")
Back to Journals » International Journal of Nanomedicine » Volume 13
Lipid surface modifications increase mesoporous silica nanoparticle labeling properties in mesenchymal stem cells
Authors Rosenbrand R , Barata D , Sutthavas P , Mohren R , Cillero-Pastor B, Habibovic P, van Rijt S
Received 2 August 2018
Accepted for publication 3 October 2018
Published 20 November 2018 Volume 2018:13 Pages 7711—7725
DOI https://doi.org/10.2147/IJN.S182428
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Roger Rosenbrand,1 David Barata,1 Pichaporn Sutthavas,1 Ronny Mohren,2 Berta Cillero-Pastor,2 Pamela Habibovic,1 Sabine van Rijt1
1Department of Instructive Biomaterials Engineering, MERLN Institute for Technology-Inspired Regenerative Medicine, Maastricht University, 6229 ER MD, Maastricht, the Netherlands; 2Maastricht Multimodal Molecular Imaging Institute (M4I), Division of Imaging Mass Spectrometry, Maastricht University, 6229 ER Maastricht, the Netherlands
Background: Nanoparticles have emerged as promising cell-labeling tools, as they can be precisely tailored in terms of chemical and physical properties. Mesoporous silica nanoparticles (MSNs), in particular, are easily tunable with regard to surface and core chemistry, and are able to confine dyes and drug molecules efficiently.
Purpose: The aim of this study was to investigate the effect of lipid and polyethylene glycol (PEG) surface modifications on MSN stem-cell-tracking abilities.
Methods: Lipid and PEG surface functionalized MSNs were synthesized and the effect of surface functionalization on cell internalization, proliferation, differentiation and cell proteomics was investigated in patient derived mesenchymal stem cells (MSCs).
Results: MSNs and lipid surface-modified MSNs were internalized by >80% of the MSC population, with the exception of nanoparticles modified with short PEG chains (molecular weight 750 [MSN-PEG750]). Lipid-modified MSNs had higher labeling efficiency with maximum uptake after 2 hours of exposure and were in addition internalized 17 times higher compared to unmodified MSNs, without negatively affecting differentiation capacity. Using a mass-spectrometry-based label-free quantitative proteomics approach, we show that MSN labeling leads to the up- and downregulation of proteins that were unique for the different surface-modified MSNs. In addition, functional enrichments were found in human MSCs labeled with MSNs, MSN-PEG750, and lipid-modified MSNs.
Summary: Here we show that organic modifications with lipids and PEGylation can be used as a promising strategy to improve MSN labeling capabilities. In particular, we show that lipid modifications can optimize such probes in three distinct ways: significantly improved signal strength, a barrier for sustained release of additional probes, and improved stem-cell-labeling efficiency.
Keywords: mesoporous silica nanoparticles, cell tracking, mesenchymal stem cells, lipid surface modification
Introduction
Stem cells have high potential for the replacement or regeneration of damaged cells and/or tissue, due to their unique ability to self-renew continuously and differentiate into mature cells of a variety of lineages. Stem cells may thus be of great importance in treating degenerative diseases, such as Parkinson’s disease, or for spinal cord repair. Indeed, there is a vast increase in new stem-cell therapies that are entering clinical trials: globally, the number of phase II–IV clinical trials regarding stem-cell therapies are in the thousands (ClinicalTrials.gov). Mesenchymal stem cells (MSCs) have been heavily researched in this context, as they are multipotent, easy to isolate, and excrete beneficial factors.1 However, difficulty in gaining relevant in vivo information on stem-cell implantation in larger animals and humans represents a significant bottleneck in the translation of new stem-cell-based therapies, including those based on MSCs. Therefore, reliable methods that can trace stem cells are needed to determine the fate of stem cells after implantation,2 and also to determine optimal parameters, such as administration sites, to maximize therapeutic benefit while minimizing negative side effects.3
Several methods have been reported to enable in vitro and in vivo stem-cell tracking, including exogenous fluorescent proteins. Although relatively inexpensive, these methods generally require genetic modification of stem cells via viral vectors for transfection.2 In addition, interference from tissue due to autofluorescence makes them less suitable for in vivo imaging. Nanoparticles have emerged as a promising toolbox for stem-cell tracking because of their unique magnetic and/or optical properties, which can be used in combination (ie, multimodal), and are stable over a long period for the tracking of stem cells in live imaging, either in vitro or in vivo.4 Among others, mesoporous silica nanoparticles (MSNs) are widely studied as theranostic nanoplatforms, due to their ease of bioconjugation, potential for multiple functionalisations,5,6 and ability to efficiently harbor cargo and confine fluorescent dyes within their matrix.7,8 Furthermore, MSNs are considered biocompatible and are resistant to biodegradation in the cellular environment.9 A few studies have investigated MSNs as stem-cell-labeling tools.10–13 For example, Huang et al observed no differences in proliferation, cell viability, or immunophenotypic profiles of surface markers from human MSCs (hMSCs) that internalized MSNs conjugated with fluorescein isothiocyanate (FITC), while their capacity to differentiate into osteocytes, chondrocytes, and adipocytes after MSN internalization was preserved.11
Here, we investigated the effect of organic surface modifications on the stem-cell-labeling properties of MSNs. Surface modification of nanoparticles is a promising method to improve their biocompatibility and cell-internalization rate and enable cell targeting or allow controlled drug release.14,15 In this regard, the addition of supported lipid (Lip)-bilayer shell, which in turn can be further modified with targeting/trafficking ligands and polyethylene glycol (PEG) groups, is particularly interesting. The MSN-Lip complex combines the advantages of liposomes, namely low toxicity, immunogenicity, and longer circulation,16 with those of MSNs, which possess high loading capacity, stability, and gatekeeping properties for controlled drug delivery.17,18 Furthermore, liposome instability and leakage can be overcome by this system, as the adhesion energy between the Lip and MSNs suppresses fluctuations in the membrane bilayer.17 Additional PEGylation can be used to improve the stability of MSN–Lip complexes in biological fluids to increase biocompatibility and prevent particle agglomeration.19 In short, here we show that modular design of MSNs allows for manipulation of their biological behavior, which can be optimized for (stem) cell tracking purposes.
Methods
Synthesis of MSNs
Synthesis of MSNs with functional (−SH) groups at the particle’s core and functional (−NH2) groups at the particle’s surface was based on the cocondensation method as previously reported.20 In short, tetraethyl orthosilicate (TEOS; 1.63 g, 7.82 mmol; Sigma-Aldrich, #131903, 98%), (3-mercaptopropyl)trimethoxysilane (112 mg, 0.48 mmol; Sigma-Aldrich #175617, 95%), and triethanolamine (14.3 g, 95.6 mmol; Sigma-Aldrich #90279, 99.5%) were heated for 20 minutes at 90°C under static conditions (solution 1). A second mixture of cetyltrimethylammonium chloride (2.41 mL, 1.83 mmol; Sigma-Aldrich #292737, 25% weight in H2O) and NH4F (100 mg; 2.70 mmol; Sigma-Aldrich #338869, 99.9%) in Milli-Q (21.7 g, 1.21 mmol, bidistilled) was heated to 60°C (solution 2). Solution 2 was then rapidly added to solution 1, and the obtained mixture was left to stir vigorously for 30 minutes at room temperature. Then, TEOS (138.2 mg, 0.66 mmol) was added in four equal increments every 3 minutes, and the mixture was left to stir for another 30 minutes. Subsequently, TEOS (19.3 mg, 92.8 μmol) and (3-aminopropyl)triethoxysilane (20.5 mg, 92.5 μmol; Sigma-Aldrich #A3648, 99%) were added to the mixture and left to stir overnight. Next, after two steps of washing with ethanol, organic template extraction was performed by refluxing the colloidal suspension at 90°C in a solution of 2 g ammonium nitrate (NH4NO3; Sigma-Aldrich #221244, 99.9%) dissolved in 100 mL absolute ethanol. After 45 minutes, particles were washed and redispersed in a solution of 10 mL concentrated HCl (37%; Sigma-Aldrich) and 90 mL absolute ethanol for a second reflux. MSNs were then collected by centrifugation, washed twice with absolute ethanol, and redispersed in 20 mL absolute ethanol. Particles were stored at −20°C until further use.
Synthesis of MSN-PEG2,000 and MSN-PEG750
MSNs (10 mg) were collected by centrifugation and washed once with Milli-Q water. Then they were redispersed in a 2 mL sodium bicarbonate buffer (pH 8.3). Afterwards, 10 mg O-[(N-succinimidyl)succinyl-aminoethyl]-O′-methylpolyethylene glycol with a molecular weight (MW) of either 750 kDa (OMe-PEG750-NHS; Sigma-Aldrich #712531) or 25 mg of the same linker with 2,000 kDa MW (Sigma-Aldrich #41214) was added and the solution left to stir for 2 hours at room temperature. Subsequently, particles were collected by centrifugation and washed three times with Milli-Q water. Particles were stored in a small volume (2 mL) of Milli-Q water at 4°C until further use. For particle labeling, 1 mg MSN-SHinNH2out was dissolved in 1 mL absolute ethanol. Then, 1 μL ATTO633-maleimide stock solution (5 μg ATTO633-maleimide/μL anhydrous DMF) was added to the particles and left to stir overnight. Next, particles were collected by centrifuging and washed three times with absolute ethanol, redispersed in 1 mL absolute ethanol, and stored at −20°C until further use.
