Back to Journals » Drug Design, Development and Therapy » Volume 10
Liposomal flucytosine capped with gold nanoparticle formulations for improved ocular delivery
Authors Salem H, Ahmed S, Omar MM ![]()
Received 3 July 2015
Accepted for publication 31 July 2015
Published 13 January 2016 Volume 2016:10 Pages 277—295
DOI https://doi.org/10.2147/DDDT.S91730
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Heba F Salem,1 Sayed M Ahmed,2 Mahmoud M Omar3
1Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt; 2Department of Industrial Pharmacy, Faculty of Pharmacy, Assiut University, Assiut, Egypt; 3Department of Industrial Pharmacy, Faculty of Pharmacy, Deraya University, El-Minia, Egypt
Abstract: Nanoliposomes have an organized architecture that provides versatile functions. In this study, liposomes were used as an ocular carrier for nanogold capped with flucytosine antifungal drug. Gold nanoparticles were used as a contrasting agent that provides tracking of the drug to the posterior segment of the eye for treating fungal intraocular endophthalmitis. The nanoliposomes were prepared with varying molar ratios of lecithin, cholesterol, Span 60, a positive charge inducer (stearylamine), and a negative charge inducer (dicetyl phosphate). Formulation F6 (phosphatidylcholine, cholesterol, Span 60, and stearylamine at a molar ratio of 1:1:1:0.15) demonstrated the highest extent of drug released, which reached 7.043 mg/h. It had a zeta potential value of 42.5±2.12 mV and an average particle size approaching 135.1±12.0 nm. The ocular penetration of the selected nanoliposomes was evaluated in vivo using a computed tomography imaging technique. It was found that F6 had both the highest intraocular penetration depth (10.22±0.11 mm) as measured by the computed tomography and the highest antifungal efficacy when evaluated in vivo using 32 infected rabbits’ eyes. The results showed a strong correlation between the average intraocular penetration of the nanoparticles capped with flucytosine and the percentage of the eyes healed. After 4 weeks, all the infected eyes (n=8) were significantly healed (P<0.01) when treated with liposomal formulation F6. Overall, the nanoliposomes encapsulating flucytosine have been proven efficient in treating the infected rabbits’ eyes, which proves the efficiency of the nanoliposomes in delivering both the drug and the contrasting agent to the posterior segment of the eye.
Keywords: computed tomography imaging, fungal endophthalmitis, intraocular inflammation, nanoliposomes
Introduction
Eye infections may lead to severe ocular morbidity and blindness.1,2 In developing countries, corneal blindness is associated with infections.3 The applied drug should reach the posterior segment of the eye in order to treat vitreoretinal diseases. Intraocular inflammation of the posterior and/or anterior chamber of eye is known as endophthalmitis. Fungal endophthalmitis may originate from endogenous infection (due to systemic fungal infection) or from exogenous infection (due to trauma or surgery). Candida albicans is the most common causative agent of fungal endophthalmitis.4–6 Treatment of fungal endophthalmitis can be achieved using one or more of the following drugs: amphotericin B, fluconazole, ketoconazole, miconazole, itraconazole, caspofungin, voriconazole, and flucytosine.6–12
Flucytosine has a broad-spectrum antifungal activity against the Candida sp., Saccharomyces cerevisiae, and specific forms of dematiaceous molds.13 The use of flucytosine within ophthalmic preparations was limited due to the development of drug resistance and weak intraocular penetration.13 The majority of ocular drug preparations should effectively permeate across the tissue barriers of the eyes in order to reach the therapeutic targets within the globe.14 Liposomes have been extensively used as drug carriers.15 The use of liposomes as drug delivery carriers is an excellent means to improve the uptake of topically applied ocular drugs.16
Gold nanoparticles have an antimicrobial effect.17 Gold induces a strong X-ray attenuation, which makes it an ideal candidate for computed tomography (CT). It is known that the bone and tissue absorption are higher and the toxicity is lower with gold nanoparticles compared to iodine. Those characteristics enable gold nanoparticles to be more suitable as a CT contrasting agent. Furthermore, gold nanoparticles have been confirmed to be nontoxic and biocompatible in vivo.18,19
Topical ocular drugs cannot penetrate to the posterior segment of the eye. For treating the posterior segment of the eye, the drug was intravenously administered at a high dose. Intravitreal administration was also used. Unfortunately, only wide therapeutic drugs can be used in a high dose. Enhancing of the intraocular penetration of topically applied drugs may be the most favorable and safest choice.
The main aim of the current study was to enhance intraocular penetration and evaluate the therapeutic efficacy of the topically applied flucytosine capped with gold nanoparticles-loaded nanoliposomes for treating the symptoms associated with intravitreal endophthalmitis in rabbits.
Materials and methods
Materials
Flucytosine was purchased from Hangzhou Uniwise International Co., Ltd. (Hangzhou, People’s Republic of China). Soy bean phosphatidylcholine (PC), cholesterol (Ch), stearylamine (SA), gold(III) chloride trihydrate (HAuCl4·3H2O), L-ascorbic acid, and dicetyl phosphate (DCP) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Methyl alcohol was obtained from BDH Ltd (Liverpool, UK). Sodium Pentothal® was purchased from Abbott Laboratories (Abbott Park, IL, USA). The Spectra/Por® dialysis membranes (molecular weight cutoff of 12–14 and 40 kDa) were obtained from Spectrum Laboratories Inc. (Rancho Dominguez, CA, USA). C. albicans was supplied from Mycological Center, Faculty of Science, Assiut University (Assiut, Egypt). All clinical isolated Candida samples were subcultured on Sabouraud dextrose agar for 5 days and maintained aerobically at 30°C. All chemicals were used according to the manufacturer’s instructions. All reagents were of analytical grade.
Preparation of gold nanoparticles
The preparation of gold nanoparticle was carried out using the simple reduction method, as reported by Frens.20 In brief, the concentration of the prepared Gold(III) chloride solution (HAuCl4) (50 mL) ranged from 4×10−6 M to 5×10−5 M. Sucrose (5% w/v) was added to serial dilutions of gold(III) chloride, and the prepared mixtures were continually boiled. HAuCl4 at 1 mM was the selected concentration which was used in conducting the experiments. Fifty mL of aqueous solution consisting of L-ascorbic acid (1.6 mM) was added to the boiling solution and stirred for approximately 10 minutes, until wine-red color was produced.21–23 To get uniformity of size, the prepared solution of colloidal gold nanoparticles was centrifuged for 15 minutes at 10,000 rpm. The supernatant was kept for further use.
Preparation of the nanoliposomes
The nanoliposomes were prepared using the thin film hydration technique.24 PC and Ch were dissolved in a mixture of chloroform and ethanol (3:2). As shown in Table 1, the liposomal formulations F1, F2, and F3 were prepared using PC, Ch, and Span 60 at a ratio of 3:1:0.1, 2:1:0.1, and 1:1:0.1, respectively. The liposomal formulations F4, F5, and F6 were formulated using PC, Ch, Span 60, and SA at a molar ratio of 1:1:0.1:0.05, 1:1:0.1:0.1, and 1:1:0.1:0.15, respectively. The liposomal formulations F7, F8, and F9 were prepared using PC, Ch, Span 60, and DCP at a molar ratio of 1:1:0.1:0.05, 1:1:0.1:0.1, and 1:1:0.1:0.15, respectively. Table 1 represents the composition of the liposomal formulations (F1–F9).
 | Table 1 Composition of both positively and negatively charged liposomal formulations in molar ratio |
The organic solvents were vaporized under vacuum conditions within a rotary evaporator at a constant temperature of 40°C for a period of 20 minutes. Gold nanoparticles were added to 15 mg of flucytosine within a phosphate buffer (10 mL, 0.1 M, pH 7.4) to hydrate the prepared film. The preparations were sonicated for a period of 2 hours. To separate the non-entrapped flucytosine and gold nanoparticles, the liposomal dispersion was subjected to centrifugation using a refrigerated centrifuge at 21,000 rpm for 120 minutes at a constant temperature of 15°C (High-Speed Refrigerated Centrifuge, CR22N; Hitachi Ltd., Tokyo, Japan).
Following the centrifugation, the supernatant was carefully siphoned off to separate the unencapsulated flucytosine and gold nanoparticles. The liposomes remained as sediment containing the entrapped drugs. The sediment was resuspended in 5 mL of isotonic phosphate buffer (pH 7.4) in order to be evaluated. The liposomal dispersion (free from the unencapsulated flucytosine and gold nanoparticles) was stored at a constant temperature of 4°C within glass vials.
Determination of liposomes entrapment efficiency
A specific volume (5 mL) of the liposomal dispersion was subjected to a high-speed centrifugation at 21,000 rpm at 15°C for 120 minutes. The sediment was redispersed in 5 mL of isotonic phosphate buffer. The centrifugation was repeated twice. The liposomes were separated from the supernatant. One mL of the prepared liposomes was diluted and adjusted to a volume of 5 mL with methanol in a 10 mL volumetric flask, and the amount of the encapsulated drug was determined spectrophotometrically at a λ of 276 nm, respectively. The amount of the encapsulated gold nanoparticles was determined using an atomic absorption spectrometer (AAnalyst 200; PerkinElmer Inc., Waltham, MA, USA) at a wavelength of 243 nm, where the samples were processed at 120°C in diluted aqua regia in an oil bath for 1 hour.25
The percentage of entrapment efficiency was calculated using the following equation:
|
|
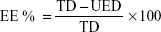
where EE % is the encapsulation efficiency percentage, TD is the total drug amount, and UED is the amount of unencapsulated drug.
Particle size analysis and zeta potential measurements
The produced liposomes and the prepared solution of colloidal gold nanoparticles were characterized for their sizes and polydispersity index using the Malvern PCS4700 instrument (Malvern Instruments, Malvern, UK).
The liposomal formulations were diluted with HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer (10 mM, pH 7.4) to reduce the turbidity of the dispersions. The zeta potential of the prepared liposomal formulations and the prepared solution of colloidal gold nanoparticles were measured using a Zetasizer Nano-ZS, Model ZEN 3600 (Malvern Instruments).
Transmission electron microscope
Transmission electron microscope (TEM) was used to investigate the nanoliposomes to show their morphology and detect the possible defects within their lamellar lipid bilayer.26 TEM was also used to characterize the prepared solution of colloidal gold nanoparticles to show their size, morphology, and shape.
The prepared liposomes and the prepared solution of colloidal gold nanoparticles were processed by mounting each one under investigation onto copper grids in order to adsorb the liposome particles from the suspension. The samples were stained with 2.5% uranyl acetate for 30 seconds and then dried. The specimens were observed under a TEM (TEM-1010 T; JEOL, Tokyo, Japan) operated at 120 kV.
In vitro release study of the liposomes
Dissolution of the liposomes was performed via the method of dialysis according to Hao’s method with minor modifications using an isotonic phosphate buffer (pH 7.4) as a dissolution medium.27 The experiments were carried out in triplicate. The equivalent of 5 mg of flucytosine-loaded liposomal suspension was introduced into the dialysis bags with a molecular weight cutoff of 12 kDa. The in vitro gold nanoparticle release of the liposomal formulations was estimated using dialysis tubing (molecular weight cutoff of 40 kDa). The dialysis bags were suspended in 25 mL of isotonic phosphate buffer (pH 7.4) and placed within the dissolution flask of the dissolution apparatus with a constant temperature of 37°C±0.5°C.
