Back to Journals » International Journal of General Medicine » Volume 19
Lipoprotein(a) in Residual Cardiovascular Risk: Measurement Challenges, Assay Standardisation, and Clinical Implications
Authors Lu Y ![]() , Jiang D, Zhang J, Liang Y
, Jiang D, Zhang J, Liang Y
Received 24 February 2026
Accepted for publication 28 May 2026
Published 4 June 2026 Volume 2026:19 605053
DOI https://doi.org/10.2147/IJGM.S605053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Redoy Ranjan
Yiwen Lu,1,* Dongyang Jiang,1,* Jinfeng Zhang,2 Ying Liang1
1School of Clinical Medicine, Shandong Second Medical University, Weifang, People’s Republic of China; 2School of Clinical and Basic Medicine, Shandong First Medical University, Jinan, 250117, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ying Liang, Email [email protected]
Abstract: Lipoprotein(a) [Lp(a)] is a largely genetically determined, independent contributor to residual atherosclerotic cardiovascular disease (ASCVD) risk, but its clinical interpretation is constrained by measurement complexity. Variation in apolipoprotein(a) [apo(a)] KIV2 copy number, glycosylation, lipid composition, and isoform co-expression complicates mass-based reporting and may cause isoform-sensitive bias in immunoassays. This review examines how these metrological limitations affect Lp(a) reporting, assay comparability, LDL-C estimation, and clinical risk stratification. Current evidence supports preferential use of validated molar reporting (nmol/L) and discourages fixed conversion between mg/dL and nmol/L. Automated immunoassays remain practical for screening and population studies, whereas ELISA and LC-MS/MS have complementary roles in assay validation, reference-method development, and standardisation. Clinically, high Lp(a) may distort calculated LDL-C, and fixed 30% Lp(a)-cholesterol correction can misclassify risk or treatment response. Harmonised molar measurement, ancestry-inclusive validation, and clearer distinction between intact Lp(a) particles and free apo(a) are essential for improving risk assessment and implementing Lp(a)-targeted therapies.
Keywords: lipoprotein(a), Lp(a), apoprotein(a), apo(a), cardiovascular diseases, heart disease risk factors, analysis
Introduction: Residual Risk of Cardiovascular Disease and the Rise of Lp(a)
The Global Burden of Cardiovascular Disease and the Concept of Residual Risk
Cardiovascular disease (CVD) is the leading cause of death worldwide, significantly impacting healthy life expectancy and imposing an excessive economic burden on healthcare systems.1,2 With an increasingly large population base and the progression of an ageing society, the incidence of cardiovascular disease continues to rise, presenting a formidable challenge in controlling its risk factors.3,4
Systematic analyses based on real-world evidence indicate that even among patient groups achieving target control for certain common risk factors, cardiovascular events cannot be entirely avoided. Despite the effective implementation of guideline-directed management and therapy (GDMT) and high patient adherence to treatment measures, residual risks of adverse cardiovascular events persist.5 The Residual Risk Reduction Initiative (R3i) defines this residual cardiovascular risk as residual risk: the risk of cardiovascular events that persists even after patients have achieved current treatment targets for low-density lipoprotein cholesterol (LDL-C), blood pressure, and blood glucose control, along with therapeutic lifestyle modifications.6
Evidence for Lp(a) as an Independent Residual Risk Factor and Current Guideline Recommendations
Against the backdrop of residual risk in cardiovascular disease, research has identified a significant increase in novel candidate risk factors associated with residual risk. These factors hold substantial predictive value for CVD and its complications. The association between lipoprotein(a) [Lp(a)] and coronary heart disease (CHD),7–9 stroke8,9 and aortic valve stenosis (AVS)9,10 has been well-established. Management guidelines for cardiovascular disease in countries such as Europe,11,12 and China,13 as well as the United States,14 and the expert panel of the National Academy of Clinical Biochemistry (NACB),15,16 have all designated Lp(a) as an independent residual risk factor for cardiovascular diseases including coronary heart disease, stroke, peripheral artery disease, coronary artery calcification, and calcific aortic valve stenosis. Accumulated research demonstrates that elevated Lp(a) levels drive residual risk through a triple mechanism: promoting atherosclerosis (by carrying oxidised phospholipids), promoting thrombosis (by inhibiting fibrinolysis), and promoting inflammation.17 Lipoprotein(a) has emerged as a novel therapeutic target in residual cardiovascular risk management. However, current clinical quantification of lipoprotein(a) remains limited, potentially constraining clinical control.
Molecular Structure and Polymorphism of Lp(a): The Root Cause of Detection Challenges
Structure and Composition: LDL-Like Particles and Apolipoprotein(a)
Under electron microscopy, Lp(a) appears spherical with a diameter of approximately 21 nm and a density of about 1.05–1.10 g/mL. The core lipid structure of Lp(a) resembles that of low-density lipoprotein (LDL) in that it is also composed of cholesterol esters and triglycerides, surrounded by a membrane structure formed by phospholipids and free cholesterol. Its protein component consists of a single apolipoprotein B-100 (apoB).18 Within Lp(a) particles, apolipoprotein B100 (apoB) and apolipoprotein(a) [apo(a)] coexist stably in a 1:1 molar ratio. The LDL-C-like lipid core is enveloped by the kringle domains of apo(a). A disulfide bond covalently links apo(a) to the Apo B100 within the LDL-C-like lipid core. Apo(a) contains a plasminogen-like kringle V domain, variable numbers of plasminogen-like kringle IV domains (types 1–10), and an inactive protease region.19
Notably, apo(a) shares structural homology with plasminogen in evolutionary terms. Both contain characteristic kringle domains, but a significant difference exists: plasminogen contains five kringle domains (KI-KV), while apo(a) retains only KIV and KV types. The KV domain exists as a single copy in apo(a). In contrast, the KIV domain of apo(a) can be classified into 10 highly homologous subtypes (KIV1-KIV10), which exhibit 75–85% amino acid sequence homology with the KIV kringle domain of plasminogen,20 this confers apo(a) with plasminogen-like properties, enabling it to bind to exposed lysine-rich vascular endothelium—a function similar to and competitively overlapping with that of plasminogen.21–23 Within the KIV domain of apo(a), the KIV-1 and KIV-3 to KIV-10 subtypes exist as single copies. The KIV2 subtype exhibits polymorphism due to variations in the number of gene-encoded repeat sequences, ranging from 2 to 40 copies,24,25 this represents the primary cause of inter- and intra-individual heterogeneity in apo(a) molecular weight, significant size heterogeneity in Lp(a) particles, and the crucial molecular basis for inter-individual and inter-ethnic variations in Lp(a) levels.17,26 Glycosylation modifications of apo(a) exhibit significant site heterogeneity, with modification sites distributed not only within the core Kringle domain but also extending to the Linker sequences connecting these domains,27 this similarly contributes to the size heterogeneity observed in Lp(a) particles (Table 1).20,25,28–34 The complex molecular structure of lipoprotein(a) and the molecular weight polymorphism of its apo(a), such as the differing immunoreactivity of various apo(a) isoforms, have consistently been major constraints in developing Lp(a) detection methods.35
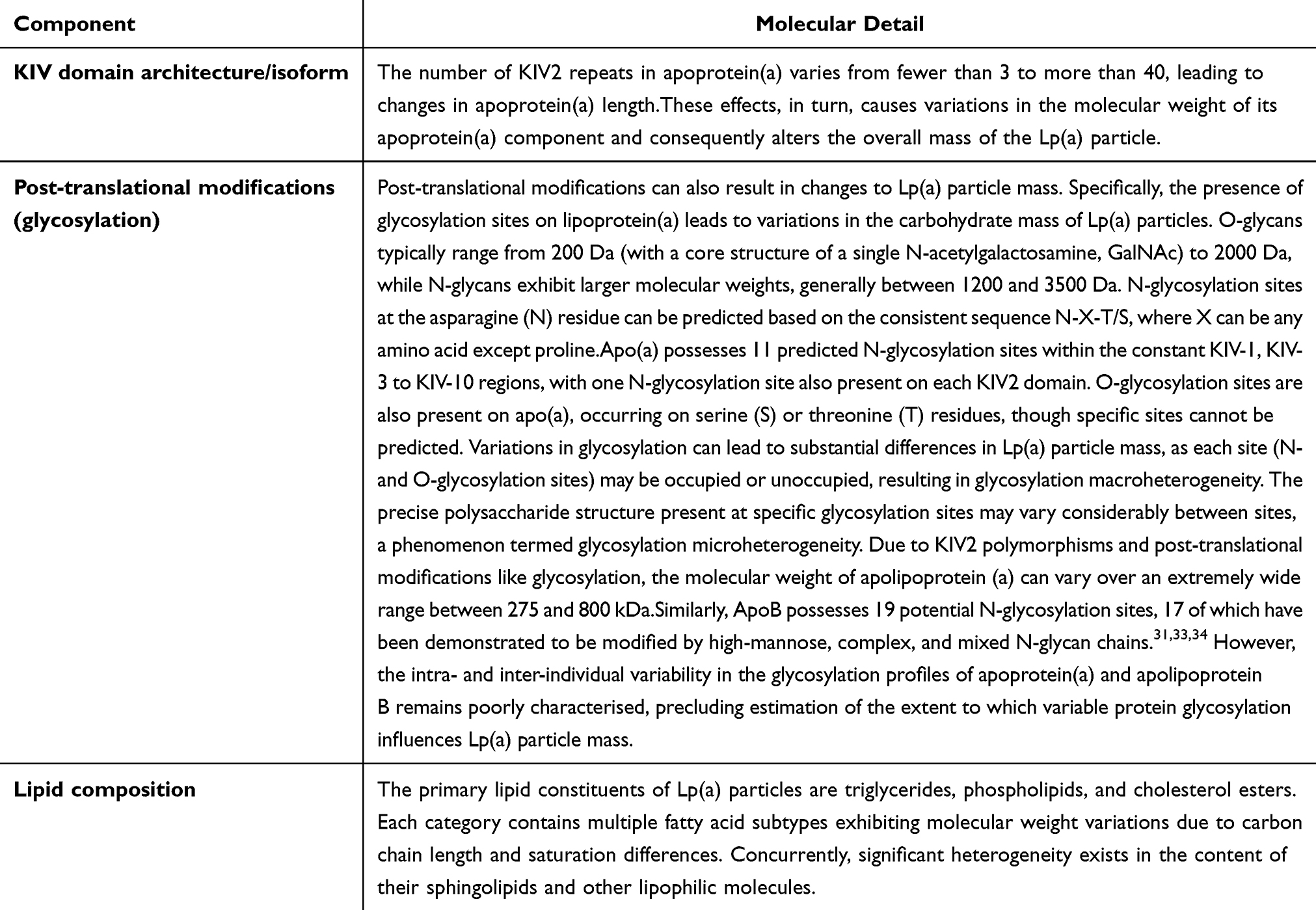 |
Table 1 Structural Sources of Lp(a) Particle-Mass Heterogeneity and Assay Implications |
Synthesis and Assembly: Liver Specificity and the “Two-Step” Mechanism
The synthesis of lipoprotein(a) neither originates from the conversion of very low-density lipoprotein (VLDL) nor depends on the metabolic breakdown of other lipoproteins, and it itself lacks the ability to convert into other lipoproteins.36,37 The biosynthesis of lipoprotein(a) (Lp(a)) occurs almost entirely in the liver. Plasma levels of each Lp(a) subtype are primarily determined by their respective synthesis efficiencies rather than their clearance rates from circulation.38,39 Generally, the molecular weight of apo(a) isoforms is inversely correlated with secretion efficiency.40,41 Accordingly, smaller apo(a) isoforms, which contain fewer Kringle IV-2 repeats, usually have higher plasma Lp(a) concentrations, thereby contributing to inter-individual and inter-ethnic heterogeneity in Lp(a) levels.42 This phenomenon suggests that smaller, more numerous lipoprotein(a) particles dominate biological effects through a mechanism of preferential expression, a notion supported by relevant studies.43,44 It is now widely accepted in the scientific community that Lp(a) assembly follows a two-step cascade mechanism: In the first step, a lysine residue at a specific site on apolipoprotein B100 (apoB100) undergoes conformational prepositioning through non-covalent interactions with a weakly binding lysine binding site (LBS) in the kringle IV7,8 domain of apo(a).45–47 In the second step, Cys4057 in the KIV9 domain23,48 forms a stable covalent disulfide bond with Cys4326 on apoB100.49,50 At the individual level, over 80% of individuals inherit two distinct molecular weight apo(a) subtypes from their parents, exhibiting co-expression patterns of dual low molecular weight, high-low molecular weight, or dual high molecular weight subtypes.23,51