Synthesis of MSN-Lip and MSN-Lip-PEG2,000
MSNs were functionalized with a lipid bilayer either using 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) to prepare MSN-Lip or a 95:5 ratio of DOPC:1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(PEG)-2,000] (PE-PEG2,000) to prepare MSN-Lip-PEG2,000 in accordance with previously reported methods.21,22 This method is based on the fact that lipids dissolve as monomers in ethanol, while they self-assemble into bilayer liposomal structures as the water content of the solution is increased.
Lipid preparation
For MSN-Lip, 2.5 g DOPC (Avanti Polar Lipids #850375) and for MSN-Lip-PEG2,000 particles, 2.1 g DOPC and 0.4 g PE-PEG2,000 (Avanti Polar Lipids #880160) were dissolved in 4 mL chloroform (Sigma-Aldrich #C2432, 99.8%). The chloroform was then evaporated using a rotary evaporator to create a lipid film. The lipid film was further dried in a vacuum oven at 40°C overnight. Then, the lipid film was dissolved in a 1 mL mixture of 40% ethanol and 60% Milli-Q water to obtain a 2.5 g/mL lipid solution.
MSN-Lip synthesis
MSNs (1 mg) were collected by centrifugation and redispersed in 100 μL lipid solution (DOPC for MSN-Lip, 95:5 DOPC:PE-PEG2,000 for MSN-Lip-PEG2,000). Then, 700 μL Milli-Q water was added to the solution to induce the formation of supported lipid bilayers on the external surface of the particles. After 10 minutes, particles were collected by centrifugation, and lipid-functionalized MSNs were redispersed in 800 μL Milli-Q water. Both MSN-Lip and MSN-Lip-PEG2,000 were stored at 4°C until further use.
MSN characterization
Morphological characterization of the synthesized MSNs was performed by transmission electron microscopy (TEM) using an FEI Tecnai and scanning electron microscopy (SEM) using an FEI Teneo in the secondary electron mode. Size and electrokinetic potential (ζ-potential) were measured by dynamic light scattering at 25°C at an angle of 90° using the Zetasizer Nano ZSP (Malvern Instruments, Malvern, UK). Fluorescence microscopy (Nikon Eclipse Ti) was used to evaluate dual labeling by two probes: Atto 633–maleimide (Atto-Tec, Siegen, Germany) and FITC-NHS ester for MSNs and Atto 633–maleimide and DOPC:PC TopFluor 488 (99.96:0.04) for MSN-Lip.
Cell culture
hMSCs isolated from human bone marrow from donors with written informed consent (full approval for their use from the Ethical Committee at Maastricht University, in accordance with the Declaration of Helsinki) were used. hMSCs from donor 133A and donor 313 were used at passages 4–6. hMSCs were grown in αMEM (with ribonucleosides and deoxyribonucleosides; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS, 1% penicillin–streptomycin, 1% L-glutamine, and 1% L-ascorbic acid 2–phosphate magnesium salt (basic medium). Cells were cultured in a 5% CO2 incubator at 37°C. Culture medium was changed every 2–3 days. For cell-culture-differentiation assays, basic and osteogenic (basic medium supplemented with 10 nM dexamethasone) media were used.
Metabolic activity assay
MTT assays were performed to assess cell metabolism after MSN-hMSC labeling. Cells were exposed to MSNs, MSN-Lip, MSN-Lip-PEG2,000, MSN-PEG750, and MSN-PEG2,000 (measured in triplicate) in concentrations of 10–250 μg/mL. Fifteen control wells were included per 96-wells plate. Cells were seeded so that the plate was 60%–80% confluent when performing the MTT assay. Generally, cells were seeded on day 1 in a 96-well plate and exposed to MSNs on day 2. After exposure, the culture medium was aspirated and 90 μL fresh culture medium and 10 μL MTT solution (5 mg MTT/mL sterile PBS, 98%; Sigma-Aldrich) were added to each well and incubated for 3 hours in a 5% CO2 incubator at 37°C. Afterward, medium was aspirated and 100 μL isopropanol (VWR, 99,70%), 0.1% triton-X100 (VWR, proteomic grade) were added to the wells, fixating the cells and solubilizing the purple precipitate that had formed during incubation. The plate was left for 45 minutes on a plate shaker to dissolve all crystals. Finally, absorbance was read at 570 nm (Clariostar; BMG Labtech). The average of the control was set to 1, and metabolic activity was calculated as cell viability compared to this value.
Flow cytometry
Flow cytometry was performed to assess quantitatively the uptake of different MSN types by hMSCs. hMSCs were exposed to ATTO633-maleimide labeled MSNs (MSNs, MSN-Lip, MSN-Lip-PEG2,000, MSN-PEG750, and MSN-PEG2,000) at a concentration of 25 μg/mL. Cells were seeded in six-well plates at different cell densities, depending on the time point of flow-cytometry analysis. Generally, cells were seeded on day 1 in a six-well plate and exposed to MSNs on day 2. Flow cytometry was performed at 2, 24, and 48 hours after MSN exposure. To prepare the samples, cells were washed with PBS, trypsinized, and redispersed in culture medium. Cell suspensions were then centrifuged and redispersed in 300 μL PBS and kept on ice for flow-cytometry analysis. Flow cytometry was performed using a BD Accuri C6. A total of 10,000 cells (gated) were collected for each measurement. FlowJo version 10 was used for data analysis.
Microscopy
For fluorescence-microscopy experiments, hMSCs were seeded on sterilized coverslips in six-well plates and exposed to ATTO 647–maleimide-labeled MSNs (25 μg/mL). For fluorescence imaging, hMSC culture was stopped after 2 hours (5,500 cells seeded/cm2) and 24 hours (3,000 cells seeded/cm2) of MSN exposure. Cells were fixed directly with 4% paraformaldehyde (VWR, technical grade) in PBS for 15 minutes at room temperature (RT) and then washed twice with PBS. Afterwards, hMSCs were permeabilized with 0.1% Triton X-100 in PBS for 5 minutes at room temperature and washed four times with PBS. Cells were stained against actin (phalloidin–Alexa Fluor 647, 1:500, Thermo Fisher Scientific) and DAPI to stain the nuclei (1 μg/mL, 95%; Sigma-Aldrich). The samples were mounted with Mowiol medium and stored at 4°C until use. For live-cell experiments, cells were grown for 1 day and stained with CellTracker green (10 mM solution in DMSO, 1:400; Thermo Fisher Scientific). After 30 minutes, cell culture was exposed to MSNs while using fluorescence microscopy (Nikon Eclipse TI-E, with environmental control). Acquired images were processed with NIS-Elements AR 4.51.01, 64-bit.
Alkaline phosphatase-activity quantification
hMSCs were seeded at a density of 10,000 cells/cm2. After 24 hours, medium was replaced by basic or osteogenic MSN-loaded media at final dilutions of 0, 25, and 75 μg/mL. Subsequent media exchange did not include further particle load. ALP activity was measured using CDP-Star assay kit (Roche) according to the manufacturer’s instructions for cell-culture samples at days 1, 7, and 14. ALP activity was quantified by measurement of luminescence in white 96-microwell plates using a microplate reader (Clariostar).
Proteomics
For proteomics experiments, cells were washed twice with cold PBS, then collected in 5 M urea in 50 mM ammonium bicarbonate (ABC) in 1.5 mL Eppendorf tubes. Cell lysis was performed with three freeze–thaw cycles using a warm-water bath and liquid nitrogen. Protein concentration was assessed by Bradford assay. The lysate was then reduced with 20 mM dithiothreitol for 45 minutes and alkylated with 40 mM iodoacetamide for 45 minutes in the darkness. The alkylation was terminated by 20 mM DDT to consume any excess iodoacetamide. Digestion was performed with a mixture of LysC and trypsin, which was added at a ratio of 1:25 (enzyme:protein). After 2 hours of digestion at 37°C in a water bath, the lysate was diluted with 50 mM ammonium bicarbonate to 1 M urea and further digested at 37°C overnight. The digestion was terminated by addition of formic acid to a total of 1%. Peptide separation was performed on a Thermo Fisher Scientific Dionex Ultimate 3000 rapid separation ultrahigh-performance liquid-chromatography system equipped with an Acclaim PepMap C18 analytical column (2 μm, 100 Å, 75 μm × 150 mm). Peptide samples were first desalted on an online-installed C18 trapping column. After desalting, peptides were separated on the analytical column with a 90-minute linear gradient from 5% to 35% acetonitrile with 0.1% formic acid at 300 nL/min flow rate. The ultrahigh-performance liquid-chromatography system was coupled with a mass spectrometry (MS) device (Q Exactive HF; Thermo Fisher Scientific). Data-dependent acquisition settings were full MS scan of 375–1,500 m/z at a resolution of 120,000, followed by MS/MS scans of the 15 most intense ions at a resolution of 30,000.
