with MSU adjuvant was likely the result of a synergistic effect of both liposome NPs and MSU adjuvant. Further reduction in size of the needle-shaped MSU crystals might elicit even better immune response as nanoparticles interact with immune cells earlier and more efficiently than microparticles after intranasal administration, but this needs to be investigated further.81,82 In this study of analyzing the adjuvant role of MSU delivered intranasally in pigs, we used 5 mg of the adjuvant. Further studies are needed to analyze the dose-dependent immune response induced by the MSU adjuvant in pigs administered with liposomal peptide flu vaccine as well as other vaccine formulations. Also, it would be interesting in the future to investigate the impact of size of liposomes on inducing immune responses. A previous study using polystyrene particles, indicated that size and shape of NPs have an immune-modulatory effect, and that smaller (193 nm) and spherical NPs can generate stronger Th1 and Th2 immune responses.83
Conclusion
In conclusion, our vaccine trial using liposome and MSU adjuvant-based subunit flu vaccine as intranasal mist in a pig model showed promise. We found the vaccine formulation safe and that the vaccine boosted both mucosal humoral and cellular responses, reduced the clinical flu signs, virus load, and pneumonic lesions. Future pre-clinical studies are aimed at analyzing the liposome-based IAV peptide vaccine dose-dependent response, dose titration of MSU adjuvant, comparison of adjuvant effect of MSU with other commercial and state-of-the-art adjuvants, and comparison of liposome-based vaccine formulation with commercially available swine flu vaccines to further improve its cross-protective efficacy against variant IAV in a pig model.
Acknowledgments
We extend our sincere thanks to Dr Juliette Hanson, Megan Strother, Sara Tallmadge, Hyesun Jang, John M Ngunjiri, and Mohamed Elaish for their help in animal studies. Dr Stephen Opiyo, Biostatistician, Molecular and Cellular Imaging Center, OARDC, helped us with statistical analyses of the data. This work was supported by OARDC OSU industry match grant and Agriculture and Food Research Initiative Competitive grant no 2013-67015-20476 from the USDA National Institute of Food and Agriculture.
Disclosure
The authors report a pending patent for a liposomal subunit flu vaccine formulation: US patent application no 15/723,793, 2017. The authors report no other conflicts of interest in this work.
References
Lambert LC, Fauci AS. Influenza vaccines for the future. N Engl J Med. 2010;363(21):2036–2044. | ||
WHO (homepage on the internet). Influenza (Seasonal) - Fact Sheet. World Health Organization; 2018. Available from: http://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal). Accessed October 2, 2018. | ||
Dykhuis HC, Painter T, Fangman TDH. Assessing production parameters and economic impact of swine influenza, PRRS and Mycoplasma hyopneimoniae on finishing pigs in a large production system. Paper presented at: Proceedings of AASV Annual Meeting; March 10–13 in Denver, Colorado. 2012. | ||
Ito T, Couceiro JN, Kelm S, et al. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J Virol. 1998;72(9):7367–7373. | ||
Dawood FS, Iuliano AD, Reed C, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. 2012;12(9):687–695. | ||
Schicker RS, Rossow J, Eckel S, et al. Outbreak of Influenza A(H3N2) Variant Virus Infections Among Persons Attending Agricultural Fairs Housing Infected Swine – Michigan and Ohio, July-August 2016. MMWR Morb Mortal Wkly Rep. 2016;65(42):1157–1160. | ||
