Back to Journals » OncoTargets and Therapy » Volume 9
Lipopolysaccharide-induced α-catenin downregulation enhances the motility of human colorectal cancer cells in an NF-κB signaling-dependent manner
Authors Cheng G, Yang S, Zhang G, Xu Y, Liu X, Sun W, Zhu L
Received 6 October 2016
Accepted for publication 18 November 2016
Published 14 December 2016 Volume 2016:9 Pages 7563—7571
DOI https://doi.org/10.2147/OTT.S123986
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Guoping Cheng,1,2 Shifeng Yang,1 Gu Zhang,1 Yanxia Xu,3 Xiaoling Liu,3 Wenyong Sun,1,2 Liang Zhu3
1Department of Pathology, Zhejiang Cancer Hospital, 2Cancer Research Institute, Zhejiang Cancer Hospital and Key laboratory Diagnosis and Treatment Technology on Thoracic Oncology of Zhejiang Province, 3School of Medicine, Hangzhou Normal University, Hangzhou, Zhejiang, People’s Republic of China
Abstract: α-Catenin is an important molecule involved in the maintenance of cell–cell adhesion and a prognostic marker in cancer since its expression is essential for preventing cancer metastasis. However, the mechanism that leads to the downregulation of α-catenin in cancer progression remains unclear. The present study revealed that lipopolysaccharide (LPS)-induced NF-κB signaling activation suppressed α-catenin expression and motility in SW620 colorectal cancer (CRC) cells, using real-time polymerase chain reaction, Western blotting, and transwell migration assays. LPS treatment reduced both the mRNA and protein expression of α-catenin and thereby enhanced cell motility. Conversely, incubating cells with an NF-κB inhibitor disrupted these effects. Furthermore, the ectopic expression of p65 alone mimicked the effects of LPS stimulation. In CRC tissues, the presence of enteric bacterial LPS-related neutrophil-enriched foci was correlated with α-catenin downregulation. Collectively, these findings suggest that LPS-induced NF-κB signaling is related to α-catenin suppression and enhanced cell motility in CRC. Therefore, NF-κB is a novel potential therapeutic target for CRC metastasis.
Keywords: lipopolysaccharide, colorectal neoplasms, α-catenin, neoplasm metastasis
Introduction
Colorectal cancer (CRC) is one of the most commonly diagnosed malignancies, and metastasis critically reduces patient prognosis. The median survival of patients with metastatic CRC is <1 year.1 However, tumor metastasis is a complex multistep process, of which only very limited details are understood. Thus, further investigations are essential to increase our understanding of its molecular mechanisms and develop novel therapies.
In addition to E-cadherin, α-catenin is an indispensable component of the cadherin–catenin protein complex. It functions as an interface between the cadherin–catenin protein complex and the actin cytoskeleton, where it maintains the integrity of intercellular adherens junctions by directly binding to actin filaments.2 Therefore, loss of α-catenin, which has been reported in several malignancies, can weaken cell–cell adhesion and promote abnormal cellular polarity, epithelial–mesenchymal transition (EMT), and ultimately tumor metastasis.2–5 In addition, α-catenin is an inhibitor of multiple signaling pathways involved in carcinogenesis and development, including the Wnt/β-catenin, Hippo-YAP, hedgehog, and NF-κB pathways.6–10 Although α-catenin is closely related to cancer progression, little is known about its regulation in cancer cells.
The NF-κB pathway, which can be activated by the binding of lipopolysaccharide (LPS) to its receptor TLR4, promotes multiple cancer behaviors such as proliferation, survival, angiogenesis, and metastasis.11 The EMT may serve as a key linkage between NF-κB activation and cancer metastasis. Several studies demonstrated that the EMT depends on the ability of the Snail-related zinc-finger transcription factors Snail and Slug, ZEB family members ZEB1/2, and the basic helix–loop–helix (bHLH) transcription factor Twist to suppress the expression of E-cadherin.12–14 All of these factors can be regulated either directly or indirectly by NF-κB.14–17 The present study demonstrated that α-catenin is another potential target through which NF-κB can promote the EMT and disturb the adhesion and morphologic stability of CRC cells.
Materials and methods
Reagents and antibodies
Monoclonal primary antibodies against human α-catenin (Cat#2028-1), β-catenin (Cat#1247-1), and E-cadherin (Cat#1702-1) were purchased from Epitomics (Burlingame, CA, USA). The primary monoclonal antibodies against the HA tag (Cat#ab9134) and human β-actin (Cat#sc-130300) were purchased from Abcam (Cambridge, UK) and Santa Cruz Biotechnology (Santa Cruz, CA, USA), respectively. The pcDNA3.1-HA-p65 and pcDNA3.1 vectors were kindly provided by Dr Jun Cui (Zhongshan School of Medicine, Sun Yat-sen University). The NF-κB inhibitor Bay 11-7082 was obtained from Sigma-Aldrich (St Louis, MO, USA).
Cell lines and cell culture
SW620 and SW480 human CRC cells and the HEK293T cells were purchased from ATCC (Manassas, VA, USA) and maintained in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco). The cells were cultured at 37°C and 5% CO2.
Patients and specimens
Forty CRC tissue blocks were collected from the Department of Pathology, Zhejiang Cancer Hospital between 2008 and 2015, and written informed consent was provided by the patients from whom the CRC tissue blocks were taken for use in this research. The hematoxylin and eosin (H&E)-stained slides were reviewed under a microscope to ensure each case with obvious neutrophil infiltration but without dramatic tissue necrosis. Tumor staging was performed according to the criteria of the International Union against Cancer tumor node metastasis system. This study was approved by the ethics committee of Zhejiang Cancer Hospital (no IRB-2016-90).