Samples of 1 mL were withdrawn from the dissolution medium at predetermined time intervals for a period of 12 hours and then diluted to 1:10 in distilled water. The dissolution medium was replaced with equal volumes of a fresh dissolution medium of isotonic phosphate buffer (pH 7.4) to maintain the sink condition. The withdrawn samples were measured spectrophotometrically at a λmax of 276 nm. The release of flucytosine from liposomes was determined under sink conditions. The bags were fully immersed below the surface while the release of gold nanoparticles was determined using an atomic absorption spectrometer (AAnalyst 200; PerkinElmer Inc.) at a wavelength of 243 nm. The dissolution profile of pure flucytosine dispersion was achieved by dispersing an equivalent amount of flucytosine powder in 2 mL of isotonic phosphate buffer (pH 7.4). The release of flucytosine was determined using the following equation:
|
|
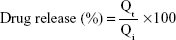
where Qi and Qt are the initial amounts of flucytosine or gold nanoparticles encapsulated in the liposomes, and the amounts of flucytosine or gold nanoparticles released at time t, respectively. All experimental procedures were repeated three times.
Stability studies of flucytosine liposomes
Physical stability study of the gold nanoparticle-loaded liposomes was carried out to evaluate the extent of leakage of the gold nanoparticles from liposomes (in a liquid form) which were stored at different conditions. Signs of sedimentation, creaming (if any), or any changes in color of the examined dispersions were also evaluated. Finally, the examined dispersions were analyzed for their particle size using a Zetasizer Nano-ZS, Model ZEN 3600 (Malvern Instruments). A protocol developed by du Plessis et al28 was adopted with some modifications. The gold nanoparticle-loaded liposomes and colloidal gold nanoparticle solution were stored in tightly sealed 20 mL glass vials at 4°C in a refrigerator, and at 25°C±1°C and 37°C±1°C in an incubator, for 3 months. Samples of liposomal dispersions and colloidal gold nanoparticle solution were examined at regular time intervals between 0 and 3 months. One milliliter of the stored samples of each formulation was centrifuged in a refrigerated centrifuge at 14,000 rpm for 1 hour to separate the free drug which leaked out from the liposome during the storage period. The amount of gold nanoparticles was determined using an atomic absorption spectrometer (AAnalyst 200; PerkinElmer Inc.) at a wavelength of 243 nm, where the samples were processed at 120°C in diluted aqua regia in an oil bath for 1 hour.
In vitro antifungal activity of flucytosine-loaded liposomes
Preparation of fungal strains
The fungal strain was identified and prepared in a sterile saline as follows: C. albicans was obtained from the Sabouraud dextrose agar to prepare cell suspensions in sterile normal saline which were adjusted to McFarland number 1 turbidity, and the suspensions were inoculated to give a final concentration of 5×105 cells/mL. These suspensions were used for in vitro antifungal activity test.
In vitro antifungal evaluation
The antifungal activity study of the prepared nanoliposomes (F3, F6, and F9) was compared with that of both the flucytosine solution (20 μL, 0.8% w/v) and the prepared solution of colloidal gold nanoparticles (20 μL, 150 μM). The disc diffusion method was used to evaluate antifungal activity of the tested preparations where the mean diameter of growth inhibition zones (in millimeters) was determined.
This was performed by disc diffusion method where sterile filter paper discs (4.8 mm in diameter) were impregnated with 20 μL of flucytosine-loaded liposomes, flucytosine solution (20 μL, 0.8% w/v), or the prepared solution of colloidal gold nanoparticles (150 μM) and were placed on the surface of the inoculated Sabouraud dextrose agar in triplicate, for each tested preparation. The Sabouraud dextrose agar plates were inoculated with the prepared C. albicans solutions (1 mL, 5×105 cells/mL). Discs without antifungal agents were placed on inoculated agar for positive growth control results. All inoculated plates where incubated at 30°C for 5 days and examined for the inhibition zone. The diameter of the inhibition zone around each disc was measured and the mean value of three experiments was calculated.29
Preparation of animals and CT imaging
Animals
Within the current study, nine adult New Zealand White rabbits with an average weight of 2 kg were provided by the animal house faculty of medicine at Assiut University. The rabbits were randomly divided into three groups (n=3), with each group including three rabbits. The 1964 Helsinki protocols for humane handling and treatment of animals were afforded to all experimental rabbits within the present study.30 All of the rabbits’ eyes were initially examined with a handheld torch. Only animals devoid of any signs of ocular pathogens were included in this study. Liposomal formulations (F3, F6, and F9) and the prepared gold nanoparticle solution were used throughout the study.
Application technique
For the first group, two drops of phosphate buffer solution were instilled into the right eyes, while two drops of F3 were instilled into the left eyes. In the second group, two drops of F6 was instilled into the right eyes, while two drops of F9 was instilled into the left eyes. In the third group, nothing was instilled into the right eyes, while two drops of the prepared solution of colloidal gold nanoparticles were instilled into the left eyes. CT imaging was carried out at 0.5, 1, 2, and 6 hours after the different formulations were applied.
CT imaging
The rabbits were injected with lorazepam (0.5 mL, 1 mg/mL) to be sedated during the scanning process of CT imaging. The tube voltage was set at 120 kV. The scanning field within the rabbits’ eyes was carried out at the axial plane (0.75 mm collimation helical scan, with a 5.1 mm table feed). The CT imaging was carried out using the Siemens SOMATOM Sensation 64 CT Scanner (Siemens CO, Erlangen, Germany). The protocols of CT imaging approved by the Animal Ethical Committee of the Faculty of Medicine, Assiut University, were employed at all times.31
In vivo aqueous humor flucytosine concentration study
Animals
As mentioned earlier, 12 New Zealand White rabbits with an average weight of 2 kg were used within this study. This study was also approved by the Animal Ethical Committee of the Faculty of Medicine, Assiut University. All animal experiments were carried out in accordance with the UK Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments. The rabbits were randomly divided into four groups where each group included three rabbits (n=3).
In the first group, left eyes were instilled with two drops of flucytosine solution (100 μL, 0.3 mg/mL) into the lower conjunctival sac of the rabbits’ left eye. In the second group, left eyes were instilled with two drops of liposomal formulation (F3). In the third group, left eyes were instilled with two drops of liposomal formulation (F6). In the fourth group, left eyes were instilled with two drops of liposomal formulation (F9). The study was carried out by instilling each aforementioned preparation (equivalent to 0.3 mg flucytosine).
To anaesthetize the rabbits, ketamine was intramuscularly injected (30 mg/kg). From the anterior chamber of the eye, 80 μL samples of aqueous humor were withdrawn at different time intervals. The samples were stored at −20°C until high-performance liquid chromatography analyses were performed. The area under the curve, the maximum flucytosine concentration in aqueous humor (Cmax), and the time consumed to reach this concentration (Tmax) were calculated.
In vivo antifungal activity of flucytosine and gold-loaded liposomes
Animals
Thirty-two New Zealand White rabbits with an average weight of 2 kg were used within this study. They were provided by the animal house of the Faculty of Medicine at Assiut University. The rabbits were randomly divided into four groups where each group included eight rabbits (n=8). All of the rabbits’ eyes were initially examined with a handheld torch. All experimental animals were free from any signs of ocular pathology.
Inoculation technique
The procedure was based on a model of experimental keratomycosis.33 The intraperitoneal injection of 0.5 mL sodium pentothal (Abbott Laboratories) was used to sedate the rabbits. Local anesthetic was not applied to avoid the presence of antimicrobial agents. Both eyes of all rabbits were intrastromally injected with 15 μL inoculum (C. albicans, containing 2.5×105 cells/mL) by inserting a sterile 27-gauge needle into the central corneal stroma tangential to the corneal surface to a depth of one-half of the corneal thickness. If penetration of the inoculum into the anterior chamber or reflux of the inoculum was observed during the experiment, those rabbits were excluded. The rabbits were examined with a slit lamp. Only rabbits with the following symptoms were included in the experiment: widespread inflammation, white fluffy corneal ulcers, the redness and edema of eyelids, a hypopyon, excessive tearing, or discharge.
Treatment procedure
This study included 32 rabbits; they were randomly divided into four equal groups. For the first group, two drops of flucytosine solution (0.2% w/w) were instilled into the right eyes. For the second group, two drops of liposomal formulation (F3) were instilled into the right eyes. For the third group, two drops of liposomal formulation (F6) were instilled into the right eyes. Finally, for the fourth group, two drops of liposomal formulation (F9) were instilled into the right eyes. All formulations were prepared under aseptic conditions. They were applied to the right eyes. The left eyes were considered to be the control.
Application of the eye drops and the follow-up
Fifty microliters of previously mentioned formulations was applied topically as eye drops. Application of eye drops into the conjunctival sac of rabbits commenced 48 hours after the inoculation procedure. For the first 3 days, the application was completed four times daily, and was reduced to three times daily during the next period of treatment which last for 28 days. After application, the rabbits’ eyes were manually closed for approximately 2 minutes. The rabbits’ eyes were examined daily over a 28-day period using a handheld torch and with a slit lamp for signs of infection. To detect gradual healing of the infected eyes, both the right and the left eyes of the four groups were examined. Examination was initially carried out at the beginning of application and after each week. The severity of inflammatory reaction was determined (hypopyons, iritis). Photographs were captured after the induction of keratitis, and also they were captured after every week of the treatment period (4 weeks). Signs of inflammation were followed-up for each rabbit and recorded according to their enumeration. Occurrence of any complications were also recorded such as corneal perforation and resistant corneal ulcer.
Statistical analysis
For evaluating the significance of the differences between the two sets of values, two-tailed Student’s t-tests were applied. Statistical significance was calculated at 95% confidence intervals (P>0.05). A one-way analysis of variance (ANOVA) was applied to the diameters of inhibition zone of in vitro antifungal study and also to the mean ocular penetration of in vivo CT imaging.
Results and discussion
Entrapment efficiency
The entrapped flucytosine concentration was expressed as a loading efficiency percent. Loading efficiency is generally defined as the percent fraction of the total input drug encapsulated (in lipid bilayers and/or aqueous compartments) in the liposomes at a particular phospholipids concentration, and is expressed as a weight percentage and calculated using the following equation:
|
|
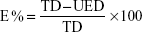
To clarify the formulation within this equation, E% is the percentage of encapsulation or the loading efficiency, TD is the total drug amount, and UED is the amount of unencapsulated drug.
As shown in Table 2, it can be concluded that flucytosine has been successfully incorporated into liposomes. Encapsulation efficiency of flucytosine ranged from 70.52%±1.86% to 85.52%±1.64%. The EE % increased as a result of the Ch content increase. As a result of increasing the Ch content within the lipid bilayer of liposomes, the rigidity of the lipid bilayer was increased.34 Consequently, the raised stability was reported.