For protein identification and quantification, data-dependent acquisition spectra were analyzed with Proteome Discoverer version 2.2. Within this software, the search engine Sequest was used with the Swiss-Prot human database (Homo sapiens, TaxID 9606). The database search was performed with the following settings: enzyme was trypsin, maximum two missed cleavages, minimum peptide length six, precursor mass tolerance 10 ppm, fragment mass tolerance 0.02 Da, dynamic modifications of methionine oxidation and protein N-terminus acetylation, static modification of cysteine carbamidomethylation. Only proteins with a false-discovery rate ≤1% were taken into account in the analysis. Normalization was performed based on total peptide amount. To analyze the statistical significance of changes observed in protein abundance, ANOVA was used. The Benjamini–Hochberg method was used to correct P-values for multiple testing. Principal component analysis (PCA) was performed in Proteome Discoverer using abundance of all quantified proteins. STRING (https://string-db.org) was used to assess functional enrichments within the samples and UniProtKB to investigate the role of the proteins.
Results
Synthesis of surface-modified MSNs
Surface- and core-modified MSNs (amines on the surface and thiols in the core) were synthesized through a multistep, delayed cocondensation method.20 The thiol groups were incorporated in the core to allow covalent coupling of fluorescent dyes in the core of the particle without interfering with surface chemistry. The surface amines were used for further coupling with PEG linkers. To characterize the MSNs, SEM, TEM, fluorescence microscopy, and dynamic light-scattering analyses were performed. Monodisperse, spherical, and evenly shaped MSNs were confirmed by SEM (Figure 1A), and with TEM the mesoporous structure of the MSNs was visualized (Figure 1B). The presence of the amine (surface) and thiol (core) groups within the MSNs was confirmed by ζ-potential measurements (Figure 1C) and fluorescent labeling with ATTO 633–maleimide and FITC-NHS coupled to the thiol and amine groups of MSNs, respectively (Figure S1A). From these MSNs, four surface-functionalized MSNs were synthesized (Figure 1D): MSNs with supported lipid bilayers (MSN-Lip), MSNs with PEGylated supported lipid bilayers (MSN-Lip-PEG2,000), MSNs surface-functionalized with PEG (MSN-PEG2,000), and MSNs surface-functionalized with short PEG chains (MSN-PEG750).
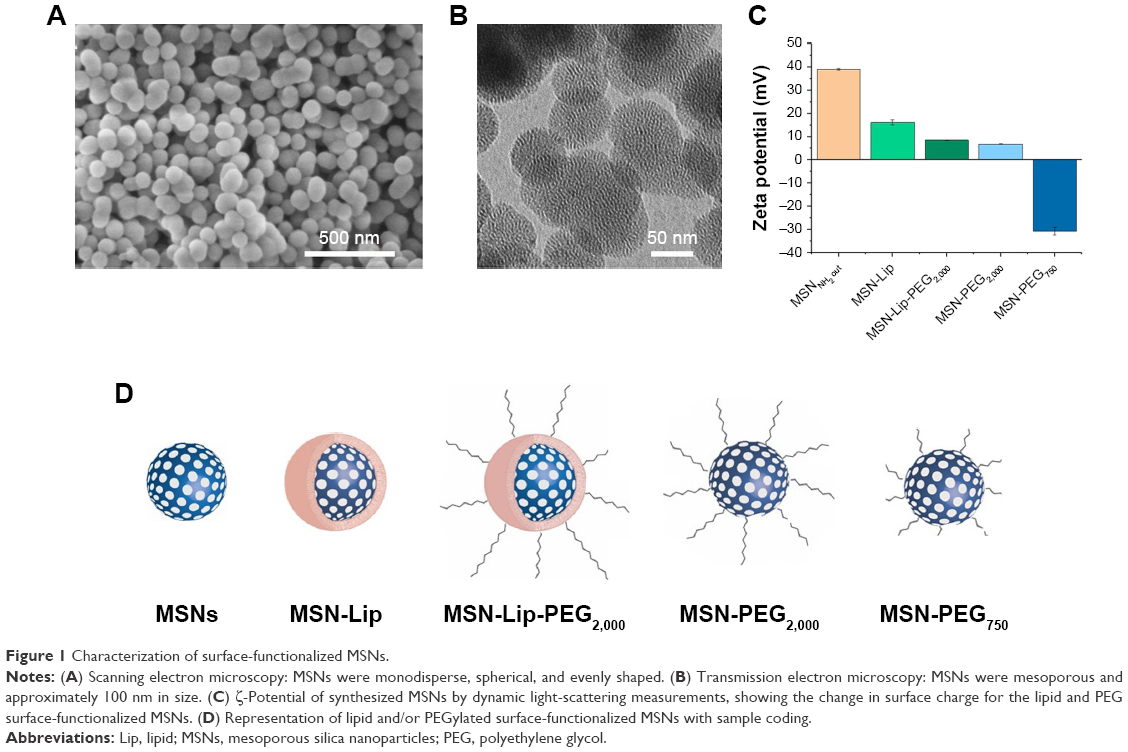 | Figure 1 Characterization of surface-functionalized MSNs. |
To synthesize MSN-Lip, previously reported solvent-exchange methods were used.21 Lipids consisted of either 100% DOPC or a combination of DOPC with PC-PEG2,000 in a 95:5 ratio. To prove that MSNs were indeed functionalized with the lipid bilayer, MSNs were labeled in the core using maleimide ATTO 633 and a fluorescently labeled lipid was included in the bilayer consisting of DOPC:PC TopFluor 488 (99.96:0.04).
MSN-Lip-PEG2,000 were synthesized using the same methods, and included PC-PEG2,000 lipids in the bilayer (1:6 ratio; PC-PEG2,000:DOPC). With fluorescence microscopy, colocalization of the MSNs and surrounding lipid bilayer were observed, thus confirming the success of the MSN-Lip formation (Figure S1B). In addition, surface ζ-potential changed significantly after lipid functionalization from 39±0.4 for unfunctionalized to 16.2±1.1 for MSN-Lip and 8.5±0.04 for MSN-Lip-PEG2,000 (Figure 1C), and their sizes increased (Table 1), further proving successful functionalization.
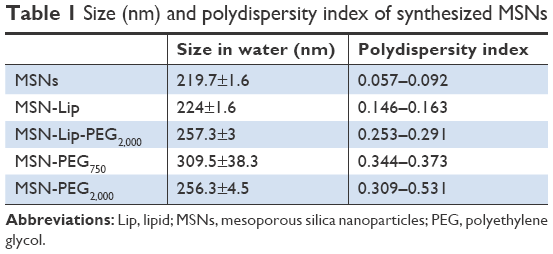 | Table 1 Size (nm) and polydispersity index of synthesized MSNs |
Finally, to functionalize the MSNs with short PEG chains (ie, 750 MW) and long PEG chains (ie, 2,000 MW), surface amines were coupled with NHS-functionalized PEG linkers. All synthesized MSNs efficiently bound fluorescent dyes via maleimide coupling. PEGylation of the MSNs led to a significant reduction in ζ-potential of the MSNs (Figure 1C). MSNs functionalized with PEG2,000 chains had a ζ-potential of 6.8±0.1, a similar value to that observed for MSN-Lip-PEG2,000, whereas PEGylation with the shorter chains led to a negative ζ-potential of −30.8±1.6. This may have been due to higher surface PEGylation efficiency with the lower-MW PEG compared to the more sterically hindered PEG2,000 chains. The effect of surface functionalization with PEG chains on lowered ζ-potential has also previously been observed by He et al.23 Also, their size increased significantly compared to the unfunctionalized MSNs, further confirming their successful functionalization (Table 1). SEM was used additionally to study the morphology of MSN-Lip, MSN-Lip-PEG2,000, MSN-PEG750, and MSN-PEG2,000 (Figure S2). No noticeable differences were observed between PEG- and Lip-functionalized MSNs compared to non-surface-functionalized MSNs. Polydispersity-index values for MSNs and Lip- and PEG-functionalized MSNs showed that all synthesized particles were monodisperse (Table 1).
In summary, MSNs containing thiol groups in the core and amine groups on the surface were successfully synthesized. These MSNs confined fluorescent dyes in their core and were surface functionalized with lipid bilayers (MSN-Lip, MSN-Lip-PEG2,000) or PEG modifications (MSN-PEG750 and MSN-PEG2,000), to create four surface-modified labeled MSNs.
MSNs did not influence hMSC metabolic rate
Possible effects of the MSNs on primary hMSC-proliferation rates were investigated using the MTT metabolic activity assay to measure the growth rate of cells. Cells from two donors (D313 and D133A) were used to account for possible donor variability. hMSCs were exposed to MSNs for either 24 or 72 hours at concentrations of 10–250 μg/mL. No dose–response effects on metabolic activity were observed for either of the donors after 24 or 72 hours of exposure to the MSNs (Figures 2 and S3). However, for donor 133A, a significant mild reduction in metabolic activity was observed for MSN-PEG750 (Figure S3A). Furthermore, the addition of MSNs to hMSCs did not influence cell morphology (Figure S4). From these data, it can be concluded that non-surface-modified MSNs and lipid and PEG2,000-modified MSNs did not influence cell-proliferation rates of hMSCs up to high concentrations in two donors. For further labeling studies, we chose to expose cells to 25 μg/mL MSNs, as this dose was atoxic and provided proficient MSNs to be taken up by the hMSCs.
