Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Lipid nanoparticles for cyclosporine A administration: development, characterization, and in vitro evaluation of their immunosuppression activity
Authors Guada M, Sebastián V, Irusta S, Feijoó E, Dios-Viéitez, Blanco-Prieto MJ
Received 20 June 2015
Accepted for publication 1 September 2015
Published 16 October 2015 Volume 2015:10(1) Pages 6541—6553
DOI https://doi.org/10.2147/IJN.S90849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Carlos Rinaldi
Melissa Guada,1,2 Victor Sebastián,3,4 Silvia Irusta,3,4 Esperanza Feijoó,1 María del Carmen Dios-Viéitez,1 María José Blanco-Prieto1,2
1Department of Pharmacy and Pharmaceutical Technology, School of Pharmacy, University of Navarra, Pamplona, 2Instituto de Investigación Sanitaria de Navarra, IdiSNA, Pamplona, 3Chemical and Environmental Engineering Department and Nanoscience Institute of Aragon, University of Zaragoza, Zaragoza, 4Networking Research Center on Bioengineering, Biomaterials and Nanomedicine, CIBER-BBN, Madrid, Spain
Abstract: Cyclosporine A (CsA) is an immunosuppressant commonly used in transplantation for prevention of organ rejection as well as in the treatment of several autoimmune disorders. Although commercial formulations are available, they have some stability, bioavailability, and toxicity related problems. Some of these issues are associated with the drug or excipients and others with the dosage forms. With the aim of overcoming these drawbacks, lipid nanoparticles (LN) have been proposed as an alternative, since excipients are biocompatible and also a large amount of surfactants and organic solvents can be avoided. CsA was successfully incorporated into LN using the method of hot homogenization followed by ultrasonication. Three different formulations were optimized for CsA oral administration, using different surfactants: Tween® 80, phosphatidylcholine, taurocholate and Pluronic® F127 (either alone or mixtures). Freshly prepared Precirol nanoparticles showed mean sizes with a narrow size distribution ranging from 121 to 202 nm, and after freeze-drying were between 163 and 270 nm, depending on the stabilizer used. Surface charge was negative in all LN developed. High CsA entrapment efficiency of approximately 100% was achieved. Transmission electron microscopy was used to study the morphology of the optimized LN. Also, the crystallinity of the nanoparticles was studied by X-ray powder diffraction and differential scanning calorimetry. The presence of the drug in LN surfaces was confirmed by X-ray photoelectron spectroscopy. The CsA LN developed preserved their physicochemical properties for 3 months when stored at 4°C. Moreover, when the stabilizer system was composed of two surfactants, the LN formulations were also stable at room temperature. Finally, the new CsA formulations showed in vitro dose-dependent immunosuppressive effects caused by the inhibition of IL-2 levels secreted from stimulated Jurkat cells. The findings obtained in this paper suggest that new lipid nanosystems are a good alternative to produce physicochemically stable CsA formulations for oral administration.
Keywords: cyclosporine A, lipid nanoparticles, oral administration, stability, immunosuppressive activity, Jurkat cells
Introduction
Cyclosporine A (CsA) is a well-known immunosuppressive agent widely used in the prevention of allograft organ rejection and several autoimmune disorders, such as psoriasis, rheumatoid arthritis, dry eye, and ulcerative colitis. CsA was an important discovery in the immunotherapy field since it was the first immunosuppressant with selective action on lymphocyte inhibition avoiding myelotoxicity. The molecule was isolated from the fungal extract of Tolypocladium inflatum.1 CsA is a neutral cyclic peptide consisting of eleven amino acid residues. As a result of this peculiar structure and its high molecular weight (1,203 Da), CsA presents poor biopharmaceutical properties, including hydrophobicity, and low permeability through biological barriers (ie, gastrointestinal tract, skin, and cornea). These characteristics mean that CsA is classified as Class IV according to the Biopharmaceutics Classification System.2 Nonetheless, this molecule has also been classified as Class II into the same system.3
CsA was initially marketed as a conventional oil-based form for oral administration (Sandimmune®; oral solution or soft gelatin capsules). This formulation presented some inconveniences associated with low and unpredictable drug bioavailability, leading to an erratic relationship between oral dose and total exposure. Subsequently, with the aim of achieving a more consistent pharmacokinetic profile, a reformulated product consisting of a microemulsion was developed (Sandimmune Neoral®; oral solution or soft gelatin capsules). This product has made it possible to enhance oral absorption and reduce variability compared to the first mentioned formulation.4 However, Sandimmune Neoral® is not capable of sustaining constant levels of the drug in blood within the narrow therapeutic window, and therefore CsA monitoring is still required.5 In addition, there are some other safety issues that remain unsolved: the dose-dependent nephrotoxicity attributed to the pronounced initial peak blood drug concentration, gastrointestinal disorders caused by Cremophor RH40, and the ethanol content, which is contraindicated in a certain patient population. Along with these, pharmaceutical issues associated with the microemulsion dosage forms have also been raised. High concentrations of emulsifying agents and organic solvents lead to incompatibility with the shells of soft gelatin capsules as well as precipitation of components when stored at certain temperatures.6,7
During the last decade, lipid nanoparticles (LN), which consist of a solid lipid matrix stabilized by surfactants, have gained considerable interest as suitable oral delivery systems for drugs that exhibit poor and variable gastrointestinal absorption, not only because of their adequate in vivo performance but also as a result of their versatility in manufacturing processes. Their numerous advantages combine those presented by oil-based formulations and polymeric colloidal carriers. Within the benefits offered by LN we may mention the physiological and biocompatible excipients in their composition, low surfactant quantities required for their stabilization, avoidance of organic solvents, enhancement of physicochemical stability by lyophilization or spray drying, scale-up feasibility, and relatively low cost production. In addition, LN enable us to enhance drug absorption, protect the drug from possible biological fluid degradation, and allow controlled drug release and drug targeting.8,9 Considering the aforementioned attributes, LN seem an attractive alternative to design a suitable CsA oral delivery system.
Therefore, the main purpose of this study was to develop and characterize safe and stable LN for CsA oral administration. The influence of different surfactants in the properties of the nanosystems and the physicochemical stability of the new CsA formulations developed were also investigated. The biological activity of the optimized lipid nanosystems was studied in vitro by measuring the inhibition of IL-2 production of Jurkat cells after treatment with the nanoparticles and Con A stimulation.
Materials and methods
Reagents
CsA and Tween® 80 (Tw) were provided by Roig Farma S.A. (Barcelona, Spain). Precirol® ATO 5 was a gift from Gattefossé (Lyon, France). L-α-phosphatidylcholine (Lec) from egg yolk, taurocholic acid sodium salt hydrate (TC), Pluronic® F127 (PL), D-(+)-trehalose dihydrate, formic acid 98% for mass spectroscopy, chloroform (HPLC grade), dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Con A were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Methanol (HPLC gradient grade) was supplied by Merck & Co., Inc. (Whitehouse Station, NJ, USA). Ammonium acetate (HPLC grade) was purchased from Scharlau (Sentmenat, Spain). Roswell Park Memorial Institute 1640 cell culture media, heat-inactivated fetal bovine serum, and penicillin/streptomycin antibiotics were obtained from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals and solvents were analytical grade.