Core Metrological Issues in Lp(a) Quantification: Limitations of Mass Concentration and the Shift Toward Molar Concentration
Inherent Limitations of Mass Concentration: Why mg/dL Cannot Accurately Capture Lp(a) Particle Burden and Cardiovascular Risk
Current evidence indicates that individuals with higher circulating Lp(a) particle concentrations are at greater cardiovascular risk.52,53 However, as noted above, Lp(a) particles vary substantially in mass and size because of apo(a) KIV2 repeat-number polymorphism, post-translational modifications such as glycosylation, heterogeneity in lipid composition, and the co-expression of multiple apo(a) isoforms. Consequently, two individuals with the same total Lp(a) mass concentration may have substantially different numbers of circulating Lp(a) particles. One individual may predominantly carry fewer large particles, whereas another may carry more small particles. As a result, Lp(a) mass concentration does not necessarily correspond consistently to the patient’s true cardiovascular risk. This is a fundamental limitation of reporting Lp(a) in mg/dL: the value reflects the total mass of a heterogeneous particle population rather than the number of particles per litre of plasma. Reliance on mass concentration alone may therefore obscure clinically relevant heterogeneity in risk driven by differences in particle number.
Beyond this biological heterogeneity, mass-based reporting also has intrinsic analytical and metrological limitations. Even if all components of Lp(a) could be measured precisely and their summed mass used for calibration, the resulting value would still not necessarily represent the true plasma particle concentration. This persistent bias arises because the composition of purified Lp(a) particles does not necessarily represent the overall particle population in plasma, and because the relative proportions of the two Lp(a) isoforms in heterozygous individuals may shift during purification.20 These factors can introduce error into estimates of Lp(a) particle mass, especially given the difficulty of accurately quantifying its lipid, protein, and carbohydrate components. Therefore, precise determination of total Lp(a) particle mass—and, by extension, reliable inference of particle concentration from mass concentration—is difficult to achieve in practice.
A further problem arises in assays that use antibodies directed against the repetitive KIV2 region. Depending on whether the sample contains smaller or larger apo(a) isoforms than the calibrator, Lp(a) concentrations may be underestimated or overestimated.31,54 Importantly, both the direction and magnitude of this bias depend on the degree of matching in KIV2 epitope copy number between the calibrator and the patient sample. Specifically, when the apo(a) isoform in the test sample is larger than that in the calibrator and contains more KIV2 repeats, each Lp(a) particle may generate a stronger immunoreactive signal, leading to potential overestimation of Lp(a) concentration. Conversely, when the apo(a) isoform in the test sample is smaller than that in the calibrator and contains fewer KIV2 repeats, each particle may produce a weaker signal, leading to potential underestimation.
By contrast, molar concentration expressed in nmol/L, which is based on Lp(a) particle number, is increasingly regarded as the preferred reporting approach. Its advantages extend beyond metrology to risk characterisation and clinical interpretation. By directly quantifying particle concentration, this approach avoids mass-calculation bias introduced by variability in protein, lipid, and carbohydrate composition. More importantly, it is biologically closer to the true carrier of Lp(a)-related risk: the circulating Lp(a) particle burden.52,53 Accordingly, current consensus statements increasingly recommend expressing Lp(a) results as particle concentration in nmol/L.35,55,56 However, for immunoassays to report Lp(a) particle concentration reliably in nmol/L, they should use specific monoclonal antibodies that recognise non-repetitive epitopes outside the KIV2 repeat region and do not cross-react with plasminogen.
Apo(a) Isoform Size: Uncertain Incremental Risk Information Beyond Particle Concentration
It should be noted that neither mass concentration nor particle concentration directly captures the apo(a) isoform size expressed by an individual. A substantial body of evidence suggests that the association between smaller apo(a) isoforms and increased cardiovascular risk is largely explained by the tendency of low-molecular-weight apo(a) isoforms to be associated with higher Lp(a) particle concentrations.57,58 After adjustment for Lp(a) concentration, the independent association between isoform size and cardiovascular risk is markedly attenuated or may even disappear, suggesting that the circulating burden of Lp(a) is the predominant determinant of risk.59–61 In other words, the risk may be driven primarily by Lp(a) particle burden rather than by apo(a) isoform size itself.
However, the evidence is not entirely consistent, and some studies suggest that smaller apo(a) isoforms may have greater pathogenic potential. Using mutually adjusted phenotypic analyses, isoform-specific genetic instruments, and Mendelian randomisation, Saleheen et al62 provided evidence that smaller apo(a) isoform size may exert a pathogenic effect independent of Lp(a) concentration. Another possible exception may involve specific pathological processes, such as pro-inflammatory oxidised phospholipid burden, in which smaller isoforms carrying greater amounts of oxidised lipids may be particularly atherogenic.63–65 In addition, experimental studies have shown that recombinant apo(a) isoforms of different sizes differ in their ability to stimulate collagen-primed monocytes. Specifically, 10K and 18K isoforms significantly increased ROS and MMP-9 production, whereas the 34K isoform had little effect; the magnitude of this response was inversely related to apo(a) size.66
Nevertheless, the current evidence is neither sufficiently consistent nor sufficiently strong to justify moving away from a risk-assessment framework centred on Lp(a) particle concentration. In particular, it has not yet been clearly established that apo(a) isoform size provides additional clinically meaningful risk information beyond particle burden.53,67 Taken together, Lp(a) particle concentration remains the more direct, interpretable, and clinically implementable measure for risk characterisation in current practice.68
Challenges in Reporting Units and Fixed Conversion Factors: Clinical Transition from mg/dL to nmol/L
At present, serum/plasma Lp(a) concentrations are still reported using two unit systems: molar concentration, expressed as nanomoles per litre (nmol/L), and mass concentration, expressed as milligrams per decilitre (mg/dL). In clinical practice, a commonly used approach has been to apply an average conversion factor of 2.4, whereby the mass concentration in mg/dL is multiplied by this factor to derive an estimated value in nmol/L. This average conversion factor was derived from statistical relationships between molar and mass concentrations reported in previous studies and is, in essence, only an approximation.20,69 Other conversion factors, including 2.02 and 1.67, have also been proposed.37,70 Because different conversion coefficients can substantially alter the resulting nmol/L values, the use of fixed conversion factors inevitably propagates and may amplify measurement uncertainty; therefore, such conversion should be avoided.20
Because much of the earlier epidemiological evidence, many risk thresholds, and existing clinical laboratory workflows were established using mg/dL, the transition from mass-based to molar reporting should not be interpreted as an immediate discontinuation of mg/dL. Rather, it should be viewed as a gradual process of metrological standardisation and clinical implementation.53,71,72 The 2024 ADLM guidance document on the measurement and reporting of lipids and lipoproteins71 recommends that Lp(a) should be reported in molar units where feasible; however, when validated molar-based assays are not available, reporting in mass units remains acceptable. The 2024 focused update to the National Lipid Association (NLA) scientific statement53 takes a similar position: from a technical perspective, apo(a) isoform size heterogeneity supports the preferential use of nmol/L, but if a healthcare setting only has access to assays reported in mg/dL, measuring Lp(a) is still preferable to not measuring it at all.
