Back to Journals » International Journal of Nanomedicine » Volume 14
Lipid–polymer hybrid nanoparticles as a next-generation drug delivery platform: state of the art, emerging technologies, and perspectives
Authors Mukherjee A ![]() , Waters AK, Kalyan P
, Waters AK, Kalyan P ![]() , Achrol AS, Kesari S
, Achrol AS, Kesari S ![]() , Yenugonda VM
, Yenugonda VM
Received 15 December 2018
Accepted for publication 22 January 2019
Published 19 March 2019 Volume 2019:14 Pages 1937—1952
DOI https://doi.org/10.2147/IJN.S198353
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Venkata Yenugonda
Views: 10010
Anubhab Mukherjee,1,2 Ariana K Waters,1,2 Pranav Kalyan,3 Achal Singh Achrol,2 Santosh Kesari,2 Venkata Mahidhar Yenugonda1,2
1Drug Discovery and Nanomedicine Research Program, 2Department of Translational Neurosciences and Neurotherapeutics, John Wayne Cancer Institute, Pacific Neuroscience Institute, Providence Saint John’s Health Center, Santa Monica, CA, USA; 3Agoura High School, Agoura Hills, CA, USA
Abstract: Lipid–polymer hybrid nanoparticles (LPHNPs) are next-generation core–shell nanostructures, conceptually derived from both liposome and polymeric nanoparticles (NPs), where a polymer core remains enveloped by a lipid layer. Although they have garnered significant interest, they remain not yet widely exploited or ubiquitous. Recently, a fundamental transformation has occurred in the preparation of LPHNPs, characterized by a transition from a two-step to a one-step strategy, involving synchronous self-assembly of polymers and lipids. Owing to its two-in-one structure, this approach is of particular interest as a combinatorial drug delivery platform in oncology. In particular, the outer surface can be decorated in multifarious ways for active targeting of anticancer therapy, delivery of DNA or RNA materials, and use as a diagnostic imaging agent. This review will provide an update on recent key advancements in design, synthesis, and bioactivity evaluation as well as discussion of future clinical possibilities of LPHNPs.
Keywords: lipid–polymer hybrid nanoparticle, lipid-based nanoparticle, polymer-based nanoparticles, drug delivery, gene delivery
Introduction
Nanotechnology is a compelling medicinal platform with the potential to greatly impact the delivery of a plethora of therapeutics, encompassing small molecule therapeutics, genes, RNAs, peptides, and diagnostic imaging agents, as well as holding great promise for improving the therapeutic index and pharmacokinetics of several drugs under systemic settings.1–4 In general, these payloads are encapsulated within or covalently grafted on the surface of the nanocarriers, and after being systemically incorporated, their release is monitored by factors such as formulation of the matrix, pH of the microenvironment, and temperature of the surroudings.5–7 The inherent potential of nanoparticles (NPs) for therapeutic cargo delivery is primarily attributable to few key parameters, including average nanometric size, homogeneity, surface potential, and drug loading, among others.8,9 Surface-coated immuno-inert NPs can also skillfully bypass the reticuloendothelial system yielding increased bioavailability of encapsulated drugs.10 The plausible advantages of nanocarriers are summarized as follows: 1) improvement to a drug’s overall pharmacokinetic and pharmacodynamic properties without alteration of its molecular structure; 2) enhanced effective tissue targeting, cellular targeting, and molecular targeting; 3) the ability to circumvent many inherent biological impediments; 4) targeted and nontargeted drug delivery to their respective site of action (cytosol, nucleus, etc) and enhanced therapeutic index of the drug; 5) delivery of multiple drugs with differing chemical properties.11,12
In the last few decades, an increasing amount of nanotechnology-based products have begun clinical trials – including liposomes, polymeric NPs, albumin NPs, and inorganic NPs – of which a small number have already been accepted for clinical use.13–17 Among these nanocarrier types, the two best characterized are liposomes and polymeric NPs.
Liposomes are artificial “fat bubbles”, ie, vesicles, characterized by having one or more bilayers spontaneously achieved by dispersal of natural or synthetic amphiphatic lipids in water. Since their discovery, they have largely been exploited as delivery vehicles because of their biocompatibility and advantageous safety profile. Their surface can be modified by attaching polyethylene glycol (PEG) which, in turn, prolongs circulation half-life.18,19 Doxil and Myocet, two leading doxorubicin liposomes, received Food and Drug Administration (FDA) approval in 1995 and 1999, respectively, followed later by others of the same category.20 To date, around 16 liposomal drugs are clinically approved and a few of those are marketed, such as AmBisome (amphotericin B), DaunoXome (daunorubicin), DepoCyt (cytarabine), DepoDur (morphine), and Visudyne (verteporfin).15 Despite clinical approval, until recently no FDA-approved liposomal drugs showed significant overall survival (OS) improvement over the parent drug.21 In a 2017 study, phase III outcomes of liposomal combination drugs such as cytarabine–daunorubicin (Vyxeos; CPX-351), as contrasted with their individual counterpart cytarabine and daunorubicin (“7+3”) in 60- to 75-year-old patients with high-risk acute myeloid leukemia, revealed enhanced OS of 9.56 vs 5.95 months.22
Polymeric NPs, on the contrary, can be manufactured (via nanoprecipitation or the double emulsion method) by self-assembly of biodegradable amphiphilic block copolymers with varying hydrophobicities and are appropriate for systemic administration. The core–shell structure of polymeric NPs facilitates encapsulation of hydrophobic drugs and sustained drug release and extends circulation time. Their surfaces can also be decorated with ligands for targeted drug delivery.23,24 For instance, Genexol-PM is a polymer-based NP formulation of paclitaxel (PCX) and poly (D,L-lactide)-b-polyethylene glycol-methoxy (PLGA-mPEG), which has been approved for metastatic breast cancer therapy in Korea and the European Union.25,26
In order to utilize the unique attributes of liposomes and polymeric NPs that led to their initial clinical success, but overcome limitations like structural disintegration, limited circulation time, and content leakage, a new progeny of delivery system has been developed: lipid–polymer hybrid nanoparticles (LPHNPs).27 The hybrid system can be a sturdy drug delivery rostrum with high encapsulation efficiency, well-defined release kinetics, well-tolerated serum stability, and well-triggered tissue, cellular, and molecular targeting properties. In this article, we will review the emerging innovations of LPHNPs, incorporating new developments in their production strategies and drug delivery applications in cancer therapy.