Development and optimization of LN
LN preparation
LN were prepared by the hot homogenization followed by ultrasonication method. Firstly, 200 mg of lipid (Precirol® ATO 5) and different amounts of drug (CsA) were melted at 70°C (slightly above the lipid melting point). Then, 10 mL of an aqueous solution containing 2% (weight/volume [w/v]) of surfactant/co-surfactant preheated at the same temperature were added to the lipid phase and immediately homogenized by ultrasonication with a Microson™ ultrasonic cell disruptor (NY, USA) for 4 minutes at 10–12 W. The emulsion formed was cooled in an ice bath to obtain a nanoparticle suspension by lipid solidification. Then, the excess of surfactant aqueous solution and free drug was removed by diafiltration using Amicon® Ultra-15 10,000 molecular weight cut-off filters at 4,500× g for 30 minutes and washed twice with distilled water. Finally, LN suspension was kept at −80°C and lyophilized to concentrate the LN and obtain a nanoparticulate powder. Trehalose was used as cryoprotective agent. Blank LN were prepared following the same procedure as described earlier without the drug incorporation step.
Effect of surfactant/co-surfactant on particle properties
Different types of surfactants (Tw, Lec, TC, PL) were used to prepare the LN to assess their influence on the mean particle diameter, size distribution, and drug entrapment efficiency (EE). Different surfactant combinations were also investigated to optimize the formulation quality. For this study, the amount of drug was maintained constant at 2.5% (weight/weight [w/w]) of the lipid content.
Effect of initial drug loading on particle properties
The effect of the amount of drug incorporated in the selected formulation was evaluated at different concentrations: 2.5, 3.75, 5.0, 6.25, 7.5, and 10% (w/w) of the lipid content. All other components were kept at the same concentration. Each formulation was prepared in duplicate. Particle size, size distribution, and EE were systematically analyzed.
Characterization of CsA LN
Particle size, polydispersity index, and zeta potential
The mean particle diameter and polydispersity index (PDI) of the formulations developed were measured at 25°C by dynamic light scattering (Zetasizer Nano, Malvern Instruments, Malvern, UK) at an angle of 173°. LN suspensions were diluted with ultrapure water until an appropriate particle concentration was achieved. Each sample was measured in triplicate. Values were expressed as a mean ± standard deviation.
The surface charge of the nanoparticles was investigated by zeta potential measurements using laser Doppler velocimetry (Zetasizer Nano) at 25°C. Analysis was carried out in triplicate and each measurement was an average over at least 12 runs.
Drug EE
EE was determined by quantifying the amount of CsA incorporated in the lyophilized LN using an ultra-high-performance liquid chromatography tandem mass spectrometry (UHPLC–MS/MS) method previously validated.10 Briefly, 500 μL of chloroform were added to 5 mg of LN and vortexed for 30 seconds, then 1.5 mL of methanol were added to the mixture and vortexed for 1 minute. After centrifuging at 15,000× g for 10 minutes, the supernatant was diluted with methanol (1:10) and a 2 μL aliquot was injected into the UHPLC system for drug analysis.
Morphological characterization
The morphological examination of LN formulations was performed by transmission electron microscopy (TEM). TEM images were taken on a FEI Tecnai T20 microscope (Hilsboro, ON, USA) at the Institute of Nanocience of Aragon, Advanced Microscopy Laboratory (Zaragoza, Spain). To prepare the LN samples for TEM observation, the LN suspension was first dispersed for 30 seconds in an ultrasonic bath. A drop of this suspension was applied to a copper grid (200 mesh) coated with carbon (C) film. Then, samples were air-dried for 30 minutes at room temperature (RT) after removing the excess of sample with filter paper. The microscope was operated at 80 kV to preserve the LN morphology and diminish radiation damage.
Crystallinity studies
X-ray powder diffraction analysis (XRD) was performed to study the crystalline properties of blank and CsA LN. Lyophilized LN formulations were analyzed using RIKAGU D/Max-2500 (Tokyo, Japan). The XRD analysis range was scanned at 2.5°–50° over 2θ with a step angle of 0.03° and a count time of 1 second at a constant temperature of 25°C, step =0.03°, with 40 kV voltage and current intensity level of 80 mA.
Pure lipids and pure CsA were studied and those XRD spectra were used as references in evaluating the LN formulations. Bragg spacing was determined by the Bragg equation which relates the wavelength of the X-ray beam to both the angle of incidence and the interatomic distance.
Thermal analysis
Temperature-dependent structure and crystallinity changes in the lipids were analyzed using differential scanning calorimetry (DSC). DSC was performed using accurately weighed samples of bulk lipids and drug loaded and unloaded LN. These accurately weighed samples were sealed in aluminum pans (50 μL) and heating curves were recorded with a scan rate of 10°C/minute in the 25°C–300°C temperature range using Differential Scanning Calorimeter 822 (Mettler Toledo, Japan).
Surface elemental analysis
The surface composition of the LN as well as the individual components were analyzed by X-ray photoelectron spectroscopy (XPS). The analysis was performed with an Axis Ultra DLD (Kratos Tech., Manchester, UK). The spectra were excited by the monochromatized AlKα source (1,486.6 eV) run at 15 kV and 10 mA. For the individual peak regions, pass energy of 20 eV was used. Peaks were analyzed with the CasaXPS software, using a weighted sum of Lorentzian and Gaussian components curves after background subtraction. The binding energies were referenced to the internal C 1s (285.1 eV) standard.
Physicochemical stability studies of CsA LN
After lyophilization, approximately 100 mg of each formulation were stored in closed glass vials at three different conditions: 4°C±2°C (refrigeration), 25°C±2°C (RT), and 40°C±2°C (accelerated conditions). The physical stability of the nanosystems was evaluated by periodically measuring the mean particle diameter, size distribution, and zeta potential over a period of 3 months. Just after lyophilization and every 30 days, 10 mg of the dried powder were resuspended in 1 mL of distilled water and sonicated for 10–15 seconds. Then, samples were analyzed in triplicate as described earlier. The data are expressed as mean values ± standard deviation. Chemical stability of the formulations was studied by quantifying CsA in the dried powder over the same period of time using the UHPLC–MS/MS.10
In vitro biological activity of CsA LN
Cell culture
Jurkat cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) cultured in suspension at a concentration between 1×105 and 1×106 viable cells/mL in Roswell Park Memorial Institute 1640 cell culture medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. Cells were subcultured every 3–4 days depending on cell density to an initial concentration of 2×105 viable cells/mL.
Cell viability study
The cytotoxicity of blank and CsA LN was determined on Jurkat cells by a colorimetric method using MTT assay. For the experiment, 100 μL of cells were seeded in a 96-well plate at a density of 4×105 cells/well in fresh culture media and were incubated with the nanosystems at increasing drug concentrations up to 1.5 μg/mL in a humidified 5% CO2 atmosphere at 37°C. After 20 hours, 20 μL of MTT solution at 5 mg/mL in complete cellular media were added to the wells and incubated for 4 hours in the same conditions. After centrifuging at 200× g for 10 minutes, supernatant was carefully removed, blue formazan crystals were dissolved with DMSO and the absorbance was measured at 540 nm with a microplate reader (Labsystems iEMS Reader MF, Vantaa, Finland). Culture medium was used as negative control (100% cell viability) and a 10% DMSO solution as a positive control (0% cell viability).
