Back to Journals » International Journal of Nanomedicine » Volume 13
Lipopolysaccharide-induced inflammation in monocytes/macrophages is blocked by liposomal delivery of Gi-protein inhibitor
Authors Tucureanu MM, Rebleanu D, Constantinescu CA, Deleanu M, Voicu G, Butoi E, Calin M ![]() , Manduteanu I
, Manduteanu I
Received 6 September 2017
Accepted for publication 26 October 2017
Published 20 December 2017 Volume 2018:13 Pages 63—76
DOI https://doi.org/10.2147/IJN.S150918
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Monica Madalina Tucureanu,1,* Daniela Rebleanu,1,* Cristina Ana Constantinescu,1,2 Mariana Deleanu,3,4 Geanina Voicu,1 Elena Butoi,1 Manuela Calin,1 Ileana Manduteanu1
1Department of Biopathology and Therapy of Inflammation, Nicolae Simionescu Institute of Cellular Biology and Pathology, Bucharest, Romania; 2Faculty of Veterinary Medicine, University of Agronomic Sciences and Veterinary Medicine, Bucharest, Romania; 3Department of Lipidomics, Nicolae Simionescu Institute of Cellular Biology and Pathology, Bucharest, Romania; 4Faculty of Biotechnologies, University of Agronomic Sciences and Veterinary Medicine, Bucharest, Romania
*These authors contributed equally to this work
Background: Lipopolysaccharide (LPS) is widely recognized as a potent activator of monocytes/macrophages, and its effects include an altered production of key mediators, such as inflammatory cytokines and chemokines. The involvement of Gi protein in mediating LPS effects has been demonstrated in murine macrophages and various cell types of human origin.
Purpose: The aim of the present work was to evaluate the potential of a Gi-protein inhibitor encapsulated in liposomes in reducing the inflammatory effects induced by LPS in monocytes/macrophages.
Materials and methods: Guanosine 5´-O-(2-thiodiphosphate) (GOT), a guanosine diphosphate analog that completely inhibits G-protein activation by guanosine triphosphate and its analogs, was encapsulated into liposomes and tested for anti-inflammatory effects in LPS-activated THP1 monocytes or THP1-derived macrophages. The viability of monocytes/macrophages after incubation with different concentrations of free GOT or liposome-encapsulated GOT was assessed by MTT assay. MAPK activation and production of IL1β, TNFα, IL6, and MCP1 were assessed in LPS-activated monocytes/macrophages in the presence or absence of free or encapsulated GOT. In addition, the effect of free or liposome-encapsulated GOT on LPS-stimulated monocyte adhesion to activated endothelium and on monocyte chemotaxis was evaluated.
Results: We report here that GOT-loaded liposomes inhibited activation of MAPK and blocked the production of the cytokines IL1β, TNFα, IL6, and MCP1 induced by LPS in monocytes and macrophages. Moreover, GOT encapsulated in liposomes reduced monocyte adhesion and chemotaxis. All demonstrated events were in contrast with free GOT, which showed reduced or no effect on monocyte/macrophage activation with LPS.
Conclusion: This study demonstrates the potential of liposomal GOT in blocking LPS proinflammatory effects in monocytes/macrophages.
Keywords: guanosine 5´-O-(2-thiodiphosphate) (GOT), MAPK activation, cytokine, monocyte adhesion, chemotaxis
Introduction
Monocytes and macrophages play critical roles in inflammatory and immune responses; therefore, the modulation of their functions is a useful strategy for anti-inflammatory therapies, including nanotherapies.1 Lipopolysaccharide (LPS) is the major component of Gram-negative bacteria cell walls and can cause an acute inflammatory response by triggering the release of a vast number of inflammatory cytokines in various cell types.2 LPS is widely recognized as a potent activator of monocytes/macrophages.3 Bacterial LPS has been conventionally used to study inflammation, due to the abundance of inflammatory effects that it generates through TLR4 signaling. Cellular activation triggered by LPS requires extracellular proteins, including LBP and CD14, which play a role in transferring LPS to a signaling complex composed of MD2 and MyD88.4 CD14 is a glycosylphosphatidyliositol-anchored plasma-membrane protein that associates physically with Gi/Go heteromeric G proteins.5 The involvement of Gi in mediating LPS effects was first demonstrated in murine macrophages, and since then it has been validated in various cell types, including cells of human origin.6
Extensive studies have shown that in monocytes/macrophages, LPS induces the production of cytokines, such as TNFα, IL1β, IL6, IL8, IL10, IL12, IL15, and TGFβ, with TNFα and IL6 being induced through activation of Gi.5,7,8 Moreover, the expression of MCP1, a potent chemotactic factor belonging to the CC-chemokine family, has been reported to be upregulated by various stimuli, including LPS, in different cell types and p38 MAPK is considered a major regulator in the production of MCP1.9–11
Considerable data support the idea that stimulation of monocytes/macrophages with LPS leads to activation of MAPK.12–14 The family of MAPKs includes ERK, JNK, and p38 MAPK. MAPKs phosphorylate different intracellular proteins and transcription factors that subsequently regulate gene expression. ERK proteins are activated by endogenous or exogenous mitogens, cytokines, and growth factors, and regulate cell proliferation, differentiation, and survival.15 JNK and p38 MAPK are predominantly activated by inflammatory cytokines (TNFα and IL1β) and bacterial LPS.16 Moreover, it has been shown that in monocytes/macrophages, LPS induces p38 MAPK activation through activation of Gi.5 Studies utilizing pertussis toxin, which specifically inhibits receptor–Gi coupling by catalyzing the adenosine diphosphate ribosylation of Gαi proteins, have demonstrated inhibition of LPS-induced transcription of TNFα and IL1 mRNA and inhibition of p38, JNK, and ERK1/2 phosphorylation.17
Considering all the evidence that Gi proteins are important regulators for the inflammatory effects of LPS in monocytes/macrophages, cells with leading roles in cardiovascular disease initiation and progression, and that a potent Gi inhibitor, guanosine 5′-O-(2-thiodiphosphate) (GOT), reduced inflammation in vascular cells, we evaluated in this study the anti-inflammatory effects of free GOT and GOT encapsulated in liposomes (Lipo/GOT) on LPS-activated human monocytes and macrophages.18
We report here that Lipo/GOT blocked the production of cytokines and chemokines, such as IL1β, TNFα, IL6, and MCP1, and also inhibited activation of MAPK induced by LPS in monocytes and macrophages. Moreover, Lipo/GOT reduced monocyte adhesion and chemotaxis. All demonstrated events were in contrast with free GOT, which showed reduced or no effect on monocyte/macrophage activation with LPS. This study demonstrates the potential of Lipo/GOT in blocking LPS proinflammatory effects in monocytes/macrophages.
Materials and methods
Materials
Enzyme-linked immunosorbent assay (ELISA) kits for IL1β, TNFα, IL6, and MCP1, and polyclonal rabbit antihuman/mouse/rat phospho-p38 (T180/Y182) antibody, polyclonal rabbit antihuman p38 antibody, polyclonal rabbit antihuman phospho-JNK (T183/Y185) antibody, monoclonal mouse antihuman JNK panspecific antibody, polyclonal rabbit antihuman phospho-ERK1/ERK2 antibody, and monoclonal mouse human ERK1/ERK2 antibody were from R&D Systems (Minneapolis, MN, USA). The transwell inserts were from Corning (Corning, NY, USA).
Egg phosphatidylcholine (EPC), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-(amino[polyethylene glycol]-2,000) (DSPE-PEG) came from Avanti Polar Lipids (Alabaster, AL, USA). Amicon centrifugal filter columns with a cutoff of 100 kDa were from EMD Millipore (Billerica, MA, USA). Deionized (18.2 MQ/cm) water was generated in-house using a Milli-Q System from Millipore. GOT, LPS from Escherichia coli serotype O111:B4, MTT, and all other reagents were from Sigma-Aldrich (St Louis, MO, USA).
Cells
THP1 human monocytic cells (purchased from ECACC) derived from an acute monocytic leukemia patient were cultivated at 37°C in RPMI medium containing 10% heat-inactivated FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μg/mL neomycin in a humidified incubator with 5% CO2. THP1 cells were differentiated into macrophages with 100 nM phorbol 12-myristate 13-acetate (PMA) for 72 hours prior to other treatments. For adhesion assays, the human endothelial cell (HEC) line EA.hy926 (purchased from ATCC) was used. Cells were grown in DMEM with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μg/mL neomycin in a humidified incubator with 5% CO2 at 37°C. All cell lines used in this study were tested regularly for Mycoplasma contamination.