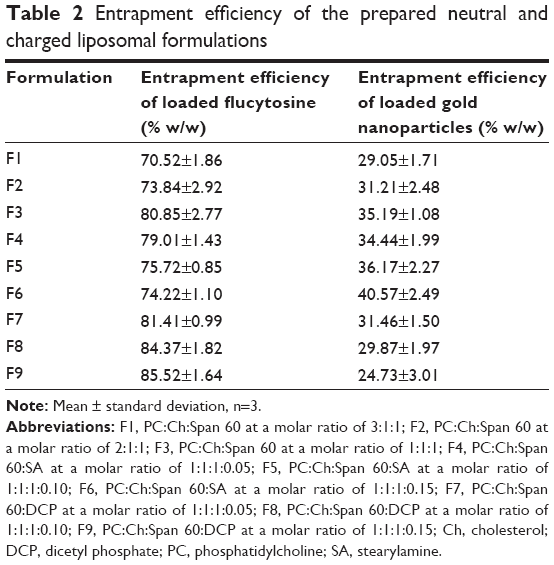 | Table 2 Entrapment efficiency of the prepared neutral and charged liposomal formulations |
The charge inducers effect on the entrapment efficiency percentages of flucytosine in liposomes was obvious with formulations F4–F9. The negatively charged liposomes, which contained PC/Ch/Span 60/DCP in a molar ratio of 1:1:0.1:0.15, exhibited the highest EE % (approaching 85.52%±1.64%), and the liposomal formulation (F3) composed of PC/Ch/Span 60 in a molar ratio of 1:1:0.1 exhibited encapsulation efficiency higher than those of the liposomal formulations F4, F5, and F6. This order of EE % may be due to flucytosine having a pKa of approximately 3.26. At this level of pKa, flucytosine presents as an ionized positive charge amino group, so an electrostatic attraction is formed between the drug and the negative charge inducer DCP. This attraction leads to an increase in the EE % when compared with the positively charged liposome. On the contrary, an electrostatic repulsion is formed between the drug and the positive charge inducer, SA, which leads to a decrease in the EE % of less than those for the neutral formulations. Similar results were determined during the incorporation of the charge inducers into indomethacin liposomes and acetazolamide liposomes.35,36 The results of encapsulation efficiency of gold nanoparticle-loaded liposomes can be attributed on the same basis, but the gold nanoparticles have the negative charge. Consequently, the encapsulation efficiency of the liposomal formulation F6 is the highest. The one-way ANOVA showed a significant difference between all pairs at P<0.05.
Particle size analysis, zeta potential measurements, and morphology
The results of particle size measurements for freshly prepared charged liposomal dispersions are shown in Table 3. The mean particle size was estimated to be 120.1±10.1 nm for neutral liposomal formulation F3. Liposomal formulations F4–F6 showed mean particle diameter measurements of 122.9±5.5 nm, 129.0±4.2 nm, and 135.1±12.0 nm, respectively, which were slightly higher than those of liposomal formulations F1–F3. Liposomal formulations F7–F9 showed a mean particle size of 150.5±14.1 nm, 158.9±4.9 nm, and 174.1±8.9 nm, respectively, which were higher than that of liposomal formulations F1–F3. These findings can be explained on the basis of inclusion of a charge inducer within the liposomes, which increases the spacing between the adjacent bilayers, resulting in the formation of larger liposomes compared with those of the neutral liposomes.37 Furthermore, the negatively charged lipids electrostatically attract the drug cation which may be expected to force the phospholipid head groups apart. Thus, the particle size is increased.38 This increase in particle size, as mentioned previously, would account for the higher encapsulation efficiency of the negative liposomes, compared with those of the neutral and positive. Moreover, the centrifuged sample of the prepared solution of colloidal gold nanoparticles had an average particle size of 13.3±4.0 nm. The results showed homogeneous vesicles in which the presence of close bilayer structures was confirmed. The prepared liposomes had a closed bilayer structure ranging in size from 80–120 nm. The shape of liposomes appeared spherical as shown in Figure 1. Those findings were consistent with the results obtained from the particle size measurements, as shown in Table 3.
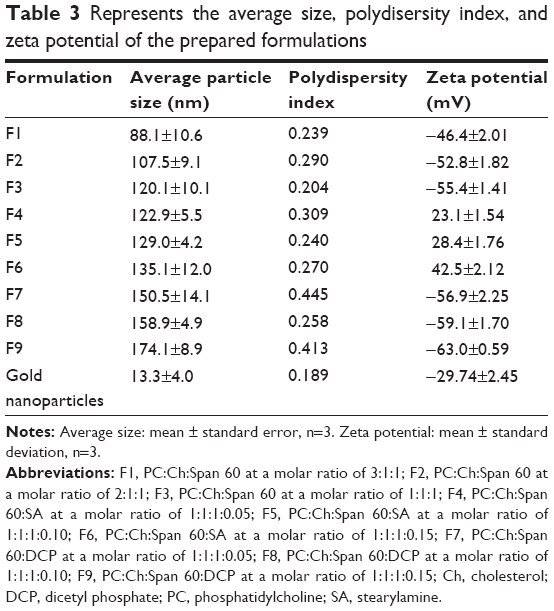 | Table 3 Represents the average size, polydisersity index, and zeta potential of the prepared formulations |
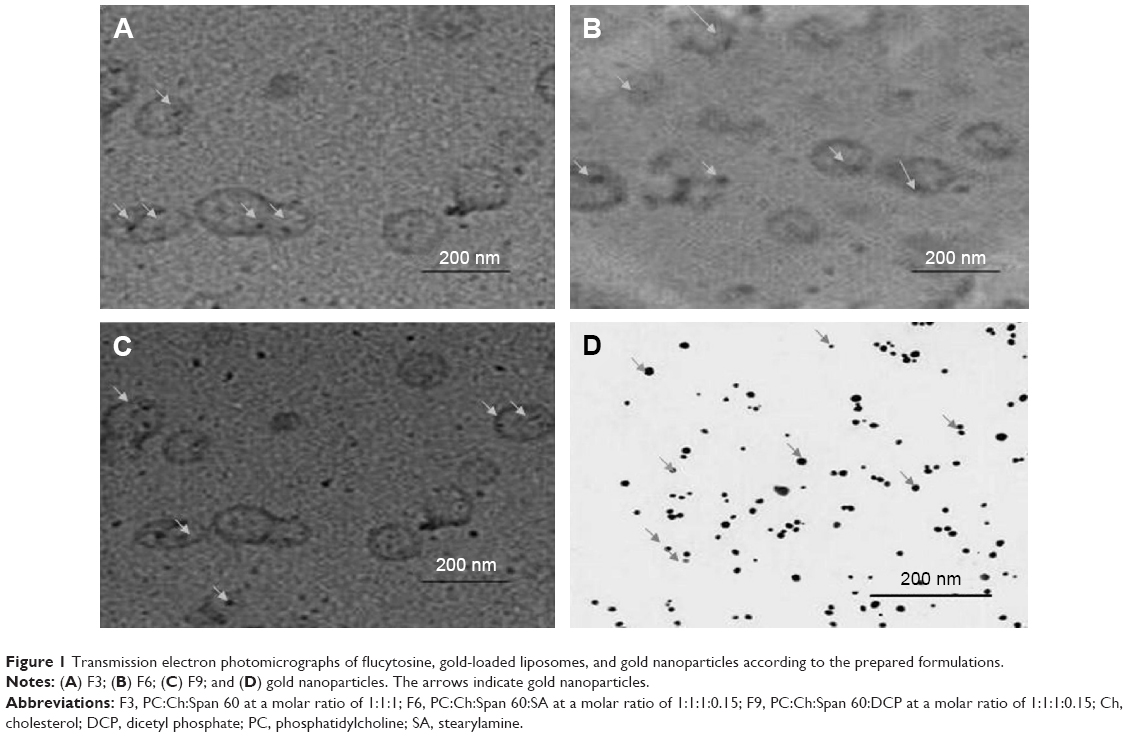 | Figure 1 Transmission electron photomicrographs of flucytosine, gold-loaded liposomes, and gold nanoparticles according to the prepared formulations. |
It is clear that the highest zeta potential has been obtained with the liposomal formulation F9, while the lowest measurement is produced with the liposomal formulation F6. The obtained results indicate that all liposomal preparations are stable according to reported literature.39
If the particles have low zeta potential values, then there is no force to prevent the particles coming together and flocculating. The general dividing line between stable and unstable colloidal formulations is generally considered at either +30 mV or −30 mV. A generally normal stable condition for suspension is considered when the zeta potential of the prepared colloidal formulations is greater than +30 mV or its negativity is more than −30 mV.40
The measurement of zeta potential is significant for characterization of gold nanoparticles. The zeta potential measurement of the prepared solution of colloidal gold nanoparticles was −29.74±2.45 mV. The high negative charge on gold nanoparticles is considered a fundamental indicator for their particle size. The negative zeta potential charge of nanoparticles indicates that their particle size is less than 100 nm.41
Liposomal formulations F1–F3 carry a strong negative charge as shown in Table 3. These results may be attributed to the influence of the liposome composition and the low ionic strength. Effect of nonionic surfactant may lead to increase the negative charge of the prepared liposomes. These findings are in agreement with results that have been found by Sureewan et al.42 Moreover, gold nanoparticles may contribute to the high negative charge of gold nanoparticle-loaded liposomes, where the zeta potential of gold nanoparticles was −29.74±2.45 mV. The ionic strength of liposomal dispersion may affect the zeta potential of the prepared liposomes. Crommelin43 has reported that the zeta potential of liposomes was larger at low ionic strength.43
Drug release in vitro
The release of flucytosine from the prepared nanoliposomes within a phosphate-buffered saline medium of pH 7.4 was investigated. The percentage of cumulative release of flucytosine and gold nanoparticles from the nanoliposomes is presented in Figures 2 and 3. From cumulative release profiles (Figures 2 and 3), it is apparent that the liposomal formulation F6 showed the fastest rate of flucytosine release (9.06 μg·cm−1·h−1/2). In contrast, the liposomal formulation F9 showed the slowest release rate of flucytosine (7.03 μg·cm−1·h−1/2).