Inhibition of IL-2 production by Con A stimulated Jurkat cells
The biological effect of CsA incorporated in LN was assessed on human T-lymphocyte cell line (Jurkat cells) and Sandimmune Neoral® was used as reference. For this study, 4×105 cells/well were seeded in a 96-well plate and cells were treated with CsA loaded LN (equivalent to 10 and 25 ng/mL of drug) and unloaded LN (equivalent to the highest concentration). Then, Con A solution was added to the wells at a final concentration of 20 μg/mL and the plate was incubated in a humidified 5% CO2 atmosphere at 37°C. Con A-stimulated and non-stimulated cells without treatments were used as positive and negative controls, respectively. After 24 hours of incubation, microplate was centrifuged at 200× g for 10 minutes and the supernatants were collected and stored at −20°C until analysis. Human IL-2 levels were measured by enzyme-linked immunosorbent assay (BD OptEIA™, BD Biosciences, San Jose, CA, USA) following the manufacturer instructions. Absorbance measurements were carried out at 450 nm on a microplate reader (PowerWave XS, Biotek, Winooski, VT, USA).
Statistical analysis
Mann–Whitney U-test was performed for statistical comparison between different groups considering statistically significant differences when P<0.05. Data analysis was conducted using GraphPad Prism version 5.00 (GraphPad Software, Inc., La Jolla, CA, USA).
Results and discussion
LN preparation and characterization
Over the years it has been a challenge to incorporate CsA in a suitable oral drug delivery system. Researchers have spent major efforts on developing alternatives, such as self-nano-emulsifying drug delivery systems,6 lipid- based nanoparticles,11,12 polymeric-based nanoparticles,13 micelles,14,15 liposomes,16,17 pH sensitive nanoparticles,7 etc. These novel design CsA carriers offer several advantages compared to the formulations commercialized previously. These include enhancement of drug bioavailability, avoidance of the blood peak concentration, lower risk of nephrotoxicity, and controlled release of the drug, among others.
The main objective of this study was to design CsA lipid- based nanoparticles for oral administration as an alternative to the currently marketed Sandimmune Neoral® in order to overcome some concerns about stability, safety, and pharmacokinetic behavior associated with the drug, excipients, or dosage forms.
CsA was successfully incorporated into LN using Precirol® ATO 5 as the lipid matrix. Precirol is a “generally recognized as safe” fatty ester with long acid chain length (palmitic acid) composed of a mixture of mono-, di-, and triglycerides. When this type of lipid is used to prepare LN, it has the ability to form less perfect crystals with many imperfections on the matrix and therefore offers more space for drug accommodation.18 The hot homogenization followed by ultrasonication method was selected for nanoparticle preparation since it is an organic solvent free melting process, easy to scale up, in which no complex equipment is needed, and which avoids the need for high concentrations of surfactants and co-surfactants. As previously mentioned, preventing the use of organic solvent and large amounts of surfactants in the final product is directly related to the safety of the dosage form, which is one of the main drawbacks of the marketed formulation.
To obtain suitable LN, the influence of different variables such as type of surfactant and their combination, and also the amount of CsA incorporated into the system, were investigated in terms of particle size, size distribution, and EE. In this work, Tw and PL were chosen as nonionic surfactants, Lec as amphoteric surfactant, and TC as anionic surfactant, all of them usually used as stabilizing agents in manufacturing LN.
In this study, two types of stabilizer systems were investigated. One set of formulations was prepared with a single surfactant (Tw, TC, Lec) and in the other set mixtures of Lec, TC and/or PL were employed to optimize LN characteristics.
Table 1 summarizes some of the physicochemical properties of the formulations developed including mean particle diameter, size distribution, and drug EE. Regarding LN 1, Tw was capable of stabilizing the lipid system producing particles of approximately 121 nm and a monodisperse size distribution (PDI 0.163) along with high CsA EE. These results are in good agreement with those obtained by Estella-Hermoso de Mendoza et al19 who developed good quality Precirol LN for oral administration containing Edelfosine and Tw as surfactant. Given its optimal characteristics, LN 1 was selected for further analysis, hereafter referred to as LN Tw-CsA.
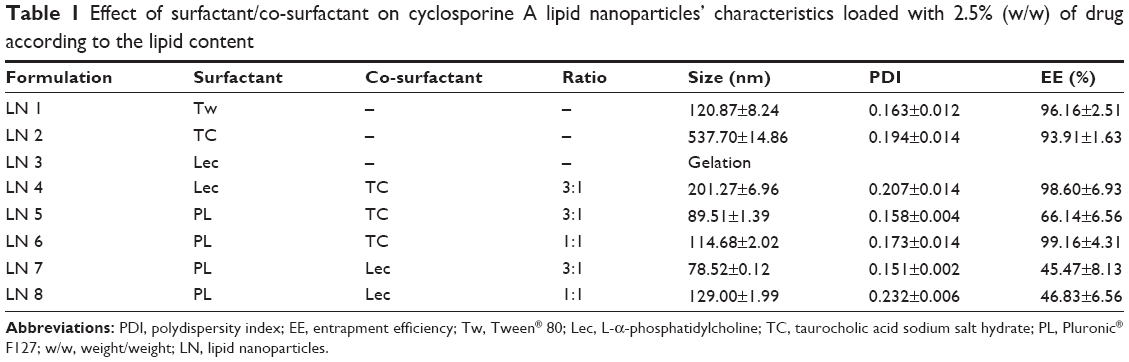 | Table 1 Effect of surfactant/co-surfactant on cyclosporine A lipid nanoparticles’ characteristics loaded with 2.5% (w/w) of drug according to the lipid content |
TC also led to a submicron particle diameter (538 nm) with a narrow size distribution and good incorporation of the drug, as can be seen in the case of LN 2 (Table 1); however, particle size was above the limit advisable for oral administration20 and thus this formulation needed to be optimized.
Where Lec was used as a surfactant (LN 3), the formulation became a gel at the cooling step of the manufacturing process. This gelation has been attributed to the limited mobility of phospholipid molecules that leads to incomplete coverage of the particle interface.21 In order to overcome this limitation, the addition of co-surfactants with high mobility (eg, bile salts) has been proposed to retard or prevent gel formation during nanoparticle preparation when using phospholipids such as Lec as stabilizing agents.22 Besides, the combination of surfactants could boost the effect of lowering the surface tension of the emulsion leading to a reduction in particle size and also may enhance long-term stability of lipid nanosystems.
It has been described that CsA is very well solubilized by mixtures of lecithin and bile salts and that this solubility is enhanced at higher lecithin concentrations due to hydrophobic interaction with phospholipid molecules.14
In this regard, LN 4 was prepared with a blend of Lec and TC at proportion 3:1. This surfactant combination led to stable nanoparticle dispersions with high drug loading capacity, and a particle size and PDI suitable for oral administration (Table 1). Therefore, this formulation was used for further studies (henceforth referred to as LN Lec:TC-CsA).
In addition, mixtures of PL:Lec and PL:TC at different ratios were also evaluated (Table 1). In both cases a high influence of PL on the mean particle diameter of the lipid nanosystems was observed (LN 5–LN 8). Higher concentrations of PL led to a decrease in particle size. It appears that this nonionic surfactant is potent for lowering surface tension. Although particle size and size distribution were optimal in all the formulations containing PL, drug EEs were low when PL:Lec was employed as emulsifier. It is possible that the presence of PL in the aqueous phase improves CsA solubility in this phase and thus decreases its incorporation in the lipid matrix. Indeed, this fact has been previously reported for encapsulation of lipophilic drugs in solid lipid nanoparticles (SLN).23 Formulations consisting of PL:TC showed higher CsA entrapment than the ones containing PL:Lec, CsA incorporation being higher when increasing TC concentration. This phenomenon could be due to the ability of bile salts to disturb the hydrophobic chains of the lipid phase, thus improving the solubility of lipophilic drugs in the system.16 Accordingly, LN 6 showed optimal characteristics for this study and was selected for further analysis. This formulation is hereafter referred to as LN PL:TC-CsA.