Real-time polymerase chain reaction (PCR)
Total RNA (800 ng) extracted from each sample was reverse transcribed into cDNA using reverse transcriptase (M-MLV; Promega, Madison, WI, USA). α-Catenin gene expression was quantified using SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA) on the StepOnePlus system (Applied Biosystems). The expression levels of all genes were normalized to that of human β-actin. The sequences of the primers were as follows: α-catenin (F: 5′-CGCACCATTGCAGACCATTG and R: 5′-GCACCACAGCATTCATCAAGT), β-catenin (F: 5′-GAATGAAGGTGTGGCGACATAT and R: 5′-CAAGTCCAAGATCAGCAGTCTC), E-cadherin (F: 5′-GAAGAAGGAGGCGGAGAAGA and R: 5′-ACACGAGCAGAGAATCATAAGG), ZEB1 (F: 5′-TGTAGAGGATCAGAATGACTC and R: 5′-CAGAATGTAATCGCATGTGT), Snail1 (F: 5′-CTGCTACAAGGCCATGTC and R: 5′-GGACTCTTGGTGCTTGTG), Fascin1 (F: 5′-TTGTGACCTCCAAGAAGAAT and R: 5′-CCCACCGTCCAGTATTTG), β-actin (F: 5′-CCTGGCACCCAGCACAAT and R: 5′-GCTGATCCACATCTGCTGGAA), and TLR4 (F: 5′-CGGAGGCCATTATGCTATGT and R: 5′-TCCCTTCCTCCTTTTCCCTA).
Migration assay
Cell migration was measured as described previously.18 Prior to the experiment, SW620 cells were treated with 0.5 μg/mL LPS for 24 h in RPMI-1640 medium containing 2% FBS to avoid any influence from LPS-stimulated cell proliferation during the migration assay.
Wound healing assay
Each 2×106 SW620 cells were seeded into the six-well culture plate and 24 h post-LPS or phosphate-buffered saline (PBS) treatment, a sterile 10 μL tip was used to scrape a straight line across the monolayer. The cells were then maintained properly for another 24 h. Wound closure measurements at 0 h and 24 h posttreatment were performed by microscopy. The wound closure percentages were calculated by comparing the wound width of 24 h to that of 0 h of each group. Three replications were performed, and the results were statistically analyzed.
Transfection
The pcDNA3.1-HA-p65 and empty pcDNA3.1 vectors were purified, and 0.5 μg of either vector was transfected into cells using Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions. SW620 cells (2×105) transfected with the HA-p65 construct or empty vector were maintained in RPMI-1640 with 10% FBS for 48 h.
Western blotting
Forty-eight hours after LPS treatment or transfection, SW620 cells were lysed in protein lysis buffer (Pierce, Rockford, IL, USA) supplemented with protease and phosphatase inhibitor cocktails (Pierce) following the manufacturer’s protocol. Protein concentrations were determined using a Bio-Rad protein assay kit and a Model 680 microplate reader (Bio-Rad Laboratories, Richmond, CA, USA). Samples were separated on sodium dodecyl sulfate–polyacrylamide gels and then transferred to polyvinylidene fluoride membranes (Bio-Rad Laboratories). After blocking, the membranes were incubated with antibodies diluted in Tris-buffered saline containing 5% skim milk and 0.1% Tween-20 overnight at 4°C. The following primary antibodies were used for Western blotting: anti-human α-catenin (1:1,000), β-catenin (1:1,500), E-cadherin (1:1,500), HA (1:2,000), and β-actin (1:4,000). The bands were quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Cell block preparation and immunohistochemistry (IHC) staining of α-catenin
Cell cultures were collected and resuspended in PBS in a 10 mL disposable centrifuge tube and then centrifuged at 1,500 rpm for 1 min. The cell pellets were then fixed in formalin and embedded in paraffin for further analysis using H&E staining or IHC. Paraffin sections (4 μm thick) of either tissue or cell blocks were deparaffinized and rehydrated in xylene and a graded alcohol series. Endogenous peroxidase activity was blocked by 0.3% hydrogen peroxide, and the sections were blocked with 10% goat serum for 20 min. The sections were then incubated with anti-human α-catenin (1:100), β-catenin (1:200), and E-cadherin (1:100) primary antibodies for 90 min at room temperature. After washing in PBS, the slides were incubated with biotinylated secondary antibodies (Polymer HRP Goat anti-Mouse & Rabbit IgG) for 30 min, and the slides were developed using 3,3′-diaminobenzidine (DakoCytomation, Carpinteria, CA, USA) according to the manufacturer’s instructions and counterstained with hematoxylin. Negative control slides were incubated with secondary antibody to verify the specificity of the staining.
Statistical analysis
Each quantitative experiment was carried out three times independently, and all data are presented as mean ± standard error of the mean. Comparisons between groups were performed using two-tailed paired Student’s t-tests. All statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
Results
LPS treatment enhanced the motility of CRC cells in vitro
The LPS-induced inflammatory response is important for promoting malignant tumor metastasis.19,20 To evaluate the influence of the LPS-induced inflammatory response on CRC cell motility, SW620 CRC cells were cultured and subjected to real-time PCR to confirm the expression of TLR-4, which is an essential LPS receptor (Figure S1). Next, the cells were pretreated with 0.5 μg/mL LPS for 24 h, and migration assays were performed. Cells treated with LPS exhibited dramatically increased migration through the transwell filter membrane compared with the control group (Figure 1A and B). Similar observations were found in wound healing assays (Figure 1C and D), confirming that LPS treatment increased SW620 cell motility.