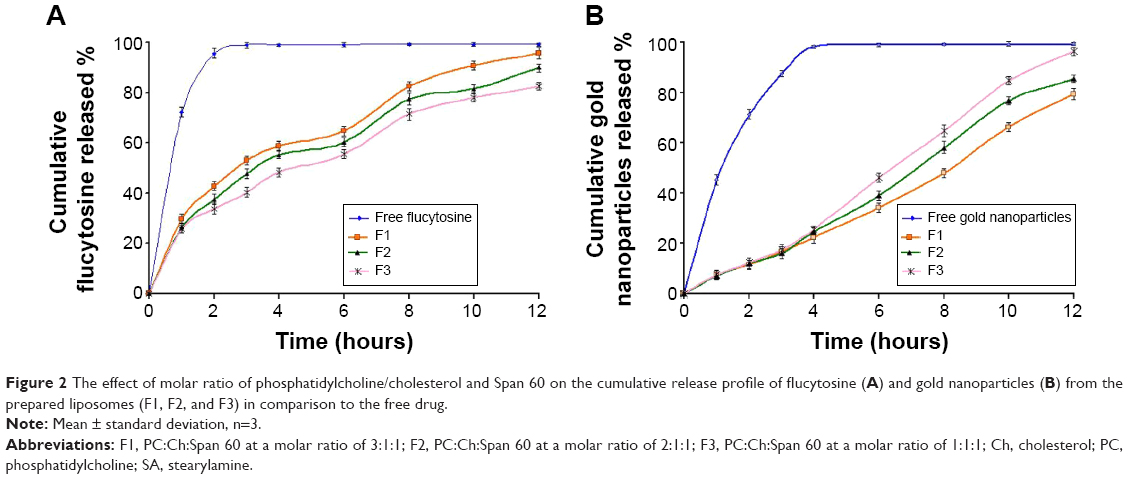 | Figure 2 The effect of molar ratio of phosphatidylcholine/cholesterol and Span 60 on the cumulative release profile of flucytosine (A) and gold nanoparticles (B) from the prepared liposomes (F1, F2, and F3) in comparison to the free drug. |
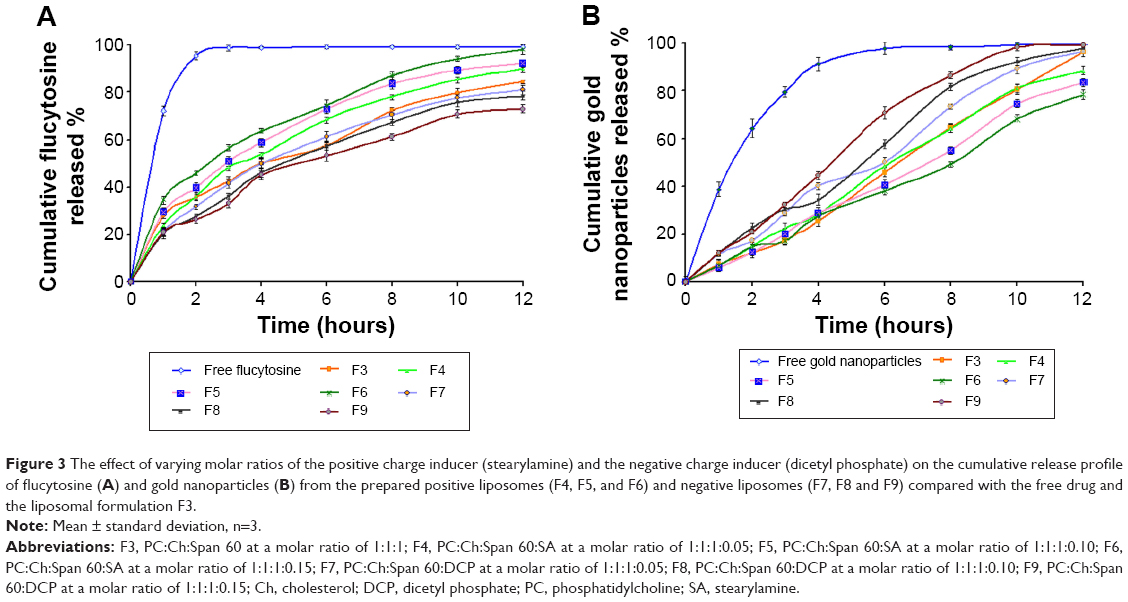 | Figure 3 The effect of varying molar ratios of the positive charge inducer (stearylamine) and the negative charge inducer (dicetyl phosphate) on the cumulative release profile of flucytosine (A) and gold nanoparticles (B) from the prepared positive liposomes (F4, F5, and F6) and negative liposomes (F7, F8 and F9) compared with the free drug and the liposomal formulation F3. |
Atomic absorption spectrophotometry was used to determine the gold nanoparticle concentration. From cumulative release profiles (Figures 2 and 3), the liposomal formulation F9 showed the fastest rate of gold nanoparticle release (2.86 μg·cm−1·h−1) while the liposomal formulation F6 showed the slowest one (2.08 μg·cm−1·h−1).
Those findings may be attributed to the forces of electrostatic attraction that may be formed between flucytosine cation and negatively charged liposomes. Thus, the extended release is achieved. However, an electrostatic repulsion which occurs between flucytosine cation and the positively charged liposomes may lead to a higher drug-release rate. However, gold nanoparticles have a strong negative charge. Thus, the electrostatic repulsion which occurs between the gold nanoparticles and the negatively charged liposomes may be the reason for the elevated release rate of gold nanoparticles. These results are in agreement with those obtained by Hosny who found that the rate of drug release was increased as result of an electrostatic repulsion between ciprofloxacin and the negatively charged liposomes.38
Linear regression analysis of the release data fitted into zero-order, first-order, and Higuchi diffusion-controlled model equations were applied to all in vitro flucytosine release results.44 The results showed that the majority of flucytosine release from the prepared liposomal formulations followed the Higuchi diffusion-controlled model, as shown in Figure 4. The obtained results are in agreement with many studies, whose findings indicate that many loaded drugs can be released from liposomes by the same mechanisms.36,45
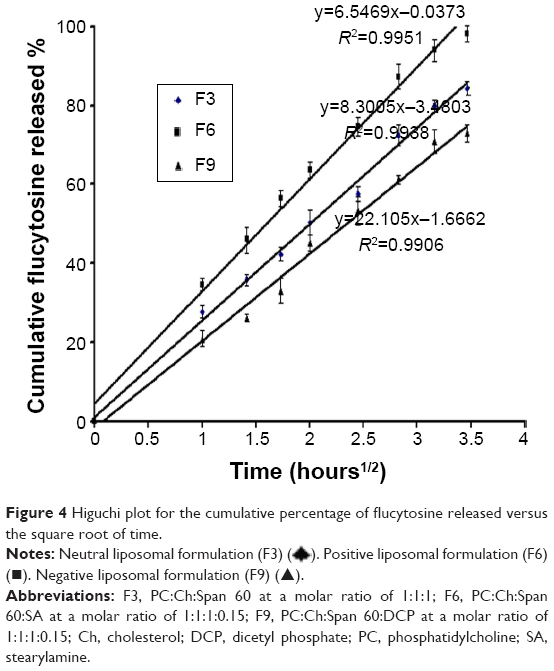 | Figure 4 Higuchi plot for the cumulative percentage of flucytosine released versus the square root of time. |
 ). Positive liposomal formulation (F6) (■). Negative liposomal formulation (F9) (▲).
). Positive liposomal formulation (F6) (■). Negative liposomal formulation (F9) (▲).However, the release rate of gold nanoparticles followed the zero-order kinetics with r2>0.990, as shown in Figure 5. The obtained results are in agreement with those found by Margalit et al who illustrated the release mechanism of metal nanoparticles.46,47
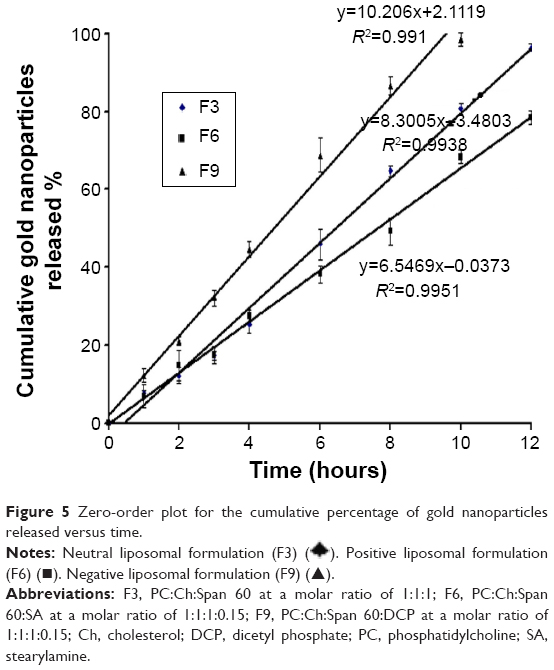 | Figure 5 Zero-order plot for the cumulative percentage of gold nanoparticles released versus time. |
Two-tailed Student’s t-tests were applied to cumulative release results. Calculated statistical significance at 95% confidence intervals (P>0.05) reveals a significant difference. Because of the highest flucytosine EE %, the liposomal formulation F9 was selected for the further examination. In addition, the liposomal formulation F6 was selected because of its highest gold nanoparticle entrapment efficiency percentage. Furthermore, the liposomal formulation F3 was selected for further examination.
Stability study of liposomes
The visual appearance and average particle size of liposomes are important parameters to be evaluated. With regard to storage of liposomes, drug leakage from the vesicles represented an important problem. The liposomal dispersions were sampled at regular time intervals between 0 and 3 months and they were examined for their physical appearance including signs of sedimentation, creaming, and any change in the color of the dispersions. The visual appearance showed that liposomal formulations F3, F6, and F9 stored at 4°C±1°C, 60%±5% relative humidity showed no sedimentation or color change throughout 3 months. However, the formulation stored at 25°C±1°C and 37°C±1°C, 60%±5% relative humidity showed a pale yellow color during the course of stability study (3 months). No sedimentation was observed.
Liposomal formulations F3, F6, and F9 exhibited a slight decrease in encapsulation efficiency (% w/w) and an increase in particle size during the course of stability study (3 months) at 4°C±1°C, as reported in Tables 4 and 5. Those changes were found to be insignificant (P>0.05). However, liposomal formulations F3, F6, and F9 stored at 25°C±1°C and 37°C±1°C showed a sharp decrease in the entrapment efficiency and a sharp increase in the particle size as shown in Tables 6 and 7. The decrease in the encapsulation efficiency and increase in the particle size were found to be significant (P<0.05). This means that for up to 3 months of storage period at 4°C, the prepared gold nanoparticle-loaded liposomes (F3, F6, and F9) were somewhat stable. However, after 3 months of storage at 25°C±1°C and 37°C±1°C, the formulations were unstable concerning the encapsulation efficiency and the particle size. The decrease in encapsulation efficiency at 25°C±1°C and 37°C±1°C may be explained on the basis of the fluidity of vesicular membrane. The fluidity of vesicular membrane is increased at higher temperature, resulting in significant leakage of loaded drug.48 Possible chemical degradation of the phospholipids at 25°C±1°C and 37°C±1°C may be another reason. It leads to defects in the vesicular membrane packing.49 These findings are in accordance with those of other studies, which found that clofazimine-loaded liposomes were stable at refrigeration temperature (2°C–8°C).50 Above absolute zero temperature, colloidal particles in bilayer of liposomes are in a dynamic state. As the temperature increases, the motion and vibration of colloidal particles increases.48 The stability of gold nanoparticles within different liposomal formulations (F3, F6, and F9) stored at 4°C±1°C, 25°C±1°C, and 37°C±1°C was monitored over 3 months. Results showed that gold nanoparticles stored at 4°C±1°C exhibited an 8.63% (F3), 9.6% (F6), and 9.23% (F9) increase in size, respectively; the increase in size of gold nanoparticles within different liposomal formulations (F3, F6, and F9) stored at 25°C±1°C was 60.43%, 64.80%, and 65.38%, respectively; and the increase in size of the gold nanoparticles at 37°C±1°C was 123.74%, 141.60%, and 133.85%, respectively. Those findings are concurrent with those reported by Rükan et al.51 This can be attributed to a better interaction between the liposomes and the gold nanoparticles at 4°C±1°C preventing particle aggregation even over long storage periods. While at higher temperature, the fluidity of vesicular membrane is increased, which results in a significant leakage of loaded gold nanoparticles,48 and/or aggregation of particle may occur.