The influence of the initial amount of drug used to prepare the nanosystems was investigated in terms of particle size, PDI, and drug EE. For this study LN Lec:TC was selected since Lec:TC are known to solubilize CsA to a greater extent. Size ranges between 195.55±5.16 and 208.55±3.18 nm with PDI below 0.219±0.011 were obtained for all the studied drug concentrations. Results showed that increasing concentration of CsA from 2.5% to 10% (w/w) added to the lipid matrix did not notably change the mean particle diameter and size distribution. However, a slight reduction in drug EE was observed from 98.60%±6.93% to 71.30%±5.76% as the CsA concentration was increased from 2.5% to 10% (w/w), respectively. The appearance of macroscopic agglomerates in the highest concentration tested was also observed, possibly due to the limited ability of the system to incorporate the drug. This phenomenon has already been reported.12 Reduction of spaces in the matrix owing to rearrangements of the lipid causes drug expulsion and thus agglomerate formation.
Another important characteristic of LN is the particle surface charge, which is measured by zeta potential. This parameter is essential to predict the dispersion stability. In general, dispersions with high absolute zeta potential values are considered stable systems due to the electric repulsion forces generated among charged particles preventing aggregation. Nonetheless, it has been reported that in the case of nonionic surfactants, the situation is more complex and stability is reached by steric repulsion.18 In this study, zeta potential values of the optimized LN, measured before lyophilization process, were −27.8±1.5 mV (LN Lec:TC-CsA), −20.6±2.5 mV (LN PL:TC-CsA), and −14.6±1.9 mV (LN Tw-CsA). Variation in zeta potential values among the developed LN may be explained by structural changes in the surface caused by differences in the emulsifier utilized to produce them. In all cases particles were negatively charged due to the fatty acid in the lipid matrix,24 those containing ionic surfactants (Lec and TC) being more negative.
Blank LN were prepared and characterized to compare the physical properties of the nanosystems (CsA loaded and unloaded LN) and to evaluate possible changes in particle size and zeta potential value caused by the drug incorporation. As can be observed in Table 2, unloaded formulations showed similar characteristics to those obtained by loaded LN regarding particle size, PDI, and surface charge. To that effect, it appears that CsA, as a lipophilic and neutral molecule, was completely solubilized in the lipid phase without producing any change in these particle properties.
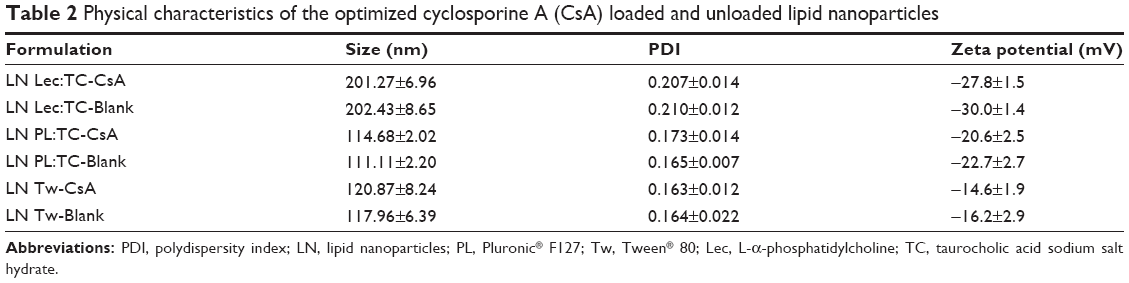 | Table 2 Physical characteristics of the optimized cyclosporine A (CsA) loaded and unloaded lipid nanoparticles |
Morphological characterization of the LN
TEM characterization was performed in order to explore the particle morphology and size distribution. The TEM images (Figure 1) revealed that the optimized LN are dispersed as individual particles with a well-defined spherical shape. Figure 1A and B depicts LN Lec:TC-Blank, whereas Figure 1D and E shows LN Lec:TC loaded with CsA. We can infer from TEM micrographs that the morphology is not altered by the presence of the drug. Particle size distribution histograms estimated from TEM images clearly prove that the particle size distribution of both formulations is governed by a Gaussian distribution (Figure 1C and F). The sizes obtained ranged from 258±20 to 261±21 nm in LN Lec:TC unloaded and loaded with CsA, respectively. Consequently, TEM analysis indicates that the presence of CsA has no effect either on the morphology or the size of nanoparticles. This statement is consistent with the results obtained from the particle size measurement by dynamic light scattering (Table 2).
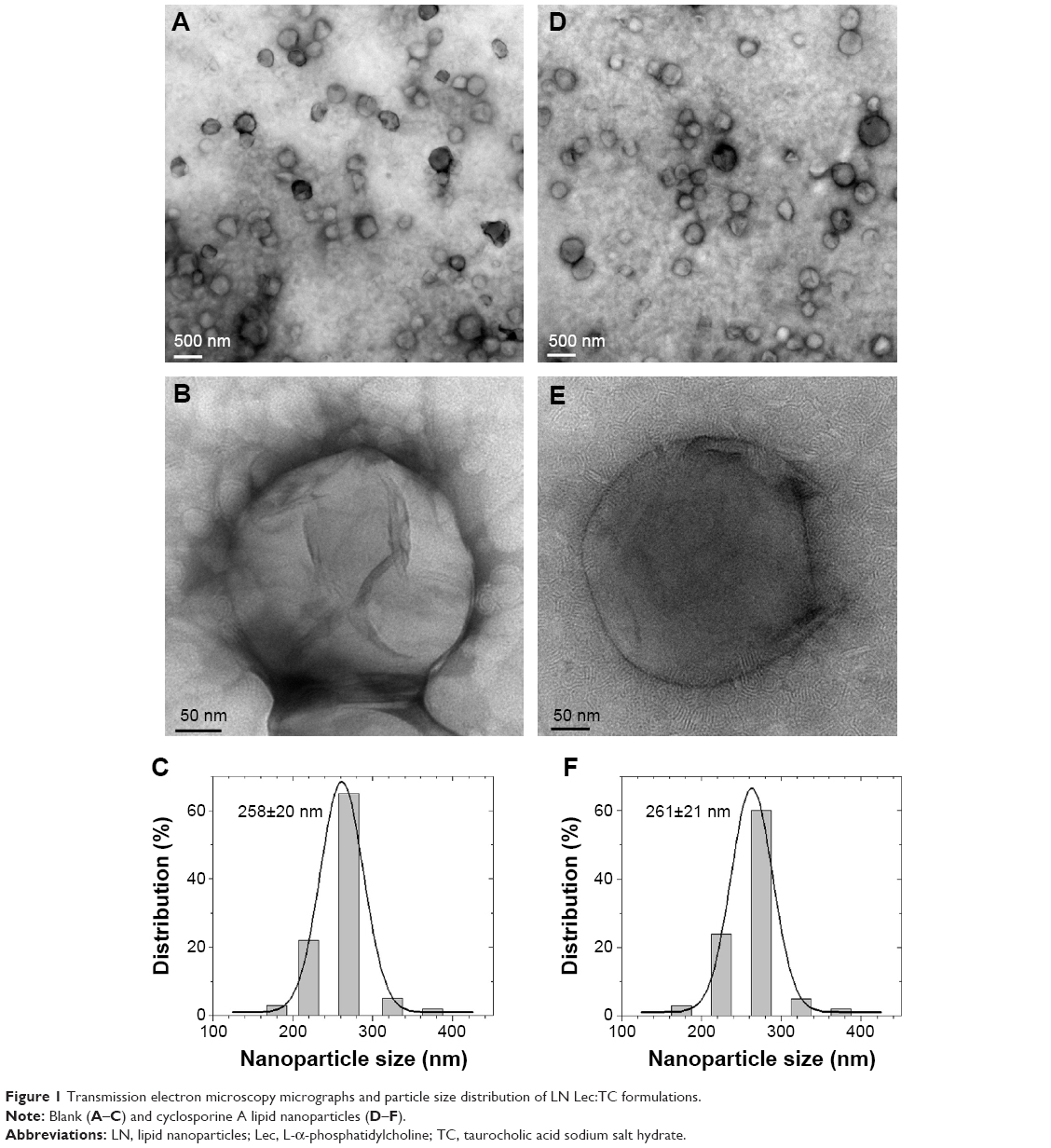 | Figure 1 Transmission electron microscopy micrographs and particle size distribution of LN Lec:TC formulations. |
Crystallinity and thermal analysis of the LN