Structure elucidation and mechanism of hybrid formation
As can be inferred from their name, LPHNPs merge the features of both polymeric NPs and liposomes. They consist of three building blocks as illustrated in Figure 1. These are 1) a polymer core encapsulating the drug, 2) a lipid monolayer surrounding the polymer core, and 3) an outer lipid–PEG layer, a steric stabilizer prolonging systemic circulation of the LPHNPs by evading immune destruction. The middle lipid monolayer behaves like a molecular barricade that mitigates the loss of entrapped drugs over the course of the LPHNP formulation and protects the core from degradation by preventing the diffusion of water into the inner core.27,28
 | Figure 1 Structure of a lipid–polymer hybrid nanoparticle (LPHNP) comprises of a polymer core encapsulating a pay load, a lipid shell, and an outer lipid–PEG layer. |
The molecular mechanics of fusion between lipid and polymer is still under investigation. It is apparent that distinguished methods of LPHNP manufacturing have different mechanisms of formation. For instance, in single-step methods, the polymer precipitates from the organic solvent when added to aqueous media containing lipids, which subsequently spontaneously self-assemble into a monolayer surrounding the core. PEGylated lipids also self-assemble during this step, wherein a lipid moiety clings onto the surface of the polymer core and the PEG chain extends externally toward the aqueous environment. During the two-step method, a plausible mechanism of LPHNP formation may involve an initial bilayer structure formation and adherence to the core, with subsequent disintegration of the bilayer owing to the hydrophobic interaction between polymer and lipid chains. The hybrid formation is thermodynamically favorable, with respect to hydrophobic, van der Waal, and electrostatic interactions.29,30
Methods for preparation of LPHNPs
Two-step method
Conventional method
During initial days of study, a typical two-step method was frequently employed to form LPHNPs, wherein preformed polymeric NPs were mixed with preformed lipid vesicles and the latter was surface-assimilated onto the former, propelled by electrostatic forces. The polymeric NPs are generally prepared by nanoprecipitation,31 emulsification–solvent evaporation (ESE),32 or high-pressure homogenization.33 As depicted in Figure 2, the two-step method can be subcategorized into two types: A) direct addition of the previously formed polymeric NPs to dried lipid film, or B) preformed NPs added to preformed lipid vesicles, made by initial hydration of the thin lipid film. In either case, the hybrids are assembled by the input of external energy via vortexing and/or ultrasonication of the suspension and heating at a temperature beyond the phase transition temperature of the lipid constituent. In purification step, free lipid and LPHNPs are separated by differential centrifugation. For instance, a method was developed for hybrid NP preparation using PLGA combined with cationic lipid vesicles (FA-OQLCS/PEG-OQLCS/Chol) under continuous stirring or bath sonication at 30°C, yielding stable LPHNPs with average sizes between 200 and 400 nm and a surface potential of (+) 20–30 mV.32,34 Using different precursors, Thevenot et al and Troutier et al utilized a similar method to make LPHNPs.31,35
 | Figure 2 The two versions of lipid–polymer hybrid nanoparticle preparation via the two-step method. |
In order to obtain a homogeneous NP suspension with unimodal particle distribution, hybrid NPs undergo sequential extrusion and/or homogenization after preparation. While extruding the sample, as normally practiced in the laboratory, the NP suspension is downsized by passing them through a porous membrane. For example, Messerschmidt et al created tumor necrosis factor (TNF)-functionalized hybrid NPs using polystyrene as the core and shell made up of egg phosphatidylcholine (egg-PC), cholesterol, DSPE–mPEG, and maleimide-DSPE–mPEG. The resultant NPs were downsized by extruding through a 200 nm porous membrane.36 This technique was also employed by Hu et al to construct a red blood cell membrane-coated NPs (~120 nm) and by Sengupta et al to produce chemotherapeutic LPHNPs (~200 nm).37,38 As a substitute, some other groups explored homogenization to formulate unimodal LPHNPs (~60 nm) made up of a maltodextrin polysaccharide core and a DPPC/Chol lipid shell.33,39
Nonconventional method
Aside from the abovementioned methodologies, few other methods going beyond convention have also been implemented to manufacture LPHNPs. For example, polymeric NPs (ie, polyglutamic acid, polylysine) of average size 400–500 nm were produced by spray drying, dispersed in DCM containing the lipids (tripalmitin, tristearin, cetyl alcohol). This suspension was later spray-dried again to prepare LPHNPs of size range 0.9–1.2 μm with aspray dryer that was inappropriate for the production of NPs.40 The recently marketed nanometric spray dryer can be used to produce smaller hybrid NPs as well.41
Additionally, in recent years, a particle molding method by soft lithography called particle replication in nonwetting templates (PRINT) was explored to prepare LPHNPs for the delivery of genetic materials.42
Optimization of formulation parameters
The physical characteristics of LPHNPs (average size, colloidal stability, polydispersity) prepared by the two-step method are majorly regulated by the following formulation parameters: 1) preformed lipid vesicle size and polydispersity index (PDI), 2) surface potential of the outer lipid shell, 3) ionic strength of the aqueous phase, 4) lipid-to-polymer ratio, and 5) PEG chain length and mol% of PEG–lipid.31,35 In particular, extrusion-produced particles are smaller and more homogeneous compared with that generated by direct lipid film hydration with polymer solution. Moreover, homogeneity of LPHNPs is critically dependent on the charge of the lipid vesicles. As such, least particle aggregation with high colloidal stability and narrow PDI was attained by using only one lipid type to form the vesicles, ie, only the zwitterionic lipid DPPC or the cationic lipid DPTAP. On the contrary, aggregation-prone LPHNPs have been formed when both DPTAP and DPPC were used, the mechanism of which could be attributed to the electrostatic interactions between the two types of lipids.35 Separately, hybrid particles’ colloidal stability was notably impacted by lipid to polymer ratio (AV/AP). At high lipid to polymer AV/AP and high cationic lipid concentration, the lipid layer may potentially act as electrostatic stabilizer. At low AV/AP and low fractions of cationic lipid, partial lipid coating of the polymer core was insufficient to stabilize the LPHNP. The anionic surface of the core of one hybrid molecule was exposed to the cationic DPTAP of another hybrid leading to the agglomeration of LPHNPs. Intriguingly, with DPPC alone, the hybrid NPs were less susceptible to agglomeration even at low AV/AP. This can again be credited to the zwitterionic nature of DPPC reducing the possibility of electrostatic interactions.35
Similar to liposomes, a drawback of LPHNPs is their weak colloidal stability relative to the ionic strength of the solution; electrostatic interactions alone cannot stabilize hybrid particles in aqueous medium with >10 mM NaCl ionic strength. Again, like liposomes, this is mitigated by incorporating conjugated PEG–lipids into the lipid mixture (DPPC/DPTAP) to impart stabilizing PEG chains onto the surfaces of the LPHNP. To envisage the most advantageous conditions to attain colloidal stability, Thevenot et al thoroughly investigated the effects of the PEG chain length (n=16, 45, and 113) and mol% (1%, 5%, and 10%) of PEG–lipid in lipid formulation, where the polymer core was made up of PLA and lipid layers were comprised of DPTAP, DPPC, and PEG–phosphoethanolamine (PEG–PE). Keeping the lipid composition unchanged (DPPC:DPTAP:PEG–PE=40:50:10% w/w), and upon gradual increase in the degree of polymerization in the PEG chain, they observed that the mol% of the PEG–lipid inhabiting the lipid shell decreased with stretching chain length from ~3% for n=16 and 45 to ~2% for n=113. The observation was ascribed to spatial repulsion between the bulky lipid–PEG shells with longer PEG chains. Importantly, thickness of the lipid layer adsorbed onto PLA particles was increased with the amount of PEG–lipids adsorbed or with the PEG chain length, from 67 Å at n=16 to 98 Å at n=113. This resulted in an increased charge screening effect, which subsequently lowered the zeta potentials from (+) 51 mV at n=16 to (+) 22 mV at n=113. This is owed to a transition from mushroom-like confirmation to outward brush-like confirmation. In accordance with the quasi-elastic light scattering (QELS) study, the n=16 PEG shell was not adequate enough to be effectively sterically stabilizing. The n=45 PEG shell showed moderate stabilization, while the n=113 PEG shell displayed the best colloidal stability for the hybrid NPs. Thickness of the lipid shell increased from ~52 Å at 1% PEG–PE to 79 Å at 10% PEG–PE, and this lowered the zeta potentials from (+) 47 to (+) 26 mV, respectively.31
Apart from average size, PDI, and surface potential (which is a measure of colloidal stability), other important formulation parameters are % drug loading (%DL), % encapsulation efficiency (%EE), and in vitro release kinetics of the drug from the LPHNPs. For example, Mieszawska et al demonstrated an %EE =85% w/w and %DL =11% w/w for their anticancer drug.32 Hasan et al also showed an %EE =32%–46% w/w for siRNA using the PRINT method.42 This is worth mentioning because for LPHNPs prepared by the two-step method, %EE of the drug is majorly determined by the formulation parameters of the polymeric core, which has been extensively explored in previous studies; similar trends are followed when the same polymer was used for the LPHNP formation.43
One-step method
The limitation of the two-step method is that preparing polymeric NPs and lipid vesicles separately makes the process inefficient in terms of energy and time spent. The commonly available and more efficient alternative is a one-step method. Preformed lipid vesicles and polymeric NPs are not prerequisites for the one-step method. The method solely requires mixing of lipid and polymer solutions that subsequently tend to self-assemble to form LPHNPs. The most common processes are nanoprecipitation and/or ESE, both of which are often implemented for the production of nonhybrid polymeric NPs. Here, the lipids/PEG–lipids used function as stabilizing agents for the hybrid produced, while ionic or nonionic surfactants (PVA, DMAB, poloxamer) are generally used as stabilizers in the preparation of regular, nonhybrid polymeric NP.