Preparation and characterization of GOT-encapsulated liposomes
Liposomes were prepared from a mixture of EPC, cholesterol and DSPE-PEG in chloroform, combined at a molar ratio of 69:30:1 mol%, and dried in a rotary evaporator. Multilamellar vesicles were formed by hydrating the lipidic film with PBS (Lipo) or with a solution containing the Gi inhibitor GOT (Lipo/GOT) at an inhibitor:lipid ratio of 1:5 to reach a final lipid concentration of 10 mM. Unilamellar Lipo were formed by successive extrusion of the multilamellar vesicles through polycarbonate membranes with pores of 200 nm and 100 nm using a miniextruder (Avanti Polar Lipids). To separate the Lipo/GOT from free GOT, centrifugation with the Amicon filtration units with 100 kDa cutoff was used.
Lipo size was determined by photon-correlation spectroscopy with a submicron-particle analyzer (Nicomp 380; Particle Sizing Systems, Port Richey, FL, USA) employing multimodal distribution. The Lipo suspension was diluted into 0.22 μm-filtrated PBS so as to reach a count rate of 250–350 kHz. Instrument parameters were set as follows: automatic choice of channel width, vesicle mode, number weighting, and automatic change from Gaussian distribution mode to multimodal mode if χ2>3.
The ζ-potential of Lipo was determined by electrophoretic light scattering, which determines electrophoretic movement of charged particles under an applied electric field, using a Zetasizer Nano (Malvern Instruments, Malvern, UK). The measurements were done at 25°C in DMEM, each result being the average of measurements. The amount of GOT encapsulated into Lipo was determined by high-performance liquid chromatography employing an Agilent 1290 Infinity with a Zorbax SB C18 column (2.1×100 mm, 1.8 μm). The mobile phase consisted of 0.05 mM ammonium acetate at pH 5.9. The flow rate was 0.2 mL/min, and GOT was detected at 254 nm.
Experimental design
THP1 monocytes or THP1-derived macrophages, differentiated as already described, were preincubated for 2 hours in culture medium containing 100 μM GOT, either free or encapsulated into Lipo. Then, the monocytes and macrophages were activated with 100 ng/mL LPS from E. coli for an additional 24 or 48 hours. Free GOT and Lipo in the presence or absence of LPS were used as controls. GOT concentration was chosen based on our previous studies that showed a blockade of monocyte transmigration induced by the cytokine resistin through inhibition of inflammatory molecule secretion (particularly the chemokines fractalkine and MCP1) when endothelial or smooth-muscle cells were treated with 100 μM GOT inhibitor.18
Cytotoxicity evaluation
The viability of monocytes/macrophages after incubation with free GOT, Lipo/GOT, or blank Lipo was assessed by MTT assay. This technique measures intracellular purple formazan produced in metabolically active cells after reduction of tetrazolium salt under the action of dehydrogenase enzymes. THP1 monocytes and THP1-derived macrophages, seeded in 96-well plates at a density of 104 cells/well, were exposed for 24 and 48 hours to culture medium containing different concentrations of free GOT, Lipo/GOT, or blank Lipo. A 0.5% Triton X-100 solution was used as positive control. At the end of incubation, the culture medium was replaced with MTT solution (0.5 mg/mL in culture medium without phenol red) for 3 hours at 37°C and 5% CO2. Then, the MTT solution was removed and the formed formazan crystals were solubilized for 4 hours at 37°C using a lysis buffer (0.1 N HCl in isopropanol). Optical absorbance was measured at 570 nm with the reference wavelength at 720 nm on a microplate reader (Tecan Genios). Results are expressed as percentages relative to results obtained with control cells (cells exposed to culture medium).
MAPK-phosphorylation analysis
To assess the effect of GOT on the expression of MAP kinases activated by LPS, THP1 monocytes were centrifuged and THP1 differentiated to macrophages were washed in ice-cold PBS. Cells were solubilized in Laemmli lysis buffer (60 mM tris-HCl pH 6.8, 2% sodium dodecyl sulfate, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue) and heated to 100°C for 10 minutes. Total protein (50 μg) was subjected to 10% polyacrylamide-gel electrophoresis and transferred using a Trans Blot semidry system to nitrocellulose membranes. The blots were blocked in tris-buffered saline containing 1% BSA and subsequently incubated overnight with antihuman antibodies for phospho-p38, p38, phospho-JNK, JNK, phospho-ERK, and ERK. After being washed, the membranes were incubated for 1 hour with the secondary antibodies, washed, and incubated with enhanced-chemiluminescence reagents and analyzed with an ImageQuant LAS 4000 (GE Healthcare, Little Chalfont, UK). Optical density was measured with ImageJ software. Quantification of phosphorylated MAPK proteins – pp38, pJNK, and pERK – was determined by normalization to total, unphosphorylated protein: p38, JNK, and ERK, respectively.
Inflammatory cytokine immunoassay
The cytokines IL1β, TNFα, and IL6 and the chemokine MCP1 released by monocytes and macrophages under the conditions already mentioned were measured using DuoSet ELISA kits (R&D Systems) according to the manufacturer’s protocol. Unstimulated cells were used as controls. Briefly, after LPS activation, the conditioned media was centrifuged and kept at −20°C until analysis. One day before performing ELISA assay, the microplate was coated with a capture antibody against the molecules. Standards, controls, and samples were incubated for 2 hours, and the detection antibody for an additional 2 hours. Streptavidin–HRP was added to the microplate and H2O2-tetramethylbenzidine used as a substrate for HRP. The reaction was stopped using 2 N H2SO4 and optical density determined using a microplate reader. The concentration of proteins released in the conditioned media was calculated using the standard curve.
Monocyte-adhesion assay
THP1 monocytes were treated for 2 hours with free GOT or Lipo/GOT, prior to their activation with 100 ng/mL LPS for 24 hours. After activation, monocytes were labeled with 10 μmol/L of the fluorescent dye 2′7′-bis(2-carboxyethyl)-5(6)-carboxyfluorescein acetoxymethyl ester at 37°C for 1 hour in RPMI 1640 culture medium and subsequently washed by centrifugation. Confluent HECs were stimulated with LPS for 24 hours and then incubated with labeled monocytes (106 cells/mL) at 37°C for 1 hour. Nonadherent cells were removed by washing with warm culture medium and fixed with 4% paraformaldehyde. Fluorescent monocytes adhered to HECs were counted in three random fields under a fluorescence microscopy (Olympus IX81) using 10× magnification.
Monocyte chemotaxis
After exposure of THP1 monocytes to different conditions – pretreatment with free GOT or Lipo/GOT and activation with LPS – cells were added to the upper compartment of a transwell system (8 μm pore size) that contained in the lower compartment 100 ng/mL resistin, a cytokine with chemoattractive effects.18 The whole system was incubated for 18 hours at 37°C, then migrated monocytes present in the lower compartment were counted in three random fields at 10× magnification using a phase-contrast inverted microscopy (Olympus IX81).
Statistical analysis
Data are presented as mean ± SEM compared to controls. Statistical significance was determined by Student’s t-test (two tailed) or analysis of variance using GraphPad Prism 7 software. Values of P<0.01 or P<0.05 were considered significant.
Results
Characterization of Lipo loaded with Gi inhibitors
PEGylated Lipo consisting of a mixture of EPC, cholesterol and a phospholipid derivative of PEG (DSPE-PEG) and encapsulating GOT were prepared. The PEGylated phospholipid was inserted into Lipo bilayers with the purpose of avoiding Lipo destabilization, and provided stability in a serum-containing medium. The average hydrodynamic diameter and ζ-potential of plain Lipo or Lipo/GOT are shown in Table 1. No difference can be seen between Lipo and Lipo/GOT in regard to average hydrodynamic diameter, which was around 150 nm with unimodal size distribution (as shown by the polydispersity index, which was always smaller than 0.2). The ζ-potential of Lipo was negative (~−15 mV) and GOT encapsulation did not change the surface charge of Lipo significantly. The amount of GOT entrapped in Lipo was determined by high-performance liquid chromatography after separation of Lipo from GOT and was about 150 μg/μmol lipids.
 | Table 1 Size and ζ-potential of Lipo and Lipo/GOT |
Lipo loaded with Gi inhibitors did not affect monocyte/macrophage viability
Evaluation of Lipo/GOT impact on monocyte/macrophage viability is an important issue when anti-inflammatory action of a liposomal GOT formulation is envisaged. The MTT assay was employed to find out the effect of different concentrations of Lipo/GOT on monocyte/macrophage viability. Free GOT or blank Lipo were used as controls. The data showed that the viability of THP1 monocytes and THP1-derived macrophages was not significantly affected by concentrations of Lipo/GOT up to 100 μM (Figure 1). A solution of 0.5% Triton X-100 was used as a positive control in these experiments. A significant decrease in macrophage viability was seen after 48 hours of treatment with either free GOT or Lipo/GOT at a concentration of 200 μM, but not after incubation with blank Lipo, showing that plain Lipo did not affect cellular balance.