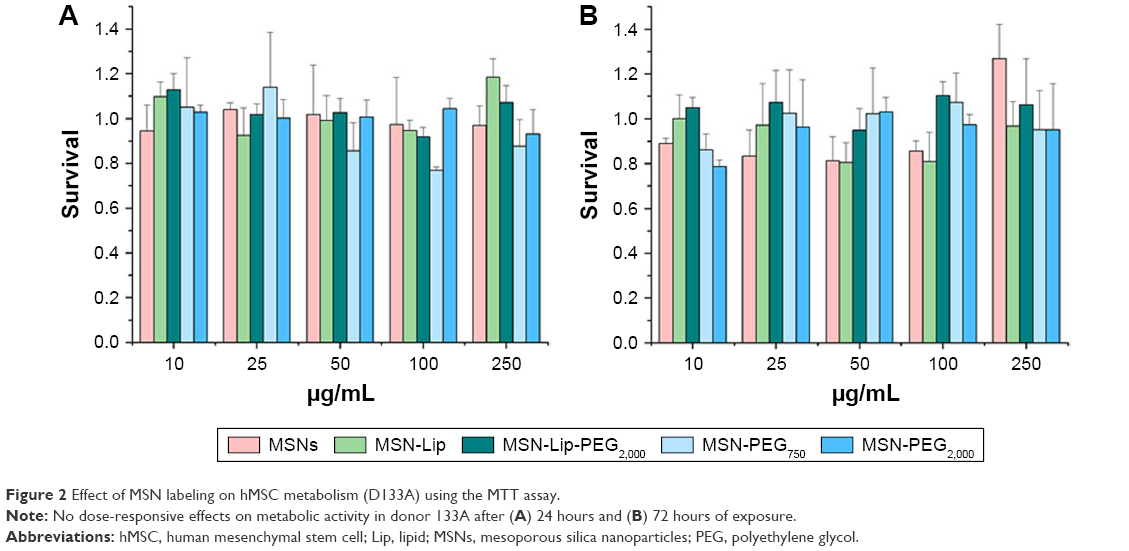 | Figure 2 Effect of MSN labeling on hMSC metabolism (D133A) using the MTT assay. |
Organic surface modifications increased MSN labeling of hMSCs
Intracellular uptake of MSNs, MSN-Lip, MSN-Lip-PEG2,000, MSN-PEG750, and MSN-PEG2,000 by hMSCs was quantitatively assessed using flow cytometry. In short, MSNs were first covalently labeled in the core with a far-red fluorescent dye (ATTO 647N), so as not to affect their surface properties. Over 90% of the hMSC population was labeled with MSNs within 24 hours, with the exception of MSN-PEG750. In particular, after only 2 hours of exposure, there was substantial MSN uptake by hMSCs: MSNs (81.3%), MSN-Lip (96.0%), MSN-Lip-PEG2,000 (91.5%), and MSN-PEG2,000 (87.0%; Figure 3A). Particle uptake increased after 24 and 48 hours for MSNs (95%) and MSN-PEG2,000 (93.0%), whereas the uptake of lipid-modified MSNs remained stable (Figure 3B). Negatively surface-charged MSN-PEG750 were only taken up by a small fraction of cells (17% after 2 hours and 22.1% after 48 hours). These observations were reproducible in the same donor (Figure 3B). A similar trend in particle uptake was observed in a second donor; however, in this donor both PEGylated MSN types were taken up to a lesser extent (D313, Figure 3B).
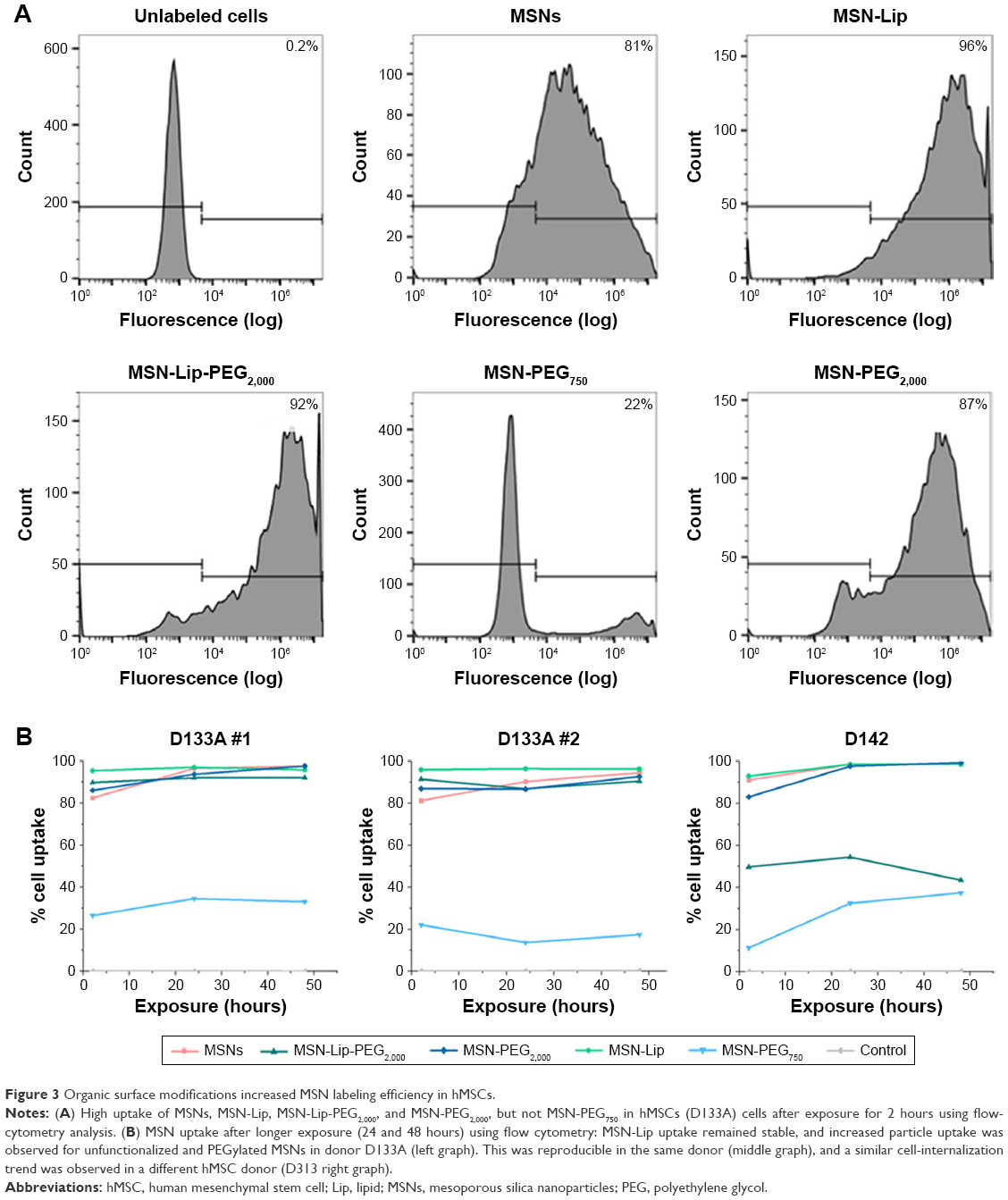 | Figure 3 Organic surface modifications increased MSN labeling efficiency in hMSCs. |
Although the degree of hMSC labeling was similar for MSNs and MSN-Lip (80% and 96%, respectively), MNS-Lip had a fluorescent signal approximately 17 times higher after 2 hours of exposure compared to non-surface-modified MSNs (Figure 4A). This suggests that the amount of MSN uptake per cell and thus signal strength was significantly higher for MSN-Lip compared to MSNs. Additionally, PEGylated MSNs (MSN-Lip-PEG2,000 and MSN-PEG2,000) were also internalized more than MSNs (Figure S5A). After 24 hours, this trend was still visible, though less noticeable (around a fourfold difference in uptake; Figure 4, right). This observation was reproducible in the same donor and in a second donor (D313; Figure S5B).