Nanoprecipitation
Traditional nanoprecipitation method requires that the drug and polymer are dissolved together in a water-miscible organic solvent (viz, acetone, EtOH) and the lipid/lipid–PEG dissolved in water. It is mandatory to heat the lipid/lipid–PEG solution beyond its gel-to-liquid transition temperature in order to achieve a homogeneously dispersed liquid crystalline phase. This is followed by drop wise addition of the polymer to the aqueous dispersion of lipid under continuous stirring. This triggers the polymer to coil into NPs with concurrent self-assemblage of the lipids surrounding the polymer owing to hydrophobic interactions, where hydrophobic tails of the lipids are directed toward the inner NP and the hydrophilic head groups face out toward the external aqueous solution. The hydrophobic lipid tails of the lipid–PEG merge into the inner lipid shell, while its PEG chains pop out to the aqueous environment, sterically stabilizing the hybrid.27 The organic media is evaporated and the LPHNPs, thus formed, are centrifuged (Figure 3). A promising noninvasive delivery of mRNA-based vaccines, developed recently, involves postinsertion of the PEGylated lipid vesicles following nanoprecipitation.44
 | Figure 3 Nanoprecipitation technique for the preparation of LPHNPs by one-step method. |
A few novel approaches are being adopted by a number of research groups to improve upon one-step methods. For example, using a bath sonication approach, Fang et al demonstrated a rapid synthesis (~5 minutes) of hybrid NPs with small and nearly uniform size.29 In 2010, Valencia et al reported the use of microfluidics, where two phases are rapidly mixed using hydrodynamic flow, directly generating homogeneous NPs with relatively narrow size distribution.45 Fang et al, again in 2012, came up with a multi-inlet vortex reactor for scale up of LPHNPs.46 To address the issues with low throughput of microfluidics, Kim et al developed a pattern-tunable microvortex platform for scale up of LPHNPs as well. The microvortex platform could restrict the average size of NP between ~30 and 170 nm with low PDI (~0.1) and high productivity (~3 g/hour) by varying flow rates (ie, Reynolds number [30–150]).47
Optimization of formulation parameters in nanoprecipitation
The unanimous formulation parameter indicated in many studies as having the most prominent effect on the characteristics of an LPHNP is the stoichiometry, ie, the lipid to polymer mass ratio (L/P ratio). In two consecutive studies performed in O C Farokhzad’s laboratory, the L/P ratio was optimized to be 15% (w/w) for optimal covering of the polymer, where PLGA was used as the polymer and lecithin plus DSPE–PEG as the lipids, to generate stable monodisperse LPHNPs (60–80 nm).27,48 This optimization was crucial as higher L/P (w/w) ratios led to lipid concentrations greater than the CMC (critical micelle concentration of the polymer), which could lead to the formation of liposomes in addition to hybrids, while lower L/P (w/w) ratios led to agglomeration. Similarly, cationic LPHNPs of average size ~65 nm were prepared by single-step nanoprecipitation of a cationic lipid (BHEM-Chol) and amphiphilic polymer mPEG–PLA for systemic delivery of siRNA, where optimal L/P ratio was reported to be 10% (w/w).49 Intriguingly, as demonstrated by Zheng et al, in the absence of lipid–PEG, a huge excess of lipid (egg-PC/DOPE lipid shells with D-α-TPGS) was needed in the formulation leading to an optimum L/P ratio of 428% (PLGA-to-lipid ratio of 3.5/15), to form hybrid NPs with relatively higher average size (150–190 nm).50
A salient feature of the lipid coating (reflected as L/P ratio), as discussed in structure elucidation, is to protect the polymer core. This indirectly regulates %EE and drug release.27 In contrast, for nonhybrid polymeric NPs, the polymer’s physicochemical property is the sole influence on %EE and release kinetics.51 At the optimal L/P ratio (15% w/w), LPHNPs showed higher %EE of the docetaxel (59%±4%) compared with its nonhybrid counterparts PLGA NPs (37%±4%) and PEGylated-PLGA NPs (19%±3%). A longer sustained release of the drug (50% in 20 hours) was shown by LPHNPs compared with that of PLGA and PLGA-PEG NPs (50% in 7 and 10 hours, respectively).27
The colloidal stability of LPHNPs (prepared either by one-step or two-step method) is contingent on the lipid–PEG present in the lipid formulation. In the absence of PEG-grafted lipid, despite reasonably high L/P ratio, lecithin-coated PLGA NPs were shown to be unstable when they formed large aggregates (~2 μm) in PBS, hinting at the inability of the lipid layer to stabilize the formulation.48 With the addition of DSPE–PEG, the LPHNPs became stable in PBS as the PEG chains conferred steric stabilization. By enhancing the PEGylated lipid in the lipid formulation, more stable hybrid NPs were yielded, where the optimal lipid–PEG amount was found at ~25% (w/w). Importantly, higher concentration of PEG on the particle surface did not alter the release kinetics or %EE.48,52,53
Nonetheless, PEG fraction present in the formulation does influence the average size of LPHNPs. For example, according to the study performed by Zheng et al, a decrease in the LPHNP size (from 230 to 150 nm) with increasing PEG fraction was observed.50 Be that as it may, other formulation parameters, viz, concentration and molecular weight of polymer, and water-to-solvent ratio have similar effects on the size of nonhybrid polymeric NPs as well as hybrid NPs.27,48,53
Emulsification–solvent evaporation
This method can further be subclassified into single and double emulsification methods as depicted in Figure 4A and B, respectively. A single ESE method (Figure 4A) is used for drugs soluble in hydrophobic solvents (oil phase). In this method, an oil-in-water (o/w) emulsion is formed when the water-immiscible oil phase containing the polymer and the drug is mixed with an aqueous phase containing dissolved lipid under ultrasonication or constant stirring. Next, the polymer core is formed by evaporation of the organic media and the lipids assemble around the polymer core concomitantly.54 As an ostensible replacement, the lipid can concurrently be dissolved in the oil phase with the polymer.55 A double ESE method (w/o/w) is applied for water-soluble drugs (Figure 4B). First, the aqueous solution of the drug is emulsified in an organic solvent (oil phase) containing polymer and lipid to form a w/o mixture. A w/o/w emulsion is generated when the mixture is emulsified again in an aqueous phase containing the lipid–PEG, followed by subsequent oil phase evaporation, to yield the LPHNPs.55 As evident from Figure 4B, the hybrids produced by the double ESE method contain certain structural anomalies. It is composed of 1) an inner aqueous core surrounded by lipid layer, 2) a polymer layer in between, and 3) an outer lipid–PEG shell. Generally, the ESE method produces LPHNPs that are larger than those produced by conventional nanoprecipitation.
 | Figure 4 LPHNPs produced by the emulsion-solvent evaporation (ESE) method. |
Optimization of formulation parameters in ESE
Resembling nanoprecipitation, L/P ratio is the most prominent among all formulation parameters to govern ESE method. Using PLGA, TPGS, and PC, Cheow and Hadinoto found a reduction in size with increase in L/P ratio and achieved a standard production yield (w/w).55 Liu et al achieved similar results with the single ESE method using PLGA and 1,2-dilauroylphosphatidylocholine (DLPC).56 Bershteyn et al demonstrated that excess lipid in the system (high L/P ratios) resulted in either multilamellarity of lipid layer or spontaneous precipitation of the lipids as liposomes.54 Here also, the L/P ratio was shown to have an influence on %EE. The optimal lipid concentration in terms of smaller size and high %EE was 0.04% (w/v).56 Interestingly, the ESE method typically resulted in higher %EE than that of nanoprecipitation due to larger particles produced. For example, polymer remaining same, nanoprecipitation yielded %EE ~20% (w/w) for PCX in ~50–60 nm LPHNPs,48 while ESE method yielded %EE ~60% (w/w) in ~200–300 nm particles.56 In 2011, a novel one-pot synthetic method was developed for ESE. Using PLGA as the core, and either cetyltrimethylammonium bromide or PEG–DSPE as an emulsifier, hybrid particles of ≥50 nm were produced, which also possessed high %EE.57 Cheow and Hadinoto, exploiting fluoroquinolone antibiotics as a model, proved that ionic interactions between the drug and the lipid are crucial in the preparation of LPHNPs by ESE method.55
Advances in drug delivery applications of LPHNPs
Given the large number of potential drug delivery applications that exist, there is great interest in exploring uses of the LPHNP platform in future clinical studies. Here, we discuss few important recent advancement made in the field and areas of future interest.
Nontargeted combinatorial drug delivery approach
Besides having an array of single drug delivery strategies via LPHNPs (eg, in chemotherapy27,48,56 and antibiotic treatment58–60), significant endeavors have been directed toward development of a combinatorial approach for cancer therapeutics. Truly effective cancer treatment regimens often require multiple chemotherapeutic drugs used in tandem or chemotherapeutic drugs administered in combination with targeting therapeutic agents.61
However, a pertinent question arises as to how best to maximize the efficacy of NP-mediated delivery of therapeutic payload: codelivery from a single nanocarrier or two different carriers? It has been proposed by many that a single biocompatible nanocarrier capable of carrying more than one agent in particular stoichiometry and releasing them in a sustained way may hold the most promise. We provide a brief account of several strategies that have already been employed to accomplish successful combinatorial drug delivery exploring the potential of LPHNP platform (Table 1).