XRD analysis enables us to identify the crystalline or amorphous state of LN, as well as revealing the spacings in the solid lipid lattice.18 This is particularly important since lipids are very often polymorphic substances. XRD patterns of pure lipid and CsA were used as references for the evaluation of LN spectra (Figure 2A). Reference spectra indicated that Precirol and CsA existed in a crystalline state before being processed to give rise to the LN Lec-TC formulation. LN prepared with and without CsA and further lyophilized exhibited the same peaks with the starting lipid material, despite the hot homogenization conditions. On the other hand, the intensity peaks that belong to CsA could not be identified in the diffractograms of CsA loaded LN, suggesting the presence of an amorphous CsA payload. The Bragg-spacing values calculated for the reflections show the presence of two types of spacings: long spacings (depending on the fatty acid chain length and the angle tilt) and short spacings (non-dependent on fatty acid chain length). Short spacings correspond to reflections at high angles, originating from the packing of lipids, and are related with the presence of polymorphs. The most stable form, the β polymorph, has a triclinic subcell with a characteristic spacing at 4.6 Å. The β′ form has an orthorhombic subcell structure with characteristic spacings at 3.8 Å and 4.2 Å. Finally, the α polymorph has a hexagonal subcell with a characteristic spacing at 4.15 Å.25 Figure 2A shows the existence of 4.61, 4.22, and 3.86 Bragg-spacings, which implies that both the bulk lipid and the nanoscale counterpart are a mixture of β and β′ polymorphs. The polymorphs differ in stability, melting point, density, and melting enthalpy. The β polymorph is the most stable and has the highest melting point and melting enthalpy. XRD results can be corroborated by DSC analysis. The DSC thermogram of pure Precirol exhibited a melting endothermic peak at 58°C, while pure CsA, Lec, and TC peaked at 140°C, 225°C, and 140°C, respectively (Figure 2B). The observed melting peak of LN, with and without CsA payload, was found to be 53°C. This depression cannot be attributed to any polymorph transition, since XRD did not reveal any Bragg-spacing modification. On the other hand, it is stated that the presence of surfactants in the melted lipid phase during the production process could distort crystals resulting in a lower melting energy.26 This fact can also be explained by the Kelvin effect, where a reduced particle size and increased surface area led to a decrease in the melting enthalpy compared to the bulk lipid.26 The CsA melting endothermic peak of loaded LN disappeared, indicating the existence of amorphous CsA or that it has been molecularly dispersed within the Precirol matrix, confirming the results obtained by XRD.
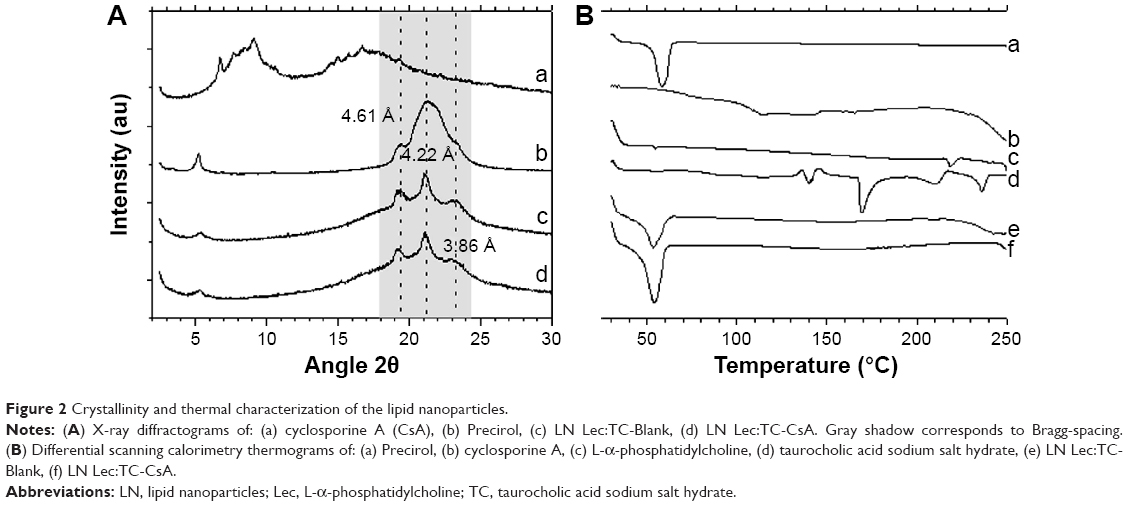 | Figure 2 Crystallinity and thermal characterization of the lipid nanoparticles. |
Surface analysis of the LN
The presence of CsA in the LN surfaces was studied by XPS. Surfaces of loaded and unloaded nanoparticles as well as some of the components were characterized. Beside C and oxygen (O), phosphorus (P) was detected in formulations containing Lec. Nitrogen (N) is a component of CsA, Lec, and TC and was detected in samples containing them, except for the unloaded LN PL:TC-Blank, probably because N concentration was below the detection limit of the technique. The absences of sulfur (S) signals also present in the TC would support the low concentration of TC on this sample surface. The N/C atomic ratio obtained from C 1s and N 1s peaks is shown in Table 3. For samples loaded with CsA there is an important increase in N content compared to the unloaded ones due to the presence of the drug. The most important increase in N signal was observed in the LN Lec:TC-CsA (Table 3). For this sample two different N 1s signals were identified, with binding energies of 402.8 and 400.0 eV (Figure 3A). The peak at higher binding energy would be associated with quaternary ammonium cations,27,28 while the peak at lower binding energy could be assigned to hydrogen-bonded amines.29 The intensity ratio between the low- and the high-binding-energy peaks increases from 0.38 in the unloaded LN to 0.95 for the loaded LN, due to the presence of CsA.