Zhang H, Wang L, Compans RW, Wang BZ. Universal influenza vaccines, a dream to be realized soon. Viruses. 2014;6(5):1974–1991. | ||
Noah DL, Noah JW. Adapting global influenza management strategies to address emerging viruses. Am J Physiol Lung Cell Mol Physiol. 2013;305(2):L108–L117. | ||
Butler D. Mutations explain poor showing of 2012 flu vaccine; 2014. Available from: http://www.nature.com/news/mutations-explain-poor-showing-of-2012-flu-vaccine-1.14940. Accessed September 20, 2018. | ||
Vincent AL, Perez DR, Rajao D, et al. Influenza A virus vaccines for swine. Vet Microbiol. 2017;206:35–44. | ||
Gauger PC, Vincent AL, Loving CL, et al. Enhanced pneumonia and disease in pigs vaccinated with an inactivated human-like (δ-cluster) H1N2 vaccine and challenged with pandemic 2009 H1N1 influenza virus. Vaccine. 2011;29(15):2712–2719. | ||
Vincent AL, Lager KM, Janke BH, Gramer MR, Richt JA. Failure of protection and enhanced pneumonia with a US H1N2 swine influenza virus in pigs vaccinated with an inactivated classical swine H1N1 vaccine. Vet Microbiol. 2008;126(4):310–323. | ||
Rajao DS, Sandbulte MR, Gauger PC, et al. Heterologous challenge in the presence of maternally-derived antibodies results in vaccine-associated enhanced respiratory disease in weaned piglets. Virology. 2016;491:79–88. | ||
Fiers W, de Filette M, Birkett A, Neirynck S, Min Jou W. A “universal” human influenza A vaccine. Virus Res. 2004;103(1–2):173–176. | ||
Lee YT, Kim KH, Ko EJ, et al. New vaccines against influenza virus. Clin Exp Vaccine Res. 2014;3(1):12–28. | ||
Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov. 2015;14(3):167–182. | ||
Purcell AW, Mccluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov. 2007;6(5):404–414. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Lü JM, Wang X, Marin-Muller C, et al. Current advances in research and clinical applications of PLGA-based nanotechnology. Expert Rev Mol Diagn. 2009;9(4):325–341. | ||
Mishra N, Goyal AK, Tiwari S, et al. Recent advances in mucosal delivery of vaccines: role of mucoadhesive/biodegradable polymeric carriers. Expert Opin Ther Pat. 2010;20(5):661–679. | ||
Usonis V, Bakasénas V, Valentelis R, Katiliene G, Vidzeniene D, Herzog C. Antibody titres after primary and booster vaccination of infants and young children with a virosomal hepatitis A vaccine (Epaxal). Vaccine. 2003;21(31):4588–4592. | ||
Woodrow KA, Bennett KM, Lo DD, Dd L. Mucosal vaccine design and delivery. Annu Rev Biomed Eng. 2012;14:17–46. | ||
Torrieri-Dramard L, Lambrecht B, Ferreira HL, van den Berg T, Klatzmann D, Bellier B. Intranasal DNA vaccination induces potent mucosal and systemic immune responses and cross-protective immunity against influenza viruses. Mol Ther. 2011;19(3):602–611. | ||
Hiremath J, Kang KI, Xia M, et al. Entrapment of H1N1 Influenza Virus Derived Conserved Peptides in PLGA Nanoparticles Enhances T Cell Response and Vaccine Efficacy in Pigs. PLoS One. 2016;11(4):e0151922. | ||
Cheng XG, Zhong GM, Mcdonough J, Macnaughton M. In vivo evaluation of MSU crystals as an adjuvant. Vaccine Development Center of San Antonio Conference. 2014;2014. Available from: http://savaccine.org/conference/2016conference/. Accessed October 8, 2018. | ||
Yassine HM, Khatri M, Zhang YJ, et al. Characterization of triple reassortant H1N1 influenza A viruses from swine in Ohio. Vet Microbiol. 2009;139(1–2):132–139. | ||
Ali A, Khatri M, Wang L, Saif YM, Lee CW. Identification of swine H1N2/pandemic H1N1 reassortant influenza virus in pigs, United States. Vet Microbiol. 2012;158(1–2):60–68. | ||