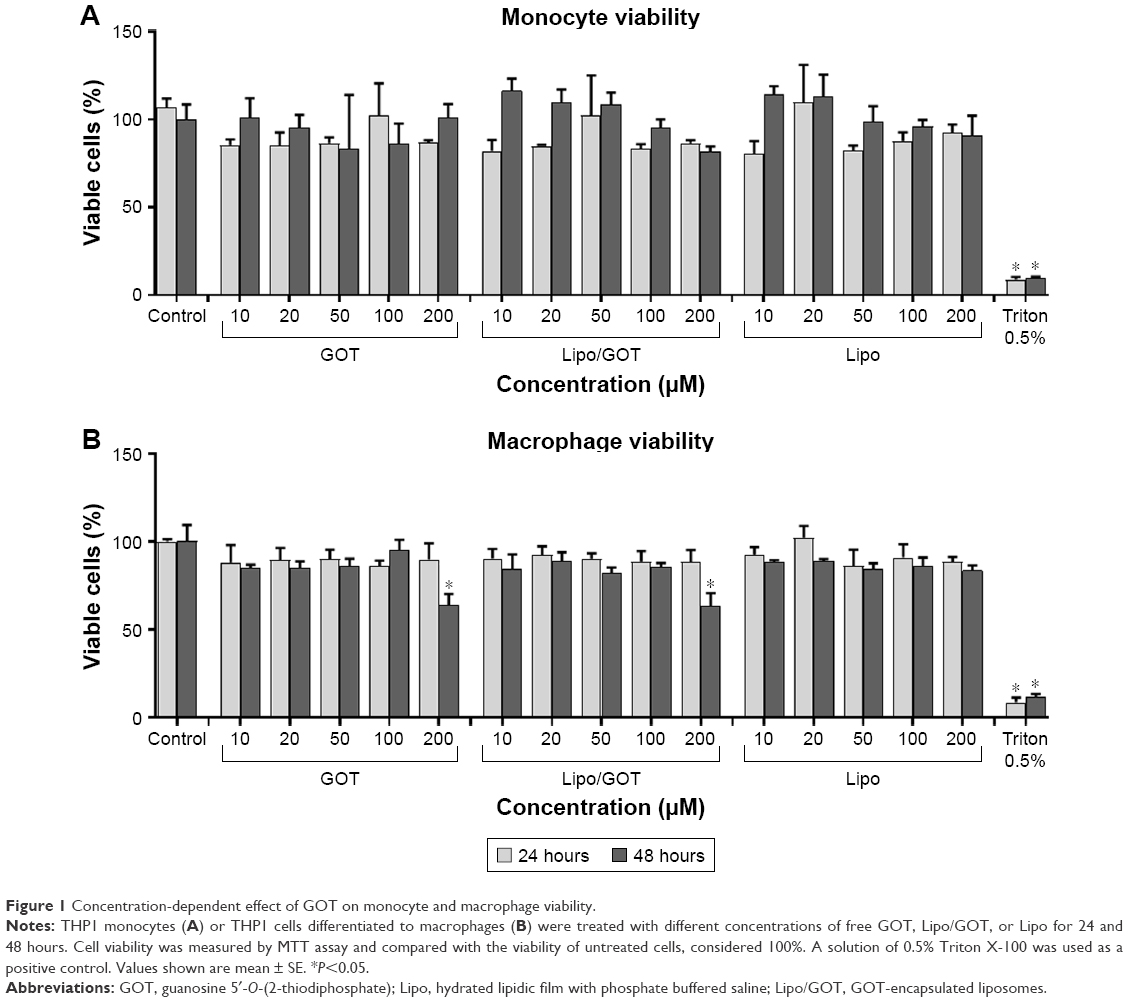 | Figure 1 Concentration-dependent effect of GOT on monocyte and macrophage viability. |
Treatment with Lipo/GOT reduced the activation of MAPK-signaling pathways induced by LPS in monocytes
To investigate whether free GOT or Lipo/GOT had a role in regulating the molecular pathways induced by LPS in monocytes, we analyzed the activation of p38, JNK, and ERK MAPK by Western blot. Our data showed that LPS significantly increased the expression of phospho-p38 and phospho-JNK after 24 or 48 hours’ activation in monocytes, showing ~1.5-fold increase for both molecules compared with control cells, while phospho-ERK was not identified. Furthermore, free GOT significantly decreased pp38 expression in monocytes only after 48 hours, while Lipo/GOT completely blocked p38 phosphorylation after 24 and 48 hours’ activation (Figure 2A).
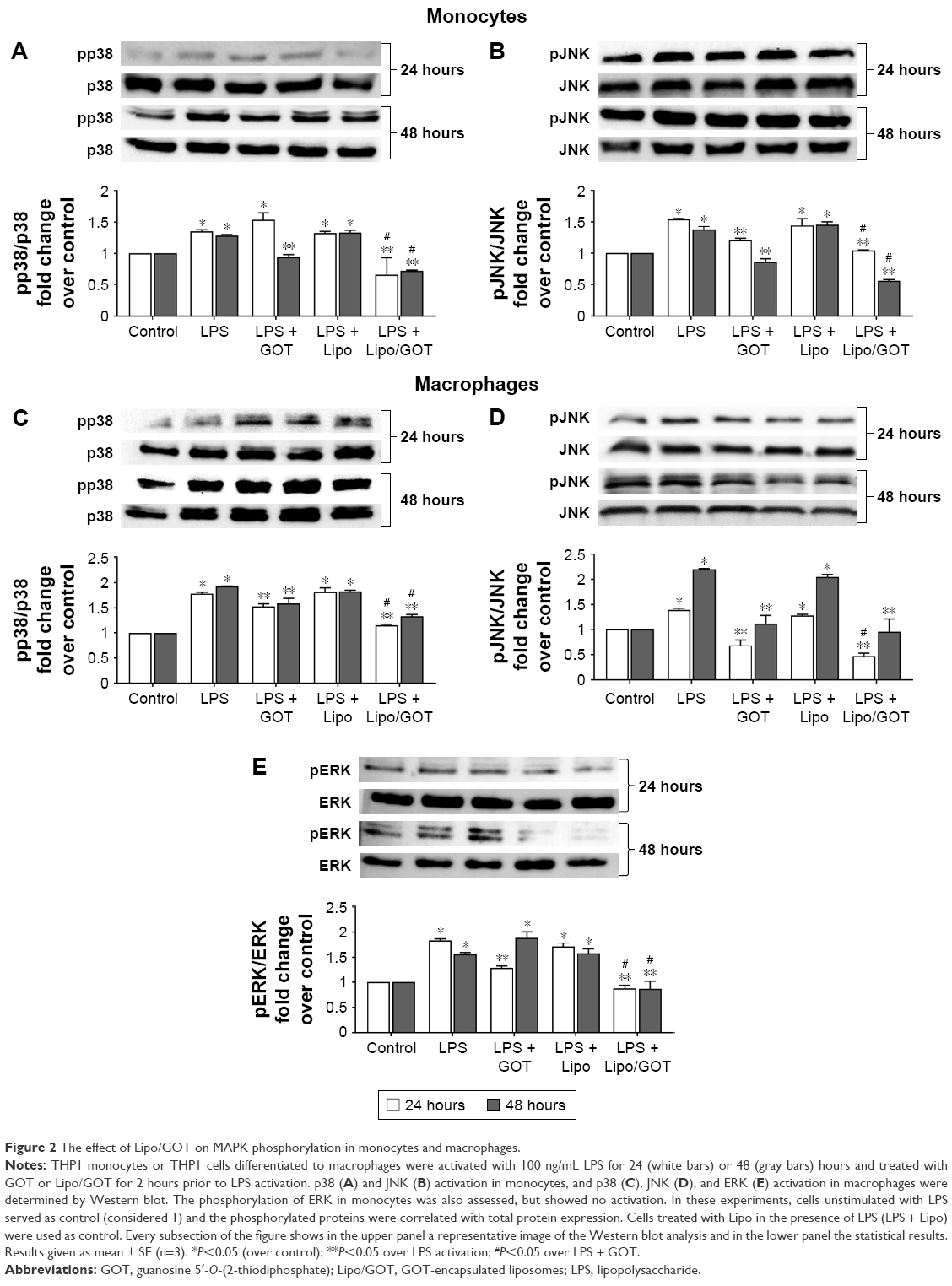 | Figure 2 The effect of Lipo/GOT on MAPK phosphorylation in monocytes and macrophages. |
pJNK expression was significantly decreased after treatment of monocytes with free GOT only after 48 hours’ activation with LPS and not after 24 hours’ stimulation, whereas Lipo/GOT significantly inhibited JNK phosphorylation, the expression of pJNK decreasing to control levels after 24 and 48 hours of LPS activation (Figure 2B). To confirm that the observed effect was due to Lipo/GOT, blank Lipo was used as control, and the experiments showed that Lipo without encapsulated GOT did not modify the activation of p38 or JNK induced by LPS, as shown in Figure 2A and B. Moreover, blank Lipo and GOT alone did not influence the expression of pp38 or pJNK by monocytes in the absence of LPS (data not shown).