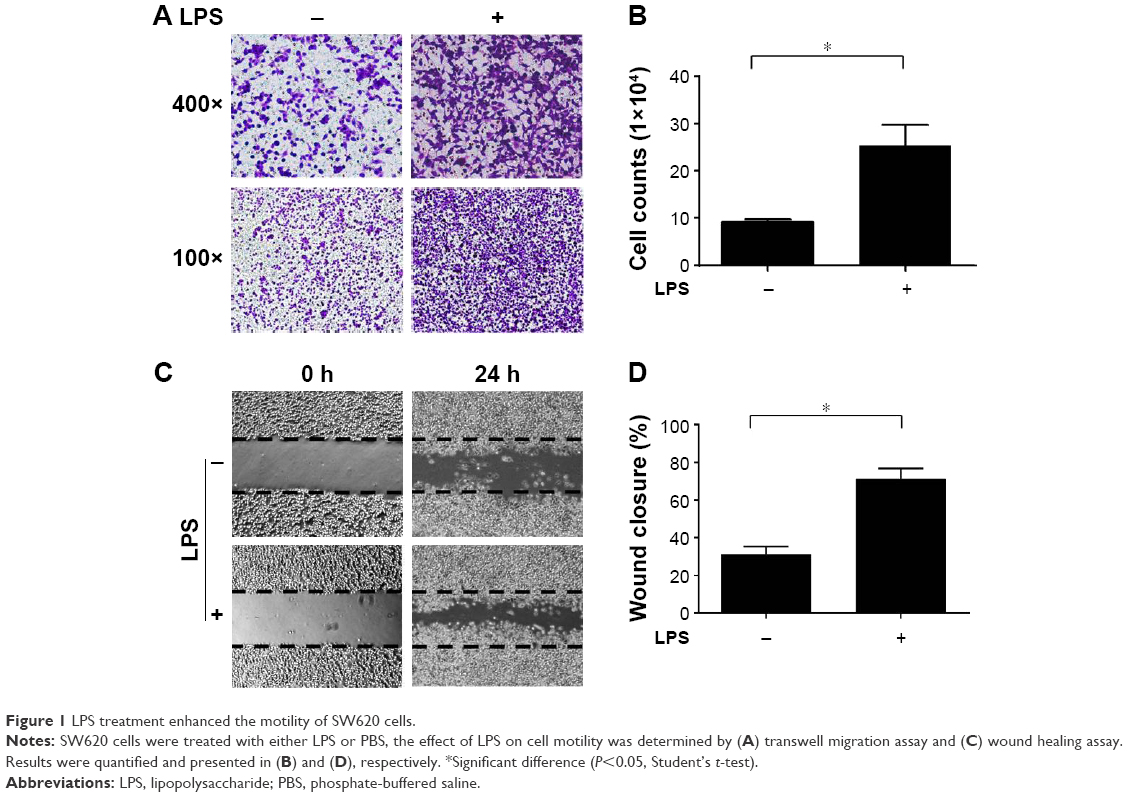 | Figure 1 LPS treatment enhanced the motility of SW620 cells. |
LPS disturbs the cadherin/catenin apparatus by inhibiting α-catenin expression
α-Catenin, which maintains cell adhesion, can be used as a prognostic factor in CRC patients, and decreased α-catenin expression leads to a morphological transition in CRC cells, promoting their motility.18 Therefore, the potential mechanism behind these effects and the ability of α-catenin to mediate the effects of LPS in CRC cells were investigated. The expression levels of key molecules in the cadherin/catenin apparatus as well as ZEB1, Snail, and Fascin1, which play roles in the EMT, were measured. LPS-treated SW620 cells were harvested, and RNA and protein samples were prepared. PCR, real-time PCR, and Western blotting demonstrated that α-catenin expression was reduced by LPS, whereas there was no difference in β-catenin or E-cadherin expression. In contrast, real-time PCR did not show any significant change in Snail or Fascin1 expression, and ZEB1 was undetectable due to its low expression (Figure 2A–D). IHC revealed a similar trend in α-catenin expression (Figure 2E). Therefore, cells treated with LPS exhibited a more independent morphology (Figure 2E), which may be caused by disruption of the cadherin/catenin apparatus. Our research presented that another CRC cell line, the SW480, possess the similar character with SW620 (Figure S2).