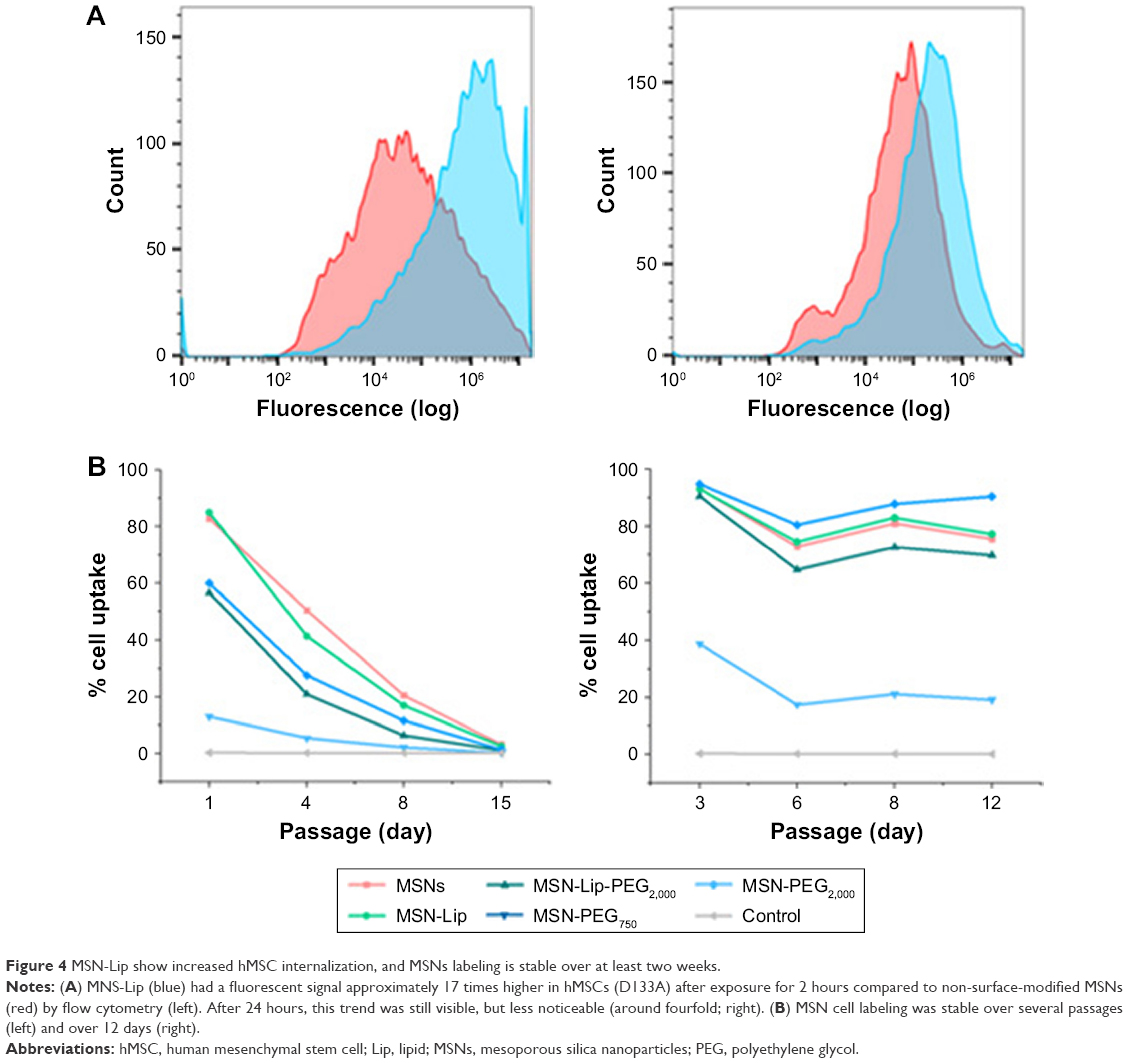 | Figure 4 MSN-Lip show increased hMSC internalization, and MSNs labeling is stable over at least two weeks. |
In summary, MSNs and surface-modified MSNs were internalized to a high extent by hMSCs, with the exception of MSN-PEG750. Lipid-modified MSNs were taken up more efficiently (96% of the population within 2 hours), and in addition were taken up several fold more than non-surface-functionalized MSNs.
MSN labeling stable over at least 2 weeks
Next, we examined the labeling capabilities of the nanoparticles in hMSCs (donor 133A) over several cell passages. For this, hMSCs were exposed to the different surface functionalized MSNs for 2 hours, the nanoparticles were removed from the medium, and flow cytometry was performed on days 1, 4, 8, and 15. Half the cells were used for flow cytometry, while the other half were used for further cell seeding. Therefore, half the nanoparticles were lost at every seeding. Using flow cytometry, we indeed observed that at each passage, the signal was halved due to the sample division, resulting in the loss of signal after about 15 days (Figure 4B); however, no additional nanoparticles were lost. These data were compared to the labeling capabilities of the MSNs when hMSCs were not being passaged. In this setup, hMSCs were plated so that they would reach confluence after 3, 6, 8, or 12 days. After exposure for 2 hours, the remaining MSNs were again washed away. Apart from the relatively small fluctuations in cell uptake, we observed that the nanoparticles were able to remain in the hMSCs for up to 12 days after exposure (Figure 4B) without any loss of signal. Even though the cells were plated at different seeding densities, the percentage of labeled cells remained the same throughout the different time points, indicating that proliferating labeled cells passed the labels on to their daughter cells. In summary, these two experiments showed that MSN-labeled hMSCs were stable over several weeks and that the signal strength of the labeled cells was proportional to the extent of nanoparticle uptake.
hMSC internalization of MSNs
Confocal and live-cell fluorescence microscopy were utilized to visualize the intracellular presence of MSNs in hMSCs. Live-cell imaging of labeled MSNs in hMSCs confirmed efficient cell uptake within several hours of exposure. As soon as after 40 minutes, active MSN-Lip uptake by the hMSCs was observed (Figures 5A and S6A). A z-stack image of hMSCs exposed to MSNs showed the intracellular presence of small MSN aggregates (Figure S6B). This was confirmed when reconstructing a 3-D image of an hMSC-labeled cell using confocal microscopy (Figure 5B). Therefore, MSNs were actively taken up by live cells after 40 minutes of incubation and allowed live-cell imaging.
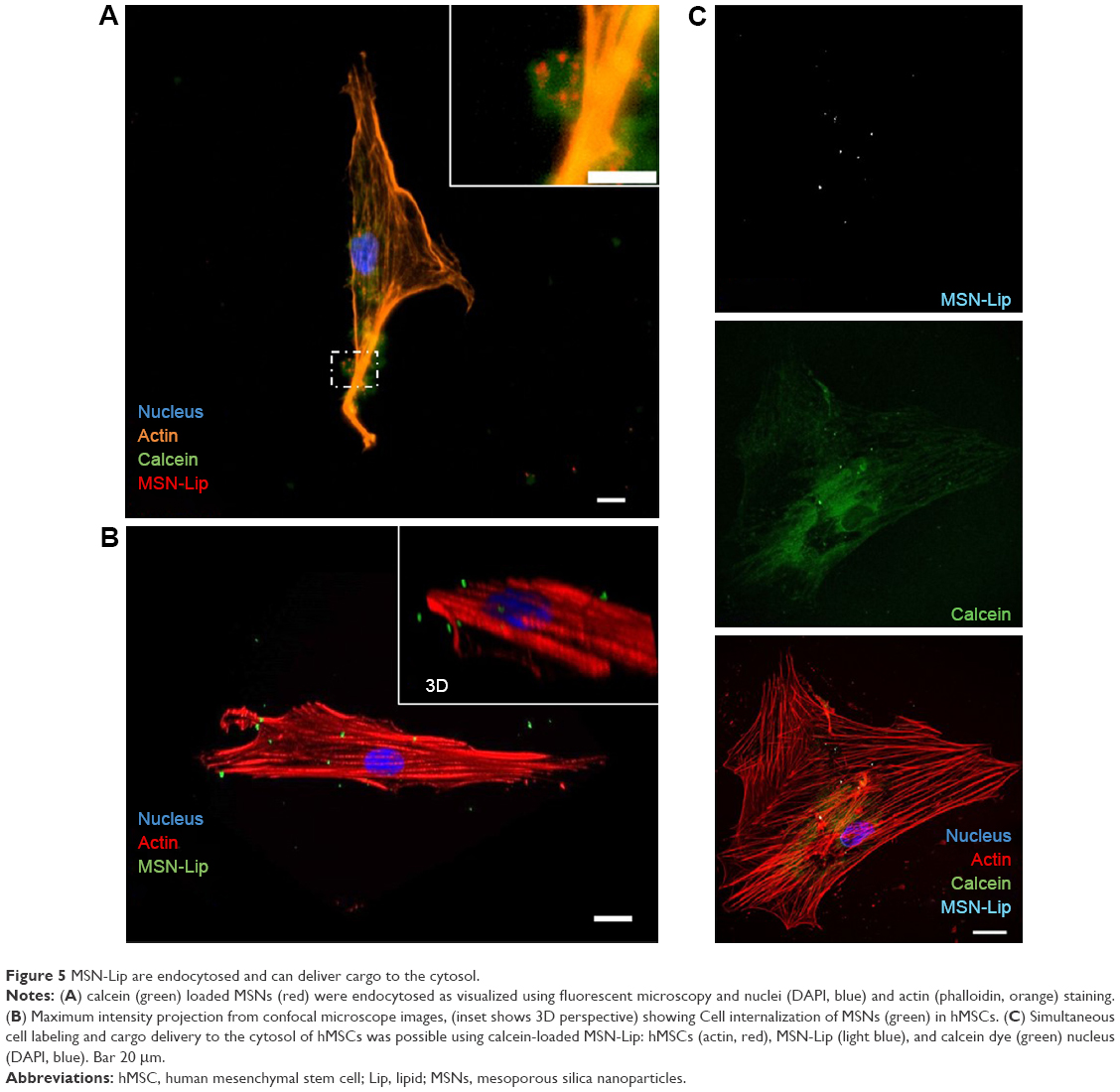 | Figure 5 MSN-Lip are endocytosed and can deliver cargo to the cytosol. |
MSN-Lip as cell-tracking and cargo-delivery platforms
Next, we assessed whether synthesized MSN-Lip could be used as multifunctional probes for the tracking of stem cells and simultaneous delivery of cellular probes. The lipid layer around the MSNs has been used as an efficient capping agent to prevent diffusion of encapsulated drugs out of mesoporous particles for controlled drug delivery to cancer cells.17 Overlay of lipid-labeled and MSN core-labeled particles revealed that the lipid layer remained intact upon hMSC MSN-Lip uptake (Figure S6C), showing that the lipid layer could thus function as a capping agent. To test whether MSN-Lip could also carry and intracellularly release additional cargo in mesopores without interfering with core labeling, a fluorescent dye (calcein) was encapsulated in the pores of labeled MSN-Lip and exposed to hMSCs. Intracellular calcein release with local diffusion within the cytoplasm was observed within 24 hours of exposure (Figure 5C), showing that these nanoparticles could be used to carry additional probes or other biomolecules into MSCs. In summary, the lipid layer remained intact after MSN-Lip uptake and was able to deliver cargo into hMSCs.