Activation of p38, JNK, and ERK MAPK was reduced after treatment of LPS-activated macrophages with GOT-loaded Lipo
The effect of free GOT and Lipo/GOT on MAPK activation was also assessed in macrophages. To this end, THP1 cells differentiated with PMA for 3 days were treated with GOT or Lipo/GOT for 2 hours prior to LPS activation for 24 or 48 hours. After stimulation, the expression of pp38, pJNK, and pERK was analyzed by Western blot. The protein expression of total p38, JNK, and ERK was used to quantify the expression of phosphorylated proteins. Our experiments showed that phospho-p38 expression was increased 1.8-fold after 24 hours’ and 1.9-fold after 48 hours’ activation with LPS. Free GOT significantly decreased the expression of pp38 induced by LPS after 24 and 48 hours, while Lipo/GOT totally inhibited p38 phosphorylation (Figure 2C).
The expression of pJNK was significantly increased compared with control cells by 1.4-fold after 24 hours’ and 2.2-fold after 48 hours’ activation with LPS. Our results showed that free GOT inhibited the activation of JNK induced by LPS after 24 and 48 hours, the level of phospho-JNK being compared with control cells. Lipo/GOT additionally decreased the expression of pJNK induced by LPS after 24 hours compared with free GOT. Likewise, the expression of pJNK was inhibited at the control level after 48 hours’ activation with LPS and treatment with Lipo/GOT (Figure 2D).
In contrary to monocytes, LPS activated ERK phosphorylation in macrophages. Activation of differentiated THP1 cells with LPS increased the expression of pERK by 1.8-fold after 24 hours’ and 1.6-fold after 48 hours’ activation. Free GOT significantly decreased the expression of pERK induced by LPS only after 24 hours and not 48 hours. Furthermore, Lipo/GOT decreased ERK activation to control levels at both tested time points (Figure 2E). In addition, blank Lipo did not modify the activation of p38, JNK, or ERK induced by LPS, as shown in Figure 2C–E. Moreover, blank Lipo and GOT alone in the absence of LPS did not influence the phosphorylation of MAPK proteins in macrophages (data not shown).
Treatment of monocytes with GOT-loaded Lipo blocked the production of IL1β, TNFα, IL6, and MCP1 induced by LPS
To investigate the effect of Lipo/GOT in LPS-stimulated monocytes, THP1 cells were activated with LPS for 24 or 48 hours and treated with GOT or Lipo/GOT. Free GOT and blank Lipo in the presence or absence of LPS were used as controls. After stimulation, IL1β, IL6, TNFα, and MCP1 were quantified by ELISA in the conditioned media.
The stimulation of THP1 monocytes with LPS resulted in IL1β production of 14±1.5 pg/mL after 24 hours and 151±70 pg/mL after 48 hours, significantly more then controls (unstimulated cells), which showed no IL1β secretion. Treatment of monocytes with GOT did not influence LPS-induced IL1β secretion, but treatment with Lipo/GOT blocked the production of IL1β by monocytes after 24 hours’ or 48 hours’ activation with LPS (Figure 3A). THP1 cells secreted IL6 only after 48 hours’ stimulation with LPS (911±154 pg/mL). IL6 production was not influenced by treatment of monocytes with GOT, but in contrast was blocked by treatment of cells with Lipo/GOT (Figure 3B).
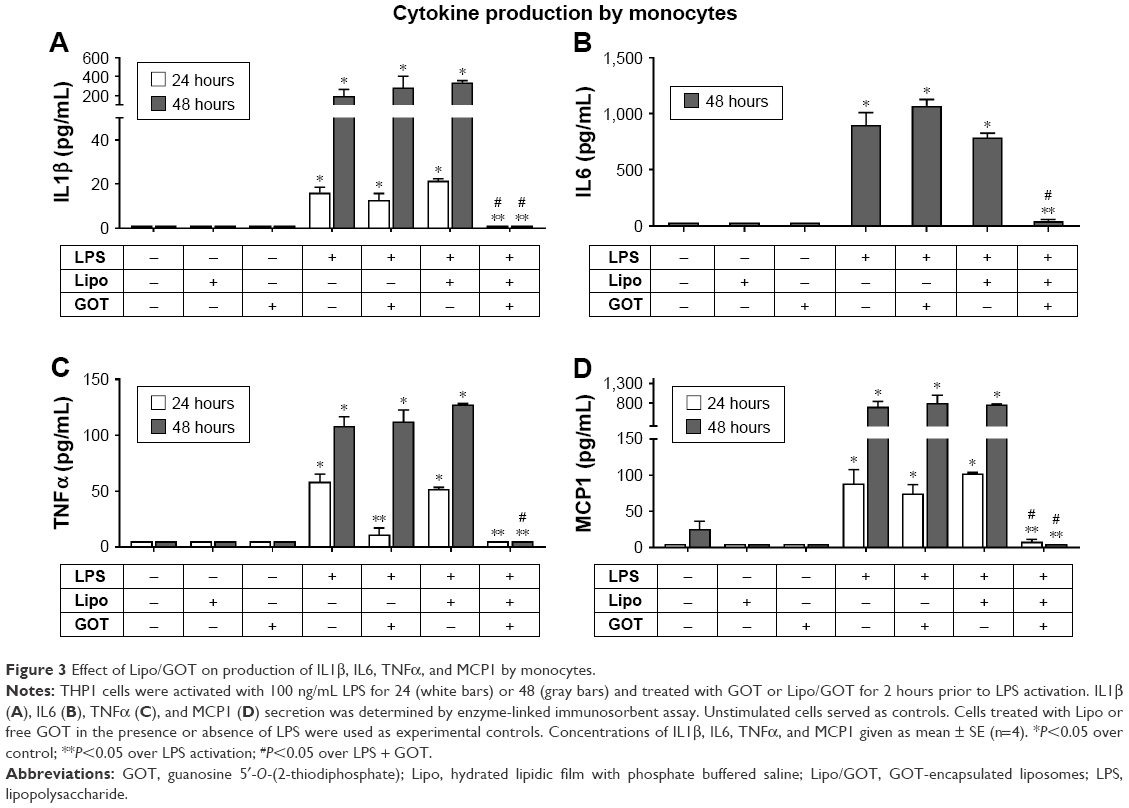 | Figure 3 Effect of Lipo/GOT on production of IL1β, IL6, TNFα, and MCP1 by monocytes. |
TNFα production was significantly increased over controls (which showed no TNFα secretion) after monocyte activation with LPS for 24 hours (63±6 pg/mL) and 48 hours (107±11 pg/mL). After 24 hours’ activation, GOT and Lipo/GOT completely inhibited the production of TNFα induced by LPS. In contrast, after activation of 48 hours with LPS, TNFα release was blocked only by Lipo/GOT and not GOT (Figure 3C).
With regard to MCP1 production, THP1 monocytes showed secretion of 81±25 pg/mL after 24 hours’ stimulation with LPS, whereas control cells did not secrete MCP1. Lipo/GOT completely blocked the release of MCP1, while GOT alone had no effect. After 48 hours in culture, THP1 monocytes activated with LPS produced increasing amounts of MCP1 (672±167 pg/mL) compared with control cells (61±23 pg/mL), the secretion of MCP1 being completely blocked by Lipo/GOT, but not by GOT alone (Figure 3D). In addition, blank Lipo did not reduce the secretion of cytokines induced by LPS. Moreover, blank Lipo and GOT alone in the absence of LPS did not stimulate the secretion of cytokines.
Production of IL1β, TNFα, IL6, and MCP1 significantly decreased in macrophages activated by LPS and treated with Lipo/GOT
To determine the effect of Lipo/GOT on the secretion of cytokines by macrophages, THP1 cells were differentiated with PMA for 3 days, activated with LPS for 24 or 48 hours, and treated with GOT or Lipo/GOT for 2 hours prior LPS activation. Free GOT and blank Lipo in the presence or absence of LPS were used as controls. Our data showed that secretion of IL1β by LPS-activated macrophages significantly increased after 24 hours (370±4 pg/mL in LPS-activated cells vs 18±2 pg/mL in control cells) and after 48 hours (920±2 pg/mL in LPS-activated cells vs 287±10 pg/mL in control cells). The treatment of macrophages with Lipo/GOT inhibited the secretion of IL1β the concentration being 215±23 pg/mL after 24 hours’ activation with LPS and 266±21 pg/mL after 48 hours’ activation. As in the case of monocytes, treatment of macrophages with GOT did not influence LPS-induced IL1β secretion (Figure 4A).