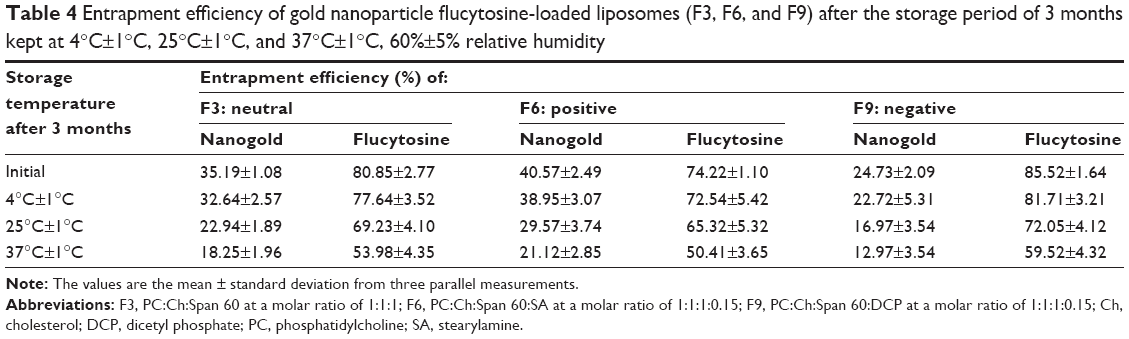 | Table 4 Entrapment efficiency of gold nanoparticle flucytosine-loaded liposomes (F3, F6, and F9) after the storage period of 3 months kept at 4°C±1°C, 25°C±1°C, and 37°C±1°C, 60%±5% relative humidity |
 | Table 5 Particle size of the prepared liposomes (F3, F6, and F9) and the released gold nanoparticles after the storage period of 3 months kept at 4°C±1°C, 25°C±1°C, and 37°C±1°C, 60%±5% relative humidity |
 | Table 6 Intraocular penetration distance of colloidal gold nanoparticle solution and neutral, positively, and negatively charged gold nanoliposomes across rabbits’ eyes was measured using computed tomography |
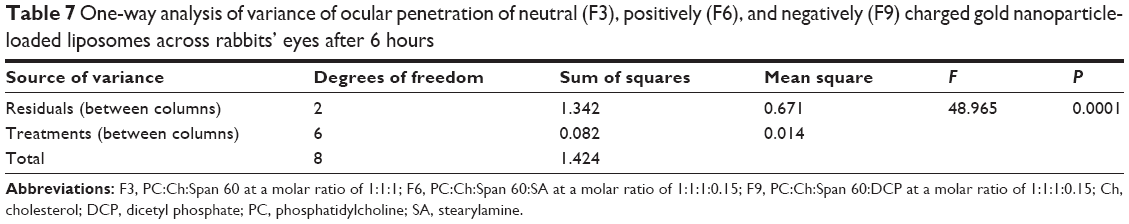 | Table 7 One-way analysis of variance of ocular penetration of neutral (F3), positively (F6), and negatively (F9) charged gold nanoparticle-loaded liposomes across rabbits’ eyes after 6 hours |
In vitro antifungal activity of gold nanoliposomes
In vitro antifungal activity study of the prepared nanoliposomes (20 μL) (F3, F6, and F9) was compared with that of both the flucytosine solution (20 μL, 0.8% w/v) and the prepared solution of colloidal gold nanoparticles (20 μL, 150 μM) using the disc diffusion method as shown in Figure 6. In vitro antifungal activity study was carried out using C. albicans of fungi isolated from the eyes and it was evaluated by measuring the mean diameter of growth inhibition zones in millimeters. It was found that all tested formulations produce a marked inhibition effect as shown in Table 8. Mean diameter of growth inhibition zone by using gold nanoparticle-loaded liposome (F3) was 31.63±0.603 mm. The effect of incorporation of SA on the antifungal activity of gold nanoparticle-loaded liposomes was the significant increase in the mean diameter of the inhibition zone (34.66±0.851 mm) as shown in Table 8. However, incorporation of DCP resulted in a significant decrease in the mean diameter of the inhibition zone (29.52±0.451 mm) (P=0.0003 using one-way ANOVA test).
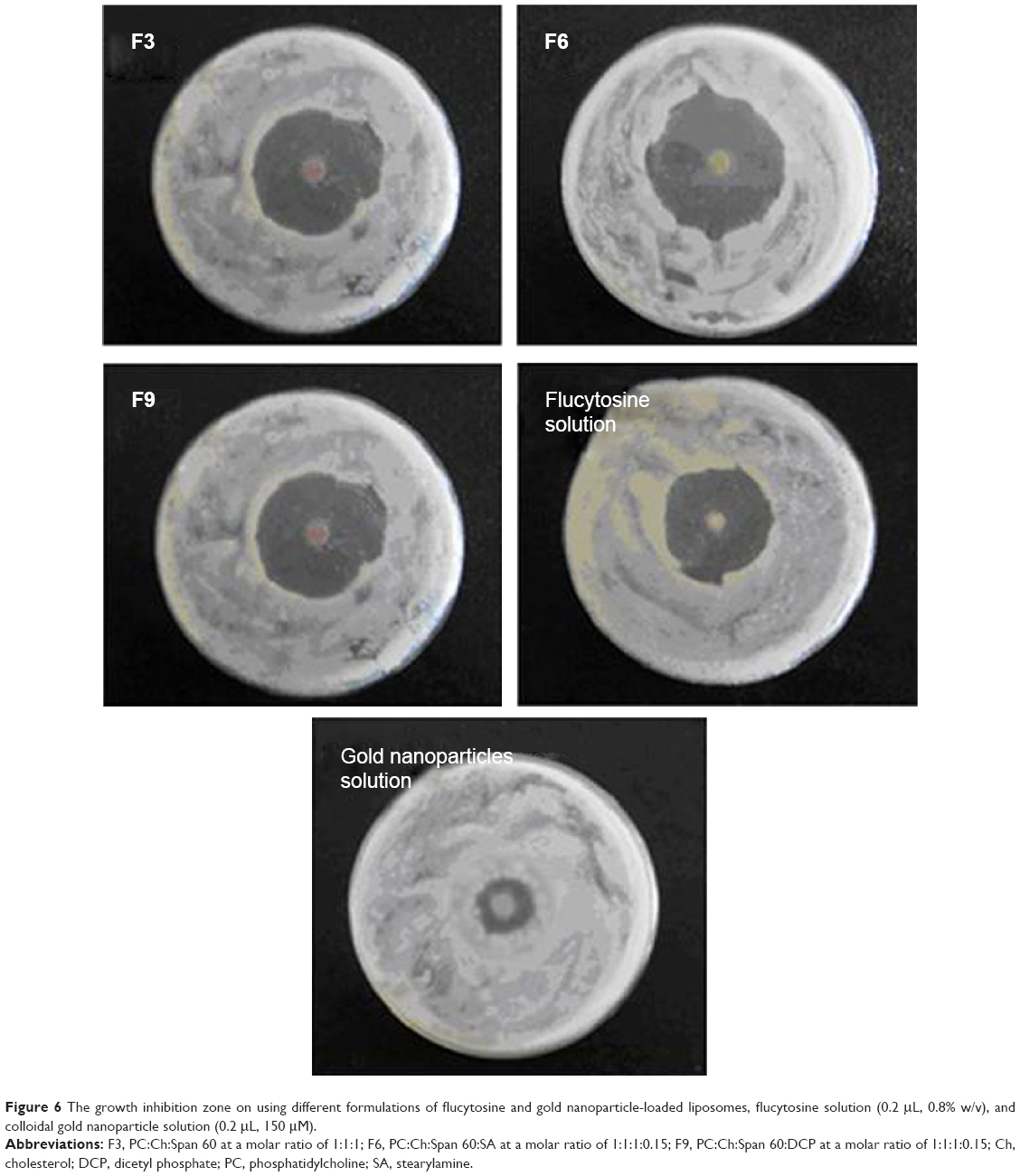 | Figure 6 The growth inhibition zone on using different formulations of flucytosine and gold nanoparticle-loaded liposomes, flucytosine solution (0.2 μL, 0.8% w/v), and colloidal gold nanoparticle solution (0.2 μL, 150 μM). |
 | Table 8 In vitro susceptibility test of different liposomal formulations and control gold nanoparticle solution by disc diffusion method |
This may be interpreted such that the incorporation of SA decreases the stability and the rigidity of the liposomal bilayer leading to an increase in the permeability of the liposomal bilayers of the loaded flucytosine. However, incorporation of DCP increases the stability and rigidity of the liposomal bilayer leading to a decrease in the permeability of the liposomal bilayers of the loaded flucytosine. Gold nanoparticles were released augmenting the antifungal effect of flucytosine. The antifungal effect of gold nanoparticles may be attributed to the property that they deactivate both transmembrane energy metabolism and membrane electron transport chain as a result of insoluble compound formation in the fungal cell wall as a consequence of inactivation of the cell wall sulfhydryl group. Additionally, gold nanoparticles may cause fungal DNA mutation and dissociate the fungal enzyme complexes.52 Those enzymes are very important for both respiratory chain and membrane permeability, where the cell lysis is caused, as a result of deactivation of the respiratory chain and membrane permeability. Furthermore, the antifungal effect of the gold nanoparticles may be explained on the basis of genome islands encoding a lot of toxins.52
In vivo CT imaging
The anatomy of the eye has two well-defined areas: 1) the anterior segment and 2) posterior segment. Topical absorption of drugs occurs either through a corneal or a non-corneal route. The cornea is composed of the epithelium, stroma, and endothelium tissues. Different corneal tissues have different drug permeation. The non-corneal absorption route takes place through the sclera and conjunctiva tissues. Hence, the penetration of the drug molecules from ophthalmic preparations through ocular tissues is considered a sophisticated process.53 Success of topical ophthalmic preparation for treatment of fungal endophthalmitis of the ocular vitreous chamber depends on attaining therapeutic drug concentrations in the posterior segment. Therefore, there was a need to measure the influx of the drug into the eye. A CT is considered to be one of the most accurate tools for measuring the size of many different tissues.54 CT imaging was used to trace the drug when loaded onto gold nanoparticles and detect the biodistribution of them across the rabbits’ eyes.55 The results represented in Figure 7 show CT scans of the rabbits’ eyes following the application of the gold nanoparticle-loaded liposomes. The penetration of the gold nanoparticles in liposomal formulations increased within the eyes during this 6 hours period. Liposomal formulation F6 gave the highest penetration distance of gold nanoparticles (10.22±0.113 mm), while liposomal formulation F3 gave the lowest penetration distance of gold nanoparticles (9.34±0.160 mm). The important question of whether the penetration of gold nanoparticles to the ocular tissues was due to the physicochemical characteristics of the nanoparticles or to the physicochemical properties of the liposomal formulation should be answered. On instillation of colloidal gold nanoparticle solution (left eyes of group 3, Figure 7), weak penetration of gold nanoparticles (1.96±0.085 mm) was reported (Table 6). The physicochemical properties of the prepared liposomal formulations may be considered in penetration of gold nanoparticles. The penetration of nanoliposomes in the eyes can be explained on the basis of the negatively charged retina, which may attract the liposomal formulation F6 to a greater extent than the other formulations. Consequently, the prolonged contact periods of the preparation on the retina may be achieved.56 It was expected that the penetration ability of the prepared neutral liposomal formulation (F3) would be the highest because of the smallest average particle size, but the results obtained show insignificant difference.57 This can be explained on the basis of the small differences in the average particle size among the formulated liposomes. The important factor that affects the drug ocular penetration rate is its physicochemical properties such as the particle size of the loaded drug and also the physicochemical properties of its delivery system.58 In the case of liposomal formulations of the drug, lipid vesicles can cross cell membranes. Consequently, they can alter the extent and rate of penetration, in addition to the drug disposition.