Effect of MSNs on osteogenic differentiation of hMSCs
To assess whether there was an effect of MSNs on hMSC ability to differentiate into the osteogenic lineage, ALP levels were quantified over time. Accordingly, hMSCs were exposed to low (25 μg/mL) and high levels (75 μg/mL) of MSNs and MSN-Lip and cultured in either basic or osteogenic medium. ALP activity was measured after 1, 7, and 14 days of culture. ALP activity was significantly increased for all conditions after 14 days for MSCs grown in osteogenic medium. There was no significant difference in ALP activity between MSN-Lip-labeled MSCs and control MSCs for either concentration (Figure 6). There was, however, a significant increase in ALP activity for MSCs exposed to unfunctionalized MSNs that was concentration dependent: a 1.3-fold increase was observed for MSCs exposed to MSNs at the low concentration (25 μg/mL, P=0.04) and a 1.5-fold increase in ALP activity for MSCs exposed to MSNs at the high concentration (75 μg/mL, P=0.02) compared to control cells. Therefore, MSN-Lip labeling did not affect ALP activity, whereas non-surface-modified MSNs led to a significant concentration-dependent increase in ALP activity after 14 days (Figure 6).
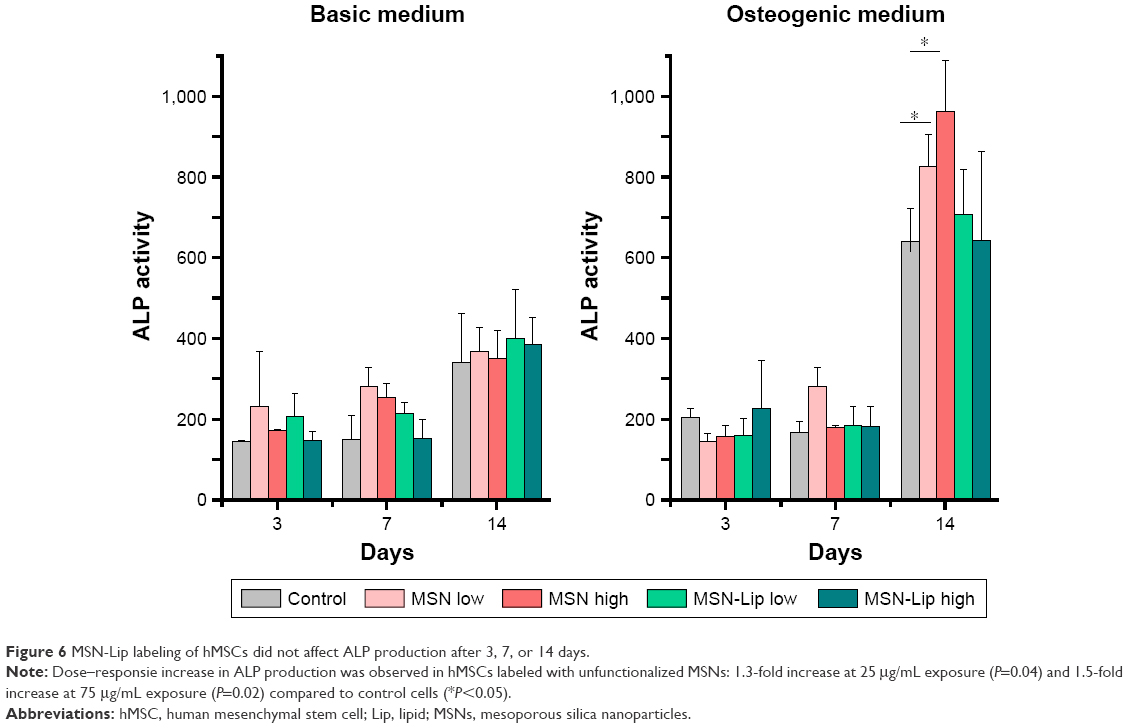 | Figure 6 MSN-Lip labeling of hMSCs did not affect ALP production after 3, 7, or 14 days. |
Effect of MSNs on hMSC proteome
To investigate the effect of MSN labeling on MSC biological processes, an MS-based label-free quantitative proteomic approach was used. For this, the proteome of hMSCs exposed for 24 hours to unfunctionalized and functionalized MSNs (MSN-Lip, MSN-Lip-PEG2,000, MSN-PEG2,000, and MSN-PEG750) was analyzed. PCA was used to assess the similarity of protein profiles across samples and determine abundance variation between samples (Figure 7). The PCA plot revealed that although no separation of MSN-labeled cells and control cells was clearly visible in PC1, a clear distinction between MSN, MSN-Lip- and MSN-PEG750-labeled cells and control cells was visible on PC2. Indeed, MSN labeling resulted in significant up- and downregulation of several proteins, which were mostly specific for the differently surface-modified MSNs (Figure 7B and C and Tables S1–S4). For example, fibromodulin, involved in fibrin formation and extracellular matrix production (UniProtKB), was upregulated only in MSN-Lip treated cells (2.6-fold). Furthermore, endophilin A1, an endosomal protein involved in clathrin-mediated endocytosis, was also upregulated in lipid-modified MSNs (2-1-fold), which may explain the increased uptake for this nanoparticle. These data indicate that MSN-Lip may be taken up by actin-dependent mechanisms. Indeed, functional enrichment analysis (STRING) showed that hMSCs labeled with MSN-Lip were enriched for cytoskeletal, cytosol, and extracellular region proteins (Gene Ontology [GO] cellular component, Table S2) compared to unlabeled control cells. Furthermore, two GO molecular functions were enriched in these cells: structural molecular activity and structural constituents of cytoskeleton (Table S5).
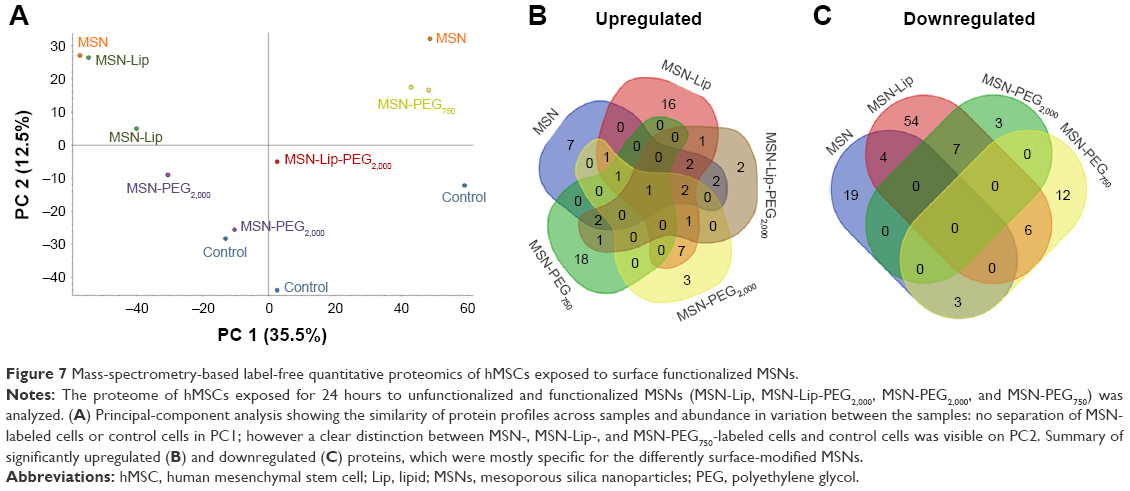 | Figure 7 Mass-spectrometry-based label-free quantitative proteomics of hMSCs exposed to surface functionalized MSNs. |
Functional enrichments were also identified in cells exposed to MSNs and MSN-PEG750 (STRING, Tables S6 and S7, respectively). Surprisingly, hMSCs labeled with non-surface-modified MSNs resulted in the downregulation of several ribosomal constituents, as well as rRNA- and RNA-binding proteins that were intricately connected (GO molecular function, Tables S5 and S6 and Figure S7). The ribosome pathway (Kyoto Encyclopedia of Genes and Genomes) and the involvement of several metabolic, transport, and biosynthetic processes were identified (GO biological process). On the other hand, MSCs labeled with MSN-PEG750 led to the downregulation of mostly extracellular region proteins and exosomes (GO cellular component, Table S6), which were associated with several biological processes such as skin and tissue development (GO biological processes).