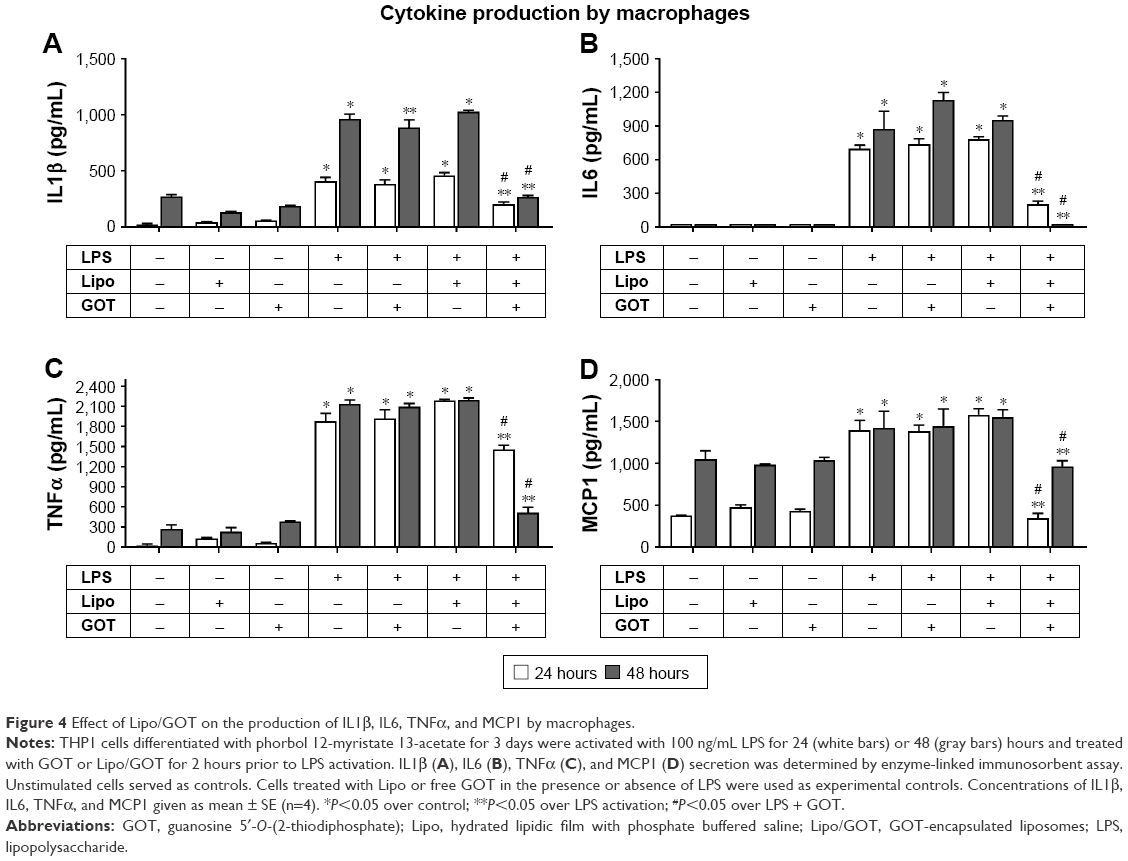 | Figure 4 Effect of Lipo/GOT on the production of IL1β, IL6, TNFα, and MCP1 by macrophages. |
The cytokine IL6 was not produced in control macrophages, but its release was significantly increased upon activation with LPS for 24 and 48 hours (668±10 pg/mL and 855±200 pg/mL, respectively). Macrophages treated with free GOT showed IL6 secretion that was comparable to LPS-activated macrophages, whereas cells treated with Lipo/GOT showed a significant reduction in IL6 after 24 hours’ (201±40 pg/mL) and a blockade of IL6 secretion after 48 hours’ activation (Figure 4B). Macrophages secreted TNFα after 24 hours’ activation with LPS at 1,764±12 pg/mL compared with 30±3 pg/mL in the conditioned media of control cells. Treatment of macrophages with free GOT did not have any effect on the secretion of TNFα induced by LPS, whereas Lipo/GOT significantly reduced the production of TNFα (1,460±67 pg/mL) compared with LPS-activated macrophages. After 48 hours’ activation, LPS stimulated the production of TNFα (2,084±12 pg/mL) compared with control cells (332±18 pg/mL). Also, Lipo/GOT significantly decreased TNFα secretion at a level comparable to control cells (443±47 pg/mL), whereas free GOT did not show any effect on TNFα production induced by LPS (Figure 4C).
MCP1 secretion in conditioned macrophage media was significantly increased upon activation with LPS for 24 hours (1,283±19 pg/mL) compared with control cells (372±8 pg/mL). Free GOT did not modify the concentration of secreted MCP1, but Lipo/GOT significantly decreased MCP1 production to control levels (373±75 pg/mL). After 48 hours’ activation with LPS, MCP1 production was significantly increased (1,253±135 pg/mL) compared with control cells (954±51 pg/mL), but was inhibited to a level comparable with control cells after treatment with Lipo/GOT (902±5 pg/mL), while free GOT did not modify MCP1 production induced by LPS (Figure 4D). To confirm that the observed effects were due to the liposomal delivery of GOT, blank Lipo was used as control, and experiments showed that Lipo without encapsulated GOT did not reduce the secretion of cytokines induced by LPS. Moreover, blank Lipo and GOT alone did not promote cytokine secretion in the absence of LPS.
Adhesion of monocytes activated with LPS blocked after treatment with GOT-loaded Lipo
Previous work has shown that adhesion of monocytes to the endothelium, the initial step in the development of atherosclerotic plaque, is increased by LPS stimulation of either human monocytes or HECs.19,20 In this study, we assessed the effect of Lipo/GOT on monocyte adhesion to HECs induced by LPS. To this end, HEC and THP1 monocytes were first activated separately for 24 hours with LPS before starting the adhesion assay. Moreover, THP1 cells were preincubated for 2 hours with free GOT or Lipo/GOT prior to LPS activation. Unstimulated HECs or monocytes were used as controls. After LPS stimulation, monocytes were marked with a fluorophore and incubated with confluent HECs for 1 hour. Then, unadhered monocytes were washed out and adhered monocytes counted under fluorescence microscopy.
Our experiments showed that adhesion of LPS-activated monocytes to control HECs was significantly increased (~3×) compared to unstimulated monocytes. Moreover, LPS-activated HECs enhanced the adhesion of activated monocytes compared to control HECs (~1.4×). GOT inhibitor decreased significantly (~2×) the number of adhered monocytes, but Lipo/GOT totally inhibited monocyte adhesion, the number of adhered monocytes activated with LPS being comparable with that of unstimulated monocytes (Figure 5). These data suggested that activation of monocytes was sufficient for enhancement of their adherence to endothelia, regardless of the state of endothelial activation. Furthermore, Lipo/GOT blocked the adhesion of monocytes, while free GOT only reduced the number of adhered monocytes to endothelia, suggesting that GOT was internalized more efficiently when incorporated into Lipo.