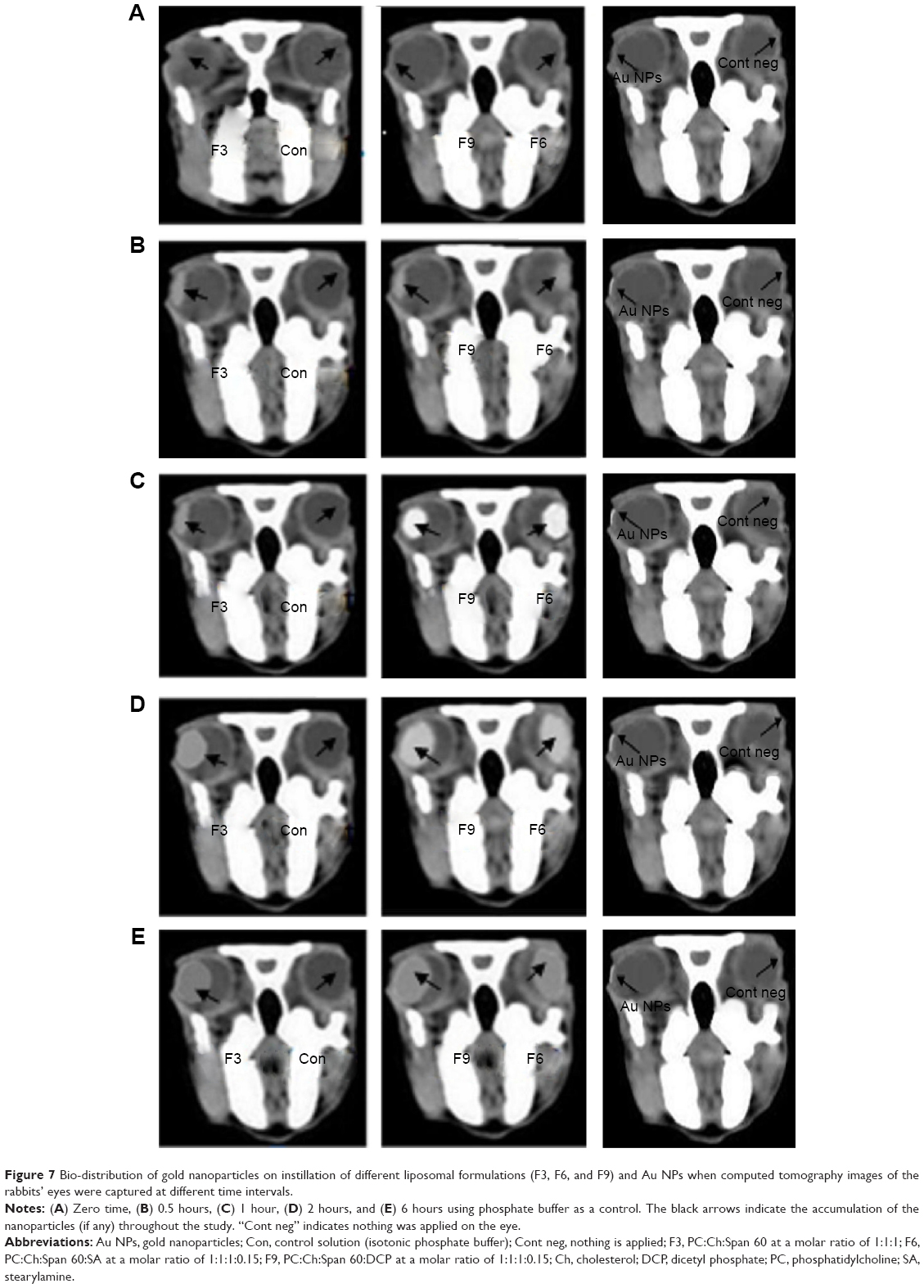 | Figure 7 Bio-distribution of gold nanoparticles on instillation of different liposomal formulations (F3, F6, and F9) and Au NPs when computed tomography images of the rabbits’ eyes were captured at different time intervals. |
The mechanism of liposome interaction with the cornea is relatively unknown, even to the present day. Many hypotheses for these mechanisms are used as an explanation of liposome interaction with the cells. They include lipid exchange, stable adsorption, endocytosis, and fusion.59
Fusion of liposomes with the cornea may exist to some extent; however, the major mechanism may be the adsorption and/or the surface lipid exchange. In light of the fusion mechanism, intact liposomes containing the drug were absorbed; consequently, the ocular drug concentration would be similar regardless of the nature of the drug entrapped and the penetration process would only be concerned with the type of liposomes, their size, and surface charge.60
The statistical analysis of the results were carried out using one-way ANOVA of means at the 5% significance level using ExcelStat Pro software, Microsoft Office 2007. Ocular penetration was taken as the parameter for ANOVA analysis. The P-value was determined and the result is shown in Table 7. One-way ANOVA at the 5% significance level and disintegration time as a parameter yielded a P-value of 0.0001, so it can be concluded that all the examined formulations were found to be different (P<0.0001). Consequently, the addition of the positive charge inducer SA and the negative charge producer DCP has influenced the ocular penetration of the liposome formulations.
In vivo aqueous humor flucytosine concentration study
Flucytosine concentrations in aqueous humor post instillation of 0.3 mg of flucytosine as solution and liposomal formulations F3, F6, and F9 are shown in Figure 8. In comparison to the other examined preparations, liposomal formulation F6 showed the highest Cmax and shortest Tmax values (Figure 8). This finding indicates that the highest rate of absorption of flucytosine occurs when liposomal formulation F6 is applied. Eye drops of flucytosine solution showed the lowest Cmax. This can be attributed to the rapid drainage, and consequently decreased residence time, of the instilled flucytosine solution. Liposomal formulation F6 showed the highest area under the curve value (97.82±17.3 μg/g/h) for first 12 hours after flucytosine instillation. Compared to flucytosine solution, the relative bioavailability of liposomal formulation F6 was 11.75 as shown in Table 9. These findings can be attributed to the improved residence time of flucytosine-loaded liposome F6 and, consequently, the greatest extent of flucytosine absorption was achieved. These findings are in accordance with that which was reported by Hosny who has found that an electrostatic force is generated between opposite charges of liposomal formulation and cornea.38 As a result of the adsorption of liposomes on the cornea surface, the liposomal drug cargo is directly transferred to the membranes of the corneal epithelial cell. Consequently, flucytosine transport across the cornea is facilitated.61 Moreover, endocytosis or fusion of liposomal membrane with the plasmalemma may be expected mechanisms in enhancing the transportation of liposomal cargo.15 Against C. albicans, Torulopsis, and Cryptococcus spp., and against specific forms of dematiaceous molds (Phialophora and Cladosporium) from ocular sources, the reported 90% minimum inhibitory concentration (MIC90) of flucytosine is 0.25 μg/mL.13,62 The eye drops of the flucytosine solution only achieve a concentration equal to or higher than the MIC90 for the first 4 hours after application as shown in Figure 8. However, flucytosine-loaded liposomal formulations (F3, F6, and F9) achieved concentrations higher than the MIC90 for 12 hours. The highest Cmax value (17.5 μg/mL) was achieved by liposomal formulation F6 1 hour after application. Liposomal formulations effectively achieve the aqueous humor flucytosine concentration above MIC90 for up to 12 hours in rabbits. Consequently, the frequent application of the antifungal drug is eliminated leading to more patient compliance. According to the aforementioned results, formulation of flucytosine as liposomal preparations successfully achieves the objectives of the study.
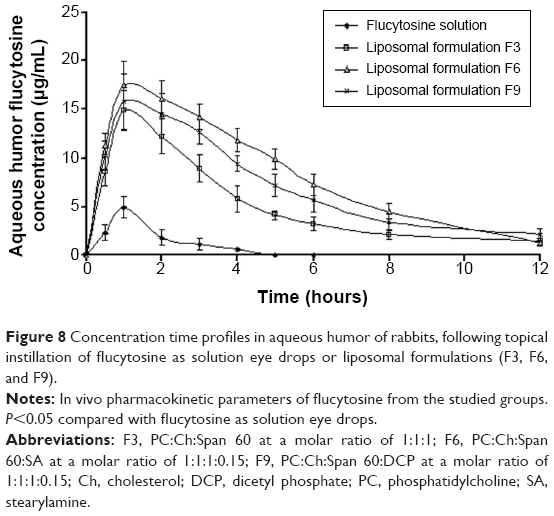 | Figure 8 Concentration time profiles in aqueous humor of rabbits, following topical instillation of flucytosine as solution eye drops or liposomal formulations (F3, F6, and F9). |
 | Table 9 Pharmacokinetic parameters of flucytosine in aqueous humor, post-topical instillation of flucytosine solution (0.3% w/v) or flucytosine-loaded liposomal formulations F3, F6, and F9 |
In vivo antifungal activity of gold nanoliposomes
The fungicidal activity of flucytosine is a result of its conversion to 5-fluorodeoxy uridine monophosphate, which frustrates the synthesis of fungal DNA. The net result is the death of the fungal cell. The antifungal activity of gold ions could be elucidated for the formation of insoluble gold compounds in the cell wall, which disrupt both the transmembrane energy metabolism and the membrane electron transport chain. This may be due to the absence in activation of the cell wall sulfhydryl group. Moreover, the enzyme complexes, which are essential for the respiratory chain and membrane permeability, become dissociated by the gold ions causing disruption of the membrane-bounded enzymes and lipids, which could lead to fungal cell death.63,64 The important question of how can the living body, especially the eyes, be affected by gold nanoparticles should be answered.
Chen et al have reported that 21 nm spherical gold nanoparticles caused undetectable organ or cell toxicity in a mouse model.65 Connor et al and Shenoy et al have reported that gold nanoparticles can be taken by cells without cytotoxic effect.18,66 The size of gold nanoparticles may be an important factor in the clearance of them from the body. Gold nanoparticles show no toxicity effects; this may be attributed to rapid clearance of gold nanoparticles from the body. However, aggregation of gold nanoparticles may occur; consequently, gold nanoparticles become trapped in the body.67 Niidome et al have reported that poly(ethylene glycol) (PEG)-modified gold nanoparticles did not show cytotoxicity.68 Kim et al have reported that all layers of the retina could be disrupted by 20 nm gold nanoparticles 24 hours after intravenous injection.69 Furthermore, the cellular viability of retinoblastoma cells, the retinal endothelial cells, and the astrocytes was not affected by intraretinal gold nanoparticles. de la Zerda et al have reported that gold nanorods can be used as optical coherence contrast imaging in living mouse eyes with low toxicity and high sensitivity.55 Kim et al have reported that neither toxic effects nor structural damages to the retinal structures were detected after intravenous injection of two different sizes (20 nm and 100 nm) of gold nanoparticles.69 Finally, the researches conducted on gold nanoparticles as drug in the eye were few until now. The long-term biological effects of gold nanoparticles need further studies.