A more appropriate approach is therefore to use nmol/L as the primary reporting unit when an assay system has been calibrated and methodologically validated for molar reporting. When laboratories continue to use validated mass-based methods, reporting in mg/dL remains acceptable, provided that the assay platform and calibration system are clearly stated in the laboratory report. During the transition period, a dual-reporting framework may also be considered, with nmol/L as the preferred unit and mg/dL as a supplementary unit. However, it should be emphasised that, whether single-unit molar reporting or dual-unit reporting is used, one unit should not be derived from the other by applying a fixed conversion factor to a single measurement result. Instead, both reported values should be generated through reporting procedures validated by the manufacturer or the laboratory.53,71
Clinical and Metrological Considerations in Lp(a) Measurement: Testing Strategy, Ancestry-Related Interpretation, and LDL-C Estimation
Lifetime Testing, Repeat Measurement, and Ancestry-Related Interpretation of Lp(a)
Approximately 90% of interindividual variation in Lp(a) concentration is genetically determined, with metabolic status and other secondary factors exerting only a limited influence.24,73,74 Consequently, Lp(a) concentrations tend to remain relatively stable over the life course. Accordingly, lipid-management guidelines and related evidence from China, Europe, the United States, and other regions generally support a once-in-a-lifetime Lp(a) measurement for most clinical purposes. These documents emphasise that Lp(a) testing is generally reproducible and that a single measurement is usually sufficient to improve cardiovascular risk prediction and determine whether an individual exceeds established risk thresholds12–14,53,68,75,76 (Figure 1B). Nevertheless, because Lp(a) measurement remains affected by persistent methodological limitations, a single result may still be vulnerable to false-positive or false-negative classification. In clinical practice, repeat testing with the same assay method is therefore advisable when the initial value lies close to a clinical decision threshold. Repeat measurement should also be considered in specific settings in which Lp(a) concentrations may change, including suspected reversible secondary causes of Lp(a) elevation, such as nephrotic syndrome, or after initiation of Lp(a)-lowering therapy, such as antisense oligonucleotides or lipoprotein apheresis, when serial monitoring is required to assess treatment response.32
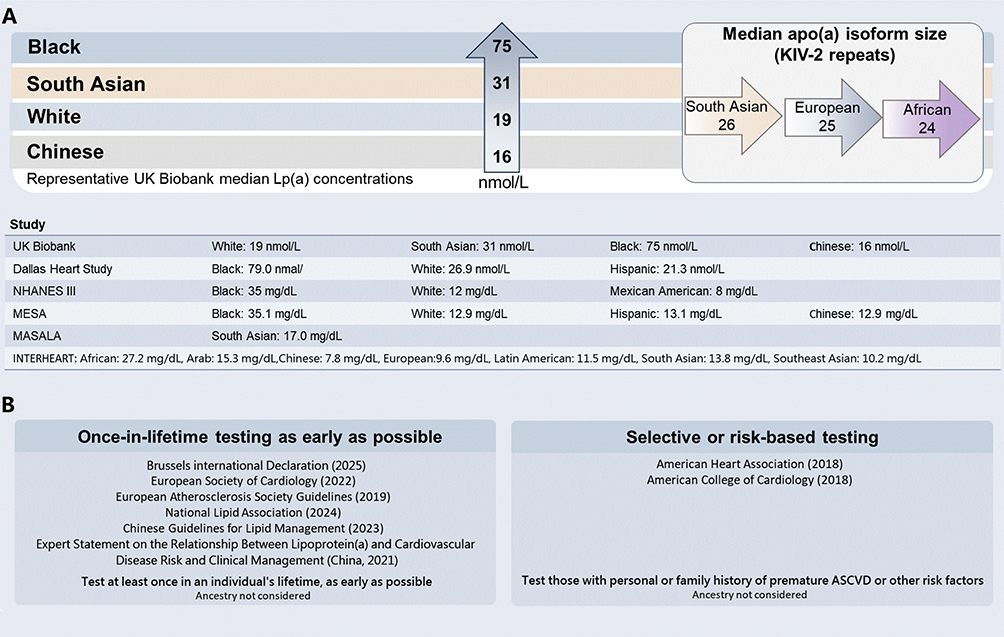 |
Figure 1 Ethnic differences in median lipoprotein(a) concentrations and guideline recommendations for Lp(a) testing. (A) Representative median Lp(a) concentrations reported across ethnic or ancestry groups in major population-based studies, with median apo(a) isoform size shown as KIV-2 repeat number. Values are presented in the units reported by the original studies and were not converted between nmol/L and mg/dL. (B) Summary of selected guideline and consensus recommendations for Lp(a) testing. Abbreviations: apo(a), apolipoprotein(a); ASCVD, atherosclerotic cardiovascular disease; KIV-2, kringle IV type 2; Lp(a), lipoprotein(a). |
Existing evidence indicates that Lp(a) levels and their population distributions differ across racial, ethnic, and ancestry groups77 (Figure 1A). However, these distributional differences do not imply that validated and widely accepted ancestry-specific clinical risk thresholds have already been established. The relative increase in ASCVD risk associated with each 50 nmol/L increment in Lp(a) appears broadly similar across populations, although the estimate for the Chinese subgroup could not be reliably assessed because of the limited number of events.78 Similarly, the 2022 European Atherosclerosis Society (EAS) consensus statement on Lp(a) concluded that the causal and continuous association between Lp(a) and cardiovascular outcomes is generally observed across ethnic groups, while emphasising that evidence in non-White populations remains comparatively limited and requires further expansion.76 The 2024 NLA focused update further noted that, although Lp(a) distributions differ across racial and ethnic groups, current evidence is still insufficient to support race-based definitions of “elevated” Lp(a).53
A more appropriate interpretation is therefore that the relative risk gradient associated with elevated Lp(a) may be broadly consistent across populations, whereas the performance of fixed thresholds for risk identification, their relationship with absolute risk, and the population-attributable burden may still vary by ancestry.53,76 Guan et al, in an analysis from the Multi-Ethnic Study of Atherosclerosis (MESA) cohort,79 suggested that traditional mg/dL cut points do not perform identically for CHD risk discrimination across racial and ethnic groups. More recent analyses using disaggregated ethnicity categories have also shown that, even within broad groups such as “Asian” or “Hispanic,” the prevalence of elevated Lp(a) and the associated clinical risk profiles remain heterogeneous.80 Grant et al81 using data from the Atherosclerosis Risk in Communities (ARIC) cohort, further showed that although the relative ASCVD risk associated with Lp(a) did not differ significantly between Black and White participants, the higher prevalence of elevated Lp(a) in Black individuals may translate into a greater population-attributable burden. Taken together, currently used uniform thresholds remain practical for clinical use, but they should not be interpreted as resolving all issues related to risk stratification across ancestry groups.53,76,81 Future research should therefore use standardised nmol/L-based assays and large prospective cohorts with broader ancestry representation, sufficient event numbers, and harmonised outcome definitions to determine whether ancestry-specific Lp(a) thresholds can meaningfully improve ASCVD risk stratification and clinical decision-making.
Lp(a) Interference in LDL-C Estimation: Pseudo-Statin Resistance and Risk Misclassification
Elevated LDL-C is the principal modifiable risk factor for ASCVD and remains the primary therapeutic target in lipid management.82 The Friedewald, Sampson-NIH, and Martin-Hopkins equations, which are widely used to estimate LDL-C, include cholesterol carried by Lp(a) and intermediate-density lipoprotein (IDL).83 Although IDL-C contributes little in most fasting plasma samples, Lp(a) is a cholesterol-rich lipoprotein.84 A commonly cited approximation assumes that cholesterol accounts for 30% of Lp(a) mass, but this fraction shows substantial interindividual and assay-dependent variability.85,86 Consequently, individuals with high baseline Lp(a) levels will have overestimated calculated LDL-C values. Furthermore, the cholesterol content within lipoprotein(a) contributes to total cholesterol measurements used in cardiovascular risk scoring systems (eg, QRISK3 and Pooled Cohort Equations). The atherogenic effects of Lp(a) may be partially attributed to LDL-C, leading to collinearity bias that underestimates or even obscures the risk associated with elevated Lp(a).87 Additionally, this may result in patients with elevated Lp(a) being misclassified as having familial hypercholesterolemia (FH).88–90
Some studies indicate statins exert a neutral effect on Lp(a) concentration.91 However, some patients (especially those with baseline Lp(a)>30mg/dL) may experience a decrease in LDL-C levels but an increase in Lp(a) levels after statin therapy.87,92 Early research reported that Lp(a) concentrations could rise by up to 45% from baseline and suggested that this represented a true increase in concentration, not an artefact due to altered immunoreactivity of the lipoprotein(a) particles.93 Cell culture studies demonstrating time- and dose-dependent increases in Lp(a) mRNA expression and apo(a) production by statins further corroborate this.92
The resulting increase in the Lp(a)-C contribution to calculated LDL-C may mask the true reduction in LDL-C, creating the false impression of statin ineffectiveness, often referred to as pseudo-resistance; at the same time, higher Lp(a) itself may contribute to residual cardiovascular risk.69,92 This creates an apparent paradox: although statins may increase Lp(a) in some patients, they remain first-line lipid-lowering therapy for patients at elevated cardiovascular risk. The 2025 Focused Update of the 2019 ESC/EAS Guidelines for the management of dyslipidaemias recommends interpreting Lp(a) elevation in the context of overall cardiovascular risk.94 Accordingly, current guidance supports at least one lifetime Lp(a) measurement and selective re-measurement in specific contexts, rather than routine serial monitoring for all patients >30 mg/dL. Fixed-fraction “Lp(a)-corrected LDL-C” calculations should not be used mechanically for routine decision-making. Instead, apoB and/or non-HDL-C should be considered to better characterise atherogenic particle burden, while Lp(a)-adjusted LDL-C approaches may be used as interpretive adjuncts when they have been analytically validated. In high-risk patients who remain above LDL-C goals, adding a PCSK9 inhibitor is appropriate and also lowers Lp(a) modestly.95 Lp(a) measurement may be considered in patients with recurrent or progressive ASCVD despite optimal risk-factor control, including those with target-vessel restenosis or bypass graft failure; however, routine serial monitoring is not universally recommended.69,94
The most straightforward correction method uses the measured Lp(a) mass concentration in mg/dL, multiplies it by an assumed average cholesterol mass fraction to estimate Lp(a)-cholesterol [Lp(a)-C], and subtracts this value from LDL-C estimated by standard equations, such as Friedewald, Martin-Hopkins, or Sampson-NIH. A major limitation of this approach is the marked interindividual variability in the actual cholesterol mass fraction of Lp(a) particles, which can introduce error into the Lp(a)-C estimate. Direct measurements indicate that the cholesterol fraction of Lp(a) mass ranges from 5.8% to 57.3% across individuals.96 The clinical acceptability of this potential error should therefore be carefully considered when such estimates are used for decision-making. At present, for Lp(a) reported in particle concentration units (nmol/L), there is no universally validated, large-scale correction formula to convert particle concentration to Lp(a)-C for routine clinical use; recently proposed molar approaches remain investigational and require independent external validation before widespread adoption.97 Part of the reported “statin pseudo-resistance” may therefore reflect Lp(a)-C embedded in measured or calculated LDL-C, rather than true on-treatment LDL-C. This interpretation is consistent with consensus statements emphasising the independent and often under-recognised contribution of Lp(a) to atherosclerotic risk and the need to interpret LDL-C in this context.53,76,94,98
Although a 30% Lp(a)-C fraction is commonly used as a pragmatic average for LDL-C “correction”,76,99 interindividual variability is substantial and assay standardisation remains incomplete.76,96 In the absence of individualised Lp(a)-C data, a fixed proportion may represent a practical but imperfect compromise. However, applying a uniform 30% subtraction can introduce clinically meaningful error in patients with markedly elevated Lp(a), even when triglyceride concentrations are within an acceptable range.