Yassine HM, Al-Natour MQ, Lee CW, Saif YM. Interspecies and intraspecies transmission of triple reassortant H3N2 influenza A viruses. Virol J. 2007;4:129. | ||
Khatri M, Dwivedi V, Krakowka S, et al. Swine influenza H1N1 virus induces acute inflammatory immune responses in pig lungs: a potential animal model for human H1N1 influenza virus. J Virol. 2010;84(21):11210–11218. | ||
de Filette M, Fiers W, Martens W, et al. Improved design and intranasal delivery of an M2e-based human influenza A vaccine. Vaccine. 2006;24(44–46):6597–6601. | ||
Vergara-Alert J, Argilaguet JM, Busquets N, et al. Conserved synthetic peptides from the hemagglutinin of influenza viruses induce broad humoral and T-cell responses in a pig model. PLoS One. 2012;7(7):e40524. | ||
Zhang N, Qi J, Feng S, et al. Crystal structure of swine major histocompatibility complex class I SLA-1 0401 and identification of 2009 pandemic swine-origin influenza A H1N1 virus cytotoxic T lymphocyte epitope peptides. J Virol. 2011;85(22):11709–11724. | ||
Cheng X, Tsao CM, Saul J, Sylvia V, Cornet D, Christy R. Comparison of Two Nanoparticle Formulations for Localized Delivery of Platelet-Derived Growth Factor (PDGF) from Aligned Collagen Fibers. Pharmaceutical Nanotechnology. 2013;1(2):105–114. | ||
Fry DW, White JC, Goldman ID. Rapid separation of low molecular weight solutes from liposomes without dilution. Anal Biochem. 1978;90(2):809–815. | ||
Cheng X, Haggins DG, York RH, Yeni YN, Akkus O. Analysis of crystals leading to joint arthropathies by Raman spectroscopy: comparison with compensated polarized imaging. Appl Spectrosc. 2009;63(4):381–386. | ||
Dhakal S, Hiremath J, Bondra K, et al. Biodegradable nanoparticle delivery of inactivated swine influenza virus vaccine provides heterologous cell-mediated immune response in pigs. J Control Release. 2017;247:194–205. | ||
Dhakal S, Goodman J, Bondra K, et al. Polyanhydride nanovaccine against swine influenza virus in pigs. Vaccine. 2017;35(8):1124–1131. | ||
Dwivedi V, Manickam C, Patterson R, et al. Cross-protective immunity to porcine reproductive and respiratory syndrome virus by intranasal delivery of a live virus vaccine with a potent adjuvant. Vaccine. 2011;29(23):4058–4066. | ||
Dwivedi V, Manickam C, Dhakal S, et al. Adjuvant effects of invariant NKT cell ligand potentiates the innate and adaptive immunity to an inactivated H1N1 swine influenza virus vaccine in pigs. Vet Microbiol. 2016;186:157–163. | ||
Ludewig B, Barchiesi F, Pericin M, Zinkernagel RM, Hengartner H, Schwendener RA. In vivo antigen loading and activation of dendritic cells via a liposomal peptide vaccine mediates protective antiviral and anti-tumour immunity. Vaccine. 2000;19(1):23–32. | ||
Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. 1994;12:635–673. | ||
Rousset F, Garcia E, Defrance T, et al. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A. 1992;89(5):1890–1893. | ||
de Bruin TG, van Rooij EM, de Visser YE, Bianchi AT. Cytolytic function for pseudorabies virus-stimulated porcine CD4+ CD8dull+ lymphocytes. Viral Immunol. 2000;13(4):511–520. | ||
Binjawadagi B, Dwivedi V, Manickam C, Ouyang K, Torrelles JB, Renukaradhya GJ. An innovative approach to induce cross-protective immunity against porcine reproductive and respiratory syndrome virus in the lungs of pigs through adjuvanted nanotechnology-based vaccination. Int J Nanomedicine. 2014;9:1519–1535. | ||
Tao W, Hurst BL, Shakya AK, et al. Consensus M2e peptide conjugated to gold nanoparticles confers protection against H1N1, H3N2 and H5N1 influenza A viruses. Antiviral Res. 2017;141:62–72. | ||