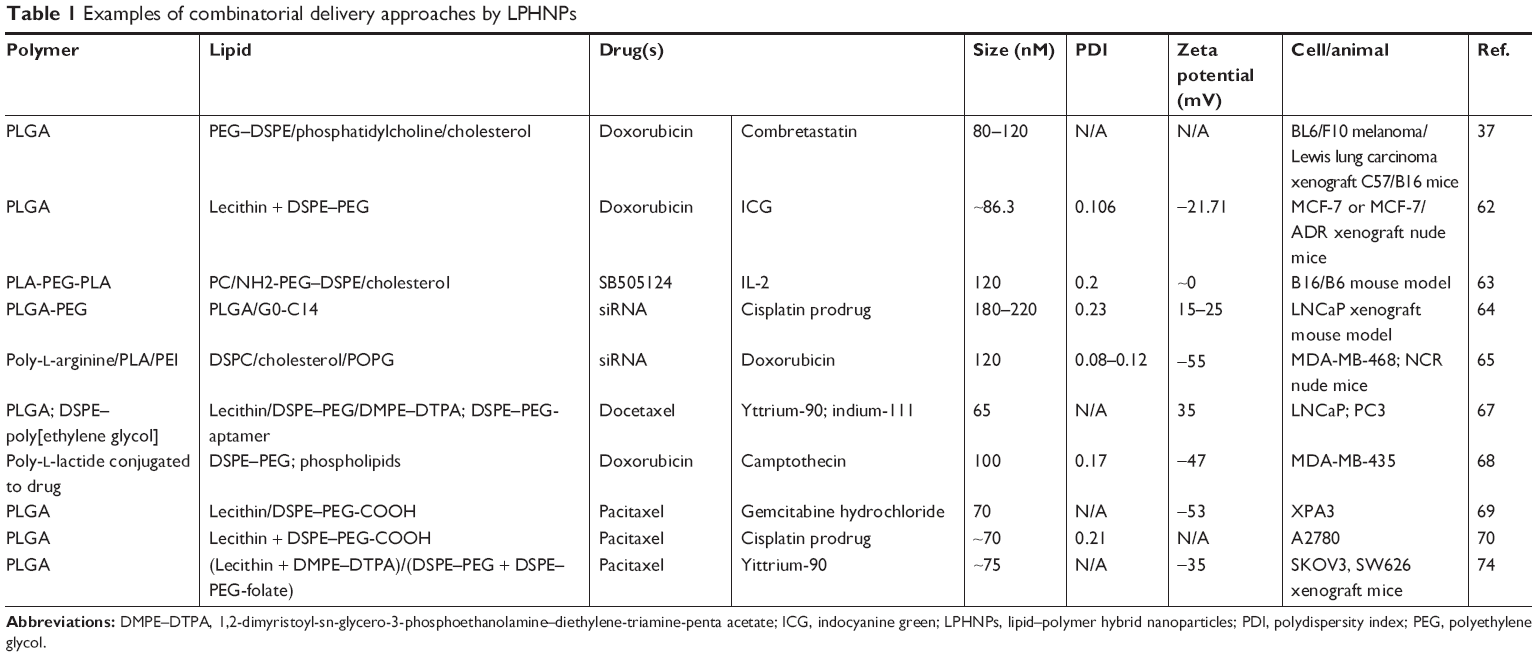 | Table 1 Examples of combinatorial delivery approaches by LPHNPs |
Two major obstacles of antiangiogenic cancer therapy include 1) not allowing tumor to obtain an effective concentration of the chemotherapeutics and 2) induction of tumor hypoxia, resulting in chemoresistance and enhanced invasiveness. To address these issues, Sengupta et al came up with a hybrid nanocell where the core was comprised of DOX-conjugated PLGA and the envelope was made up of PC-Chol-DSPE–PEG entrapping combretastatin-A4 (for vascular closure) and validated its therapeutic efficacies in the murine model of melanoma and Lewis lung carcinoma.37 In 2013, Zheng et al, to accomplish combined chemo-photothermal therapy, successfully synthesized PLGA-lecithin-PEG hybrid NPs by a single-step sonication method, which could simultaneously deliver DOX and indocyanine green (ICG) to the tumor microenvironment. It induced apoptotic cell death to DOX-sensitive MCF-7 or DOX-resistant MCF-7/ADR in vitro and inhibited MCF-7 or MCF-7/ADR tumor growth and inhibited tumor recurrence under systemic settings.62 To surmount the immunoinhibitory property of the tumor microenvironment, Park et al synthesized hybrid NPs comprised of SB505124-entrapped cyclodextrins and cytokine-encapsulating PLGA within a PC-Chol-DSPE–PEG shell, which can simultaneously deliver both agents to the tumor microenvironment. As reported, sustained release of TGF-β receptor-I inhibitor and IL-2-inhibited tumor growth significantly enhanced survival of tumor-bearing mice and enhanced the natural killer cell activity and CD8+ T-cell infiltration.63 In order to sensitize intrinsically (and acquired) cisplatin-resistant tumors, Xu et al prepared a hybrid NP with a cationic lipid-like molecule and PLGA-PEG for codelivery of cisplatin prodrug and siRNAs targeting the REV1, REV3L genes, which are engaged in the modification-susceptible translesion DNA synthesis pathway. Importantly, after a single dose, a remarkably sustained (up to 3 days) downregulation of both the genes was measured by real-time qPCR. Systemic administration of these NPs displayed synergistically inhibited tumor growth in a human metastatic lymph node carcinoma of the prostate xenograft mouse model, outcompeting Pt monotherapy.64 Again in 2013, Deng et al designed and synthesized layer-by-layer hybrid NPs to codeliver an MRP1 siRNA that downregulates a drug-resistant pathway and DOX to treat triple negative breast cancer in an MDA-MB-468 xenograft model. It turned out to be a potent combination therapy in their study, emphasizing the potential of layer-by-layer NPs as a multitherapeutic platform for combinatorial therapy.65 In another study, to obtain improved synergistic effect by sequential and site-specific delivery, Jiang et al reported a nanodepot, where the core is a liposome encapsulating DOX in the aqueous interior, and an outer shell comprised of cross-linked hyaluronic acid entrapping a TNF-related apoptosis-inducing ligand. Substantial tumor growth inhibition was found in the MDA-MB-231 murine xenograft model.66
To achieve a concurrent treatment of chemotherapy and radiotherapy, using nanoprecipitation method, Wang et al developed small LPHNPs named ChemoRad coencapsulating chemotherapeutic (docetaxel) in the PLGA core and radiotherapy agents (indium-111 or yttrium-90) chelated to a 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-diethylene-triamine-penta acetate (DMPE–DTPA) lipid shell. As per their report, absorption of radioactive isotopes was not detrimental toward encapsulation and release of the drug. Within 45 minutes, particles were taken up by LNCaP prostate cancer cells and showed enhanced cytotoxicity over their single counterparts.67 Concurrent incorporation of two chemotherapeutic drugs (doxorubicin and camptothecin) into a single LPHNP system was also achieved by covalent grafting of the drugs with the polymer. Aryal et al synthesized DOX-PLA and CPT-PLA conjugates, adjusted their molar ratio, and enveloped them by egg-PC-DSPE–PEG-COOH using a nanoprecipitation method.68 In a similar way, PCX-gemcitabine69 and PCX-cisplatin70 conjugates were prepared and loaded onto LPHNPs.
Active-targeted drug delivery
In recent years, cancer research has increasingly focused on receptor-mediated active-targeted drug delivery to decrease off-site chemotherapy toxicities and increase drug accumulation in target tumor cells. This has been achieved by functionalizing NPs (drug delivery systems) with the ligand (targeting moieties) for a particular receptor (eg, folate, integrin, transferrin) overexpressed in specific cancer cells.71 In the following section, we will discuss different methods utilized for surface modification followed by active targeting (Table 2).