Despite the potential superiority of apoB concentration over LDL-C in predicting cardiovascular risk,100 LDL-C remains the primary therapeutic target for ASCVD prevention. For patients with high Lp(a) and formula-derived LDL-C values near treatment thresholds, apoB and/or non-HDL-C should be used to contextualise risk and treatment decisions. A validated Lp(a)-adjusted LDL-C estimate, preferably based on molar Lp(a), may provide additional interpretive value where available, but it should not replace established LDL-C targets in routine practice. This approach aligns with guideline trends advocating at least once-in-a-lifetime Lp(a) testing and more refined risk reclassification near treatment thresholds.76,89,101
Formula-derived LDL-C, regardless of the equation used, cannot separate cholesterol carried by Lp(a) from cholesterol carried by LDL particles. Even after excluding samples with triglycerides ≥400 mg/dL, residual triglyceride-related imprecision in calculated LDL-C may persist. These caveats do not alter the core message: although fixed-fraction Lp(a)-C adjustments, such as the commonly used 30% correction, have long been used as pragmatic tools, routine decision-making should prioritise apoB and/or validated molar Lp(a)-adjusted LDL-C estimates where available. Direct LDL-C measurement should be reserved primarily for reducing equation-related imprecision rather than for correcting Lp(a)-C contamination, especially when Lp(a) is high or LDL-C values lie near treatment thresholds.
Assay Standardisation and the Evolution of Detection Technologies
Moving Toward Molar Reporting: The WHO/IFCC SRM-2B Reference Material
The limited comparability of earlier Lp(a) immunoassays was partly attributable to the absence of standardised calibration materials and value-transfer systems. Immunoassays often use calibrators with high Lp(a) concentrations; however, these calibrators may predominantly contain isoforms with smaller apo(a) molecules. When apo(a) isoforms in patient samples are larger than those in the calibrator, this mismatch may lead to systematic overestimation of Lp(a) concentrations.20 In addition, commercial assays and research laboratories have historically assigned target values to calibration materials using different procedures. These assignments were often based on different in-house reference materials and on poorly defined “primary calibrators”, without a unified metrologically traceable reference-material system.20,35 As a result, the same serum or plasma sample may yield different Lp(a) mass or particle concentrations when measured using different assays.35 Together, these factors represent important additional sources of inaccuracy in Lp(a) measurement and poor comparability among commercial methods.20
To address this problem, the World Health Organization (WHO) designated the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) SRM-2B as the secondary reference material for Lp(a) immunoassays in 2003, now commonly referred to as WHO/IFCC SRM-2B. Its assigned value is metrologically traceable to a primary reference measurement procedure. SRM-2B has played a central historical role in harmonising Lp(a) testing. As the first internationally recognised reference reagent for Lp(a) immunoassays, it provided a common calibration anchor across analytical systems, enabled value assignment and traceability in molar units, and substantially reduced inter-method variation arising from inconsistent calibration. Previous IFCC studies showed that SRM-2B had good stability, limited matrix effects, and acceptable commutability, supporting its use as a reference material for improving the harmonisation of Lp(a) measurement. Accordingly, guidelines in many countries recommend that calibrators used in clinical laboratories should be traceable to the WHO/IFCC SRM-2B.11,102
However, reference-material traceability should not be interpreted as evidence that an assay method is entirely free from bias. The principal value of SRM-2B lies not in eliminating all measurement bias, but in providing a common basis for accuracy-oriented value assignment, value transfer to manufacturers’ master calibrators, and cross-method comparison. Parallelism testing showed that SRM-2B was parallel to frozen serum control C (FSC C) in 71% of assay systems and to an in-house serum pool (IHSP) in 63% of systems,103 indicating that SRM-2B still has practical limitations. As a lyophilised human serum preparation, SRM-2B also raises, through commutability testing, the possibility that its analytical behaviour may differ from that of fresh serum. After reconstitution, its matrix may differ from fresh patient serum in protein conformation, lipid microenvironment, and other properties, which may in turn affect antibody-binding efficiency. These findings suggest that reference materials should continue to be refined to more closely mimic native serum and further improve harmonisation across Lp(a) immunoassay platforms.
In addition, improvements in calibration systems must be accompanied by rigorous control of assay performance itself. To reduce the influence of apo(a) isoform size on measurement results, manufacturers of calibration materials and assay developers should further optimise calibrator matrices so that they more closely resemble human serum. At the same time, assay systems should adequately evaluate their linear range and the risk of a high-dose hook effect, particularly for samples with extremely high Lp(a) concentrations.104 Testing platforms should therefore have reliable sample-dilution capability. Samples exceeding the linear range or suspected of being affected by the hook effect should be appropriately diluted and retested to ensure accurate results.
Immunoassay Methods: Practical Utility and Standardisation Challenges
Lp(a) has been quantified using a range of immunochemical methods. Early radioimmunoassays (RIA), radial immunodiffusion (RID) assays, and some subsequent routine immunoturbidimetric or nephelometric assays commonly used anti-apo(a) polyclonal antibodies. In addition, several early immunoassay studies did not clearly specify whether the antibodies used were monoclonal or polyclonal, nor did they adequately define the recognised epitopes or domain specificity.105 This impression was also supported by the 2000 IFCC standardisation study: among the 22 systems included, most used anti-apo(a) polyclonal antibodies, whereas only a small number used latex-coated monoclonal antibodies.106 If antibody reactivity or the calibration system is influenced by the KIV2 repeat region, apo(a) isoform-sensitive bias may occur.107,108
Compared with earlier routine immunoassays, which were relatively fixed in format, ELISA offers a more adaptable assay platform. It offers greater methodological flexibility in the choice of capture and detection antibody pairs, epitope selection, experimental conditions, and calibration strategies. However, the ELISA format itself does not automatically eliminate bias arising from apo(a) size polymorphism. Whether an Lp(a) assay is isoform-independent depends not on the assay format per se, but on whether the analytical signal is affected by the copy number of KIV2 repeat epitopes and whether quantification can be based on non-repeated epitopes that approximate a “one-recognition-event-per-Lp(a)-particle” principle.
This methodological principle was systematically demonstrated in the 1995 study by Marcovina et al.105 The sandwich ELISA developed in that study later became one of the long-standing reference methods used by the Northwest Lipid Metabolism and Diabetes Research Laboratory (NLMDRL). This method used monoclonal antibodies directed against defined epitopes. An antibody targeting the a-6 epitope on the KIV-2 domain, which is present as a variable number of identical repeats, was used as the capture antibody to improve the capture efficiency of apo(a)-containing particles; another antibody targeting the single a-40 epitope on the non-repeated kringle IV type 9 (KIV-9) domain of apo(a) was used as the detection antibody.20,105,108 This design allowed each apo(a)-containing particle, as far as possible, to contribute only once to the quantitative signal. In performance validation, the assay showed within-assay coefficients of variation of 2.3–4.0% and between-assay coefficients of variation of 4.0–6.9%. In 723 samples with a single apo(a) band, the method showed excellent agreement with an apoB-based size-insensitive assay, with a correlation coefficient of 0.986 and a mean bias of only 2.4 nmol/L, or 5.5%.105 The 2000 IFCC standardisation study further noted that this method had been extensively evaluated and that its measurement accuracy was not affected by apo(a) size polymorphism. In the reference value-assignment process, the within-assay and between-assay coefficients of variation were 3.6% and 7.0%, respectively. Together, these findings support the methodological reliability and accuracy of this assay as an isoform-independent reference method.106 It has subsequently been regarded as an accurate reference method in later studies of Lp(a) measurement.109
At the same time, other research groups have expanded the practical applications of Lp(a) immunoassay platforms from different perspectives. Bustos et al110 developed a monoclonal antibody-based sandwich ELISA using the anti-apo(a) monoclonal antibody 2F4E7 as the capture antibody and a biotinylated anti-apoB-LDL monoclonal antibody as the detection antibody. The assay showed low intra-assay and inter-assay coefficients of variation, suggesting that monoclonal antibody combinations can be used to construct stable immunoquantitative platforms for Lp(a). This study also again highlighted the importance of apo(a) epitope selection and the apo(a) isoform composition of calibration sera for measurement results. Wang et al111 developed a sandwich ELISA for detecting oxidised Lp(a) and found that ox-Lp(a) levels were significantly higher in patients with coronary heart disease than in controls, suggesting that immunoassays can also be used to study oxidative modification of Lp(a) and its potentially pathogenic phenotypes.