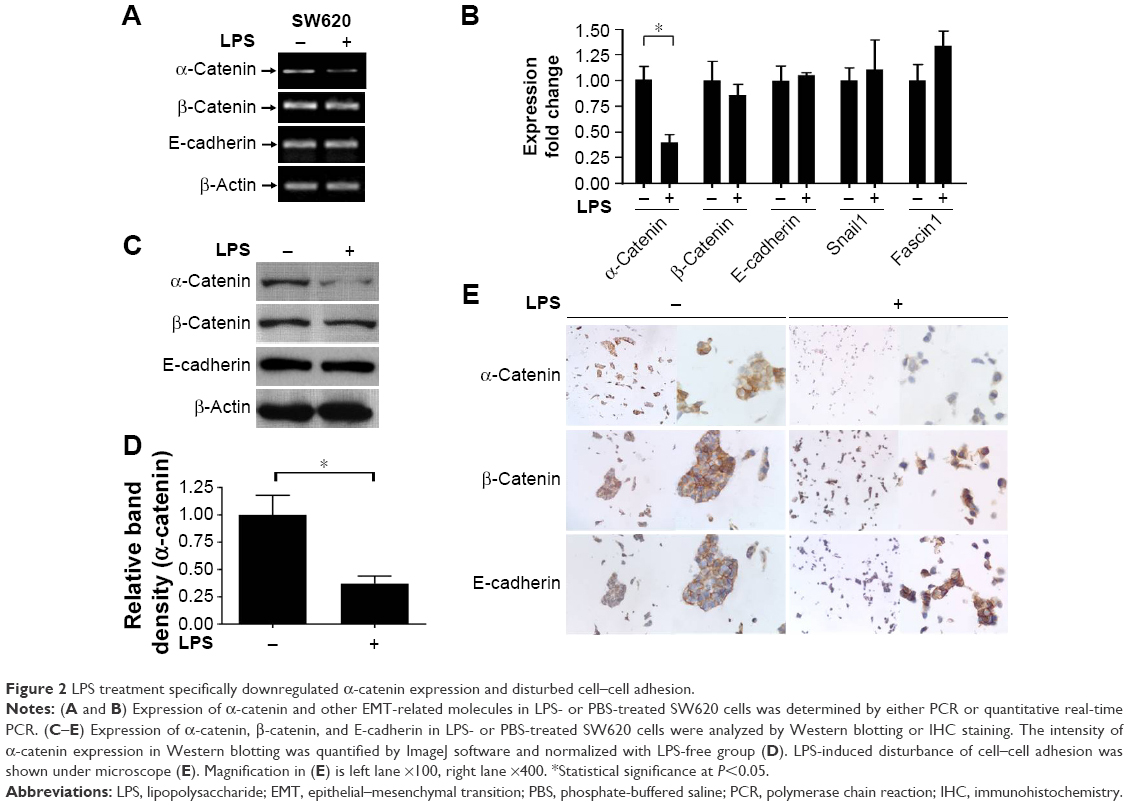 | Figure 2 LPS treatment specifically downregulated α-catenin expression and disturbed cell–cell adhesion. |
LPS-induced α-catenin downregulation is dependent on the NF-κB pathway
Since LPS is a strong activator of the NF-κB pathway and NF-κB activation is an essential event during cancer metastasis,11,21 we next investigated the mechanism by which LPS promotes CRC migration. SW620 cells that had been pretreated with LPS were incubated with the NF-κB inhibitor Bay 11-7082 (100 μM). To evaluate the effectiveness of Bay 11-7082, SW620 cells were harvested 30 min after treatment with LPS and Bay 11-7082. Western blotting demonstrated that I-κB degradation, which is induced by NF-κB activation, was effectively attenuated by Bay 11-7082 compared with the control (Figure 3A). Next, cells were harvested 24 h or 48 h poststimulation, and analyses revealed that inhibiting the NF-κB pathway diminished LPS-induced cell motility and α-catenin downregulation (Figure 3B and C). We next investigated the specificity of NF-κB-induced α-catenin downregulation. p65, a key nuclear transcription factor involved in LPS-activated NF-κB signaling, was exogenously expressed in SW620 cells via transfection of the pcDNA3.1-HA-p65 construct. The cells were lysed 48 h after transfection and analyzed by Western blotting. As shown in Figure 3D, HA-p65 expression was accompanied by α-catenin downregulation. Taken together, these data suggest that NF-κB activation is a key molecular event involved in increased cancer cell motility.
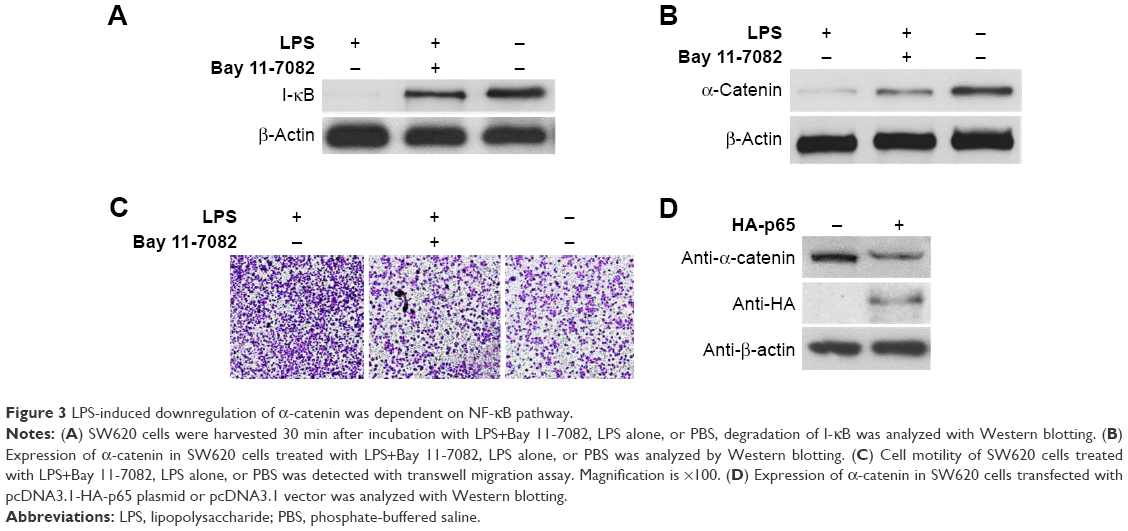 | Figure 3 LPS-induced downregulation of α-catenin was dependent on NF-κB pathway. |
α-Catenin expression is negatively correlated with neutrophil infiltration in CRC tissues
Billions of bacteria inhabit the colorectal cavity; therefore, CRC cells are inevitably exposed to bacterial components such as LPS, which leads to the recruitment of immune cells including neutrophils.22 Thus, neutrophil infiltration is indirect evidence of bacterial LPS exposure in tissue areas devoid of necrosis. To assess the relationship between α-catenin downregulation and neutrophil infiltration in vivo, H&E-stained slides from 40 CRC cases (the clinicopathological features of the patients are listed in Table 1) were reviewed under a microscope to identify the regions with neutrophil infiltration but no dramatic tumor necrosis. IHC was performed to analyze α-catenin expression. For each of the tissue slides, 200 CRC cells in either neutrophil-enriched area or that with few neutrophils were observed under a microscope. The ratio of cells that strongly express α-catenin (with moderate or stronger membrane staining that shows clear and entire border of the cells) was calculated using a 20× objective