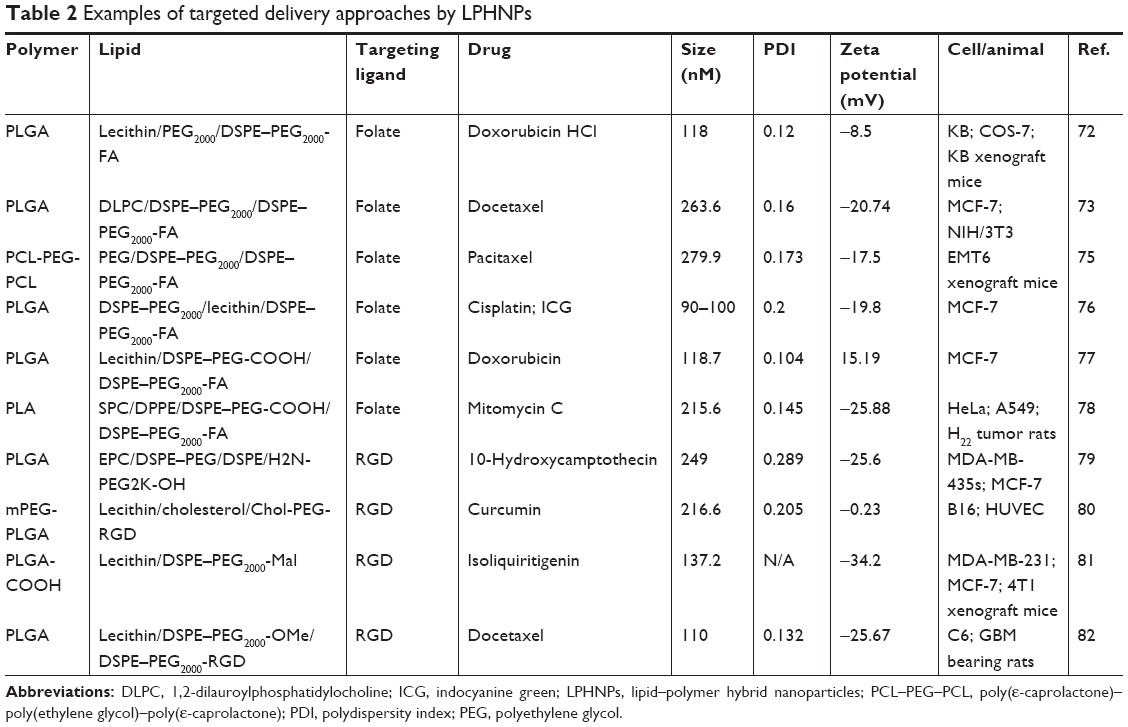 | Table 2 Examples of targeted delivery approaches by LPHNPs |
Folate-mediated delivery
In 2015, Wu et al synthesized a reduction-sensitive biodegradable hybrid NP, targeted with a folate ligand, to deliver DOX. The NP was comprised of a PLGA core, a soybean lecithin and monomethoxy-poly(ethylene glycol)-S-S-hexadecyl (mPEG-S-S-C16) monolayer, and DSPE–PEG-folate and prompted a faster release of DOX in the presence of 10 mM dithiothreitol. Owing to receptor-mediated endocytosis, these particles enhanced cellular uptake and cytotoxicity in folate-overexpressing human oral cavity squamous carcinoma cells (KB cells) and showed greater tumor accumulation and appreciable antitumor efficacy in KB cells xenografted into BALB/c nude mice.72 Folic acid was also selected as a ligand for targeted delivery of docetaxel to certain breast cancer and ovarian cancer cells by Liu et al in 2010, where their LPHNP was composed of a PLGA core, DLPC to envelop, DSPE–PEG2000 for stealth and DSPE–PEG5000-folate for active targeting. NPs were tuned for stoichiometric management of the targeted delivery, were stable with desired surface property, and showed long half-life in plasma.73 With a view to harness a combined outcome of chemotherapy and radiotherapy, another folate-decorated LPHNP was designed by Werner et al and synthesized using nanoprecipitation technique to simultaneously encapsulate PCX and yittrium-90. Monodisperse hybrid NP contains a PLGA core and a shell of soybean lecithin, DMPE–DTPA, DSPE–PEG, and DSPE–PEG-folate. From in vivo efficacy studies using an ovarian peritoneal metastasis model, it is clearly evident that folate-targeted NPs containing dual chemoradiotherapeutics were noticeably more effective compared with nontargeted and single drug NPs.74 In 2015, using a thin-film hydration and ultrasonic dispersion method, Zhang et al prepared PCX-loaded LPHNPs with a poly(ε-caprolactone)–poly(ethylene glycol)–poly(ε-caprolactone) (PCL-PEG-PCL) core, DSPE–PEG2000 corona, and DSPE–PEG2000-folate active targeting ligand. In BALB/c mice bearing EMT6 (mammary carcinoma cells) tumors, intratumoral delivery of PTX-loaded folate-targeted hybrid NPs showed lower toxicity but similar antitumor efficacy compared with Taxol® and greater therapeutic efficacy than nontargeted NPs.75 To circumvent the inherent limitations of cisplatin (chemotherapeutic) and ICG (photothermal therapeutic) delivery, in 2016, Gu et al derived a folate-fabricated, cisplatin and ICG-loaded, homogeneous, stable LPHNPs using PLGA, lecithin, DSPE–PEG2000, and DSPE–PEG2000-FA by a single-step sonication method. Targeting efficiency of the folate-modified NPs was greater in folate receptor (FR) overexpressing MCF-7 cells than in FR-negative A549 cells. Moreover, treatment of targeted NPs with 808 nm near infra-red laser irradiation induced substantial cell death by apoptosis and necrosis of MCF-7 cells compared with standard care therapy.76 Using an ESE technique, a DOX-encapsulated LPHNP was constructed with PLGA-lecithin-DSPE–PEG and further decorated with DSPE–PEG-folate. It had been shown that folate-targeted NPs confer higher uptake and cytotoxicity in MCF-7 cells compared with nontargeted NPs.77 In order to bypass the limitations of mitomycin C (a water-soluble antibacterial and antitumor agent), it was complexed with soy-PC and loaded onto a folate-functionalized LPHNP system having a PLA core and DPPE/DSPE–PEG/DSPE–PEG-folate shell using an ESE technique. Importantly, folate-decorated NPs markedly improved pharmacokinetic profile (compared with free drug) by extending circulation time and showed better in vitro and in vivo therapeutic efficiency.78
Targeted delivery by RGD
Yang et al, in 2013, used a modified single ESE method to produce an LPHNP with PLGA-EPC/DSPE–PEG and DSPE–PEG-c(RGDyk) to deliver 10-hydroxycamptothecin (HCPT) via targeting integrin α∨β3-positive cancer cells. They optimized DSPE–PEG:EPC molar ratio to be 5:5 and the lipids:PLGA mass ratio to be 1:15 with a zeta potential of about −26 mV and with an average size of 230 nm. They found an improved cytotoxicity profile of HCPT against MDA-MB-435s cells for the targeted NPs.79 Using a double ESE technique (w/o/w), RGD-functionalized polymer-core lipid-shell hybrid NPs were synthesized by Zhao et al in 2013. By the virtue of integrin recognition, these particles enhanced cytotoxicity of curcumin in vitro and demonstrated tumor growth inhibition and prolonged survival in a subcutaneous B16 murine tumor model.80 To amend the poor bioavailability of isoliquiritigenin (ISL), a natural antibreast cancer dietary compound, Gao et al recently developed iRGD (tumor-homing peptide) decorated LPHNPs by a modified nanoprecipitation method using PLGA-lecithin/DSPE–PEG-Mal. A postinsertion approach (iRGD covalently grafted onto the PEG) was adopted to decorate the surface with iRGD peptides. Surface-functionalized ISL-loaded NPs demonstrated better cytotoxicity and apoptotic cell death of different types of breast cancer cells, prolonged in vivo circulation, and exhibited higher tumor growth inhibition efficacy in 4T1-bearing breast tumor murine models compared with the free drug and nontargeted counterparts.81 In a separate study, Shi et al developed RGD-modified LPHNPs for the delivery of docetaxel to glioblastoma multiforme, from the precursors PLGA-soy lecithin and DSPE–PEG (containing DSPE–PEG2000-RGD and DSPE–PEG2000 at a molar ratio of 8.5:1.5). After a series of experiments including cellular uptake, tumor spheroid penetration, and tumor growth inhibition, a rat model of brain GBM was used to evaluate antitumor efficacy of the RGD-functionalized docetaxel-loaded NPs where the median survival times for the rats treated with these particles were prolonged by 57 days.82
Other methods for active targeting
Besides folate and RGD-mediated delivery, few attempts have also been made using transferrin, antibodies, and aptamers to target cancer cells. For example, Zheng et al followed a postinsertion method to develop a transferrin (Tf-DOPE) conjugated LPHNPs encapsulating aromatase inhibitor (7a-APTADD). Tf-conjugated NPs showed higher inhibition of aromatase in breast cancer cells (SKBR-3) than that by the nontargeted NPs.50 Again, Zhang et al covalently grafted amine-terminated A10 RNA aptamer to carboxyl functionalized lipid–PEG conjugates in order to target prostate-specific membrane antigen (PSMA) overexpressed in LNCaP cells. Aptamer-conjugated NBD-encapsulated hybrid NPs were successfully taken up by LNCaP via active-targeted mechanism but not by PC3 cells, which do not express PSMA.27 Utilizing single-step nanoprecipitation with the precursor PLGA-soy lecithin-DSPE–PEG/DSPE–PEG-Mal, Hu et al prepared a PTX-loaded LPHNP platform. A half-antibody displaying anticarcinoembryonic activity was conjugated to the hybrid NP via postinsertion to target carcinoembryonic antigen (CEA)-overexpressed pancreatic cancer cells. Antibody-conjugated NPs, thus synthesized, were shown to selectively target CEA-positive BxPC-3 pancreatic cancer cells resulting in an elevated cellular uptake and higher in vitro cytotoxicity compared with nontargeted NPs.83
siRNA delivery
RNAi is an evolutionarily conserved unique cellular surveillance mechanism, with sequence-specific post-transcriptional gene silencing capability. Ever since its disclosure in cultured mammalian cells and mammals, studies aimed at demonstrating the clinical potential of siRNAs have been reported in mouse models as well as in nonhuman primates and more recently in humans.84 Global efforts have been witnessed in developing vectors for in vivo delivery of therapeutic siRNAs.85 Most recently, a lipid-based RNAi therapeutic (ONPATTRO™, Alnylam Pharmaceuticals Inc.) received FDA approval, which is the first of its category, for the treatment of polyneuropathy of hereditary transthyretin-mediated amyloidosis in adults.86 Nevertheless, a few cellular as well as preclinical studies have already been performed for LPHNP-mediated RNAi. Here, we summarize a few of them.