Recent method-comparison studies indicate that systematic bias and sample-specific differences remain evident across commercial immunoassays, even when some assays are traceable to WHO/IFCC SRM-2B or use multi-point calibration. For example, Dikaios et al112 compared an IFCC candidate mass spectrometry-based reference measurement procedure with eight commonly used immunoassays. They found that immunoassays reporting results in nmol/L generally correlated well with the candidate reference method, although substantial sample-specific differences persisted. Previous comparisons among six commercial immunoassays also reported relative biases of approximately −8% to 22%.113 These discrepancies likely reflect unresolved analytical and metrological issues in current assay systems, including antibody epitope selection, KIV2 repeat-related reactivity, antibody-pair design, calibrator value assignment, multi-point calibration strategies, and reference-material commutability.112,113
In addition, assay results obtained under specific metabolic conditions require cautious interpretation. Cross-reactive substances capable of binding both antibodies may produce false-positive signals. For example, studies have shown that, in patients with hypertriglyceridemia (HTG), 10–78% of Lp(a) can form stable complexes with triglyceride-rich lipoproteins (TRLs), compared with approximately 1% in healthy individuals. These complexes resist ultracentrifugation and are difficult to dissociate during the incubation and washing steps of ELISA. This phenomenon provides a mechanistic explanation for the negative association between plasma Lp(a) levels and triglycerides in patients with HTG: Lp(a)-TRL complexes may accelerate the metabolic clearance of Lp(a), thereby lowering its true concentration.114 It also suggests that sandwich ELISA methods based on an “apo(a)-capture/apoB-detection” strategy may overestimate actual Lp(a) concentrations because of cross-reactivity with apoB within these complexes. Although current studies generally tend to support non-fasting sampling,114–117 fasting sampling may still have practical relevance for patients tested with this type of ELISA method.
However, these analytical challenges do not mean that Lp(a) ELISA, immunocapture platforms, or routine automated immunoassays lack practical value. From a clinical-practice perspective, the key issue in evaluating existing Lp(a) immunoassays is not whether they eliminate all analytical error, but whether, within clearly defined reporting units, calibration systems, and intended-use boundaries, they provide reproducible and interpretable information for risk screening, risk re-stratification, and therapeutic decision-making. For routine clinical screening and risk stratification, automated immunoturbidimetric and nephelometric platforms are well suited to large-scale implementation because they are widely accessible, offer relatively high throughput, and can be readily incorporated into routine clinical laboratory quality-control systems. The main value of ELISA and related immunocapture platforms is not to replace routine automated testing, but to provide controllable and verifiable experimental tools for establishing reference methods, validating assay performance, evaluating pharmacodynamic effects in clinical trials, and quantifying Lp(a) particle concentration, oxidative modification status, and Lp(a)-associated analytes through antibody-pair selection, epitope restriction, and optimisation of experimental and calibration conditions.53,76,100
Large-scale clinical and epidemiological studies also support the practical value of routine automated immunoassay platforms and ELISA-based methods. A UK Biobank study78 measured Lp(a) concentrations using a uniformly calibrated immunoturbidimetric assay in 460,506 middle-aged participants and showed an approximately linear association between Lp(a) concentration and incident ASCVD risk. This finding provides practical support for the use of automated platforms in large-scale population studies and risk-stratification applications. The study by Saleheen et al, based partly on the Pakistan Risk of Myocardial Infarction Study (PROMIS), also illustrates this functional division.62 In that study, Lp(a) concentrations were measured in 9015 patients with acute myocardial infarction and 8629 controls using a Denka Seiken high-sensitivity immunoturbidimetric assay, whereas an in-house ELISA that did not recognise the KIV2 domain was used for supplementary measurement in a subset of samples. Lp(a) concentrations measured by the two methods were highly correlated, suggesting that automated immunoturbidimetric assays are suitable for large-sample testing, whereas ELISA can serve as a complementary tool for methodological validation and specific research questions. Updates in guidelines and consensus documents further indicate that Lp(a) testing has gradually moved from a purely research-based tool into the framework of clinical risk assessment and preventive management. This shows that, although assay standardisation still requires continued improvement, Lp(a) measurement already has clear clinical and public-health value.53,68,76
Therefore, the purpose of current standardisation efforts is not to negate the clinical utility of existing immunoassays, but to further improve the comparability of values across platforms, the consistency of risk classification near clinical thresholds, and the reliability of eligibility screening and treatment monitoring in the future era of Lp(a)-targeted therapy.112,118 In this context, newly developed monoclonal antibody-based sandwich ELISAs, including the LPA4/LPA-KIV9 ELISA recently developed by Marcovina et al,109 provide a methodological pathway for further reducing measurement bias while preserving the clinical accessibility of immunoassay-based testing.
LC-MS/MS in Lp(a) Standardisation: Principles, Reference-Method Role, and Limitations
Targeted liquid chromatography–tandem mass spectrometry (LC-MS/MS) can accurately quantify proteins in complex biological matrices and has therefore become an important higher-order tool in standardised biomarker measurement systems.119 Unlike ELISA and other antigen–antibody-based methods, LC-MS/MS selects specific quantitative peptides outside the KIV2 repeat region,120 thereby minimising the influence of apo(a) size polymorphism on measurement. This analytical principle gives LC-MS/MS a degree of specificity and additional functional capability that conventional immunoassays, including ELISA, cannot fully provide. In particular, LC-MS/MS can avoid interference related to apo(a) size heterogeneity, glycosylation, and potential cross-reactive analytes in immunoassays. More importantly, with calibration strategies such as isotope dilution, LC-MS/MS results can be made traceable to the International System of Units (SI), which is a key element of the reference framework for assay standardisation.121
Recent IFCC-related work has further strengthened the central role of LC-MS/MS in the standardisation framework for Lp(a). Diederiks et al122 reported an ISO 17511:2020-compliant, LC-MS-based, IFCC-endorsed reference measurement procedure (RMP) that supports the standardisation of Lp(a) in molar units by targeting apo(a)-specific peptides. However, this RMP is technically demanding and requires highly trained personnel. To facilitate the transition of in vitro diagnostic manufacturers from mass-based to molar-unit reporting, the authors further developed a semi-automated, higher-throughput LC-MS-based designated comparison method. In direct method-comparison studies, this method showed excellent agreement with the RMP, with a median regression slope of 0.997 and a median bias of −0.2 nmol/L (−0.2%). It has therefore been positioned as a higher-order designated comparison method for guiding the restandardisation of Lp(a) assay results by IVD manufacturers.
Nevertheless, the high cost of LC-MS instrumentation, operational complexity, and the continued need for batch-mode sample preparation substantially limit its clinical accessibility and routine practicality, and may also reduce patient-level acceptability. Therefore, unlike automated immunoassays, which are better suited to clinical screening because of their high throughput, lower cost, and easier integration into routine quality-control workflows, the main value of LC-MS/MS lies in reference material value assignment, recalibration of commercial assays, inter-laboratory comparability assessment, improvement of classification consistency near clinical decision thresholds, and standardised support for enrolment screening and treatment monitoring in the future era of Lp(a)-targeted therapy. Its core role is therefore closer to that of a standardisation anchor and method-comparison tool than that of a routine front-line clinical screening assay.
In addition, current measurement approaches still require caution in defining the measurand. It is now well recognised that proteins do not exist as single uniform molecular entities but may exhibit conformational polymorphism. This complexity indicates that defining the analytical target either at the particle level, such as Lp(a), or at the monomeric protein level, such as apo(a), has inherent limitations.31 Accordingly, a persistent and non-negligible issue is that LC-MS/MS quantifies Lp(a) indirectly by detecting characteristic apo(a) peptides. In this respect, it shares a key limitation with apo(a)-based assays, apo(a)-capture sandwich ELISAs, and other immunoassays: these methods cannot distinguish free apo(a) from intact Lp(a) particles.83
This distinction may have clinical relevance. Because free apo(a) and intact Lp(a) particles may differ in their pathogenic mechanisms, their biological effects may also differ. Therefore, when free apo(a) is markedly elevated, its inclusion in apo(a)-based quantification may lead to overestimation of the intact Lp(a) particle burden. In other words, what these methods measure may be closer to an “apo(a)-related antigen burden” or an “apo(a) amount-of-substance” rather than a strict concentration of intact Lp(a) particles. In most populations, free apo(a) usually accounts for less than 5% of total plasma apo(a),123 and therefore does not generally have a major impact on clinical interpretation. However, the proportion of free apo(a) in total plasma apo(a) can increase substantially in certain conditions, including nephrotic syndrome,124 the post-renal transplantation state,124 end-stage renal disease (ESRD) treated with hemodialysis or peritoneal dialysis,124–127 and hyperhomocysteinemia.124 In these settings, NLMDRL-type apo(a)-based assays, apo(a)-capture sandwich ELISAs, and related immunoassays may overestimate the true concentration of intact Lp(a) particles. These considerations suggest that further research is needed to determine whether modified apo(a)-related cardiovascular risk prediction algorithms are needed for precision medicine.
Conclusions
Lp(a) is an established independent contributor to residual ASCVD risk, but its clinical utility depends on accurate, comparable, and interpretable measurement. Structural heterogeneity—particularly apo(a) KIV2 copy-number variation, glycosylation, lipid composition, and isoform co-expression—limits the reliability of mass-based values and can introduce assay-dependent bias. Validated molar reporting in nmol/L should therefore be preferred, whereas fixed conversion between mg/dL and nmol/L should be avoided.
Current immunoassays remain useful for routine screening and risk stratification when their calibration systems, reporting units, and analytical limitations are clearly defined. ELISA and LC-MS/MS provide additional value for reference-method development, assay validation, and standardisation. In patients with high Lp(a), calculated LDL-C should be interpreted cautiously because fixed Lp(a)-cholesterol corrections may misclassify residual risk or apparent treatment response. Future work should prioritise globally harmonised molar measurement, commutable reference materials, ancestry-inclusive validation, and improved distinction between intact Lp(a), free apo(a), and biologically relevant Lp(a) subspecies. These advances will be essential for more precise cardiovascular risk stratification and for the clinical implementation of emerging Lp(a)-lowering therapies.
Abbreviations
Lp(a), Lipoprotein(a); Apo(a), Apolipoprotein(a); ApoB, Apolipoprotein B-100.