lens. Generally, compromised α-catenin expression was more frequently observed in CRC tissue areas with enriched neutrophil infiltration (Figure 4A). Interestingly, CRC cells in these regions tended to form more irregular, cribriform glands, which may be morphologically related to poor cell–cell attachment. The result of statistical analysis indicated that a significantly higher proportion of CRC cells expressed high α-catenin levels in regions with few neutrophil infiltration within the cases (P<0.001, Figure 4B). These results suggested that neutrophil infiltration correlated with reduced α-catenin expression, which provides in vivo evidence that LPS exposure inhibits α-catenin expression.
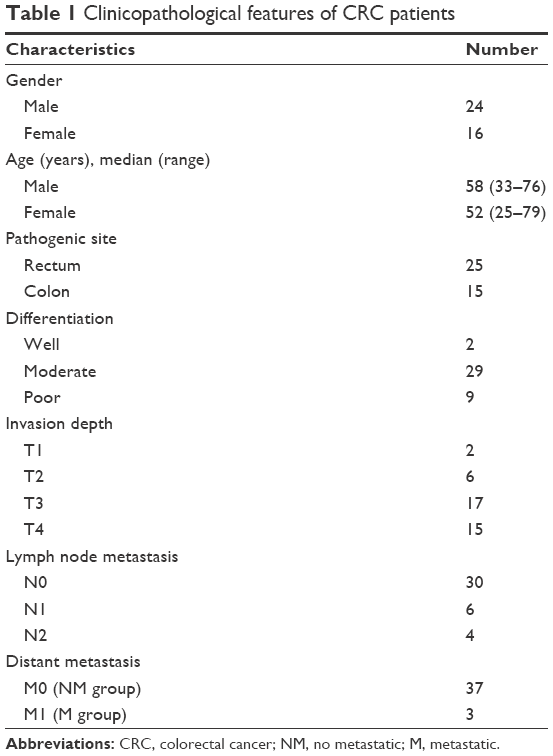 | Table 1 Clinicopathological features of CRC patients |
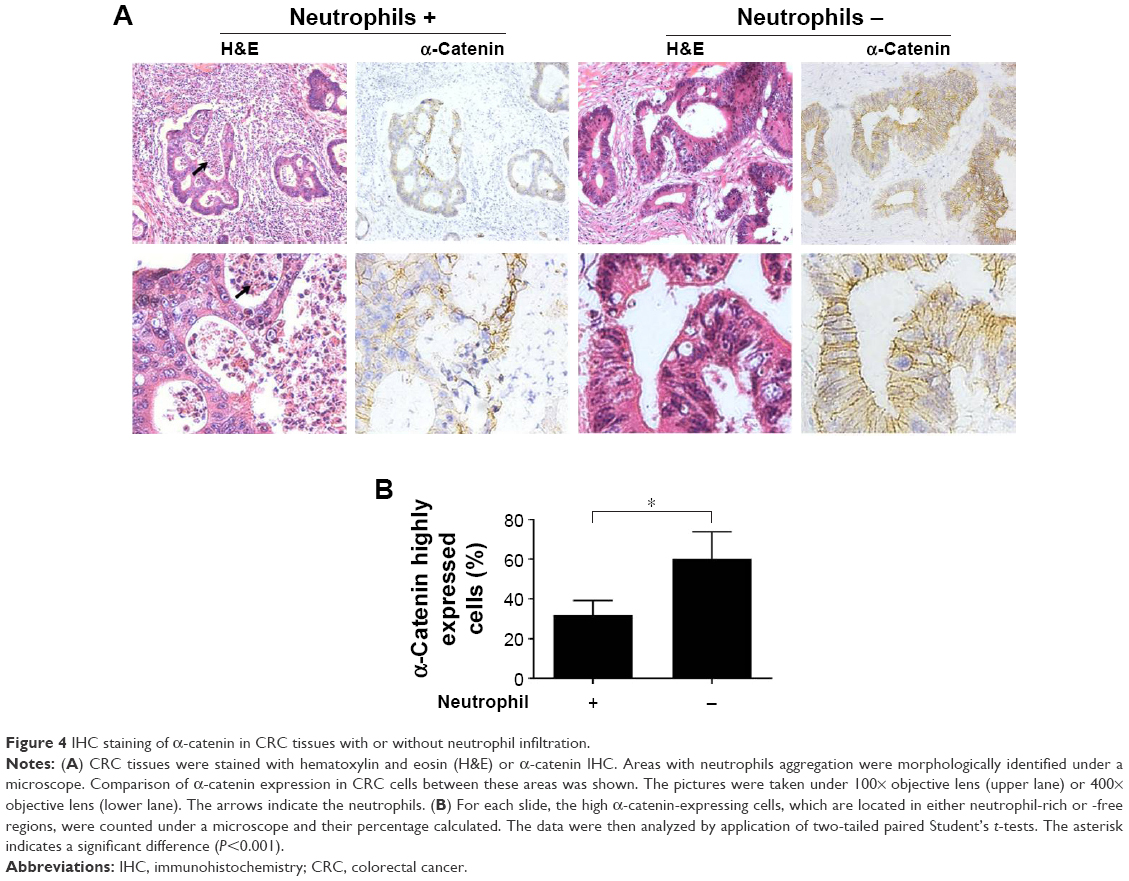 | Figure 4 IHC staining of α-catenin in CRC tissues with or without neutrophil infiltration. |
Discussion
A detailed understanding of the mechanism of cancer metastasis will improve the prognosis of patients with CRC. One hypothesis is that disruption of the cadherin–catenin protein complex results in loss of cell–cell adhesion and promotes tissue reorganization in cancer.5
Although its role in restraining metastasis has been less investigated compared with those of E-cadherin and β-catenin, α-catenin is downregulated in multiple cancers, including CRC, and its expression is significantly correlated with poor differentiation, a higher metastatic potential, and unfavorable prognosis.2,18,23–25 In breast cancer, reduced α-catenin expression is often observed in advanced-stage tumors.26 It is possible that decreased α-catenin expression directly impairs cell–cell adhesion, thereby releasing the cancer cells. This hypothesis is supported by the current data (Figure 2E). Despite its critical role in cancer progression, the mechanism behind α-catenin downregulation is unclear. Some reports have suggested that DNA methylation, genetic alterations, and hypoxia in the cancer microenvironment play a role.4,23,27,28 Here, we provided novel insights into the regulation of α-catenin in CRC by focusing on the innate immune component NF-κB signaling.
Abnormal NF-κB activity in some cancer cells could be related to poor prognosis.29 Although the NF-κB pathway includes various intrinsic negative regulators, recent studies in basal-like breast cancer cells have suggested that NF-κB activity is inhibited by the direct binding of α-catenin to I-κB. This protects I-κB from lysine 48-linked ubiquitination and degradation, thereby keeping I-κB kinases (IKKs) in an inactive state. The depletion of α-catenin promotes cancer cell growth both in vitro and in vivo, whereas the knockdown of RelA directly inhibits this effect.10 Moreover, α-catenin knockout mice developed