The proteome of MSN-PEG2,000-labeled hMSCs could not be separated from the proteome of control cells in PC1 or PC2, but did separate from the proteome of MSN-, MSN-Lip- and MSN-PEG750-labeled hMSCs. In total, 15 proteins were upregulated and eleven proteins downregulated in MSN-PEG2,000-labeled cells and no functional enrichments could be identified. This was also observed for MSN-Lip-PEG2,000-labeled hMSCs (Figure 7), indicating that nanoparticle-surface PEGylation prevented functional enrichment of certain types of proteins or pathways.
Several identical proteins were identified across MSN-labeled hMSC proteomes (Table S7). For example, in all MSN-treated cells, nuclear receptor corepressor 2, a transcriptional corepressor, was significantly upregulated. Isoform 2 of FHL2, an inhibitor of transcriptional activity of FOXO1 and its apoptotic function (UniProtKB), was significantly upregulated in MSN-, MSN-Lip-, and MSN-Lip-PEG2,000-exposed cells (2.6-, 3.7-, and 2.1-fold, respectively). Involved in microtubule activation and a positive regulator of proliferation and protein transport, KIF20B was upregulated in all samples, except MSN-Lip-PEG2,000 (1.8-fold to double). There were several proteins that were significantly upregulated in cells labeled with lipid or PEG2,000 surface-modified MSNs, which were significantly downregulated in MSN-PEG750-treated cells. These included inactive PTK7, which is involved in the Wnt-signaling pathway and plays a role in multiple cellular processes, including polarity and adhesion, and JAKMIP1, a transporter protein. Also, in the PCA plot, MSN-PEG750-labeled hMSCs were clearly separated from MSN-Lip-labeled hMSCs in PC1 and MSN-PEG2,000 in PC2. These data indicated that in addition to being internalized to a much lower extent, MSN-PEG750 were also differently processed compared to MSN-Lip and MSN-PEG2,000.
In summary, MSN labeling resulted in the up- and downregulation of proteins that were mostly specific for the differently surface-modified MSNs. In addition, functional enrichments were found in hMSCs labeled with MSNs, MSN-PEG750, and MSN-Lip, but not in hMSCs labeled with MSN-PEG2,000. This shows that surface modifications played a role in the biological processing of these nanoparticles and may be an important parameter in nanoparticle design.
Discussion
Surface coating and modification of nanoparticles is a popular strategy to improve nanoparticle stability, biocompatibility, or assign a desired bioactivity, such as improved cell internalization. The labeling efficiency of non-surface-functionalized MSNs in hMSCs has previously been demonstrated.11,24 Mesoporous silica coating has even been used to improve cell internalization and consequently the signal strength of magnetic nanoparticles in magnetic resonance imaging25 and gold nanorods in photoacoustic imaging.26 Here, we showed that lipid-bilayer surface modifications of MSNs led to increased signal strength compared to non-surface-modified MSNs, with up to 17-fold difference in particle uptake after 2 hours. This is an important observation, as gradual loss of signal due to cell division has been described as one of the biggest drawbacks of superparamagnetic iron oxide nanoparticles for magnetic resonance imaging.27 Also in our experiments, we observed that cell proliferation resulted in signal loss, as the internalized nanoparticles were passed on to their daughter cells after each division. Since no loss of nanoparticles without passaging of cells was observed, signal strength was directly related to extent of nanoparticle internalization.
Apart from higher uptake, lipid-surface modifications also led to higher labeling efficiency (maximum labeling within 2 hours) compared to non-surface-modified MSNs. In recent studies, it was shown that both the clathrin-mediated endocytic pathway and actin-dependent endocytosis are pivotal uptake mechanisms in MSN internalization by hMSCs.11,28 The upregulation of endosomal protein endophilin A1 in MSN-Lip-labeled cells may explain the increased uptake rate of MSN-Lip compared to non-surface-modified MSNs. Therefore, our results indicate that organic surface modifications with lipids are an effective strategy to significantly improve the signal strength and labeling efficiency of MSN-based cellular probes.
In our nanoparticle-based imaging platform, the fluorescent probes were confined to the core of the MSNs, leaving the possibility of delivering cellular probes or biomolecules via the porous silica shell. This can be used for the delivery of fluorescent probes for imaging of intracellular targets or signaling peptides and small-molecule drugs to modulate stem-cell fate.29 The lipid-bilayer surface modifications on the MSNs can be used as efficient gatekeepers to prevent cargo diffusion out of the particles. Indeed, the adhesion energy between the lipids and MSN surface suppresses fluctuations in the membrane bilayer, resulting in slow release of cargo.17,30 Here, we showed that fluorescently labeled MSN-Lip could escape endosomes and deliver an additional dye into the cytosol of hMSCs. Endosomal escape was also reported for unfunctionalized MSNs.31 The gatekeeping capabilities of the supported lipid bilayers thus open up the possibility of tracking stem cells while at the same time delivering biomolecules or an additional dye in a controllable fashion. Therefore, MSN-Lip represent a multifunctional platform that can combine drug or probe delivery together with live-cell imaging.
For stem-cell tracking, the biological effects of internalized nanoparticles are a very important consideration for their development, especially with regard to possible effects on the regenerative capabilities of labeled cells. Several studies have shown that nanoprobes can have a negative effect on the differentiation capabilities of stem cells. For example, it was reported that although polymer-coated superparamagnetic iron oxide nanoparticles did not affect cell viability, proliferation, adipogenic, or osteogenic differentiation, the nanoparticles did significantly reduce chrondrogenesis in hMSCs.32 Another study showed that quantum dots suppressed the expression of osteocyte-specific marker genes and ALP activity, but not cell growth or the morphology and mineralization of hMSCs.33 Here, we showed that MSN-Lip did not affect cell growth or ALP production when hMSCs were grown in osteogenic medium, even at higher nanoparticle concentrations. Concentration-dependent increased ALP production was observed at the protein level for non-surface-functionalized MSN-labeled hMSCs in osteogenic medium. Huang et al similarly showed that FITC-conjugated MSNs induced a significant increase of ALP mRNA levels after 1 hour’s osteogenic induction; however, they did not observe an effect on the long-term osteogenic capabilities of MSN-labeled hMSCs vs unlabeled hMSCs.34 This is in line with our observations and several other studies that also showed that MSNs did not have significant deleterious effects on the morphology, cell proliferation, cell viability, or differentiation efficiency of stem cells.10–13
A proteomic approach to characterize possible shifts in stem-cell activities upon MSN labeling has until now not been done. Here, we showed that several proteins were significantly up- and downregulated in labeled cells vs unlabeled cells. Most of the identified proteins were specific for each surface modification, supporting the notion that nanoparticle-surface chemistry plays an important role in their biological processing. In particular, in MSN-Lip-labeled hMSCs, cytoskeletal and extracellular region proteins were enriched and structural molecular activity and structural constituents of cytoskeleton molecular functions identified. The alteration of structural proteins may have been due to increased uptake of MSN-Lip compared to the other MSNs. In an earlier study, a link between actin organization and MSC internalization of MSNs was suggested.28 This did not lead to a significant difference in osteogenesis compared to control cells, which is also in line with our ALP findings. Surprisingly, hMSC labeling with non-surface-modified MSNs led to significant downregulation of several ribosomal units that were closely connected (Figure S7). This was not observed for any of the other MSN-labeled hMSCs or reported previously.
A few upregulated proteins were identical across samples. The most notable was HL2, which was significantly upregulated across MSN-, MSN-Lip-, and MSN-Lip-PEG2,000-labeled hMSCs and is known to inhibit the transcriptional activity of FOXO1 and its apoptotic function by enhancing the interaction of FOXO1 with SIRT1 and FOXO1 deacetylation.35 Moreover, KIF20B, involved in microtubule activation and a positive regulator in proliferation and protein transport, and PTK7, involved in the Wnt-signaling pathway, which plays an important role in the regulation of MSC proliferation and differentiation,36 were both significantly upregulated in MSN-Lip- and MSN-PEG2,000-labeled hMSCs.
Therefore, surface modifications are important in determining the biological processing of MSN-based particles and can lead to significant alterations in protein expression, and in some cases enrichment of certain proteins or molecular pathways. Whether the upregulation of these proteins also leads to altered stem-cell behavior needs to be further investigated. In terms of proliferation or differentiation capabilities, we and others did not observe altered stem-cell behavior for MSN-labeled stem cells. PEG2,000 modifications did not result in any functional enrichment of protein sets, and may be a good strategy to avoid alterations to cellular pathways without greatly affecting signal strength. PEGylation of the lipid bilayer did lead to lower labeling efficiency and reduced internalization in one donor.
PEGylation with short PEG chains (MW 750), however, did not improve MSN-labeling capabilities. The different observed behavior was likely due to the negative surface charge of PEG750-coated MSNs. High positive charge has been correlated with fast cellular uptake that is in part independent of normal clathrin- and actin-dependent mechanisms.28 Also, the cellular processing of MSN-PEG750 differed significantly compared to unlabeled particles and also the other MSN-labeled hMSCs. In particular, extracellular protein components were enriched. PEGylation is a widely used surface coating to prevent protein adhesion and activation of the immune system, to enhance stability both in vitro and in vivo, but also to reduce cell internalization.37,38 Here, we show that PEGylation length is crucial in the biological processing of MSNs, where shorter PEG chains lead to lower metabolic activity of cells, reduced cell internalization, and enrichment of extracellular components.