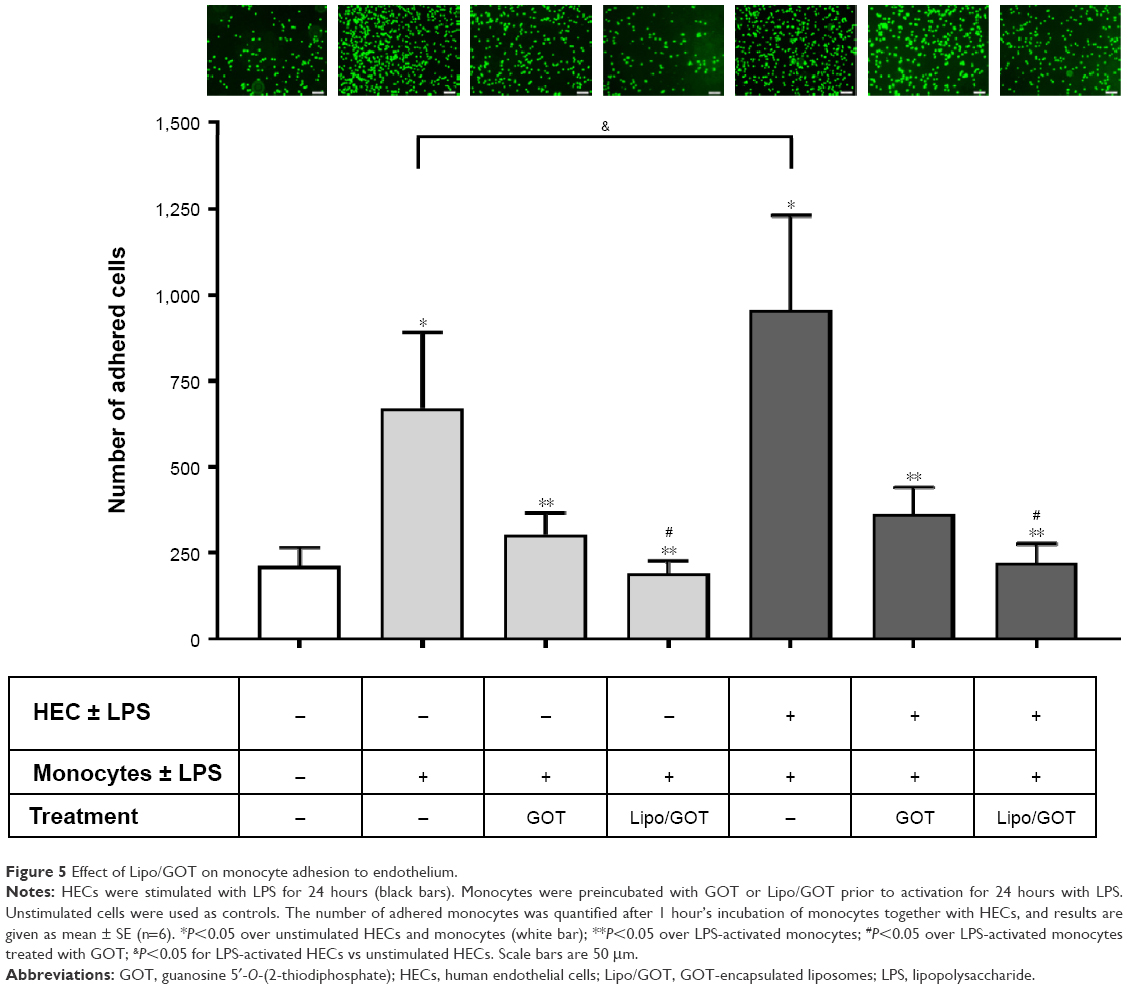 | Figure 5 Effect of Lipo/GOT on monocyte adhesion to endothelium. |
Treatment with GOT-loaded Lipo inhibited chemotaxis toward resistin of monocytes activated with LPS
To investigate the effect of Lipo/GOT on monocyte functionality further, we examined the chemotaxis of LPS-activated monocytes toward resistin, a cytokine that acts as a chemoattractant, as shown by our recent work.18 To this purpose, LPS-activated monocytes preincubated with free GOT or Lipo/GOT were added to a chemotaxis chamber. After 18 hours, the monocytes migrated toward resistin (added in the lower compartment of the transwell system) were counted under light microscopy. Our results showed that the number of LPS-activated monocytes migrating toward resistin increased around fourfold compared to the number of control cells. Moreover, treatment with free GOT decreased twice the number of migrated monocytes, while Lipo/GOT completely inhibited the migration of THP1 cells activated with LPS (Figure 6).
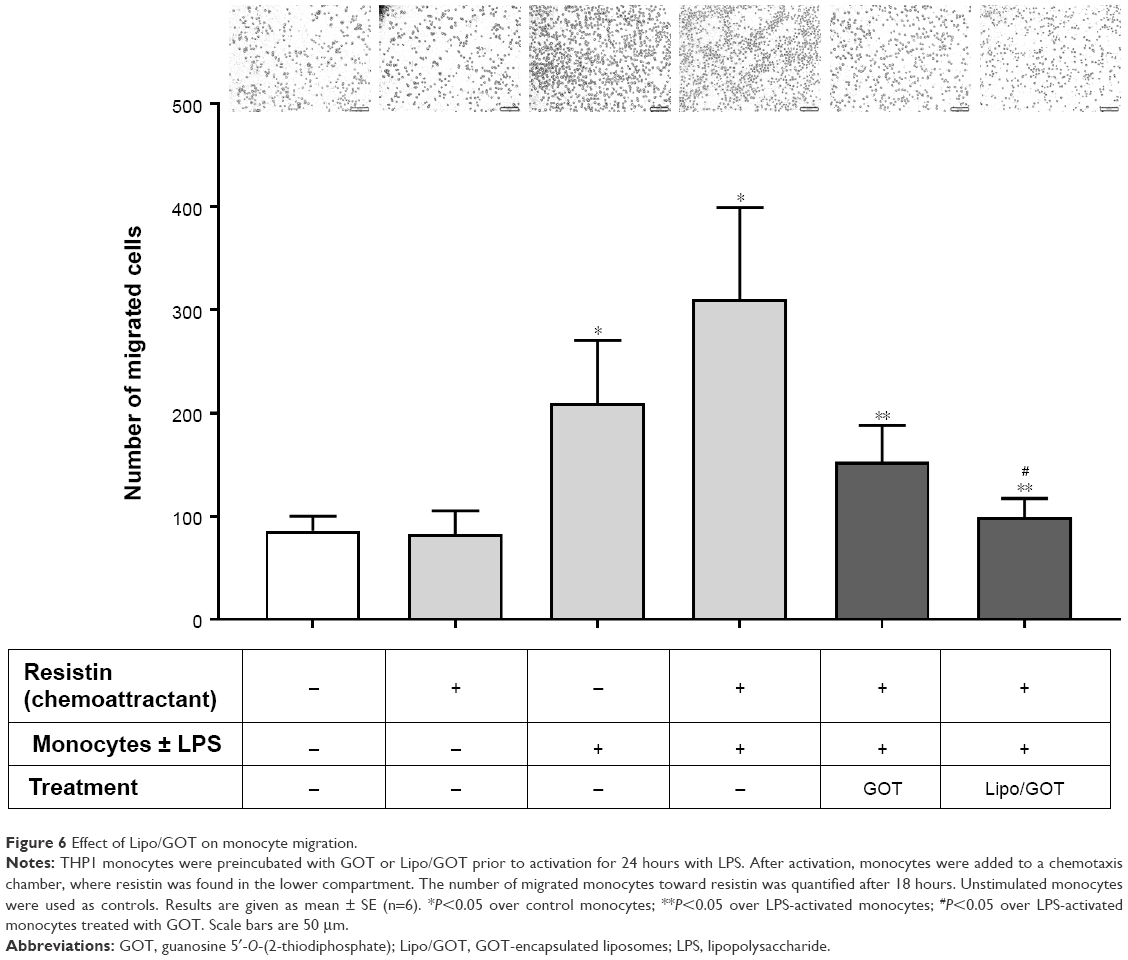 | Figure 6 Effect of Lipo/GOT on monocyte migration. |
Discussion
Monocytes and macrophages are important mediators for both innate and adaptive immunity, due to their crucial role in many inflammatory processes associated with infection, atherosclerosis, diabetes, and cancer.21 Based on the ability of monocytes and macrophages to assimilate foreign particles efficiently, the use of nanoparticles is a promising solution for the management of inflammatory diseases. Compared with free therapeutic agents, drug-loaded Lipo are the most frequently used nanocarrier for targeted drug delivery and provide several advantages, mainly because they improve the pharmacokinetics and bioavailability of these agents. Moreover, the use of PEG as a steric stabilizer of Lipo structure enhances their stability and circulation time in the bloodstream.22 Using a well-established cellular model of inflammation, the aim of the present work was to evaluate the potential of a Gi inhibitor encapsulated in Lipo in reducing the inflammatory effects induced by LPS in monocytes/macrophages, such as the activation of MAPK-signaling pathways, inflammatory cytokine secretion, monocyte adhesion, and chemotaxis.23
G proteins are heterotrimers comprising α-, β-, and γ-subunits that dissociate in response to proinflammatory stimuli after activation of G-protein-coupled receptors. The free Gα and Gβγ subunits can then activate different mediators that regulate various immune and inflammatory pathways, such as MAPK signaling.24 There are data that link Gα subunits to LPS-induced MAPK activation and cytokine production.5 This early study showed that CD14, an important molecule in LPS signaling, is physically associated with Gα subunits and that the proinflammatory effects of LPS are regulated by heterotrimeric G proteins. For this reason, targeting G proteins with pharmacological agents has since then been considered of great interest in regulating inflammation.