The results from Table 10 show that the antifungal activity of flucytosine capped with gold in liposomal formulations is better than that of the flucytosine solution. According to the percentage of healing of rabbits’ eyes infected with C. albicans, the applied formulations can be arranged in a descending order as follows: F6 > F9 > F3 > flucytosine solution. It was previously recorded that the liposomal formulation increased the lipids’ solubility as well as their ability to permeate through the cell membrane.
 | Table 10 The percentage and the time of healing of rabbits’ eyes infected with Candida albicans were measured when nanoliposomes formulations F3, F6, and F9 were applied |
The aforementioned results show that liposomal formulation enhances the antifungal effects of flucytosine. According to the results of in vivo antifungal activity, flucytosine capped with gold nanoliposomal formulations (F3, F6, and F9) is more effective than the flucytosine solution, as shown in Figure 9. This may be due to the sustained release of the liposomal formulations into the eye. The liposome may penetrate the ocular tissues and release flucytosine and gold nanoparticles to apply its effect. The penetration of the drug molecules into the eye from topical ophthalmic formulations is a sophisticated phenomenon.
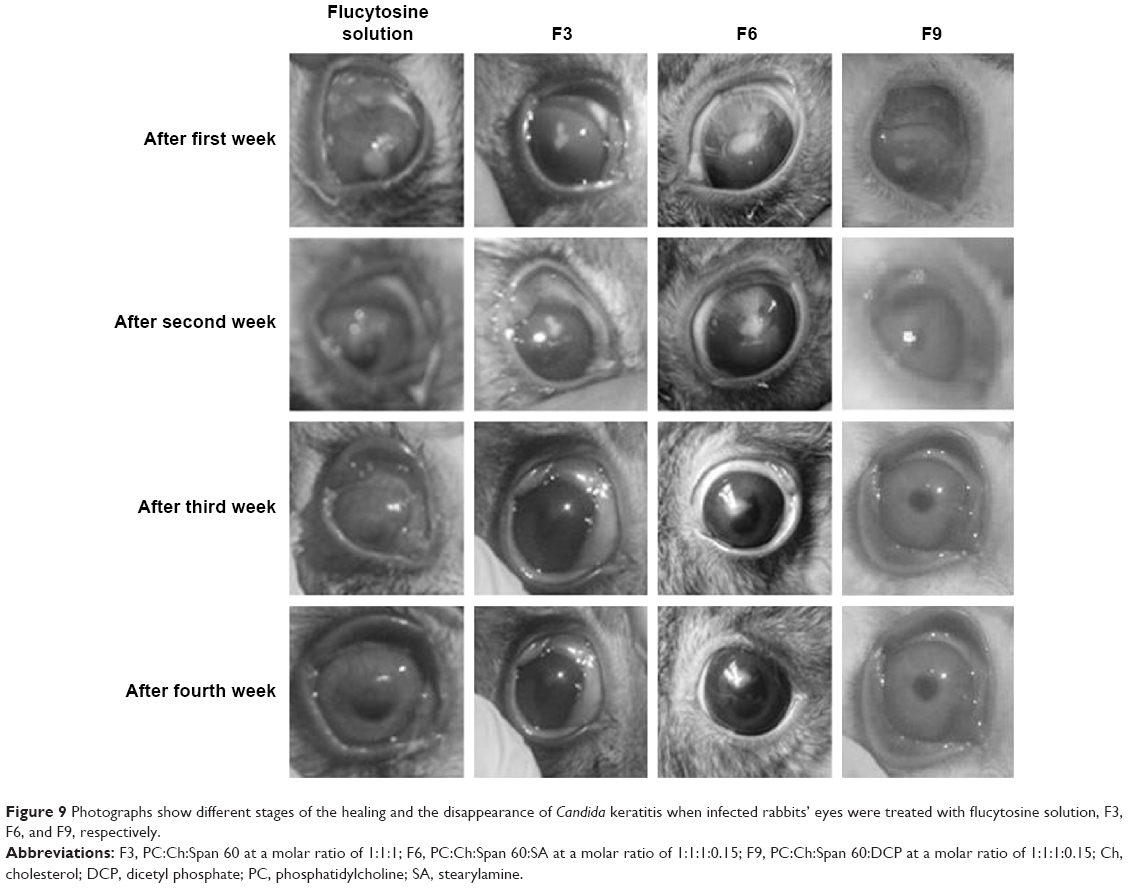 | Figure 9 Photographs show different stages of the healing and the disappearance of Candida keratitis when infected rabbits’ eyes were treated with flucytosine solution, F3, F6, and F9, respectively. |
Nonionic surfactants may be used in formulation of nanoliposomes to meet the challenges through their potential ability to increase bioavailability by increasing the solubility of drug, prolonging pre-cornea retention, and enhancing permeability. Guinedi et al have reported that no major histological changes in ocular tissues were reported after instillation of Span 60 containing niosomes for 40 days.70 Pandey et al have reported that Span 60 was added to the composition of ofloxacin-loaded niosomes to improve the ocular bioavailability of ofloxacin for the prolonged period of time.71 Pepić et al have reported that intraocular dexamethasone absorption was improved by using a nonionic surfactant-containing micelle system.72 Nonionic surfactants were used by many researchers in formulation of ophthalmic preparations.73–76
According to liposomal formulations, the drug penetration rate has not only been determined by the physicochemical characteristics of the drug itself, but also by the physicochemical characteristics of its delivery system.58 In the present study, the liposomal formulation F6 has the lowest time for healing the rabbits’ eyes. Finally, the liposomal formulation F3 had the least effective and the slowest healing rate. To understand the high effectiveness of the positively charged liposome, the charge of cornea should be clarified.
The prolonged contact time between the positively charged liposomes and the negatively charged surface of the cornea has led to a sustaining of the drug release from the liposomes.58,77 In contrast, the use of liposomal formulation F9 has led to repulsion between the negatively charged liposomes and the negatively charged surface of the cornea. The existing repulsion resulted in disruption of the lipid bilayer. This in turn promoted the release of the drug from liposomes giving a high opportunity for penetration of drug through the corneal membranes. On the other hand, the neutral liposomes have the lowest effect on transcorneal flux.
The obtained results are in accordance with a study by Schaeffer and Krohn who found that the liposome penetration uptake by the cornea was the greatest for the positively charged liposomes, less for the negatively charged liposomes, and the least for neutral liposomes, suggesting that electrostatic adsorption is the expected mechanism of interaction between the corneal surface and the different prepared charged liposomes.15
Statistical analysis of the differences between positive controls with regard to the number of residual ulcers showed a highly significant difference existing between the treated eyes and the controls (P=7.46×10−7). When comparing flucytosine nanoliposomal formulations with the flucytosine solution, there was a statistically significant difference (P=0.0052).
Conclusion
Flucytosine capped with gold nanoparticle liposomal formulations were easily prepared, stable, and capable of delivering therapeutic amounts of drugs into the posterior chamber of the eye in a short period of time. The present study revealed for the first time that using topical flucytosine capped with gold nanoparticle liposomal formulations was effective in treating the experimental C. albicans infection of the rabbit cornea.
In the present study, the percentage of the healing of rabbits’ eyes infected with C. albicans increased as the result of an increase in the positive zeta potential charge of the used liposomes. At the same time, there was a strong correlation between the average intraocular penetration of gold nanoparticles and the percentage of healed eyes.
Using CT imaging technique, the results clearly showed that the intraocular tracking of the gold nanoparticles could be achieved, when gold nanoparticles operate as a contrasting agent. Gold nanoparticles have the advantages of being an inexpensive material as well as allowing simple tracking of the drug to the posterior segment of the eye without tissue destruction.
Disclosure
The authors report no conflicts of interest in this work.
References
Juárez-Verdayes MA, Reyes-López MA, Cancino-Diaz ME, et al. Isolation, vancomycin resistance and biofilm production of Staphylococcus epidermidis from patients with conjunctivitis, corneal ulcers, and endophthalmitis. Rev Latinoam Microbiol. 2006;48(3–4): 238–246. | ||
Foster A, Sommer A. Corneal ulceration, measles, and childhood blindness in Tanzania. Br J Ophthalmol. 1987;71(5):331–343. | ||
Srinivasan R, Kanungo R, Goyal JL. Spectrum of oculomycosis in South India. Acta Ophthalmol (Copenh). 1991;69(6):744–749. | ||
Klotz SA, Penn CC, Negvesky GJ, Butrus SI. Fungal and parasitic infections of the eye. Clin Microbiol Rev. 2000;13(4):662–685. | ||
Sridhar J, Flynn HW Jr, Kuriyan AE, Miller D, Albini T. Endogenous fungal endophthalmitis: risk factors, clinical features, and treatment outcomes in mold and yeast infections. J Ophthalmic Inflamm Infect. 2013;3(1):60. | ||
Narendran N, Balasubramaniam B, Johnson E, Dick A, Mayer E. Five-year retrospective review of guideline-based management of fungal endophthalmitis. Acta Ophthalmol. 2008;86(5):525–532. | ||
Axelrod AJ, Peyman GA. Intravitreal amphotericin B treatment of experimental fungal endophthalmitis. Am J Ophthalmol. 1973;76(4):584–588. | ||
Christmas NJ, Smiddy WE. Vitrectomy and systemic fluconazole for treatment of endogenous fungal endophthalmitis. Ophthalmic Surg Lasers. 1996;27(12):1012–1018. | ||
Yoshizumi MO, Banihashemi AR. Experimental intravitreal ketoconazole in DMSO. Retina. 1988;8(3):210–215. | ||
Blumenkranz MS, Stevens DA. Therapy of endogenous fungal endophthalmitis: miconazole or amphotericin B for coccidioidal and candidal infection. Arch Ophthalmol. 1980;98(7):1216–1220. | ||
Kalavathy CM, Parmar P, Kaliamurthy J, et al. Comparison of topical itraconazole 1% with topical natamycin 5% for the treatment of filamentous fungal keratitis. Cornea. 2005;24(4):449–452. | ||
Breit SM, Hariprasad SM, Mieler WF, Shah GK, Mills MD, Grand MG. Management of endogenous fungal endophthalmitis with voriconazole and caspofungin. Am J Ophthalmol. 2005;139(1):135–140. | ||
Vermes A, Guchelaar HJ, Dankert J. Flucytosine: a review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J Antimicrob Chemother. 2000;46(2):171–179. | ||
Prausnitz MR, Noonan JS. Permeability of cornea, sclera, and conjunctiva: a literature analysis for drug delivery to the eye. J Pharm Sci. 1998;87(12):1479–1488. | ||
Schaeffer HE, Krohn DL. Liposomes in topical drug delivery. Invest Ophthalmol Vis Sci. 1982;22(2):220–227. | ||
Calvo P, Alonso MJ, Vila-Jato JL, Robinson JR. Improved ocular bioavailability of indomethacin by novel ocular drug carriers. J Pharm Pharmacol. 1996;48(11):1147–1152. | ||
Zhou Y, Kong Y, Kundu S, Cirillo JD, Liang H. Antibacterial activities of gold and silver nanoparticles against Escherichia coli and bacillus Calmette-Guérin. J Nanobiotechnology. 2012;10:19. | ||
Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1(3):325–327. | ||
Hauck TS, Ghazani AA, Chan WC. Assessing the effect of surface chemistry on gold nanorod uptake, toxicity, and gene expression in mammalian cells. Small. 2008;4(1):153–159. | ||
Frens G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature. 1973;241(105):20–22. | ||
Frens G. Particle size and sol stability in metal colloids. Colloid Polym Sci. 1972;250(7):736–741. | ||
Nguyen DT, Kim DJ, Kim KS, Controlled synthesis and biomolecular probe application of gold nanoparticles. Micron. 2011;42(3):207–227. | ||
Sun K, Qiu J, Liu J, Miao Y. Preparation and characterization of gold nanoparticles using ascorbic acid as reducing agent in reverse micelles. J Mater Sci. 2009;44(3):754–758. | ||