epidermal squamous cell carcinoma and showed upregulated NF-κB expression according to tissue microarrays.30 Although these results provide evidence that α-catenin is a negative regulator of tumor-promoting NF-κB activation, additional studies are needed to investigate whether α-catenin itself is regulated by NF-κB in a feedback loop. Our current study elucidated a novel relationship between NF-κB and α-catenin and demonstrated that LPS-induced NF-κB signaling promotes CRC cell motility by negatively regulating α-catenin expression. Conversely, treatment with an NF-κB inhibitor reversed these effects.
It was reported that postoperative Gram-negative bacterial infection and inflammation may lead to CRC recurrence and progression.16,17 To explore the potential mechanism behind these observations, we first mimicked the Gram-negative bacterium-induced immune response by exposing SW620 cells to LPS. Consistent with previous reports, in vitro cancer cell motility was increased in both transwell migration and wound healing assays. Both real-time PCR and Western blotting demonstrated a dramatic downregulation of α-catenin, whereas only mild changes in β-catenin and E-cadherin expression were observed. Although the expression of important metastasis-dependent molecules such as Snail and Fascin1 was increased, the changes were not statistically significant. The decreased α-catenin expression was accompanied by SW620 cell morphological alterations, in which the cells were more independent of each other with a scattered distribution compared with the control group, which exhibited more intact cell–cell connections. IHC staining of β-catenin and E-cadherin also displayed fine adhesion apparatus (Figure 2E), suggesting that LPS treatment disrupted the cadherin–catenin complex.
LPS may activate other signaling transduction pathways.31 Therefore, we evaluated the specificity of LPS-induced α-catenin downregulation. Bay 11-7082 is a chemical compound that specifically inhibits NF-κB signaling by targeting IKKs.32 SW620 cells were coincubated with LPS alone or in combination with Bay 11-7082, and migration assays were performed. Cells that received the combination treatment did not exhibit additional motility compared with the LPS group. Conversely, activating NF-κB by ectopic expression of p65 downregulated α-catenin expression. Together, these data identified α-catenin as a target gene specifically regulated by NF-κB signaling. Meanwhile, IHC staining of CRC tissue sections demonstrated weaker α-catenin expression in regions where tumorous components were infiltrated with prominent neutrophils, which can be recruited by bacterial LPS. These in vitro and in vivo data support the hypothesis that α-catenin expression in CRC cells is specifically regulated by NF-κB activity.