Conclusion
Accurate and noninvasive stem-cell tracking to determine stem-cell destinations and fate is vital in order to optimize stem-cell-based therapies. MSNs have emerged as promising cell labeling tools, as they are biocompatible, resistant to cellular degradation, and, MSN labeling does not require genetic manipulation. Here, we show that MSN functionalization with a lipid shell and PEGylation can be used as a promising strategy to enhance MSNs’ labeling capabilities. In particular, we show that lipid modifications can optimize such probes in three distinct ways: significantly improved signal strength, a barrier for sustained release of additional probes, and improved stem-cell-labeling efficiency. Further PEG2,000 modifications of the lipid bilayer may be a good strategy to avoid alterations to cellular pathways or enrichment of certain proteins. These properties, combined with their inherent biostability, biocompatibility, and low immunogenicity,17,21 indicate that lipid-modified MSNs are promising multifunctional nanoplatforms for use in medical applications, including for stem-cell-tracking purposes.
Acknowledgments
We thank Dr Carmen Lopez for technical assistance with TEM imaging and Denis Beurden for technical assistance in the cell-biology assays. This research was in part made possible with the support of the Dutch Province of Limburg.
Disclosure
The authors report no conflicts of interest in this work.
References
Wang X, Wang Y, Gou W, Lu Q, Peng J, Lu S. Role of mesenchymal stem cells in bone regeneration and fracture repair: a review. Int Orthop. 2013;37(12):2491–2498. | ||
Accomasso L, Gallina C, Turinetto V, Giachino C. Stem cell tracking with nanoparticles for regenerative medicine purposes: an overview. Stem Cells Int. 2016;2016(1):7920358–23. | ||
Edmundson M, Thanh NT, Song B. Nanoparticles based stem cell tracking in regenerative medicine. Theranostics. 2013;3(8):573–582. | ||
Deb KD, Griffith M, Muinck ED, Rafat M. Nanotechnology in stem cells research: advances and applications. Front Biosci. 2012;17:1747–1760. | ||
Rosenholm JM, Zhang J, Linden M, Sahlgren C. Mesoporous silica nanoparticles in tissue engineering – a perspective. Nanomedicine. 2016;11(4):391–402. | ||
Dengler EC, Liu J, Kerwin A, et al. Mesoporous silica-supported lipid bilayers (protocells) for DNA cargo delivery to the spinal cord. J Control Release. 2013;168(2):209–224. | ||
Bharti C, Nagaich U, Pal AK, Gulati N. Mesoporous silica nanoparticles in target drug delivery system: a review. Int J Pharm Investig. 2015;5(3):124–133. | ||
Vallet-Regí M. Mesoporous silica nanoparticles: their projection in nanomedicine. ISRN Mater Sci. 2012;2012(4):1–20. | ||
Huang X, Teng X, Chen D, Tang F, He J. The effect of the shape of mesoporous silica nanoparticles on cellular uptake and cell function. Biomaterials. 2010;31(3):438–448. | ||
Hsiao JK, Tsai CP, Chung TH, et al. Mesoporous silica nanoparticles as a delivery system of gadolinium for effective human stem cell tracking. Small. 2008;4(9):1445–1452. | ||
Huang DM, Hung Y, Ko BS, Bs K, et al. Highly efficient cellular labeling of mesoporous nanoparticles in human mesenchymal stem cells: implication for stem cell tracking. FASEB J. 2005;19(14):2014–2016. | ||
Accomasso L, Cibrario Rocchietti E, Raimondo S, et al. Fluorescent silica nanoparticles improve optical imaging of stem cells allowing direct discrimination between live and early-stage apoptotic cells. Small. 2012;8(20):3192–3200. | ||
Accomasso L, Gallina C, Turinetto V, Giachino C. Stem cell tracking with nanoparticles for regenerative medicine purposes: an overview. Stem Cells Int. 2016;2016:1–23. | ||
Wani A, Muthuswamy E, Savithra GH, Mao G, Brock S, Oupický D. Surface functionalization of mesoporous silica nanoparticles controls loading and release behavior of mitoxantrone. Pharm Res. 2012;29(9):2407–2418. | ||
He Q, Zhang J, Shi J, et al. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials. 2010;31(6):1085–1092. | ||
Madni A, Sarfraz M, Rehman M, et al. Liposomal drug delivery: a versatile platform for challenging clinical applications. J Pharm Pharm Sci. 2014;17(3):401–426. | ||
Butler KS, Durfee PN, Theron C, Ashley CE, Carnes EC, Brinker CJ. Protocells: modular mesoporous silica nanoparticle-supported lipid bilayers for drug delivery. Small. 2016;12(16):2173–2185. | ||
Vallet-Regí M, Colilla M, Izquierdo-Barba I, Manzano M. Mesoporous silica nanoparticles for drug delivery: current insights. Molecules. 2017;23(1):E47:47. | ||
Yagüe C, Moros M, Grazú V, Arruebo M, Santamaría J. Synthesis and stealthing study of bare and PEGylated silica micro- and nanoparticles as potential drug-delivery vectors. Chem Eng J. 2008;137(1):45–53. | ||
Cauda V, Schlossbauer A, Kecht J, Zürner A, Bein T. Multiple core-shell functionalized colloidal mesoporous silica nanoparticles. J Am Chem Soc. 2009;131(32):11361–11370. | ||
Cauda V, Engelke H, Sauer A, et al. Colchicine-loaded lipid bilayer-coated 50 nm mesoporous nanoparticles efficiently induce microtubule depolymerization upon cell uptake. Nano Lett. 2010;10(7):2484–2492. | ||
Liu J, Stace-Naughton A, Jiang X, Brinker CJ. Porous nanoparticle supported lipid bilayers (protocells) as delivery vehicles. J Am Chem Soc. 2009;131(4):1354–1355. | ||
He Q, Zhang J, Shi J, et al. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials. 2010;31(6):1085–1092. | ||
Slowing I, Trewyn BG, Lin VS. Effect of surface functionalization of MCM-41-type mesoporous silica nanoparticles on the endocytosis by human cancer cells. J Am Chem Soc. 2006;128(46):14792–14793. | ||
Liu HM, Wu SH, Lu CW, Sh W, Cw L, et al. Mesoporous silica nanoparticles improve magnetic labeling efficiency in human stem cells. Small. 2008;4(5):619–626. | ||
Jokerst JV, Thangaraj M, Kempen PJ, Sinclair R, Gambhir SS. Photoacoustic imaging of mesenchymal stem cells in living mice via silica-coated gold nanorods. ACS Nano. 2012;6(7):5920–5930. | ||
van Rijt S, Habibovic P. Enhancing regenerative approaches with nanoparticles. J R Soc Interface. 2017;14(129):20170093. | ||
Chung TH, Wu SH, Yao M, et al. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles in 3T3-L1 cells and human mesenchymal stem cells. Biomaterials. 2007;28(19):2959–2966. | ||
Zhao X, Kim J, Cezar CA, et al. Active scaffolds for on-demand drug and cell delivery. Proc Natl Acad Sci USA. 2011;108(1):67–72. | ||
Epler K, Padilla D, Phillips G, et al. Delivery of ricin toxin a-chain by peptide-targeted mesoporous silica nanoparticle-supported lipid bilayers. Adv Healthc Mater. 2012;1(3):348–353. | ||
Chiu HY, Deng W, Engelke H, Helma J, Leonhardt H, Bein T. Intracellular chromobody delivery by mesoporous silica nanoparticles for antigen targeting and visualization in real time. Sci Rep. 2016;6:25019. | ||
Kostura L, Kraitchman DL, Mackay AM, Pittenger MF, Bulte JW. Feridex labeling of mesenchymal stem cells inhibits chondrogenesis but not adipogenesis or osteogenesis. NMR Biomed. 2004;17(7):513–517. | ||
Hsieh SC, Wang FF, Lin CS, Chen YJ, Hung SC, Wang YJ. The inhibition of osteogenesis with human bone marrow mesenchymal stem cells by CdSe/ZnS quantum dot labels. Biomaterials. 2006;27(8):1656–1664. | ||
Huang DM, Chung TH, Hung Y, et al. Internalization of mesoporous silica nanoparticles induces transient but not sufficient osteogenic signals in human mesenchymal stem cells. Toxicol Appl Pharmacol. 2008;231(2):208–215. | ||
Yang Y, Hou H, Haller EM, Nicosia SV, Bai W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. Embo J. 2005;24(5):1021–1032. | ||
Ling L, Nurcombe V, Cool SM. Wnt signaling controls the fate of mesenchymal stem cells. Gene. 2009;433(1–2):1–7. | ||
Verhoef JJ, Anchordoquy TJ. Questioning the Use of PEGylation for Drug Delivery. Drug Deliv Transl Res. 2013;3(6):499–503. | ||
Otsuka H, Nagasaki Y, Kataoka K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv Drug Deliv Rev. 2012;64:246–255. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.