With the precursors PLGA-DOPC/cyclic cationic lipid and DSPE–PEG2000, Desai et al formulated an LPHNP through single-step nanoprecipitation to simultaneously deliver an anti-inflammatory drug capsaicin and siRNA against TNF-α (siTNFα) to treat skin inflammatory conditions under systemic settings. It was shown that the NPs could deliver capsaicin deep into dermal tissue (~360 μm), and its combination with siTNFα showed a synergistic effect on skin inflammation.87 In 2012, with cationic lipid (N,N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryloxycarbonyl aminoethyl) ammonium bromide, BHEM-Chol) and PLGA polymers, LPHNPs were prepared by Yang et al for systemic delivery of siRNA by a modified nanoprecipitation method. NPs effectively delivered siPlk1 (targeted against Plk1 oncogene) to BT474 cells in vitro and BT474 xenograft murine model in vivo and inhibited tumor growth.49 In another study, siRNA was encapsulated within hybrid NPs prepared via modified double emulsion solvent evaporation technique. PLGA and a newly synthesized cationic lipid-like molecule (G0-C14) were taken to constitute the core to which siRNA was added and added together to DSPE–PEG and lecithin. NP-mediated delivery of siPHB1, which outcompetes standard lipofectamine complexes, resulted in long-term silencing of the prohibitin 1 gene and therefore effective tumor growth inhibition in vivo (A549 xenograft BALB/C nude mice model).88 Intending to develop a well-capable siRNA delivery platform, Shi et al proposed differentially charged hollow core/shell LPHNPs made by modified double emulsion solvent evaporation technique. Interestingly, GL3 siRNA encapsulated within these hybrid NPs was successfully delivered to luciferase-expressing xenograft tumors of Dual-Luc HeLa cells and significantly reduced luciferase activity.89 Another siRNA delivery platform was created by Gao et al, wherein a cationic liposome made up of DOTAP:DOPE:cholesterol (25:43:25) was mixed with the anionic cholesterol-grafted poly(amidoamine)/siRNA, and the resulting LPHNP was further modified with DSPE–PEG/DSPE–PEG-T7 (where T7= HAIYPRH). Efficacy study on nude mice bearing MCF-7 tumor xenografts further asserted that T7-modified siEGFR-loaded NPs demonstrated highest tumor growth inhibition via transferrin receptor-mediated active-targeted delivery.90
Imaging agent delivery
In recent years, owing to its stability and biocompatibility, LPHNPs have increasingly been used as a delivery system for contrast agents (quantum dots, inorganic nanocrystals), commonly used in bioimaging such as MRI and CT. For instance, Mieszawska et al designed a unique method to append diagnostic features to LPHNPs prepared via nanoprecipitation. They conjugated gold nanocrystals (AuNC) and quantum dots to the PLGA by esterification reactions and then mixed with soybean lecithin and PEG. The in vitro experiments in mouse macrophage (J774A.1) further supported the suitability of AuNC and quantum dots-loaded LPHNPs as probes for image generation in both biological and medical settings.32 A parallel approach was to form the core with polymers having high fluorescence. Using nanoprecipitation, LPHNPs are synthesized where the core is composed of PFBT with an envelope of DMPE–PEG. Notably, hybrid NPs showed ~50% higher quantum yield compared with their nonhybrid counterpart.53
Future possibilities
As evident from our prior discussion, one-step method is preferred over the two-step method due to its simplicity. Irrespective of the method of preparation, L/P ratio is found to be the most crucial formulation parameter to monitor size, homogeneity, %EE, release kinetics, while colloidal stability is critically dependent on the steric stabilization provided by the PEG fragment of the PEGylated lipid component. Subcategorically, within the realm of one-step method, despite higher content loading in the ESE method, nanoprecipitation is favored as it can produce sub-100 nm sized particles. Nanoprecipitation, thus, has advanced to large-scale production by a continuous high-throughput microfluidic process. Research endeavors in the near future will require an advancement of a continuous high-throughput microfluidic process for the ESE method as well. The ensuing challenge in large-scale production of LPHNPs, in common with all other scale-up processes, will be entrenching consistent production of particles with controlled size and homogeneity. Another task required for researchers in the field is to convert their liquid LPHNP formulations to dry powder form, while keeping all physical characteristics intact, which is essential for long-term storage. This has previously been explored for liposomes and polymeric NPs.91,92 Conversion to dry powder, in general, is executed by lyophilization or spray-drying in the presence of cryoprotectants. As is to be expected, optimizations of formulation parameters are required while deciding the method as well as the cryoprotectants.
Within the last 2 years, a number of new applications for the LPHNP platform have been demonstrated. Notably, these include photoresponsive LPHNPs for controlled release of doxorubicin, insulin delivery, delivery of mRNA to lung tissue, and MRI-guided targeted delivery of doxorubicin, among others.93–97 Although the voyage is commenced, stepping out beyond the realm of oncology is still at the juvenile stage for LPHNPs. Future directions of LPHNP research remain open to translate its potential into practice for many other disease models. For the delivery of genetic material, it is essential to clarify the reduction of nonspecific binding with serum proteins. Significant work is also required to establish reliable clinical applications for the delivery of diagnostic bioimaging agents. Advancements in clinical techniques for minimally invasive targeted delivery may also further extend the application of LPHNPs by maximizing therapeutic delivery and minimizing systemic exposure. One such approach, called convection-enhanced delivery (CED), involves the use of one or more microcatheters placed stereotactically into target tissues (eg, intratumorally or within the target brain regions) through which an infusate is actively pumped while maintaining a pressure gradient over multiple hours. A number of recent clinical trials using CED have shown the technique to be an effective option for the delivery of therapeutics with significantly greater volume of distribution vs standard diffusion-dependent delivery methods. As such, CED holds great potential in current and future clinical trial designs for improving the reliability of therapeutic delivery and limiting confounding due to variable target bioavailability. The combination of advanced minimally invasive techniques like CED with the improved therapeutic design offered by LPHNPs may offer synergistic potential in future clinical applications and is an area of great interest for future studies.98–100
Conclusion
LPHNPs have displayed a remarkable range of successes in translating new clinical and drug delivery applications from bench to bedside, with a significant and lasting impact in the field of oncology. Indeed, in some cases, LPHNPs have already demonstrated superiority compared with liposomes and polymeric NPs. From the perspective of industrial production and scalability, efficient and simple large-scale production of NPs has already been developed. As such, we predict ever-expanding applications for LPHNPs with many more translational opportunities for future clinical trials, including beyond active targeting of anticancer therapies in oncology toward more broad applications in medical therapies and neurotherapeutics as well as diagnostic imaging agents.
Acknowledgments
VMY acknowledges the JWCI and Saint John’s Foundation, FFANY and ABCs foundations.