Previous studies have shown the use of G-protein inhibitors in reducing inflammation triggered by different mediators. Pertussis toxin (inhibitor of Gαi protein-receptor coupling) has been shown to block monocyte transmigration induced by the cytokine resistin by inhibiting fractalkine and MCP1 secretion.18,25 Moreover, gallein (inhibitor of Gβγ subunit-dependent signaling) suppressed macrophage chemotaxis and inhibited the activation of p38, JNK, and ERK MAPK induced by clusterin, a protein involved in inflammation and immunity.25,26
GOT, a guanosine diphosphate analog, was used in this study due to its capacity to completely inhibit G-protein activation by guanosine triphosphate and its analogs. The cytotoxic effects of different concentrations of GOT – free or encapsulated into Lipo – were determined using the MTT assay. High percentages of viable monocytes and macrophages were observed for all treatment groups (except for the macrophage treatment for 48 hours with 200 μM free or encapsulated GOT), indicating that Lipo/GOT had no cytotoxic effect on THP1 cells at the concentration used in the subsequent experiments.
LPS is considered an initiator of classical activation in monocytes and macrophages, its effects including an altered production of key mediators, such as inflammatory cytokines and chemokines.27 It is well known that LPS activation of monocytes and macrophages results in MAPK phosphorylation, but the majority of studies showed this effect after a few minutes or hours of LPS stimulation.28–30 In this study, we investigated the effect of Lipo/GOT on MAPK activation after long-term stimulation of THP1 monocytes and macrophages with LPS. Our results showed that LPS induced p38 and JNK phosphorylation in THP1 monocytes activated for 24 or 48 hours. Activation of ERK was not observed, probably because of the prolonged stimulation, ERK being phosphorylated after 30–60 minutes in THP1 monocytes.31 In accordance with other studies, we show here that Gis are involved in MAPK activation induced by LPS, demonstrated by the reduction in p38 and JNK phosphorylation after treatment of monocytes with GOT.5 Moreover, Lipo/GOT significantly reduced the phosphorylation of p38 and JNK compared with free GOT. In addition to monocytes, THP1-derived macrophages showed activation of p38, JNK, and ERK after 24 and 48 hours’ stimulation with LPS. Furthermore, free GOT and Lipo/GOT significantly decreased MAPK activation, encapsulated GOT showing a complete inhibition of MAPK phosphorylation induced by LPS.
Treatment for inflammatory diseases is based on blocking the action or secretion of critical mediators, and thus it is important to inhibit the cytokine secretion induced by LPS in monocytes and macrophages. Data obtained in the present study showed that bacterial LPS triggered significant production of IL1β, TNFα, and MCP1 by monocytes after 24 or 48 hours. IL6 secretion was induced only after 48 hours’ stimulation with LPS. These data are consistent with other research that showed nearly the same levels of IL1β and TNFα after 24 hours’ stimulation of THP1 cells with LPS and no IL6 production at 24 hours’ activation.32 There are few data on the effect of LPS on cytokine secretion after prolonged stimulation. Segura et al showed that the release of TNFα, IL1, IL6, and MCP1 was increased after 48 hours’ stimulation with 1 μg/mL LPS, the concentration for each mediator being higher than the level shown in the present study, but this difference could be explained by the difference in the concentration of LPS used for cell activation (1 μg/mL compared with 100 ng/mL in our experiments).33 Our study demonstrates that Lipo/GOT completely blocked the secretion of IL1β, IL6, TNFα, and MCP1 compared with free GOT, which did not decrease the secretion of these cytokines/chemokines induced by LPS. This effect of Lipo/GOT vs free GOT may be explained by increased cellular internalization of the compound when it is formulated into Lipo and gradual intracellular release, whereas the free compound acts very rapidly or is committed to intracellular degradation. The effect of free GOT in inhibiting cytokine secretion was observed only for TNFα after 24 hours’ stimulation (and not 48 hours’), probably due to an indirect mechanism that acts in the process of TNFα synthesis, this cytokine being processed as a membrane-bound precursor, unlike other cytokines that are released in association with exosomes in the extracellular space.34
Our data showed that LPS induced IL1β, IL6, TNFα, and MCP1 in THP1-derived macrophages after 24 and 48 hours’ stimulation, in concordance with other studies that showed a minor variation due to the activation duration and LPS concentration used.35–37 As in the case of monocytes, treatment of macrophages with Lipo/GOT significantly decreased the production of cytokines IL1β, IL6, TNFα, and MCP1 induced by LPS activation compared with free GOT, which showed no effect on the secretion of cytokines in the conditioned media of macrophages.
It is well known that adherence of monocytes to endothelia is essential for the localization of these cells to inflammation sites. Several reports have shown that adhesion and transmigration of LPS-stimulated monocytes are dependent on the monocyte-surface molecules CD11a/CD18 (LFA1) and CD11c/CD18 (CR4). Also, the importance of the adhesion molecule ICAM1, a high-affinity counterreceptor for LFA1, was demonstrated.38,39 Activation of HECs with LPS induces the expression of adhesion molecules, such as VCAM1, ICAM1, and E-selectin.40,41 Therefore, in the present study, we questioned the effect of free or Lipo/GOT on LPS-stimulated monocyte adhesion to activated endothelia. First, our findings suggest that the adhesion of activated monocytes to resting HECs is increased compared with control monocytes, but adhesion is intensified in case of LPS-activated HECs, suggesting the importance of the endothelium state in mediating monocyte adhesion. Treatment of monocytes with GOT prior to LPS stimulation resulted in a decrease of their adhesion to activated or nonactivated HECs, indicating the involvement of Gi in the LPS-induced production of adhesion molecules, but further experiments are needed to clarify this assumption. Moreover, Lipo/GOT completely blocked the adhesion of activated monocytes to endothelia, regardless of activation status, suggesting that Lipo/GOT is efficiently delivered and gradually released intracellularly.
For chemotaxis studies, we used resistin as a chemoattractant based on our previous data, where we demonstrated that resistin has similar chemoattractive effects on monocytes isolated from human blood as formylmethionylleucylphenylalanine a strong, well-known chemoattractant. Also, there is evidence that resistin has direct chemotactic effect on the THP1 cell line and that Gis are involved in resistin-induced lymphocyte migration.18,42,43 In the present study, an increased number of LPS-activated monocytes migrated toward resistin compared to unstimulated cells. The number of migrated cells was significantly decreased by the treatment of monocytes with free GOT or Lipo/GOT. Moreover, encapsulated GOT significantly decreased cell migration toward resistin to a level similar to control monocytes. These data suggest that Gis are involved in resistin chemotactic effects on LPS-activated monocytes. To the best of our knowledge, this is the first study to show the involvement of Gi in mediating monocyte adhesion and migration induced by LPS and the first to employ Lipo specifically to inhibit Gi in order to diminish or completely block the inflammatory effects of LPS.
There is evidence that there are differences between either cytokines or Toll-like receptor mRNA expression by LPS-stimulated monocytes and monocyte-derived macrophages and that monocytes and monocyte-derived macrophages respond differently to LPS, so they may have different functions in the innate immune system.44 Our data also showed that LPS activation of macrophages induced higher expression of IL1β, IL6, TNFα, and MCP1 than in LPS-activated monocytes. The data showed that macrophages constitutively secreted IL1β, TNFα, and MCP1 (with an increase in concentration after 48 hours in culture), unlike monocytes, which showed no secretion in control cells. Moreover, we found that LPS activated ERK phosphorylation only in macrophages, suggesting different pathways in LPS-induced ERK activation in macrophages versus monocytes.
Conclusion
The present findings reveal that Lipo/GOT had stronger anti-inflammatory effects than the free compound in reducing the proinflammatory effects of LPS in monocytes and macrophages. A reduction in MAPK activation and a significantly reduced production of major proinflammatory mediators, such as IL1β, IL6, TNFα, and MCP1, was observed in monocytes and macrophages treated with Lipo/GOT. Additionally, Lipo/GOT has a functional role in reducing the adhesion and chemotaxis of activated monocytes, major processes in the initiation and progression of inflammatory diseases. All these findings appoint Lipo loaded with a specific Gi inhibitor as an effective nanotherapy in preventing LPS inflammation.
Acknowledgments
The authors are indebted to Gabriela Mesca (technical assistance). This work was supported by UEFISCDI (Executive Agency for Higher Education Research Development and Innovation Funding; PN-II-ID-PCE-2011-3-0928 and PN-II-RU-TE-2014-4-0965 projects) and by the Romanian Academy.
Disclosure
The authors report no conflicts of interest in this work.