Torchilin V, Weissig V. Liposomes: A Practical Approach. 2nd ed. New York: Oxford University Press; 2003. | ||
Demers LM, Mirkin CA, Mucic RC, et al. A fluorescence-based method for determining the surface coverage and hybridization efficiency of thiol-capped oligonucleotides bound to gold thin films and nanoparticles. Anal Chem. 2000;72(22):5535–5541. | ||
Colas JC, Shi W, Rao VS, Omri A, Mozafari MR, Singh H. Microscopical investigations of nisin-loaded nanoliposomes prepared by Mozafari method and their bacterial targeting. Micron. 2007;38(8): 841–847. | ||
Hao Y, Zhao F, Li N, Yang Y, Li K. Studies on a high encapsulation of colchicine by niosome system. Int J Pharm. 2002;244(1–2):73–80. | ||
du Plessis J, Ramachandran C, Weiner N, Müller DG. The influence of lipid composition and lamellarity of liposomes on the physical stability of liposomes upon storage. Int J Pharm. 1996;127(2):273–278. | ||
Matar MJ, Ostrosky-Zeichner L, Paetznick VL, Rodriguez JR, Chen E, Rex JH. Correlation between E-test, disk diffusion, and microdilution methods for antifungal susceptibility testing of fluconazole and voriconazole. Antimicrob Agents Chemother. 2003;47(5):1647–1651. | ||
Helsinki F. Responsibility in Investigations on Human Subjects: M.R.C. Statement. Br Med J. 1964;2(5402):178–180. | ||
Milisits G, Romvári R, Zotte AD, Szendrő Z. Non-invasive study of changes in body composition in rabbits during pregnancy using X-ray computerized tomography. Ann Zootech. 1999;48:25–34. | ||
Bury RW, Mashford ML, Miles HM. Assay of flucytosine (5-fluorocytosine) in human plasma by high-pressure liquid chromatography. Antimicrob Agents Chemother. 1979;16(5):529–532. | ||
Wu TG, Wilhelmus KR, Mitchell BM. Experimental keratomycosis in a mouse model. Invest Ophthalmol Vis Sci. 2003;44(1):210–216. | ||
Perugini P, Pavanetto F. Liposomes containing boronophenylalanine for boron neutron capture therapy. J Microencapsul. 1998;15(4):473–483. | ||
Srinath P, Vyas SP, Diwan PV. Preparation and pharmacodynamic evaluation of liposomes of indomethacin. Drug Dev Ind Pharm. 2000;26(3):313–321. | ||
Hathout RM, Mansour S, Mortada ND, Guinedi AS. Liposomes as an ocular delivery system for acetazolamide: in vitro and in vivo studies. AAPS Pharm Sci Tech. 2007;8(1):1. | ||
Nagarsenker MS, Londhe VY, Nadkarni GD. Preparation and evaluation of liposomal formulations of tropicamide for ocular delivery. Int J Pharm. 1999;190(1):63–71. | ||
Hosny KM. Ciprofloxacin as ocular liposomal hydrogel. AAPS Pharm Sci Tech. 2010;11(1):241–246. | ||
Xu X, Yu Z, Zhu Y, Wang B. Effect of sodium oleate adsorption on the colloidal stability and zeta potential of detonation synthesized diamond particles in aqueous solutions. Diam Relat Mater. 2005;14(2):206–212. | ||
Riddick TM. Control of Colloid Stability through Zeta Potential. Livingston Publishing Company: Wynnewood, Pennsylvania; 1968. | ||
Amir T, Fatma A, Hakan A. Gold Nanoparticle Synthesis and Characterisation. Hacettepe J Biol Chem. 2009;37(3):217–226. | ||
Sureewan D, Boonnada P, Praneet O, et al. Role of the charge, carbon chain length, and content of surfactant on the skin penetration of meloxicam-loaded liposomes. Int J Nanomedicine. 2014;9:2005–2017. | ||
Crommelin DJ. Influence of lipid composition and ionic strength on the physical stability of liposomes. J Pharm Sci. 1984;73(11):1559–1563. | ||
Higuchi T. Mechanism of sustained-action medication: theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52(12):1145–1149. | ||
El-Samaligy MS, Afifi NN, Mahmoud EA. Increasing bioavailability of silymarin using a buccal liposomal delivery system: preparation and experimental design investigation. Int J Pharm. 2006;308(1–2):140–148. | ||
Margalit R, Okon M, Yerushalmi N, Avidor E. Bioadhesive liposomes as topical drug delivery systems: molecular and cellular studies. J Control Release. 1992;19(1–3):275–287. | ||
Margalit R, Alon R, Linenberg M, Rubin I, Roseman TJ, Wood RW. Liposomal drug delivery: thermodynamic and chemical kinetic considerations. J Control Release. 1991;17(3):285–296. | ||
Park SH, Oh SG, Mun JY, Han SS. Loading of gold nanoparticles inside the DPPC bilayers of liposome and their effects on membrane fluidities. Colloids Surf B Biointerfaces. 2006;48(2):112–118. | ||
Hernández-Caselles T, Villalaín J, Gómez-Fernández JC. Stability of liposomes on long term storage. J Pharm Pharmacol. 1990;42(6):397–400. | ||
Patel VB, Misra AN. Encapsulation and stability of clofazimine liposomes. J Microencapsul. 1999;16(3):357–367. | ||
Rükan G, Mayreli O, Ciara KO. Diffusion-controlled synthesis of gold nanoparticles: nano-liposomes as mass transfer barrier. J Nanopart Res. 2014;16(4):1–5. | ||
Tan YN, Lee KH, Su X. Study of single-stranded DNA binding protein-nucleic acids interactions using unmodified gold nanoparticles and its application for detection of single nucleotide polymorphisms. Anal Chem. 2011;83(11):4251–4257. | ||
Cunha-Vaz JG. The blood-ocular barriers: past, present, and future. Doc Ophthalmol. 1997;93(1–2):149–157. | ||
Nakano Y, Muro S, Sakai H, et al. Computed tomographic measurements of airway dimensions and emphysema in smokers. Correlation with lung function. Am J Respir Crit Care Med. 2000;162(3 Pt 1):1102–1108. | ||
de la Zerda A, Prabhulkar S, Perez VL, et al. Optical coherence contrast imaging using gold nanorods in living mice eyes. Clin Experiment Ophthalmol. 2015;43(4):358–366. | ||
Klang S, Abdulrazik M, Benita S. Influence of emulsion droplet surface charge on indomethacin ocular tissue distribution. Pharm Dev Technol. 2000;5(4):521–532. | ||
Verma DD, Verma S, Blume G, Fahr A. Particle size of liposomes influences dermal delivery of substances into skin. Int J Pharm. 2003;258(1–2):141–151. | ||
El-Gazayerly ON, Hikal AH. Preparation and evaluation of acetazolamide liposomes as an ocular delivery system. Int J Pharm. 1997;158(2):121–127. | ||
Singh K, Mezei M. Liposomal ophthalmic drug delivery system I. Triamcinolone acetonide. Int J Pharm. 1983;16(3):339–344. | ||
Singh K, Mezei M. Liposomal ophthalmic drug delivery system. II. Dihydrostreptomycin sulfate. Int J Pharm. 1984;19(3):263–269. | ||
Lee VH, Urrea PT, Smith RE, Schazlin DJ. Ocular drug bioavailability from topically applied liposomes. Surv Ophthalmol. 1985;29(5):335–348. | ||
Scholer HJ. Flucytosine. In: Speller DCE, editor. Antifungal chemotherapy. New York: Wiley; 1980:35–106. | ||
Chen Y, Zheng X, Xie Y, Ding C, Ruan H, Fan C. Anti-bacterial and cytotoxic properties of plasma sprayed silver-containing HA coatings. J Mater Sci Mater Med. 2008;19(12):3603–3609. | ||
Eid KAM, Salem HF, Zikry AAF, El-Sayed AFM, Sharaf MA. Antifungal Effects of Colloidally Stabilized Gold Nanoparticles: Screening by Microplate Assay. Nature and Science. 2011;9(2):29–33. | ||
Chen H, Dorrigan A, Saad S, Hare DJ, Cortie MB, Valenzuela SM.In vivo study of spherical gold nanoparticles: inflammatory effects and distribution in mice. PloS One. 2013;8(2):e58208. | ||
Shenoy D, Fu W, Li J, et al. Surface functionalization of gold nanoparticles using hetero-bifunctional poly(ethylene glycol) spacer for intracellular tracking and delivery. Int J Nanomedicine. 2006;1(1):51–57. | ||
Thassu D, Chader GJ. Ocular Drug Delivery Systems: Barriers and Application of Nanoparticulate Systems. CRC Press; 2012. | ||
Niidome T, Yamagata M, Okamoto Y, et al. PEG-modified gold nanorods with a stealth character for in vivo applications. J Control Release. 2006;114(3):343–347. | ||
Kim JH, Kim KW, Kim MH, Yu YS. Intravenously administered gold nanoparticles pass through the blood-retinal barrier depending on the particle size, and induce no retinal toxicity. Nanotechnology. 2009;20(50):505101. | ||
Guinedi AS, Mortada ND, Mansour S, Hathout RM. Preparation and evaluation of reverse-phase evaporation and multilamellar niosomes as ophthalmic carriers of acetazolamide. Int J Pharm. 2005;306(1–2):71–82. | ||
Pandey VP, Deivasigamani K. Preparation and characterization of ofloxacin non-ionic surfactant vesicles for ophthalmic use. J Pharm Res. 2009;2(8):1330–1334. | ||
Pepić I, Hafner A, Lovrić J, Pirkić B, Filipović-Grcić J. A nonionic surfactant/chitosan micelle system in an innovative eye drop formulation. J Pharm Sci. 2010;99(10):4317–4325. | ||
Kaur IP, Rana C, Singh M, Bhushan S, Singh H, Kakkar S. Development and evaluation of novel surfactant-based elastic vesicular system for ocular delivery of fluconazole. J Ocul Pharmacol Ther. 2012;28(5):484–496. | ||
Dubey A, Prabhu P. Development and investigation of Niosomes of Brimonidine tartrate and Timolol maleate for the treatment of glaucoma. Int J Pharmtech Res. 2014;6(3):942–950. | ||
Hu C, Rhodes DG. Proniosomes: a novel drug carrier preparation. Int J Pharm. 1999;185(1):23–35. | ||
Jiao J. Polyoxyethylated nonionic surfactants and their applications in topical ocular drug delivery. Adv Drug Deliv Rev. 2008;60(15):1663–1673. | ||
Bochota A, Mashhour B, Puisieux F, Couvreur P, Fattal E. Comparison of the ocular distribution of a model oligonucleotide after topical instillation in rabbits of conventional and new dosage forms. J Drug Target. 1998;6(4):309–313. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.