Kanekiyo M, Wei CJ, Yassine HM, et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature. 2013;499(7456):102–106. | ||
Smith G, Liu Y, Flyer D, et al. Novel hemagglutinin nanoparticle influenza vaccine with Matrix-MTM adjuvant induces hemagglutination inhibition, neutralizing, and protective responses in ferrets against homologous and drifted A(H3N2) subtypes. Vaccine. 2017;35(40):5366–5372. | ||
Deng L, Mohan T, Chang TZ, et al. Double-layered protein nanoparticles induce broad protection against divergent influenza A viruses. Nat Commun. 2018;9(1):359. | ||
Schwendener RA. Liposomes as vaccine delivery systems: a review of the recent advances. Ther Adv Vaccines. 2014;2(6):159–182. | ||
Gregoriadis G, McCormack B, Obrenovic M, Perrie Y, Saffie R. Liposomes As Immunological Adjuvants and Vaccine Carriers. Vaccine Adjuvants: Preparation Methods and Research Protocols. Totowa, NJ: Springer O’Hagan DT, editor. New York; 2000:137–150. | ||
Gregoriadis G. Immunological adjuvants: a role for liposomes. Immunol Today. 1990;11(3):89–97. | ||
Bernasconi V, Norling K, Bally M, Höök F, Lycke NY. Mucosal Vaccine Development Based on Liposome Technology. J Immunol Res. 2016;2016:5482087. | ||
Harding CV, Collins DS, Slot JW, Geuze HJ, Unanue ER. Liposome-encapsulated antigens are processed in lysosomes, recycled, and presented to T cells. Cell. 1991;64(2):393–401. | ||
Tandrup Schmidt S, Foged C, Korsholm KS, Rades T, Christensen D. Liposome-Based Adjuvants for Subunit Vaccines: Formulation Strategies for Subunit Antigens and Immunostimulators. Pharmaceutics. 2016;8(1):7. | ||
Thomas C, Rawat A, Hope-Weeks L, Ahsan F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol Pharm. 2011;8(2):405–415. | ||
Wang D, Christopher ME, Nagata LP, et al. Intranasal immunization with liposome-encapsulated plasmid DNA encoding influenza virus hemagglutinin elicits mucosal, cellular and humoral immune responses. J Clin Virol. 2004;31 Suppl 1:99–106. | ||
Babai I, Samira S, Barenholz Y, Zakay-Rones Z, Kedar E. A novel influenza subunit vaccine composed of liposome-encapsulated haemagglutinin/neuraminidase and IL-2 or GM-CSF. I. Vaccine characterization and efficacy studies in mice. Vaccine. 1999;17(9–10):1223–1238. | ||
Yu L, Zhou Y, Jiang Y, et al. Construction and identification of a recombinant live attenuated PRRSV expressing porcine GM-CSF. Zhongguo Yufang Shouyi Xuebao/Chinese Journal of Preventive Veterinary Medicine. 2014;36(4):260–264. | ||
Fritz RS, Hayden FG, Calfee DP, et al. Nasal cytokine and chemokine responses in experimental influenza A virus infection: results of a placebo-controlled trial of intravenous zanamivir treatment. J Infect Dis. 1999;180(3):586–593. | ||
Barbé F, Atanasova K, van Reeth K. Cytokines and acute phase proteins associated with acute swine influenza infection in pigs. Vet J. 2011;187(1):48–53. | ||
Jo SK, Kim HS, Cho SW, Seo SH. Pathogenesis and inflammatory responses of swine H1N2 influenza viruses in pigs. Virus Res. 2007;129(1):64–70. | ||
Kim B, Ahn KK, Ha Y, et al. Association of tumor necrosis factor-alpha with fever and pulmonary lesion score in pigs experimentally infected with swine influenza virus subtype H1N2. J Vet Med Sci. 2009;71(5):611–616. | ||
Yassine HM, Lee CW, Gourapura R, Saif YM. Interspecies and intraspecies transmission of influenza A viruses: viral, host and environmental factors. Anim Health Res Rev. 2010;11(1):53–72. | ||
Trebbien R, Larsen LE, Viuff BM. Distribution of sialic acid receptors and influenza A virus of avian and swine origin in experimentally infected pigs. Virol J. 2011;8:434. | ||