Author contributions
All authors made substantial contributions to conception and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Farokhzad O, Langer R. Nanomedicine: developing smarter therapeutic and diagnostic modalities. Adv Drug Deliv Rev. 2006;58(14):1456–1459. | ||
Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17(1):20–37. | ||
Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5(3):161–171. | ||
Kim BY, Rutka JT, Chan WC. Nanomedicine. N Engl J Med. 2010;363(25):2434–2443. | ||
Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. | ||
Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem Rev. 2016;116(4):2602–2663. | ||
Zheng Y, Wang L, Lu L, Wang Q, Benicewicz BC. pH and thermal dual-responsive nanoparticles for controlled drug delivery with high loading content. ACS Omega. 2017;2(7):3399–3405. | ||
Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14(1):1–16. | ||
Cho EJ, Holback H, Liu KC, Abouelmagd SA, Park J, Yeo Y. Nanoparticle characterization: state of the art, challenges, and emerging technologies. Mol Pharm. 2013;10(6):2093–2110. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Burgess P, Hutt PB, Farokhzad OC, Langer R, Minick S, Zale S. On firm ground: IP protection of therapeutic nanoparticles. Nat Biotechnol. 2010;28(12):1267–1270. | ||
Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. | ||
Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;83(5):761–769. | ||
Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24(10):1211–1217. | ||
Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics. 2017;9(4):12. | ||
Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23(31):7794–7803. | ||
Maier-Hauff K, Ulrich F, Nestler D, et al. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J Neurooncol. 2011;103(2):317–324. | ||
Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–478. | ||
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | ||
Barenholz Y. Doxil(R)-the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Petersen GH, Alzghari SK, Chee W, Sankari SS, La-Beck NM. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J Control Release. 2016;232:255–264. | ||
Chen EC, Fathi AT, Brunner AM. Reformulating acute myeloid leukemia: liposomal cytarabine and daunorubicin (CPX-351) as an emerging therapy for secondary AML. Onco Targets Ther. 2018;11:3425–3434. | ||
Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103(16):6315–6320. | ||
Torchilin VP. Micellar nanocarriers: pharmaceutical perspectives. Pharm Res. 2006;24(1):1–16. | ||
Kim TY, Kim DW, Chung JY, et al. Phase I and pharmacokinetic study of genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10(11):3708–3716. | ||
Ahn HK, Jung M, Sym SJ, et al. A phase II trial of cremorphor EL-free paclitaxel (genexol-PM) and gemcitabine in patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2014;74(2):277–282. | ||
Zhang L, Chan JM, Gu FX, et al. Self-assembled lipid-polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano. 2008;2(8):1696–1702. | ||
Wakaskar RR. General overview of lipid–polymer hybrid nanoparticles, dendrimers, micelles, liposomes, spongosomes and cubosomes. J Drug Target. 2018;26(4):311–318. | ||
Fang RH, Aryal S, Hu CM, Zhang L. Quick synthesis of lipid-polymer hybrid nanoparticles with low polydispersity using a single-step sonication method. Langmuir. 2010;26(22):16958–16962. | ||
Mandal B, Bhattacharjee H, Mittal N, et al. Core–shell-type lipid–polymer hybrid nanoparticles as a drug delivery platform. Nanomedicine. 2013;9(4):474–491. | ||
Thevenot J, Troutier AL, David L, Delair T, Ladavière C. Steric stabilization of lipid/polymer particle assemblies by poly(ethylene glycol)-lipids. Biomacromolecules. 2007;8(11):3651–3660. | ||
Mieszawska AJ, Gianella A, Cormode DP, et al. Engineering of lipid-coated PLGA nanoparticles with a tunable payload of diagnostically active nanocrystals for medical imaging. Chem Commun (Camb). 2012;48(47):5835–5837. | ||
Fenart L, Casanova A, Dehouck B, et al. Evaluation of effect of charge and lipid coating on ability of 60-nm nanoparticles to cross an in vitro model of the blood-brain barrier. J Pharmacol Exp Ther. 1999;291(3):1017–1022. | ||
Wang H, Zhao P, Su W, et al. PLGA/polymeric liposome for targeted drug and gene co-delivery. Biomaterials. 2010;31(33):8741–8748. | ||
Troutier AL, Delair T, Pichot C, Ladavière C. Physicochemical and interfacial investigation of lipid/polymer particle assemblies. Langmuir. 2005;21(4):1305–1313. | ||
Messerschmidt SK, Musyanovych A, Altvater M, et al. Targeted lipid-coated nanoparticles: delivery of tumor necrosis factor-functionalized particles to tumor cells. J Control Release. 2009;137(1):69–77. | ||
Sengupta S, Eavarone D, Capila I, et al. Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature. 2005;436(7050):568–572. | ||
Hu CM, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc Natl Acad Sci U S A. 2011;108(27):10980–10985. | ||
De Miguel I, Imbertie L, Rieumajou V, Major M, Kravtzoff R, Betbeder D. Proofs of the structure of lipid coated nanoparticles (SMBV) used as drug carriers. Pharm Res. 2000;17(7):817–824. | ||
Hitzman CJ, Elmquist WF, Wattenberg LW, Wiedmann TS. Development of a respirable, sustained release microcarrier for 5-fluorouracil I: In vitro assessment of liposomes, microspheres, and lipid coated nanoparticles. J Pharm Sci. 2006;95(5):1114–1126. | ||
Li X, Anton N, Arpagaus C, Belleteix F, Vandamme TF. Nanoparticles by spray drying using innovative new technology: the Büchi nano spray dryer B-90. J Control Release. 2010;147(2):304–310. | ||
Hasan W, Chu K, Gullapalli A, et al. Delivery of multiple siRNAs using lipid-coated PLGA nanoparticles for treatment of prostate cancer. Nano Lett. 2012;12(1):287–292. | ||
Barichello JM, Morishita M, Takayama K, Nagai T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev Ind Pharm. 1999;25(4):471–476. | ||
Su X, Fricke J, Kavanagh DG, Irvine DJ. In vitro and in vivo mRNA delivery using lipid-enveloped pH-responsive polymer nanoparticles. Mol Pharm. 2011;8(3):774–787. | ||
Valencia PM, Basto PA, Zhang L, et al. Single-step assembly of homogenous lipid-polymeric and lipid-quantum dot nanoparticles enabled by microfluidic rapid mixing. ACS Nano. 2010;4(3):1671–1679. | ||
Fang RH, Chen KN, Aryal S, Hu CM, Zhang K, Zhang L. Large-scale synthesis of lipid-polymer hybrid nanoparticles using a multi-inlet vortex reactor. Langmuir. 2012;28(39):13824–13829. | ||
Kim Y, Lee Chung B, Ma M, et al. Mass production and size control of lipid-polymer hybrid nanoparticles through controlled microvortices. Nano Lett. 2012;12(7):3587–3591. | ||
Chan JM, Zhang L, Yuet KP, et al. PLGA–lecithin–PEG core–shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. | ||
Yang XZ, Dou S, Wang YC, et al. Single-step assembly of cationic lipid-polymer hybrid nanoparticles for systemic delivery of siRNA. ACS Nano. 2012;6(6):4955–4965. | ||
Zheng Y, Yu B, Weecharangsan W, et al. Transferrin-conjugated lipid-coated PLGA nanoparticles for targeted delivery of aromatase inhibitor 7α-APTADD to breast cancer cells. Int J Pharm. 2010;390(2):234–241. | ||
Mittal G, Sahana DK, Bhardwaj V, Ravi Kumar MN. Estradiol loaded PLGA nanoparticles for oral administration: effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J Control Release. 2007;119(1):77–85. | ||
Clawson C, Ton L, Aryal S, Fu V, Esener S, Zhang L. Synthesis and characterization of lipid-polymer hybrid nanoparticles with pH-triggered poly(ethylene glycol) shedding. Langmuir. 2011;27(17):10556–10561. | ||
Kandel PK, Fernando LP, Ackroyd PC, Christensen KA. Incorporating functionalized polyethylene glycol lipids into reprecipitated conjugated polymer nanoparticles for bioconjugation and targeted labeling of cells. Nanoscale. 2011;3(3):1037–1045. | ||
Bershteyn A, Chaparro J, Yau R, et al. Polymer-supported lipid shells, onions, and flowers. Soft Matter. 2008;4(9):1787–1791. | ||
Cheow WS, Hadinoto K. Factors affecting drug encapsulation and stability of lipid–polymer hybrid nanoparticles. Colloids Surf B Biointerfaces. 2011;85(2):214–220. | ||
Liu Y, Pan J, Feng SS. Nanoparticles of lipid monolayer shell and biodegradable polymer core for controlled release of paclitaxel: effects of surfactants on particles size, characteristics and in vitro performance. Int J Pharm. 2010;395(1–2):243–250. | ||