Conclusion
These results provided in vitro and in vivo evidence suggesting that LPS-induced innate immune activity regulates the expression of α-catenin via NF-κB signal transduction. This can dramatically influence the biological behavior of CRC cells, such as motility. These findings combined with observations that α-catenin is downregulated in several advanced malignancies will help develop novel strategies for preventing CRC progression and metastasis.
Acknowledgments
We are grateful to Dr Jun Cui at Zhongshan School of Medicine, Sun Yat-sen University for kindly providing the p65 expressing construct. This work was supported by a grant from the Zhejiang Provincial Natural Science Foundation of China (no Q12H160052).
Disclosure
The authors report no conflicts of interest in this work.
References
Newland RC, Dent OF, Chapuis PH, Bokey EL. Clinicopathologically diagnosed residual tumor after resection for colorectal cancer. A 20-year prospective study. Cancer. 1993;72(5):1536–1542. | ||
Benjamin JM, Nelson WJ. Bench to bedside and back again: molecular mechanisms of alpha-catenin function and roles in tumorigenesis. Semin Cancer Biol. 2008;18(1):53–64. | ||
Shimoyama Y, Nagafuchi A, Fujita S, et al. Cadherin dysfunction in a human cancer cell line: possible involvement of loss of alpha-catenin expression in reduced cell–cell adhesiveness. Cancer Res. 1992;52(20):5770–5774. | ||
Ye Y, McDevitt MA, Guo M, et al. Progressive chromatin repression and promoter methylation of CTNNA1 associated with advanced myeloid malignancies. Cancer Res. 2009;69(21):8482–8490. | ||
Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11(7):502–514. | ||
Giannini AL, Vivanco M, Kypta RM. alpha-catenin inhibits beta-catenin signaling by preventing formation of a beta-catenin*T-cell factor*DNA complex. J Biol Chem. 2000;275(29):21883–21888. | ||
Ji H, Wang J, Fang B, Fang X, Lu Z. alpha-catenin inhibits glioma cell migration, invasion, and proliferation by suppression of beta-catenin transactivation. J Neurooncol. 2011;103(3):445–451. | ||
Silvis MR, Kreger BT, Lien WH, et al. alpha-catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci Signal. 2011;4(174):ra33. | ||
Lien WH, Klezovitch O, Fernandez TE, Delrow J, Vasioukhin V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science. 2006;311(5767):1609–1612. | ||
Piao HL, Yuan Y, Wang M, Sun Y, Liang H, Ma L. alpha-catenin acts as a tumour suppressor in E-cadherin-negative basal-like breast cancer by inhibiting NF-kappaB signalling. Nat Cell Biol. 2014;16(3):245–254. | ||
Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8(1):33–40. | ||
Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3(3):155–166. | ||
Eger A, Aigner K, Sonderegger S, et al. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24(14):2375–2385. | ||
Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. | ||
Kang Y, Massague J. Epithelial–mesenchymal transitions: twist in development and metastasis. Cell. 2004;118(3):277–279. | ||
Storci G, Sansone P, Mari S, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. 2010;225(3):682–691. | ||