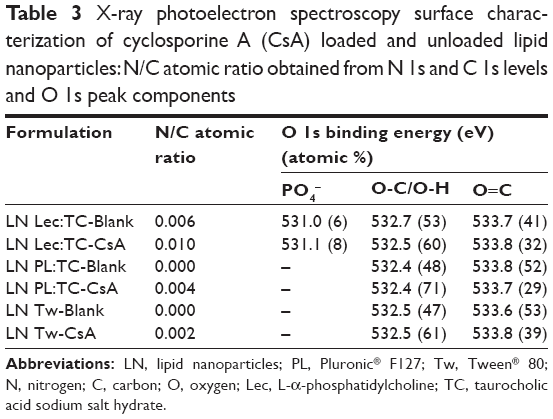 | Table 3 X-ray photoelectron spectroscopy surface characterization of cyclosporine A (CsA) loaded and unloaded lipid nanoparticles: N/C atomic ratio obtained from N 1s and C 1s levels and O 1s peak components |
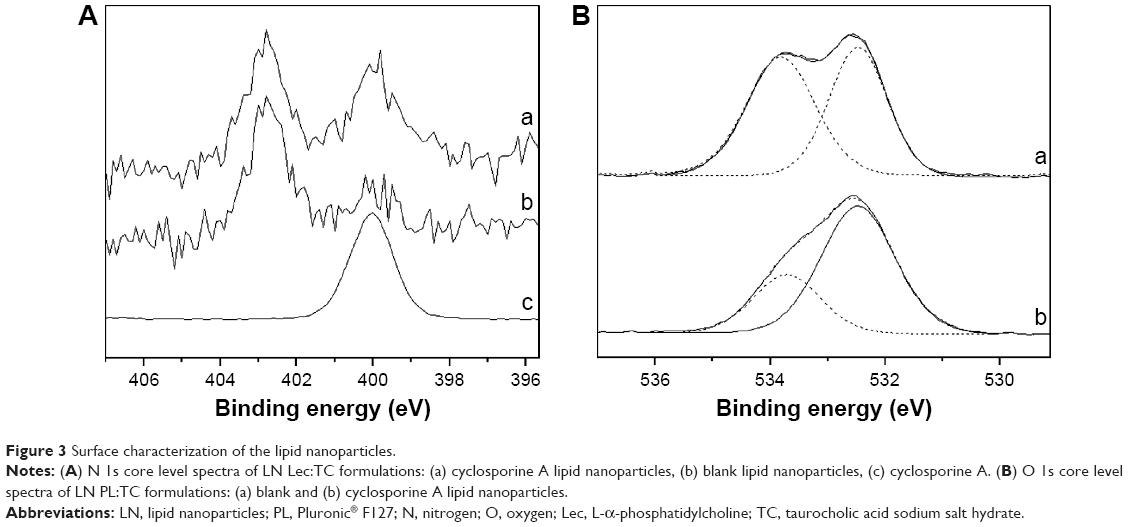 | Figure 3 Surface characterization of the lipid nanoparticles. |
Another interesting feature is the change in the O 1s peak for loaded samples (Table 3). The O 1s peak was decomposed into two peaks for samples containing PL:TC (Figure 3B) and Tw. The component at low binding energy, 532.4–532.7 eV, is attributed to O single bonded to C in C-O-H and/or in C-O-C groups while the peak at 533.6–533.8 eV would be related to O in carboxyl function.30 The third peak that appears in Lec:TC formulations would be related to the presence of PO32− groups present in the Lec.31 The atomic concentration decrease of O in carboxyl functions (peaks at 533.6–533.8 eV) in samples containing the drug compared to the unloaded ones suggests that the CsA would be interacting with these functional groups of the lipid.
Physicochemical stability studies of CsA LN
The physicochemical stability of the optimized CsA LN was studied after the lyophilization of the formulations in order to preserve their characteristics for an extended period of time. Lyophilization prolongs the physicochemical stability of lipid nanosystems by transforming the liquid nanodispersion into a dry product. Besides, a solid form allows the incorporation of the LN into capsules, tablets or pellets bearing a feasible dosage form for oral administration.8 For the lyophilization, cryoprotectant was added to the nanodispersions to reduce the LN aggregation and to obtain better particle redispersions after the freeze-drying process. Trehalose was used as the cryoprotective agent since it has been reported as being most effective in preventing particle growth in SLN.20,32
First, the effect of the freeze-drying process on the resuspension properties of the nanoparticles was studied (Figure 4). With regard to LN Lec:TC-CsA, particle characteristics remained practically unchanged after lyophilization. On the other hand, LN PL:TC-CsA and LN Tw-CsA redispersed in ultra-pure water showed a 2.35 and 1.43 fold increase in particle size, respectively. This particle growth has also been observed by other authors in SLN production.32,33 This observed variation in particle size was attributed to the different stabilizing ability of the surfactants employed in each case. In some cases, it is possible that the freeze-drying process causes a modification in the surfactant layer properties by increasing the particle concentration after water removal, leading therefore to agglomeration.8 Despite this size increase, lyophilized LN diameter was appropriate for oral delivery. Particle size distribution was also considered acceptable in the three developed LN with PDI values of approximately 0.3. In addition, slight changes in the measured zeta potential of the lyophilized formulations compared to those freshly prepared were observed. The presence of trehalose solubilized in the dispersion medium (ultra-pure water) may produce modification of its conductivity characteristics.34
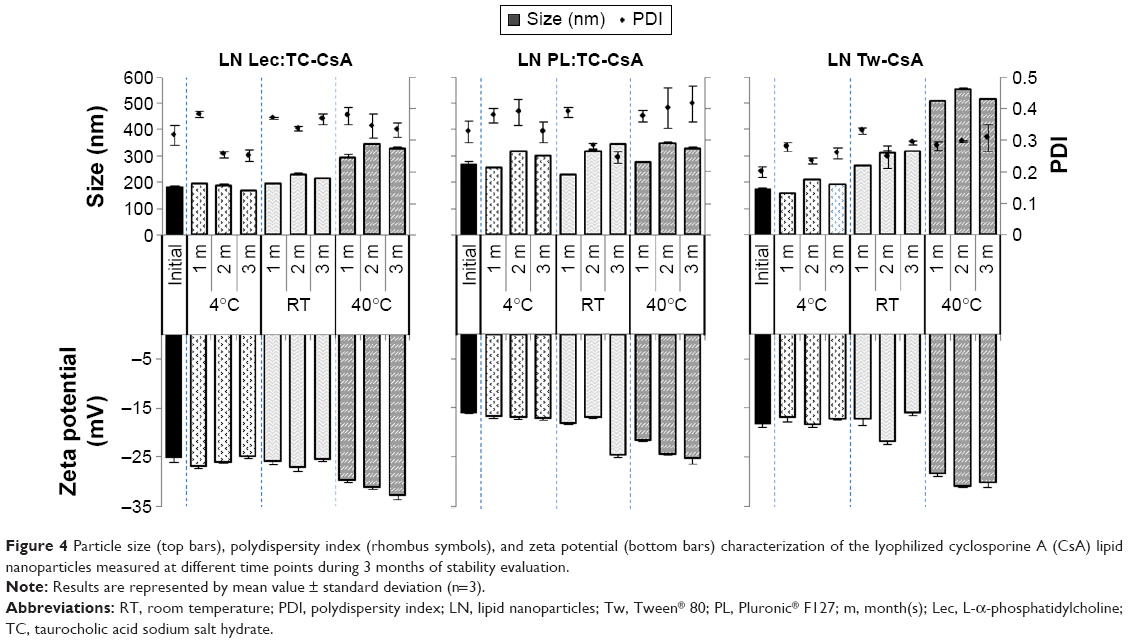 | Figure 4 Particle size (top bars), polydispersity index (rhombus symbols), and zeta potential (bottom bars) characterization of the lyophilized cyclosporine A (CsA) lipid nanoparticles measured at different time points during 3 months of stability evaluation. |
Once the redispersion properties of the LN were evaluated, the storage stability of the three developed nanosystems (LN Lec:TC-CsA, LN PL:TC-CsA, and LN Tw-CsA) was studied at various conditions (4°C, RT, and 40°C) over a period of 3 months in terms of physical and chemical properties. The formulations presented a fine, loose powder appearance in the different storage conditions, except in the case of LN Lec:TC-CsA, that after 1 month at 40°C started to lose this characteristic. This event can be promoted by larger amounts of lipid in its composition (Precirol and Lec) that are likely to melt when exposed to high temperatures.