Chu CH, Wang YC, Huang HY, Wu LC, Yang CS. Ultrafine PEG-coated poly(lactic-co-glycolic acid) nanoparticles formulated by hydrophobic surfactant-assisted one-pot synthesis for biomedical applications. Nanotechnology. 2011;22(18):185601. | ||
Cheow WS, Hadinoto K. Lipid-polymer hybrid nanoparticles with rhamnolipid-triggered release capabilities as anti-biofilm drug delivery vehicles. Particuology. 2012;10(3):327–333. | ||
Wang Y, Kho K, Cheow WS, Hadinoto K. A comparison between spray drying and spray freeze drying for dry powder inhaler formulation of drug-loaded lipid–polymer hybrid nanoparticles. Int J Pharm. 2012;424(1–2):98–106. | ||
Cheow WS, Chang MW, Hadinoto K. The roles of lipid in anti-biofilm efficacy of lipid–polymer hybrid nanoparticles encapsulating antibiotics. Colloids Surf A Physicochem Eng Asp. 2011;389(1–3):158–165. | ||
Wang Z, Ho PC. A nanocapsular combinatorial sequential drug delivery system for antiangiogenesis and anticancer activities. Biomaterials. 2010;31(27):7115–7123. | ||
Zheng M, Yue C, Ma Y, et al. Single-step assembly of DOX/ICG loaded lipid-polymer nanoparticles for highly effective chemo-photothermal combination therapy. ACS Nano. 2013;7(3):2056–2067. | ||
Park J, Wrzesinski SH, Stern E, et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11(10):895–905. | ||
Xu X, Xie K, Zhang XQ, et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci U S A. 2013;110(46):18638–18643. | ||
Deng ZJ, Morton SW, Ben-Akiva E, Dreaden EC, Shopsowitz KE, Hammond PT. Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano. 2013;7(11):9571–9584. | ||
Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Gel-liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater. 2014;24(16):2295–2304. | ||
Wang AZ, Yuet K, Zhang L, et al. ChemoRad nanoparticles: a novel multifunctional nanoparticle platform for targeted delivery of concurrent chemoradiation. Nanomedicine (Lond). 2010;5(3):361–368. | ||
Aryal S, Hu CM, Zhang L. Polymeric nanoparticles with precise ratiometric control over drug loading for combination therapy. Mol Pharm. 2011;8(4):1401–1407. | ||
Aryal S, Hu CM, Zhang L. Combinatorial drug conjugation enables nanoparticle dual-drug delivery. Small. 2010;6(13):1442–1448. | ||
Aryal S, Jack Hu C-M, Fu V, Zhang L. Nanoparticle drug delivery enhances the cytotoxicity of hydrophobic–hydrophilic drug conjugates. J Mater Chem. 2012;22(3):994–999. | ||
Kularatne SA, Low PS. Targeting of nanoparticles: folate receptor. Methods Mol Biol. 2010;624:249–265. | ||
Wu B, Yu P, Cui C, et al. Folate-containing reduction-sensitive lipid–polymer hybrid nanoparticles for targeted delivery of doxorubicin. Biomater Sci. 2015;3(4):655–664. | ||
Liu Y, Li K, Pan J, Liu B, Feng SS. Folic acid conjugated nanoparticles of mixed lipid monolayer shell and biodegradable polymer core for targeted delivery of docetaxel. Biomaterials. 2010;31(2):330–338. | ||
Werner ME, Karve S, Sukumar R, et al. Folate-targeted nanoparticle delivery of chemo- and radiotherapeutics for the treatment of ovarian cancer peritoneal metastasis. Biomaterials. 2011;32(33):8548–8554. | ||
Zhang L, Zhu D, Dong X, et al. Folate-modified lipid-polymer hybrid nanoparticles for targeted paclitaxel delivery. Int J Nanomedicine. 2015;10:2101–2114. | ||
Gu L, Shi T, Sun Y, et al. Folate-modified, indocyanine green-loaded lipid-polymer hybrid nanoparticles for targeted delivery of cisplatin. J Biomater Sci Polym Ed. 2017;28(7):690–702. | ||
Zheng M, Gong P, Zheng C, et al. Lipid-polymer nanoparticles for folate-receptor targeting delivery of doxorubicin. J Nanosci Nanotechnol. 2015;15(7):4792–4798. | ||
Li Y, Wu H, Yang X, et al. Mitomycin C-soybean phosphatidylcholine complex-loaded self-assembled PEG-lipid-PLA hybrid nanoparticles for targeted drug delivery and dual-controlled drug release. Mol Pharm. 2014;11(8):2915–2927. | ||
Yang Z, Luo X, Zhang X, Liu J, Jiang Q. Targeted delivery of 10-hydroxycamptothecin to human breast cancers by cyclic RGD-modified lipid–polymer hybrid nanoparticles. Biomed Mater. 2013;8(2):025012. | ||
Zhao Y, Lin D, Wu F, et al. Discovery and in vivo evaluation of novel RGD-modified lipid-polymer hybrid nanoparticles for targeted drug delivery. Int J Mol Sci. 2014;15(10):17565–17576. | ||
Gao F, Zhang J, Fu C, et al. iRGD-modified lipid-polymer hybrid nanoparticles loaded with isoliquiritigenin to enhance anti-breast cancer effect and tumor-targeting ability. Int J Nanomedicine. 2017;12:4147–4162. | ||
Shi K, Zhou J, Zhang Q, et al. Arginine-glycine-aspartic acid-modified lipid-polymer hybrid nanoparticles for docetaxel delivery in glioblastoma multiforme. J Biomed Nanotechnol. 2015;11(3):382–391. | ||
Hu CM, Kaushal S, Tran Cao HS, et al. Half-antibody functionalized lipid-polymer hybrid nanoparticles for targeted drug delivery to carcinoembryonic antigen presenting pancreatic cancer cells. Mol Pharm. 2010;7(3):914–920. | ||
Davis ME, Zuckerman JE, Choi CHJ, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464(7291):1067–1070. | ||
Mukherjee A, Bhattacharyya J, Sagar MV, Chaudhuri A. Liposomally encapsulated CDC20 siRNA inhibits both solid melanoma tumor growth and spontaneous growth of intravenously injected melanoma cells on mouse lung. Drug Deliv Transl Res. 2013;3(3):224–234. | ||
Alnylam Pharmaceuticals Inc. Alnylam Announces First-Ever FDA Approval of an RNAi Therapeutic, ONPATTRO™ (patisiran) for the Treatment of the Polyneuropathy of Hereditary Transthyretin-Mediated Amyloidosis in Adults; Oct 10, 2018. Available from: https://www.businesswire.com/news/home/20180810005398/en/. Accessed February 22, 2019. | ||
Desai PR, Marepally S, Patel AR, Voshavar C, Chaudhuri A, Singh M. Topical delivery of anti-TNFα siRNA and capsaicin via novel lipid-polymer hybrid nanoparticles efficiently inhibits skin inflammation in vivo. J Control Release. 2013;170(1):51–63. | ||
Shi J, Xu Y, Xu X, et al. Hybrid lipid–polymer nanoparticles for sustained siRNA delivery and gene silencing. Nanomedicine. 2014;10(5):e897–e900. | ||
Shi J, Xiao Z, Votruba AR, Vilos C, Farokhzad OC. Differentially charged hollow core/shell lipid-polymer-lipid hybrid nanoparticles for small interfering RNA delivery. Angew Chem Int Ed Engl. 2011;50(31):7027–7031. | ||
Gao LY, Liu XY, Chen CJ, et al. Core-shell type lipid/rPAA-Chol polymer hybrid nanoparticles for in vivo siRNA delivery. Biomaterials. 2014;35(6):2066–2078. | ||
Stark B, Pabst G, Prassl R. Long-term stability of sterically stabilized liposomes by freezing and freeze-drying: effects of cryoprotectants on structure. Eur J Pharm Sci. 2010;41(3–4):546–555. | ||
Holzer M, Vogel V, Mäntele W, Schwartz D, Haase W, Langer K. Physico-chemical characterisation of PLGA nanoparticles after freeze-drying and storage. Eur J Pharm Biopharm. 2009;72(2):428–437. | ||
Grigoras AG. Polymer-lipid hybrid systems used as carriers for insulin delivery. Nanomedicine. 2017;13(8):2425–2437. | ||
Silva EJ, Souza LG, Silva LAD, et al. A novel polymer-lipid hybrid nanoparticle for the improvement of topotecan hydrochloride physicochemical properties. Curr Drug Deliv. 2018;15(7):979–986. | ||
Yao C, Wu M, Zhang C, et al. Photoresponsive lipid-polymer hybrid nanoparticles for controlled doxorubicin release. Nanotechnology. 2017;28(25):255101. | ||
Kaczmarek JC, Patel AK, Kauffman KJ, et al. Polymer-lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew Chem Int Ed Engl. 2016;55(44):13808–13812. | ||
Wu B, Lu ST, Deng K, et al. MRI-guided targeting delivery of doxorubicin with reduction-responsive lipid-polymer hybrid nanoparticles. Int J Nanomedicine. 2017;12:6871–6882. | ||
Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci U S A. 1994;91(6):2076–2080. | ||
Lieberman DM, Laske DW, Morrison PF, Bankiewicz KS, Oldfield EH. Convection-enhanced distribution of large molecules in gray matter during interstitial drug infusion. J Neurosurg. 1995;2(6):1021–1029. | ||
Cserr HF, Ostrach LH. Bulk flow of interstitial fluid after intracranial injection of blue dextran 2000. Exp Neurol. 1974;45(1):50–60. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.