Julien S, Puig I, Caretti E, et al. Activation of NF-kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene. 2007;26(53):7445–7456. | ||
Zhu L, Chen H, Zhou D, et al. MicroRNA-9 up-regulation is involved in colorectal cancer metastasis via promoting cell motility. Med Oncol. 2012;29(2):1037–1043. | ||
Chen MC, Chang WW, Kuan YD, Lin ST, Hsu HC, Lee CH. Resveratrol inhibits LPS-induced epithelial–mesenchymal transition in mouse melanoma model. Innate Immun. 2012;18(5):685–693. | ||
Rousseau MC, Hsu RY, Spicer JD, et al. Lipopolysaccharide-induced toll-like receptor 4 signaling enhances the migratory ability of human esophageal cancer cells in a selectin-dependent manner. Surgery. 2013;154(1):69–77. | ||
Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10(12):1314–1319. | ||
Keshavarzian A, Haydek JM, Jacyno M, Holmes EW, Harford F. Modulatory effects of the colonic milieu on neutrophil oxidative burst: a possible pathogenic mechanism of ulcerative colitis. J Lab Clin Med. 1997;130(2):216–225. | ||
Jurjus A, Eid A, Al Kattar S, et al. Inflammatory bowel disease, colorectal cancer and type 2 diabetes mellitus: the links. BBA Clin. 2016;5:16–24. | ||
Craig DW, O’Shaughnessy JA, Kiefer JA, et al. Genome and transcriptome sequencing in prospective metastatic triple-negative breast cancer uncovers therapeutic vulnerabilities. Mol Cancer Ther. 2013;12(1):104–116. | ||
Raftopoulos I, Davaris P, Karatzas G, Karayannacos P, Kouraklis G. Level of alpha-catenin expression in colorectal cancer correlates with invasiveness, metastatic potential, and survival. J Surg Oncol. 1998;68(2):92–99. | ||
Nakopoulou L, Gakiopoulou-Givalou H, Karayiannakis AJ, et al. Abnormal alpha-catenin expression in invasive breast cancer correlates with poor patient survival. Histopathology. 2002;40(6):536–546. | ||
Hollestelle A, Elstrodt F, Timmermans M, et al. Four human breast cancer cell lines with biallelic inactivating alpha-catenin gene mutations. Breast Cancer Res Treat. 2010;122(1):125–133. | ||
Plumb CL, Adamcic U, Shahrzad S, Minhas K, Adham SA, Coomber BL. Modulation of the tumor suppressor protein alpha-catenin by ischemic microenvironment. Am J Pathol. 2009;175(4):1662–1674. | ||
Li J, Deng Z, Wang Z, et al. Zipper-interacting protein kinase promotes epithelial–mesenchymal transition, invasion and metastasis through AKT and NF-kB signaling and is associated with metastasis and poor prognosis in gastric cancer patients. Oncotarget. 2015;6(10):8323–8338. | ||
Kobielak A, Fuchs E. Links between alpha-catenin, NF-kappaB, and squamous cell carcinoma in skin. Proc Natl Acad Sci U S A. 2006;103(7):2322–2327. | ||
Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. | ||
Rauert-Wunderlich H, Siegmund D, Maier E, et al. The IKK inhibitor Bay 11-7082 induces cell death independent from inhibition of activation of NFκB transcription factors. PLoS One. 2013;8(3):e59292. |
Supplementary materials
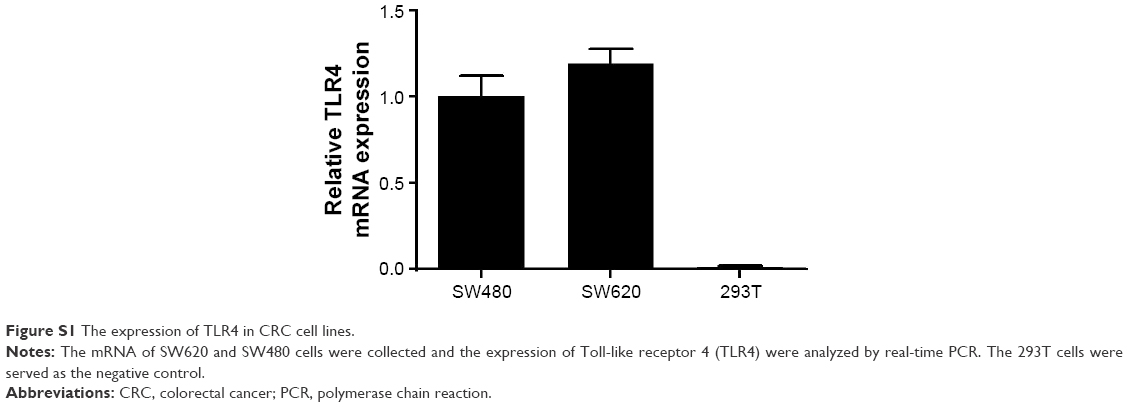 | Figure S1 The expression of TLR4 in CRC cell lines. |
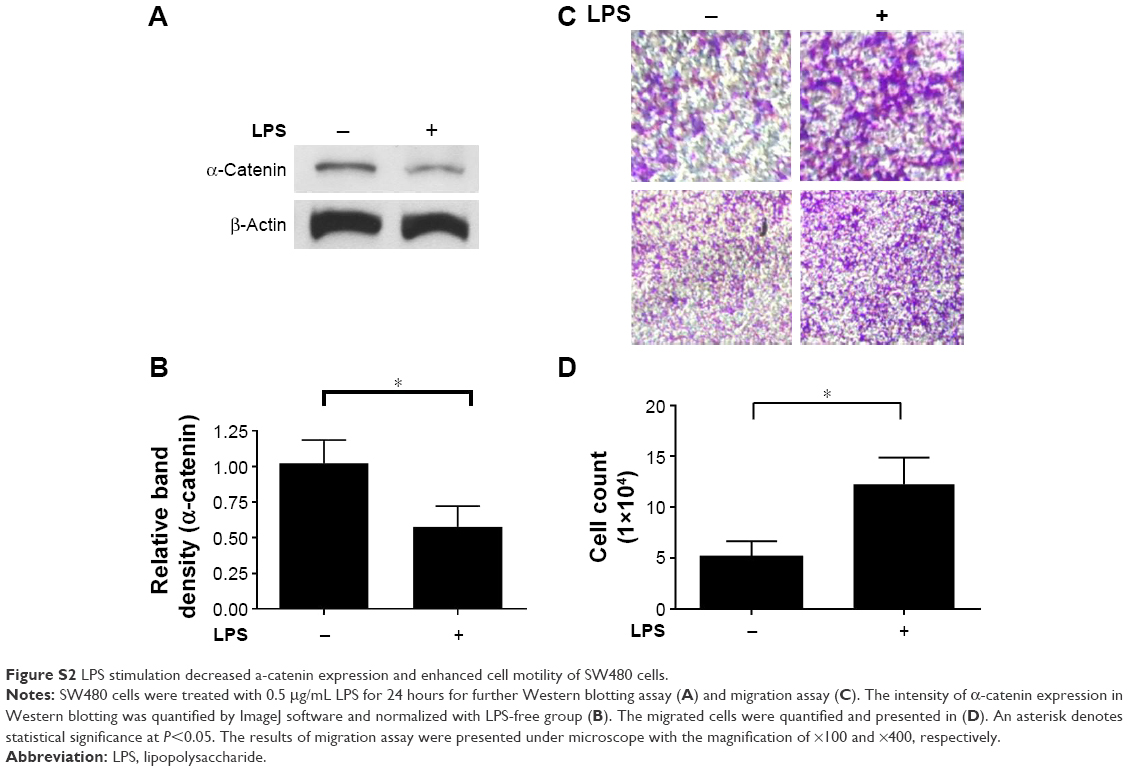 | Figure S2 LPS stimulation decreased a-catenin expression and enhanced cell motility of SW480 cells. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.