References
Calin M, Manduteanu I. Emerging nanocarriers-based approaches to diagnose and reduce vascular inflammation in atherosclerosis. Curr Med Chem. 2017;24:550–567. | ||
Ngkelo A, Meja K, Yeadon M, Adcock I, Kirkham PA. LPS induced inflammatory responses in human peripheral blood mononuclear cells is mediated through NOX4 and Giα dependent PI-3 kinase signalling. J Inflamm (Lond). 2012;9:1. | ||
Takashiba S, Van Dyke TE, Amar S, Murayama Y, Soskolne AW, Shapira L. Differentiation of monocytes to macrophages primes cells for lipopolysaccharide stimulation via accumulation of cytoplasmic nuclear factor kB. Infect Immun. 1999;67:5573–5578. | ||
Borzęcka K, Płóciennikowska A, Björkelund H, Sobota A, Kwiatkowska K. CD14 mediates binding of high doses of LPS but is dispensable for TNF-α production. Mediators Inflamm. 2013;2013:824919. | ||
Solomon KR, Kurt-Jones EA, Saladino RA, et al. Heterotrimeric G proteins physically associated with the lipopolysaccharide receptor CD14 modulate both in vivo and in vitro responses to lipopolysaccharide. J Clin Invest. 1998;102:2019–2027. | ||
Jakway JP, DeFranco AL. Pertussis toxin inhibition of B cell and macrophage responses to bacterial lipopolysaccharide. Science. 1986;234:743–746. | ||
Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60:8–26. | ||
Rossol M, Heine H, Meusch U, et al. LPS-induced cytokine production in human monocytes and macrophages. Crit Rev Immunol. 2011;31:379–446. | ||
Kunkel SL, Standiford T, Kasahara K, Strieter RM. Stimulus specific induction of monocyte chemotactic protein-1 (MCP-1) gene expression. Adv Exp Med Biol. 1991;305:65–71. | ||
Park JB, Wang TTY. Methyl (E)-(3-(3,4-dihydroxyphenyl)acryloyl)tryptophanate can suppress MCP-1 expression by inhibiting p38 MAP kinase and NF-kB in LPS-stimulated differentiated THP-1 cells. Eur J Pharmacol. 2017;810:149–155. | ||
Wong CK, Wang CB, Ip WK, Tian YP, Lam CW. Role of p38 MAPK and NF-kB for chemokine release in coculture of human eosinophils and bronchial epithelial cells. Clin Exp Immunol. 2005;139:90–100. | ||
van der Bruggen T, Nijenhuis S, van Raaij E, Verhoef J, van Asbeck BS. Lipopolysaccharide-induced tumor necrosis factor alpha production by human monocytes involves the Raf-1/MEK1-MEK2/ERK1-ERK2 pathway. Infect Immun. 1999;67:3824–3829. | ||
Junghae M, Raynes JG. Activation of p38 mitogen-activated protein kinase attenuates Leishmania donovani infection in macrophages. Infect Immun. 2002;70:5026–5035. | ||
Yang L, Guo H, Li Y, et al. Oleoylethanolamide exerts anti-inflammatory effects on LPS-induced THP-1 cells by enhancing PPARα signaling and inhibiting the NF-kB and ERK1/2/AP-1/STAT3 pathways. Sci Rep. 2016:6:34611. | ||
Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. 2009;8:1168–1175. | ||
Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2002;12:1–13. | ||
Wang J, Kester M, Dunn MJ. Involvement of a pertussis toxin-sensitive G-protein-coupled phospholipase A2 in lipopolysaccharide-stimulated prostaglandin E2 synthesis in cultured rat mesangial cells. Biochim Biophys Acta. 1988;963:429–435. | ||
Pirvulescu MM, Gan AM, Stan D, et al. Subendothelial resistin enhances monocyte transmigration in a co-culture of human endothelial and smooth muscle cells by mechanisms involving fractalkine, MCP-1 and activation of TLR4 and Gi/o proteins signaling. Int J Biochem Cell Biol. 2014;50:29–37. | ||
Bradfield PF, Johnson-Léger CA, Zimmerli C, Imhof BA. LPS differentially regulates adhesion and transendothelial migration of human monocytes under static and flow conditions. Int Immunol. 2008;20:247–257. | ||
Jiang SJ, Hsu SY, Deng CR, et al. Dextromethorphan attenuates LPS-induced adhesion molecule expression in human endothelial cells. Microcirculation. 2013;20:190–201. | ||
Lameijer MA, Tang J, Nahrendorf M, Beelen RH, Mulder WJ. Monocytes and macrophages as nanomedicinal targets for improved diagnosis and treatment of disease. Expert Rev Mol Diagn. 2013;13: 567–580. | ||
Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6:286. | ||
Porter KJ, Gonipeta B, Parvataneni S, et al. Regulation of lipopolysaccharide-induced inflammatory response and endotoxemia by β-arrestins. J Cell Physiol. 2010;225:406–416. | ||
Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122–3142. | ||
Kang BH, Shim YJ, Tae YK, et al. Clusterin stimulates the chemotactic migration of macrophages through a pertussis toxin sensitive G-protein-coupled receptor and Gβγ-dependent pathways. Biochem Biophys Res Commun. 2014;445:645–650. | ||
Lehmann DM, Seneviratne AM, Smrcka AV. Small molecule disruption of G protein βγ subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol. 2008;73:410–418. | ||
Schutte RJ, Parisi-Amon A, Reichert WM. Cytokine profiling using monocytes/macrophages cultured on common biomaterials with a range of surface chemistries. J Biomed Mater Res A. 2009;88:128–139. | ||
Hambleton J, Weinstein SL, Lem L, DeFranco AL. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci U S A. 1996;93:2774–2778. | ||
Lim W, Gee K, Mishra S, Kumar A. Regulation of B7.1 costimulatory molecule is mediated by the IFN regulatory factor-7 through the activation of JNK in lipopolysaccharide-stimulated human monocytic cells. J Immunol. 2005;175:5690–5700. | ||
Gao M, Chen L, Yang L, Yu X, Kou J, Yu B. Berberine inhibits LPS-induced TF procoagulant activity and expression through NF-kB/p65, Akt and MAPK pathway in THP-1 cells. Pharmacol Rep. 2014;66:480–484. | ||
Sun J, Shigemi H, Tanaka Y, Yamauchi T, Ueda T, Iwasaki H. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-kB signaling pathways. Biochem Biophys Rep. 2015;4:397–404. | ||
Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm. 2013;2013:697972. | ||
Segura M, Vadeboncoeur N, Gottschalk M. CD14-dependent and -independent cytokine and chemokine production by human THP-1 monocytes stimulated by Streptococcus suis capsular type 2. Clin Exp Immunol. 2002;127:243–254. | ||
Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. | ||
Lin HY, Chang KT, Hung CC, et al. Effects of the mTOR inhibitor rapamycin on monocyte-secreted chemokines. BMC Immunol. 2014;15:37. | ||
Mullen A, Loscher CE, Roche HM. Anti-inflammatory effects of EPA and DHA are dependent upon time and dose-response elements associated with LPS stimulation in THP-1-derived macrophages. J Nutr Biochem. 2010;21:444–450. | ||
Park EK, Jung HS, Yang HI, Yoo MC, Kim C, Kim KS. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res. 2007;56:45–50. | ||
Hmama Z, Knutson KL, Herrera-Velit P, Nandan D, Reiner NE. Monocyte adherence induced by lipopolysaccharide involves CD14, LFA-1, and cytohesin-1 regulation by Rho and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:1050–1057. | ||
Al-Numani D, Segura M, Dore M, Gottschalk M. Up-regulation of ICAM-1, CD11a/CD18 and CD11c/CD18 on human THP-1 monocytes stimulated by Streptococcus suis serotype 2. Clin Exp Immunol. 2003;133:67–77. | ||
Gupta H, Dai L, Datta G, et al. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ Res. 2005;97:236–243. | ||
Li C, Zhang WJ, Frei B. Quercetin inhibits LPS-induced adhesion molecule expression and oxidant production in human aortic endothelial cells by p38-mediated Nrf2 activation and antioxidant enzyme induction. Redox Biol. 2016;9:104–113. | ||
Cho Y, Lee SE, Lee HC, et al. Adipokine resistin is a key player to modulate monocytes, endothelial cells, and smooth muscle cells, leading to progression of atherosclerosis in rabbit carotid artery. J Am Coll Cardiol. 2011;57:99–109. | ||
Walcher D, Hess K, Berger R, et al. Resistin: a newly identified chemokine for human CD4-positive lymphocytes. Cardiovasc Res. 2010;85:167–174. | ||
Guo Y, Zhao G, Tanaka S, Yamaguchi T. Differential responses between monocytes and monocyte-derived macrophages for lipopolysaccharide stimulation of calves. Cell Mol Immunol. 2009;6:223–229. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.