Blish CA, Mcclelland RS, Richardson BA, et al. Genital Inflammation Predicts HIV-1 Shedding Independent of Plasma Viral Load and Systemic Inflammation. J Acquir Immune Defic Syndr. 2012;61(4):436–440. | ||
Ibrahim Z, Busch J, Awwad M, Wagner R, Wells K, Cooper DK. Selected physiologic compatibilities and incompatibilities between human and porcine organ systems. Xenotransplantation. 2006;13(6):488–499. | ||
Rogers CS, Abraham WM, Brogden KA, et al. The porcine lung as a potential model for cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295(2):L240–L263. | ||
Goco RV, Kress MB, Brantigan OC. Comparison of mucus glands in the tracheobronchial tree of man and animals. Ann N Y Acad Sci. 1963;106:555–571. | ||
Pabst R, Binns RM. The immune system of the respiratory tract in pigs. Vet Immunol Immunopathol. 1994;43(1–3):151–156. | ||
Heinen PP, Rijsewijk FA, de Boer-Luijtze EA, Bianchi AT. Vaccination of pigs with a DNA construct expressing an influenza virus M2-nucleoprotein fusion protein exacerbates disease after challenge with influenza A virus. J Gen Virol. 2002;83(Pt 8):1851–1859. | ||
Kitikoon P, Vincent AL, Janke BH, et al. Swine influenza matrix 2 (M2) protein contributes to protection against infection with different H1 swine influenza virus (SIV) isolates. Vaccine. 2009;28(2):523–531. | ||
Matsui M, Kohyama S, Suda T, et al. A CTL-based liposomal vaccine capable of inducing protection against heterosubtypic influenza viruses in HLA-A*0201 transgenic mice. Biochem Biophys Res Commun. 2010;391(3):1494–1499. | ||
Ichihashi T, Yoshida R, Sugimoto C, Takada A, Kajino K. Cross-protective peptide vaccine against influenza A viruses developed in HLA-A*2402 human immunity model. PLoS One. 2011;6(9):e24626. | ||
Meier WA, Husmann RJ, Schnitzlein WM, Osorio FA, Lunney JK, Zuckermann FA. Cytokines and synthetic double-stranded RNA augment the T helper 1 immune response of swine to porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol. 2004;102(3):299–314. | ||
Ninomiya A, Ogasawara K, Kajino K, Takada A, Kida H. Intranasal administration of a synthetic peptide vaccine encapsulated in liposome together with an anti-CD40 antibody induces protective immunity against influenza A virus in mice. Vaccine. 2002;20(25–26):3123–3129. | ||
Braga TT, Forni MF, Correa-Costa M, et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci Rep. 2017;7:39884. | ||
Ng G, Chau EM, Shi Y. Recent developments in immune activation by uric acid crystals. Arch Immunol Ther Exp. 2010;58(4):273–277. | ||
Sakamaki I, Inai K, Tsutani H. Safety of intradermal injection of monosodium urate crystals as a vaccine carrier in volunteers. Nucleosides Nucleotides Nucleic Acids. 2011;30(12):1077–1084. | ||
Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425(6957):516–521. | ||
Taus F, Santucci MB, Greco E, et al. Monosodium Urate Crystals Promote Innate Anti-Mycobacterial Immunity and Improve BCG Efficacy as a Vaccine against Tuberculosis. PLoS One. 2015;10(5):e0127279. | ||
Geiser M, Kreyling WG. Deposition and biokinetics of inhaled nanoparticles. Part Fibre Toxicol. 2010;7:2. | ||
Brenza TM, Petersen LK, Zhang Y, et al. Pulmonary biodistribution and cellular uptake of intranasally administered monodisperse particles. Pharm Res. 2015;32(4):1368–1382. | ||
Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S. Shape and size-dependent immune response to antigen-carrying nanoparticles. J Control Release. 2015;220(Pt A):141–148. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.