Data Sharing Statement
The data presented in this study are available on request from the corresponding author.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Mensah GA, Roth GA, Fuster V. The global burden of cardiovascular diseases and risk factors: 2020 and beyond. J Am Coll Cardiol. 2019;74(20):2529–17. doi:10.1016/j.jacc.2019.10.009
2. Roth GA, Mensah GA, Fuster V. The global burden of cardiovascular diseases and risks: a compass for global action. J Am Coll Cardiol. 2020;76(25):2980–2981. doi:10.1016/j.jacc.2020.11.021
3. Yang J, Zhang Y, Ma T, Tian X, Zhao Y. Epidemic status, disease burden and prediction of cardiovascular diseases in China, 1990–2019. Chin General Pract. 2024;27(02):233.
4. Maloberti A, Fabbri S, Colombo V, et al. Lipoprotein(a): cardiovascular disease, aortic stenosis and new therapeutic option. Int J Mol Sci. 2022;24(1). doi:10.3390/ijms24010170
5. Fatima K, Butler J, Fonarow GC. Residual risk in heart failure and the need for simultaneous implementation and innovation. Eur J Heart Fail. 2023;25(9):1477–1480. doi:10.1002/ejhf.3005
6. Fruchart JC, Davignon J, Hermans MP, et al. Residual macrovascular risk in 2013: what have we learned? Cardiovasc Diabetol. 2014;13:26. doi:10.1186/1475-2840-13-26
7. Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176–184. doi:10.1161/circulationaha.107.715698
8. Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302(4):412–423. doi:10.1001/jama.2009.1063
9. Larsson SC, Wang L, Li X, Jiang F, Chen X, Mantzoros CS. Circulating lipoprotein(a) levels and health outcomes: phenome-wide Mendelian randomization and disease-trajectory analyses. Metabolism. 2022;137:155347. doi:10.1016/j.metabol.2022.155347
10. Thanassoulis G, Campbell CY, Owens DS, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368(6):503–512. doi:10.1056/NEJMoa1109034
11. Cegla J, Neely RDG, France M, et al. HEART UK consensus statement on Lipoprotein(a): a call to action. Atherosclerosis. 2019;291:62–70. doi:10.1016/j.atherosclerosis.2019.10.011
12. Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111–188. doi:10.1093/eurheartj/ehz455
13. Wang Z, Liu J, Li J, et al. Chinese guidelines for lipid management (2023). Chin Circ J. 2023;38(3):237–271.
14. Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25). doi:10.1161/cir.0000000000000625
15. Wilson PWF, Rifai N, Levy D, et al. National academy of clinical biochemistry laboratory medicine practice guidelines: emerging biomarkers for primary prevention of cardiovascular disease. Clin Chem. 2009;55(2):378–384. doi:10.1373/clinchem.2008.115899
16. Davidson MH, Ballantyne CM, Jacobson TA, et al. Clinical utility of inflammatory markers and advanced lipoprotein testing: advice from an expert panel of lipid specialists. J Clin Lipidol. 2011;5(5):338–367. doi:10.1016/j.jacl.2011.07.005
17. Tasdighi E, Adhikari R, Almaadawy O, Leucker TM, Blaha MJ. LP(a): structure, genetics, associated cardiovascular risk, and emerging therapeutics. Annu Rev Pharmacol Toxicol. 2024;64(1):135–157. doi:10.1146/annurev-pharmtox-031023-100609
18. Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein (a). J Lipid Res. 2016;57(8):1339–1359. doi:10.1194/jlr.R067314
19. Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J Intern Med. 2013;273(1):6–30. doi:10.1111/j.1365-2796.2012.02592.x
20. Marcovina SM, Albers JJ. Lipoprotein (a) measurements for clinical application. J Lipid Res. 2016;57(4):526–537. doi:10.1194/jlr.R061648
21. McLean JW, Tomlinson JE, Kuang WJ, et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330(6144):132–137. doi:10.1038/330132a0
22. Guevara J Jr, Knapp RD, Honda S, Northup SR, Morrisett JD. A structural assessment of the apo[a] protein of human lipoprotein[a]. Proteins. 1992;12(2):188–199. doi:10.1002/prot.340120212
23. Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692–711. doi:10.1016/j.jacc.2016.11.042
24. Vinci P, Di Girolamo FG, Panizon E, et al. Lipoprotein(a) as a risk factor for cardiovascular diseases: pathophysiology and treatment perspectives. Int J Environ Res Public Health. 2023;20(18). doi:10.3390/ijerph20186721
25. van der Hoek YY, Wittekoek ME, Beisiegel U, Kastelein JJ, Koschinsky ML. The apolipoprotein(a) kringle IV repeats which differ from the major repeat kringle are present in variably-sized isoforms. Hum Mol Genet. 1993;2(4):361–366. doi:10.1093/hmg/2.4.361
26. Koschinsky ML, Marcovina SM. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol. 2004;15(2):167–174. doi:10.1097/00041433-200404000-00009
27. Scanu AM, Edelstein C. Learning about the structure and biology of human lipoprotein [a] through dissection by enzymes of the elastase family: facts and speculations. J Lipid Res. 1997;38(11):2193–2206.
28. Lin Y, Lubman DM. The role of N-glycosylation in cancer. Acta Pharm Sin B. 2024;14(3):1098–1110. doi:10.1016/j.apsb.2023.10.014
29. Marshall RD. The nature and metabolism of the carbohydrate-peptide linkages of glycoproteins. Biochem Soc Symp. 1974;40:17–26.
30. Rek A, Krenn E, Kungl AJ. Therapeutically targeting protein-glycan interactions. Br J Pharmacol. 2009;157(5):686–694. doi:10.1111/j.1476-5381.2009.00226.x
31. Ruhaak LR, Cobbaert CM. Quantifying apolipoprotein(a) in the era of proteoforms and precision medicine. Clin. Chim. Acta. 2020;511:260–268. doi:10.1016/j.cca.2020.10.010
32. Cegla J, France M, Marcovina SM, Neely RDG. Lp(a): when and how to measure it. Ann Clin Biochem. 2020;58(1):16–21. doi:10.1177/0004563220968473
33. Garner B, Harvey DJ, Royle L, et al. Characterization of human apolipoprotein B100 oligosaccharides in LDL subfractions derived from normal and hyperlipidemic plasma: deficiency of alpha-N-acetylneuraminyllactosyl-ceramide in light and small dense LDL particles. Glycobiology. 2001;11(10):791–802. doi:10.1093/glycob/11.10.791
34. Yang CY, Gu ZW, Weng SA, et al. Structure of apolipoprotein B-100 of human low density lipoproteins. Arteriosclerosis. 1989;9(1):96–108. doi:10.1161/01.atv.9.1.96
35. Tsimikas S, Fazio S, Viney NJ, Xia S, Witztum JL, Marcovina SM. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J Clin Lipidol. 2018;12(5):1313–1323. doi:10.1016/j.jacl.2018.07.003
36. Krempler F, Kostner G, Bolzano K, Sandhofer F. Lipoprotein (a) is not a metabolic product of other lipoproteins containing apolipoprotein B. Biochim Biophys Acta. 1979;575(1):63–70. doi:10.1016/0005-2760(79)90131-0
37. Kostner KM, März W, Kostner GM. When should we measure lipoprotein (a)? Eur Heart J. 2013;34(42):3268–3276. doi:10.1093/eurheartj/eht053
38. Rader DJ, Cain W, Ikewaki K, et al. The inverse association of plasma lipoprotein(a) concentrations with apolipoprotein(a) isoform size is not due to differences in Lp(a) catabolism but to differences in production rate. J Clin Invest. 1994;93(6):2758–2763. doi:10.1172/jci117292
39. Marcovina SM, Morrisett JD. Structure and metabolism of lipoprotein (a). Curr Opin Lipidol. 1995;6(3):136–145. doi:10.1097/00041433-199506000-00005
40. White AL, Guerra B, Lanford RE. Influence of allelic variation on apolipoprotein(a) folding in the endoplasmic reticulum. J Biol Chem. 1997;272(8):5048–5055. doi:10.1074/jbc.272.8.5048
41. White AL, Hixson JE, Rainwater DL, Lanford RE. Molecular basis for “null” lipoprotein(a) phenotypes and the influence of apolipoprotein(a) size on plasma lipoprotein(a) level in the baboon. J Biol Chem. 1994;269(12):9060–9066.
42. Ferretti G, Bacchetti T, Johnston TP, Banach M, Pirro M, Sahebkar A. Lipoprotein(a): a missing culprit in the management of athero-thrombosis? J Cell Physiol. 2018;233(4):2966–2981. doi:10.1002/jcp.26050
43. Erqou S, Thompson A, Di Angelantonio E, et al. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010;55(19):2160–2167. doi:10.1016/j.jacc.2009.10.080
44. Paultre F, Pearson TA, Weil HF, et al. High levels of Lp(a) with a small apo(a) isoform are associated with coronary artery disease in African American and white men. Arterioscler Thromb Vasc Biol. 2000;20(12):2619–2624. doi:10.1161/01.atv.20.12.2619
45. Gabel BR, Koschinsky ML. Sequences within apolipoprotein(a) kringle IV types 6–8 bind directly to low-density lipoprotein and mediate noncovalent association of apolipoprotein(a) with apolipoprotein B-100. Biochemistry. 1998;37(21):7892–7898. doi:10.1021/bi973186w
46. Becker L, Cook PM, Wright TG, Koschinsky ML. Quantitative evaluation of the contribution of weak lysine-binding sites present within apolipoprotein(a) kringle IV types 6–8 to lipoprotein(a) assembly. J Biol Chem. 2004;279(4):2679–2688. doi:10.1074/jbc.M309414200
47. Youssef A, Clark JR, Marcovina SM, Boffa MB, Koschinsky ML. Apo(a) and ApoB interact noncovalently within hepatocytes: implications for regulation of Lp(a) levels by modulation of ApoB secretion. Arterioscler Thromb Vasc Biol. 2022;42(3):289–304. doi:10.1161/atvbaha.121.317335
48. Koschinsky ML, Côté GP, Gabel B, van der Hoek YY. Identification of the cysteine residue in apolipoprotein(a) that mediates extracellular coupling with apolipoprotein B-100. J Biol Chem. 1993;268(26):19819–19825.