Figure 4 summarizes the physical characteristics (size, PDI, and zeta potential values) of the lyophilized LN after their storage at different temperature conditions and resuspension in distilled water. As can be seen in the figure, in the case of LN Lec:TC-CsA particle size was practically unaltered at 4°C and RT at the end of the 3 months. However, samples at 40°C showed a marked size increase after the first month of storage (up to 1.8 fold). With respect to LN PL:TC-CsA, a negligible progressive increase in particle size over the time (below 1.3 fold) was observed, which was slightly higher at RT and 40°C. Nonetheless, these particle size changes seem to be less influenced by temperature. In contrast, a different particle growth behavior was observed with LN Tw-CsA. In this case, particle size increase was obvious from the first month at both RT and 40°C (up to 1.8 and 3.2 fold, respectively), although at 4°C storage no evident change was observed in particle size after 3 months. The particle size enlargement may be attributed to damage of the surfactant layer causing incomplete coverage of the particle surface leading to aggregates in the system. In fact, the presence of few agglomerates in the lipid nanosystems developed could be confirmed by the size distribution with PDI mean values ranging from 0.2 to 0.4.
So far, physical stability observations sustain the hypothesis that a mixture of surfactants has a synergistic effect in extending the long-term stability of LN. These results also suggest the good performance of Lec:TC and PL:TC in stabilizing CsA lipid nanosystems, probably by the formation of a stable layer on the particle surface with an excellent repulsion effect.
Moreover, in general terms, the zeta potential of all nanosystems remained unaffected for 3 months when stored at 4°C and RT. However, a slight increment of the absolute zeta potential value was observed in samples kept at 40°C (Figure 4). This increment may be explained by a possible degradation of the lipid which occurs when the product is stored under stress conditions. This storage may cause rupture of the ester bonds, resulting in negative charge of the free fatty acid in the system along with lipid rearrangement which probably modifies the surface charge of the particles, resulting in a more negative zeta potential.
Finally, in order to assess the chemical stability of the CsA LN developed, the drug content of the formulations was quantified. During the period of the study under different conditions the three formulations conserved the amount of entrapped CsA above 92% when compared to the initial drug content (data not shown), except LN Tw-CsA kept at 40°C, which showed reduction of drug content up to 20% after the second month. This destabilization of the system can be explained by rearrangement of the lipid crystal lattice caused by interaction with the emulsifier, leading to drug expulsion.8
Summing up, the optimized LN presented good physical and chemical stability and it can be stated that the best storage condition to preserve the physicochemical properties of the developed CsA lipid nanosystems was under refrigeration at 4°C±2°C. However the nanosystems could be stable at RT for a certain period of time.
In vitro biological activity of CsA LN
The immunosuppressive activity of CsA is attributed to selective T-lymphocyte inhibition. The drug, which belongs to the calcineurin inhibitor group, forms a complex on the surface of lymphocytes with the cytosolic protein CypA impeding the T-cell activation, and consequently blocking the expression of IL-2.2,4 The biological activity of the lipid nanosystems developed was evaluated by measuring the IL-2 production of Jurkat cells, a cell line derived from human T-cell leukemia, after their stimulation with the T-cell activator Con A.
In order to ensure that the inhibition of IL-2 production was due to the effect of the drug rather than to the toxicity of the treatments, it was necessary to determine the influence of the formulations on cell viability. The MTT assay revealed negligible cytotoxicity in the range of concentrations studied (data not shown) since after 24 hours of incubation, cells exposed to the different treatments showed viabilities of over 90% compared to the negative control.
The ability of the CsA loaded LN to inhibit cytokine production was studied at concentrations equivalent to 10 and 25 ng/mL CsA. These concentrations were chosen based on previous work.35 As shown in Figure 5, IL-2 secretion was significantly suppressed by CsA LN in a dose-dependent manner compared to the positive control. The same effect was observed with Sandimmune Neoral®. Indeed, no significant differences were observed when comparing the CsA LN formulations with the reference formulation, indicating that our CsA nanosystems might be as effective as the marketed formulation. Similar inhibitory activity was observed among the different CsA loaded nanosystems, ruling out possible influence of the surfactants on the biological activity of the formulations. Moreover, since blank LN did not exhibit any significant difference in IL-2 levels compared to the stimulated control, the immunosuppressive effect of the formulations could be attributed to the incorporated CsA alone. These results are in accordance with those obtained for CsA polymeric nanoparticles using similar in vitro models35,36 and confirm that the immunosuppressive effect of the drug was conserved after its production process.
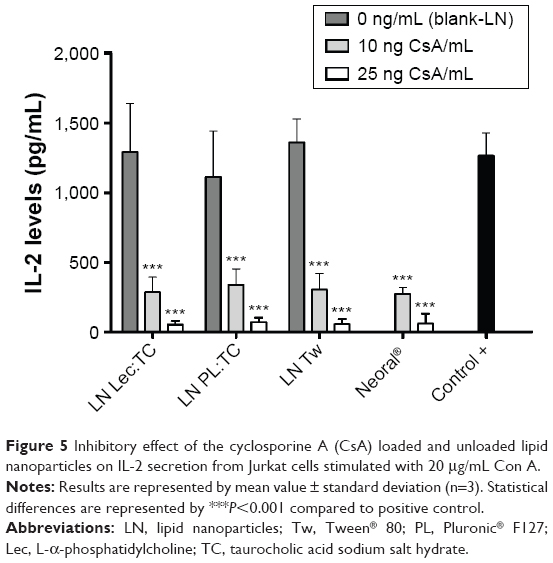 | Figure 5 Inhibitory effect of the cyclosporine A (CsA) loaded and unloaded lipid nanoparticles on IL-2 secretion from Jurkat cells stimulated with 20 μg/mL Con A. |
Conclusion
Two crucial approaches were obtained with this study. First, the CsA formulations were prepared with low surfactant concentration and avoiding organic solvents, so they are likely to have low toxicity compared to commercial formulations. And second, the CsA delivery systems were dried to obtain a powder formulation which could be easily incorporated in a conventional dosage form and also enhance the long-term stability of the final product. Interestingly, the developed formulations showed immunosuppressive effects in a stimulated human T-lymphocyte cell line. In vivo studies are in progress in order to investigate the pharmacokinetic behavior of CsA incorporated into the lipid nanosystems developed.