49. McCormick SP, Ng JK, Taylor S, Flynn LM, Hammer RE, Young SG. Mutagenesis of the human apolipoprotein B gene in a yeast artificial chromosome reveals the site of attachment for apolipoprotein(a). Proc Natl Acad Sci U S A. 1995;92(22):10147–10151. doi:10.1073/pnas.92.22.10147
50. Callow MJ, Rubin EM. Site-specific mutagenesis demonstrates that cysteine 4326 of apolipoprotein B is required for covalent linkage with apolipoprotein (a) in vivo. J Biol Chem. 1995;270(41):23914–23917. doi:10.1074/jbc.270.41.23914
51. Martín S, Ladona MG, Pedro-Botet J, Covas MI, Rubiés-Prat J. Differential expression of double-band apolipoprotein(a) phenotypes in healthy Spanish subjects detected by SDS-agarose immunoblotting. Clin Chim Acta. 1998;277(2):191–205. doi:10.1016/s0009-8981(98)00126-0
52. Razavi AC, Bhatia HS, Blumenthal RS, Shapiro MD, Mehta A. Why, how and in whom should we measure levels of lipoprotein(a): a review of the latest evidence and clinical implications. Diabetes Obes Metab. 2025;27(Suppl 8):34–46. doi:10.1111/dom.16469
53. Koschinsky ML, Bajaj A, Boffa MB, et al. A focused update to the 2019 NLA scientific statement on use of lipoprotein(a) in clinical practice. J Clin Lipidol. 2024;18(3):e308–e319. doi:10.1016/j.jacl.2024.03.001
54. Jang Y, Lee JH, Lee SG, et al. A position paper on Lipoprotein(a) from the Lipoprotein(a) task force of the Korean Society of Lipid and Atherosclerosis: current evidence, clinical applications, and future directions. Korean Circ J. 2026;56(1):9–32. doi:10.4070/kcj.2025.0388
55. Maher LL, Tokgözoğlu SL, Sanchez EJ, Underberg JA, Guyton JR. JCL roundtable: global think tank on Lipoprotein(a). J Clin Lipidol. 2021;15(3):387–393. doi:10.1016/j.jacl.2021.06.003
56. Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the national heart, lung, and blood institute workshop on Lipoprotein(a) and Cardiovascular Disease: recent advances and future directions. Clin Chem. 2003;49(11):1785–1796. doi:10.1373/clinchem.2003.023689
57. Coassin S, Kronenberg F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: the complexity of genetic variation in the LPA gene. Atherosclerosis. 2022;349:17–35. doi:10.1016/j.atherosclerosis.2022.04.003
58. Matveyenko A, Matienzo N, Ginsberg H, et al. Relationship of apolipoprotein(a) isoform size with clearance and production of lipoprotein(a) in a diverse cohort. J Lipid Res. 2023;64(3):100336. doi:10.1016/j.jlr.2023.100336
59. Ooi EM, Ellis KL, Barrett PHR, et al. Lipoprotein(a) and apolipoprotein(a) isoform size: associations with angiographic extent and severity of coronary artery disease, and carotid artery plaque. Atherosclerosis. 2018;275:232–238. doi:10.1016/j.atherosclerosis.2018.06.863
60. Hopewell JC, Seedorf U, Farrall M, et al. Impact of lipoprotein(a) levels and apolipoprotein(a) isoform size on risk of coronary heart disease. J Intern Med. 2014;276(3):260–268. doi:10.1111/joim.12187
61. Gudbjartsson DF, Thorgeirsson G, Sulem P, et al. Lipoprotein(a) concentration and risks of cardiovascular disease and diabetes. J Am Coll Cardiol. 2019;74(24):2982–2994. doi:10.1016/j.jacc.2019.10.019
62. Saleheen D, Haycock PC, Zhao W, et al. Apolipoprotein(a) isoform size, lipoprotein(a) concentration, and coronary artery disease: a mendelian randomisation analysis. Lancet Diabetes Endocrinol. 2017;5(7):524–533. doi:10.1016/s2213-8587(17)30088-8
63. Tsimikas S, Clopton P, Brilakis ES, et al. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: results from the Dallas Heart Study. Circulation. 2009;119(13):1711–1719. doi:10.1161/circulationaha.108.836940
64. Lee SR, Prasad A, Choi YS, et al. LPA gene, ethnicity, and cardiovascular events. Circulation. 2017;135(3):251–263. doi:10.1161/circulationaha.116.024611
65. Koschinsky ML, Boffa MB. Oxidized phospholipid modification of lipoprotein(a): epidemiology, biochemistry and pathophysiology. Atherosclerosis. 2022;349:92–100. doi:10.1016/j.atherosclerosis.2022.04.001
66. Sabbah N, Jaisson S, Garnotel R, Anglés-Cano E, Gillery P. Small size apolipoprotein(a) isoforms enhance inflammatory and proteolytic potential of collagen-primed monocytes. Lipids Health Dis. 2019;18(1):166. doi:10.1186/s12944-019-1106-4
67. Shah P, King S, Trabanino S, Parsa S, Chen T, Rodriguez F. Ancestral variation in Lp(a): epidemiology, isoform diversity, and testing. Curr Atheroscler Rep. 2025;27(1):117. doi:10.1007/s11883-025-01365-0
68. Kronenberg F, Bedlington N, Ademi Z, et al. The brussels international declaration on Lipoprotein(a) testing and management. Atherosclerosis. 2025;406:119218. doi:10.1016/j.atherosclerosis.2025.119218
69. Brown WV, Ballantyne CM, Jones PH, Marcovina S. Management of Lp(a). J Clin Lipidol. 2010;4(4):240–247. doi:10.1016/j.jacl.2010.07.002
70. McConnell JP, Guadagno PA, Dayspring TD, et al. Lipoprotein(a) mass: a massively misunderstood metric. J Clin Lipidol. 2014;8(6):550–553. doi:10.1016/j.jacl.2014.08.003
71. Cao J, Donato L, El-Khoury JM, Goldberg A, Meeusen JW, Remaley AT. ADLM guidance document on the measurement and reporting of lipids and lipoproteins. J Appl Lab Med. 2024;9(5):1040–1056. doi:10.1093/jalm/jfae057
72. Lyle AN, Flores EN, Coffman CC, et al. Interlaboratory comparison of serum lipoprotein(a) analytical results across clinical assays-Steps toward standardization. J Clin Lipidol. 2025;19(3):531–543. doi:10.1016/j.jacl.2025.02.010
73. Jawi MM, Frohlich J, Chan SY. Lipoprotein(a) the insurgent: a new insight into the structure, function, metabolism, pathogenicity, and medications affecting Lipoprotein(a) molecule. J Lipids. 2020;2020:1–26. doi:10.1155/2020/3491764
74. Langsted A, Kamstrup PR, Nordestgaard BG. Lipoprotein(a): fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis. 2014;234(1):95–101. doi:10.1016/j.atherosclerosis.2014.01.049
75. Association BH. Expert statement on the relationship between Lipoprotein (a) and cardiovascular disease risk and clinical management. Chin Circulation J. 2021;36(12):1158–1167.
76. Kronenberg F, Mora S, Stroes ESG, et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: a European Atherosclerosis Society consensus statement. Eur Heart J. 2022;43(39):3925–3946. doi:10.1093/eurheartj/ehac361
77. Mehta A, Jain V, Saeed A, et al. Lipoprotein(a) and ethnicities. Atherosclerosis. 2022;349:42–52. doi:10.1016/j.atherosclerosis.2022.04.005
78. Patel AP, Wang M, Pirruccello JP, et al. Lp(a) (Lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a Large National Biobank. Arterioscler Thromb Vasc Biol. 2021;41(1):465–474. doi:10.1161/atvbaha.120.315291
79. Guan W, Cao J, Steffen BT, et al. Race is a key variable in assigning lipoprotein(a) cutoff values for coronary heart disease risk assessment: the Multi-Ethnic Study of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35(4):996–1001. doi:10.1161/atvbaha.114.304785
80. Dudum R, Huang Q, Yan XS, et al. Lipoprotein(a) levels in disaggregated racial and ethnic subgroups across atherosclerotic cardiovascular disease risk levels. JACC Adv. 2024;3(6):100940. doi:10.1016/j.jacadv.2024.100940
81. Grant JK, Martin SS, Zhang S, et al. Racial differences in the burden of atherosclerotic cardiovascular disease related to elevated Lipoprotein(a) levels: the ARIC study. Circulation. 2024;150(3):250–252. doi:10.1161/circulationaha.124.069582
82. Vaduganathan M, Mensah GA, Turco JV, Fuster V, Roth GA. The global burden of cardiovascular diseases and risk: a compass for future health. J Am Coll Cardiol. 2022;80(25):2361–2371. doi:10.1016/j.jacc.2022.11.005
83. Yeang C, Witztum J, Tsimikas S. ‘LDL-C’ = LDL-C + Lp(a)-C: implications of achieved ultra-low LDL-C levels in the proprotein convertase subtilisin/kexin type 9 era of potent LDL-C lowering. (1473–6535 (Electronic)). Curr Opin Lipidol. 2015;26(3):169–178.
84. Kostner GM, Ibovnik A, Holzer H, Grillhofer H. Preparation of a stable fresh frozen primary lipoprotein[a] (Lp[a]) standard. J Lipid Res. 1999;40(12):2255–2263.