Acknowledgments
This work has been carried out in the framework of the COST Action TD1004. M Guada thanks “Asociación de Amigos de la Universidad de Navarra” for the fellowship grant. The EU CIG–Marie Curie under the REA grant agreement number 321642 is gratefully acknowledged. CIBER-BBN is an initiative funded by the VI National R&D&i Plan 2008–2011 financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
Survase SA, Kagliwal LD, Annapure US, Singhal RS. Cyclosporin A – a review on fermentative production, downstream processing and pharmacological applications. Biotechnol Adv. 2011;29(4):418–435. | ||
Italia JL, Bhardwaj V, Ravi Kumar MN. Disease, destination, dose and delivery aspects of ciclosporin: the state of the art. Drug Discov Today. 2006;11(17–18):846–854. | ||
Chiu YY, Higaki K, Neudeck BL, Barnett JL, Welage LS, Amidon GL. Human jejunal permeability of cyclosporin A: Influence of surfactants on P-glycoprotein efflux in Caco-2 cells. Pharm Res. 2003;20(5): 749–756. | ||
Beauchesne PR, Chung NS, Wasan KM. Cyclosporine A: a review of current oral and intravenous delivery systems. Drug Dev Ind Pharm. 2007;33(3):211–220. | ||
Schiff J, Cole E, Cantarovich M. Therapeutic monitoring of calcineurin inhibitors for the nephrologist. Clin J Am Soc Nephrol. 2007;2(2): 374–384. | ||
Lei Y, Lu Y, Qi J, et al. Solid self-nanoemulsifying cyclosporin A pellets prepared by fluid-bed coating: preparation, characterization and in vitro redispersibility. Int J Nanomedicine. 2011;6:795–805. | ||
Dai W, Guo Y, Zhang H, Wang X, Zhang Q. Sylysia 350/Eudragit S100 solid nanomatrix as a promising system for oral delivery of cyclosporine A. Int J Pharm. 2015;478(2):718–725. | ||
Mehnert W, Mäder K. Solid lipid nanoparticles: Production, characterization and applications. Adv Drug Deliv Rev. 2001;47(2–3):165–196. | ||
Hauss DJ. Oral lipid-based formulations. Adv Drug Deliv Rev. 2007; 59(7):667–676. | ||
Guada M, Imbuluzqueta E, Estella-Hermoso de Mendoza A, Lana H, Dios-Vieitez MC, Blanco-Prieto MJ. Ultra high performance liquid chromatography–tandem mass spectrometry method for cyclosporine a quantification in biological samples and lipid nanosystems. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;927:164–172. | ||
Müller RH, Runge S, Ravelli V, Mehnert W, Thünemann AF, Souto EB. Oral bioavailability of cyclosporine: Solid lipid nanoparticles (SLN) versus drug nanocrystals. Int J Pharm. 2006;317(1):82–89. | ||
Urban-Morlan Z, Ganem-Rondero A, Melgoza-Contreras LM, Escobar-Chavez JJ, Nava-Arzaluz MG, Quintanar-Guerrero D. Preparation and characterization of solid lipid nanoparticles containing cyclosporine by the emulsification-diffusion method. Int J Nanomedicine. 2010;5:611–620. | ||
Ankola D, Wadsworth R, Ravi Kumar M. Nanoparticulate delivery can improve peroral bioavailability of cyclosporine and match Neoral Cmax sparing the kidney from damage. J Biomed Nanotechnol. 2011;7(2): 300–307. | ||
Guo J, Wu T, Ping Q, Chen Y, Shen J, Jiang G. Solubilization and pharmacokinetic behaviors of sodium cholate/lecithin-mixed micelles containing cyclosporine A. Drug Deliv. 2004;12(1):35–39. | ||
Yu H, Xia D, Zhu Q, Zhu C, Chen D, Gan Y. Supersaturated polymeric micelles for oral cyclosporine A delivery. Eur J Pharm Biopharm. 2013; 85(3 Pt B):1325–1336. | ||
Guan P, Lu Y, Qi J, et al. Enhanced oral bioavailability of cyclosporine A by liposomes containing a bile salt. Int J Nanomedicine. 2011;6: 965–974. | ||
Chen D, Xia D, Li X, et al. Comparative study of Pluronic® F127-modified liposomes and chitosan-modified liposomes for mucus penetration and oral absorption of cyclosporine A in rats. Int J Pharm. 2013;449(1–2):1–9. | ||
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
Estella-Hermoso de Mendoza A, Campanero MA, Lana H, et al. Complete inhibition of extranodal dissemination of lymphoma by edelfosine-loaded lipid nanoparticles. Nanomedicine (Lond). 2012;7(5): 679–690. | ||
Das S, Chaudhury A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS Pharm Sci Tech. 2011; 12(1):62–76. | ||
Westesen K, Siekmann B. Investigation of the gel formation of phospholipid-stabilized solid lipid nanoparticles. Int J Pharm. 1997; 151(1):35–45. | ||
Westesen K, Siekmann B, Koch MH. Investigations on the physical state of lipid nanoparticles by synchrotron radiation X-ray diffraction. Int J Pharm. 1993;93(1–3):189–199. | ||
Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95(3):627–638. | ||
Wang K, Qi J, Weng T, et al. Enhancement of oral bioavailability of cyclosporine A: comparison of various nanoscale drug-delivery systems. Int J Nanomedicine. 2014;9:4991–4999. | ||
Marangoni AG, Narine SS. Physical Properties of Lipids. United States: Marcel Dekker; 2002. | ||
Fang J, Fang C, Liu C, Su Y. Lipid nanoparticles as vehicles for topical psoralen delivery: Solid lipid nanoparticles (SLN) versus nanostructured lipid carriers (NLC). Eur J Pharm Biopharm. 2008;70(2):633–640. | ||
Ma Q, Zhang H, Zhao J, Gong Y. Fabrication of cell outer membrane mimetic polymer brush on polysulfone surface via RAFT technique. Appl Surf Sci. 2012;258(24):9711–9717. | ||
Yeh SB, Chen CS, Chen WY, Huang CJ. Modification of silicone elastomer with zwitterionic silane for durable antifouling properties. Langmuir. 2014;30(38):11386–11393. | ||
Kristensen EM, Nederberg F, Rensmo H, Bowden T, Hilborn J, Siegbahn H. Photoelectron spectroscopy studies of the functionalization of a silicon surface with a phosphorylcholine-terminated polymer grafted onto (3-aminopropyl) trimethoxysilane. Langmuir. 2006;22(23): 9651–9657. | ||
Saad M, Gaiani C, Mullet M, Scher J, Cuq B. X-ray photoelectron spectroscopy for wheat powders: measurement of surface chemical composition. J Agric Food Chem. 2011;59(5):1527–1540. | ||
Ren J, Eckert H. Quantification of Short and Medium Range Order in Mixed Network Former Glasses of the System GeO2–NaPO3: A Combined NMR and X-ray Photoelectron Spectroscopy Study. J Phys Chem C. 2012;116(23):12747–12763. | ||
Schwarz C, Mehnert W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN). Int J Pharm. 1997;157(2):171–179. | ||
Cavalli R, Caputo O, Carlotti ME, Trotta M, Scarnecchia C, Gasco MR. Sterilization and freeze-drying of drug-free and drug-loaded solid lipid nanoparticles. Int J Pharm. 1997;148(1):47–54. | ||
Kovačević AB, Müller RH, Savić SD, Vuleta GM, Keck CM. Solid lipid nanoparticles (SLN) stabilized with polyhydroxy surfactants: Preparation, characterization and physical stability investigation. Colloids Surf A Physicochem Eng Asp. 2014;444:15–25. | ||
Hermans K, Van den Plas D, Everaert A, Weyenberg W, Ludwig A. Full factorial design, physicochemical characterisation and biological assessment of cyclosporine A loaded cationic nanoparticles. Eur J Pharm Biopharm. 2012;82(1):27–35. | ||
Takebe G, Takagi T, Suzuki M, Hiramatsu M. Preparation of polymeric nanoparticles of cyclosporin A using infrared pulsed laser. Int J Pharm. 2011;414(1–2):244–250. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.