85. Seman LJ, Breckenridge WC. Isolation and partial characterization of apolipoprotein (a) from human lipoprotein (a). Biochem Cell Biol. 1986;64(10):999–1009. doi:10.1139/o86-133
86. Jenner JL, Ordovas JM, Lamon-Fava S, et al. Effects of age, sex, and menopausal status on plasma lipoprotein(a) levels. The Framingham Offspring Study. Circulation. 1993;87(4):1135–1141. doi:10.1161/01.cir.87.4.1135
87. Trinder M, Natarajan P. Clinical utility of Lipoprotein(a) for screening does not determine clinical utility of Lipoprotein(a) for the patient-reply. JAMA Cardiol. 2021;6(9):1097. doi:10.1001/jamacardio.2021.1589
88. Chan DC, Pang J, Hooper AJ, Bell DA, Burnett JR, Watts GF. Effect of Lipoprotein(a) on the diagnosis of familial hypercholesterolemia: does it make a difference in the clinic? Clin Chem. 2019;65(10):1258–1266. doi:10.1373/clinchem.2019.306738
89. Yeang C, Willeit P, Tsimikas S. The interconnection between lipoprotein(a), lipoprotein(a) cholesterol and true LDL-cholesterol in the diagnosis of familial hypercholesterolemia. Curr Opin Lipidol. 2020;31(6):305–312. doi:10.1097/mol.0000000000000713
90. Chubykina UV, Ezhov MV, Afanasieva OI, Klesareva EA, Pokrovsky SN. Elevated Lipoprotein(a) level influences familial hypercholesterolemia diagnosis. Diseases. 2022;10(1). doi:10.3390/diseases10010006
91. de Boer LM, Oorthuys AOJ, Wiegman A, et al. Statin therapy and lipoprotein(a) levels: a systematic review and meta-analysis. Eur J Prev Cardiol. 2022;29(5):779–792. doi:10.1093/eurjpc/zwab171
92. Tsimikas S, Gordts P, Nora C, Yeang C, Witztum JL. Statin therapy increases lipoprotein(a) levels. (1522–9645 (Electronic)). Eur Heart J. 2020;41(24):2275–2284.
93. Kostner GM, Gavish D, Leopold B, Bolzano K, Weintraub MS, Breslow JL. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation. 1989;80(5):1313–1319. doi:10.1161/01.cir.80.5.1313
94. Mach F, Koskinas KC, Roeters van Lennep JE, et al. 2025 focused update of the 2019 ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2025. doi:10.1093/eurheartj/ehaf190
95. Chan DC, Watts GF. The promise of PCSK9 and Lipoprotein(a) as targets for gene silencing therapies. Clin Ther. 2023;45(11):1034–1046. doi:10.1016/j.clinthera.2023.07.008
96. Yeang C, Witztum JL, Tsimikas S. Novel method for quantification of lipoprotein(a)-cholesterol: implications for improving accuracy of LDL-C measurements. J Lipid Res. 2021;62:100053. doi:10.1016/j.jlr.2021.100053
97. Rosenson RS, López JAG, Monsalvo ML, Wu Y, Wang H, Marcovina SM. Quantification of LDL-cholesterol corrected for molar concentration of Lipoprotein(a). Cardiovasc Drugs Ther. 2024;38(1):191–197. doi:10.1007/s10557-022-07407-y
98. Thayabaran D, Tsui APT, Ebmeier S, Cegla J, David A, Jones B. The effect of adjusting LDL-cholesterol for Lp(a)-cholesterol on the diagnosis of familial hypercholesterolaemia. J Clin Lipidol. 2023;17(2):244–254. doi:10.1016/j.jacl.2023.01.006
99. Yeang C, Karwatowska-Prokopczuk E, Su F, et al. Effect of pelacarsen on Lipoprotein(a) cholesterol and corrected low-density lipoprotein cholesterol. J Am Coll Cardiol. 2022;79(11):1035–1046. doi:10.1016/j.jacc.2021.12.032
100. Reyes-Soffer G, Ginsberg HN, Berglund L, et al. Lipoprotein(a): a Genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42(1):e48–e60. doi:10.1161/atv.0000000000000147
101. Willeit P, Yeang C, Moriarty PM, et al. Low-density lipoprotein cholesterol corrected for Lipoprotein(a) cholesterol, risk thresholds, and cardiovascular events. J Am Heart Assoc. 2020;9(23):e016318. doi:10.1161/jaha.119.016318
102. Wilson DP, Jacobson TA, Jones PH, et al. Use of Lipoprotein(a) in clinical practice: a biomarker whose time has come. A scientific statement from the National Lipid Association. J Clin Lipidol. 2019;13(3):374–392. doi:10.1016/j.jacl.2019.04.010
103. Dati F, Tate JR, Marcovina SM, Steinmetz A. First WHO/IFCC international reference reagent for Lipoprotein(a) for immunoassay--Lp(a) SRM 2B. Clin Chem Lab Med. 2004;42(6):670–676. doi:10.1515/cclm.2004.114
104. Ghazal K, Brabant S, Prie D, Piketty ML. Hormone Immunoassay Interference: a 2021 Update. Ann Lab Med. 2022;42(1):3–23. doi:10.3343/alm.2022.42.1.3
105. Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem. 1995;41(2):246–255. doi:10.1093/clinchem/41.2.246
106. Marcovina SM, Albers JJ, Scanu AM, et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a). Clin Chem. 2000;46(12):1956–1967. doi:10.1093/clinchem/46.12.1956
107. Heydari M, Rezayi M, Ruscica M, Jpamialahamdi T, Johnston TP, Sahebkar A. The ins and outs of lipoprotein(a) assay methods. Arch Med Sci Atheroscler Dis. 2023;8(1):e128–e139. doi:10.5114/amsad/176653
108. Kronenberg F. Lipoprotein(a) measurement issues: are we making a mountain out of a molehill? Atherosclerosis. 2022;349:123–135. doi:10.1016/j.atherosclerosis.2022.04.008
109. Marcovina SM, Navabi N, Allen S, Gonen A, Witztum JL, Tsimikas S. Development and validation of an isoform-independent monoclonal antibody-based ELISA for measurement of lipoprotein(a). J Lipid Res. 2022;63(8):100239. doi:10.1016/j.jlr.2022.100239
110. Bustos P, Muñoz M, Ulloa N, Godoy P, Calvo C. An ELISA procedure for human Lp(a) quantitation using monoclonal antibodies. Hybrid Hybridomics. 2002;21(3):211–216. doi:10.1089/153685902760173944
111. Wang J, Zhang C, Gong J, et al. Development of new enzyme-linked immunosorbent assay for oxidized lipoprotein(a) by using purified human oxidized lipoprotein(a) autoantibodies as capture antibody. Clin Chim Acta. 2007;385(1–2):73–78. doi:10.1016/j.cca.2007.06.023
112. Dikaios I, Althaus H, Angles-Cano E, et al. Commutability assessment of candidate reference materials for Lipoprotein(a) by comparison of a MS-based candidate reference measurement procedure with immunoassays. Clin Chem. 2023;69(3):262–272. doi:10.1093/clinchem/hvac203
113. Scharnagl H, Stojakovic T, Dieplinger B, et al. Comparison of lipoprotein (a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis. 2019;289:206–213. doi:10.1016/j.atherosclerosis.2019.08.015
114. Gaubatz JW, Hoogeveen RC, Hoffman AS, et al. Isolation, quantitation, and characterization of a stable complex formed by Lp[a] binding to triglyceride-rich lipoproteins. J Lipid Res. 2001;42(12):2058–2068.
115. Chapman MJ, Ginsberg HN, Amarenco P, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32(11):1345–1361. doi:10.1093/eurheartj/ehr112
116. Ramasamy I. Update on the laboratory investigation of dyslipidemias. Clin Chim Acta. 2018;479:103–125. doi:10.1016/j.cca.2018.01.015
117. Langsted A, Freiberg JJ, Tybjaerg-Hansen A, Schnohr P, Jensen GB, Nordestgaard BG. Nonfasting cholesterol and triglycerides and association with risk of myocardial infarction and total mortality: the Copenhagen City Heart Study with 31 years of follow-up. J Intern Med. 2011;270(1):65–75. doi:10.1111/j.1365-2796.2010.02333.x
118. Ruhaak LR, Romijn F, Begcevic Brkovic I, et al. Development of an LC-MRM-MS-based candidate reference measurement procedure for standardization of serum apolipoprotein (a) tests. Clin Chem. 2023;69(3):251–261. doi:10.1093/clinchem/hvac204
119. Villanueva J, Carrascal M, Abian J. Isotope dilution mass spectrometry for absolute quantification in proteomics: concepts and strategies. J Proteomics. 2014;96:184–199. doi:10.1016/j.jprot.2013.11.004
120. Lassman ME, McLaughlin TM, Zhou H, et al. Simultaneous quantitation and size characterization of apolipoprotein(a) by ultra-performance liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2014;28(10):1101–1106. doi:10.1002/rcm.6883
121. Armbruster D, Miller RR. The Joint Committee for Traceability in Laboratory Medicine (JCTLM): a global approach to promote the standardisation of clinical laboratory test results. Clin Biochem Rev. 2007;28(3):105–113.
122. Diederiks NM, Ruhaak LR, Romijn F, Pieterse MM, Smit NPM, Cobbaert CM. An LC-MS-based designated comparison method with similar performance to the Lp(a) reference measurement procedure to guide molar Lp(a) standardization. Clin Proteomics. 2024;21(1):5. doi:10.1186/s12014-023-09446-5
123. Gries A, Nimpf J, Nimpf M, Wurm H, Kostner GM. Free and Apo B-associated Lpa-specific protein in human serum. Clin Chim Acta. 1987;164(1):93–100. doi:10.1016/0009-8981(87)90110-0
124. Herrmann W, Quast S, Ellgass A, et al. An increased serum level of free Apo(a) in renal patients is more striking than that of Lp(a) and is influenced by homocysteine. Nephron. 2000;85(1):41–49. doi:10.1159/000045628
125. Mooser V, Marcovina SM, Wang J, Hobbs HH. High plasma levels of apo(a) fragments in Caucasians and African-Americans with end-stage renal disease: implications for plasma Lp(a) assay. Clin Genet. 1997;52(5):387–392. doi:10.1111/j.1399-0004.1997.tb04358.x
126. Trenkwalder E, Gruber A, König P, Dieplinger H, Kronenberg F. Increased plasma concentrations of LDL-unbound apo(a) in patients with end-stage renal disease. Kidney Int. 1997;52(6):1685–1692. doi:10.1038/ki.1997.503
127. Shoji T, Kimoto E, Yamada A, et al. Elevated plasma free apo(a) levels in hemodialysis patients. Nephron. 2000;86(3):389–390. doi:10.1159/000045816
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.