Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
Lipid metabolism in chronic obstructive pulmonary disease
Authors Chen H, Li Z, Dong L, Wu Y, Shen H, Chen Z ![]()
Received 27 November 2018
Accepted for publication 19 March 2019
Published 13 May 2019 Volume 2019:14 Pages 1009—1018
DOI https://doi.org/10.2147/COPD.S196210
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Haipin Chen,1 Zhouyang Li,1 Lingling Dong,1 Yinfang Wu,1 Huahao Shen,1,2 Zhihua Chen1
1Key Laboratory of Respiratory Disease of Zhejiang Province, Department of Respiratory and Critical Care Medicine, Second Affiliated Hospital of Zhejiang University School of Medicine, Institute of Respiratory Diseases, Hangzhou, Zhejiang, People’s Republic of China; 2State Key Lab of Respiratory Disease, Guangzhou, Guangdong, People’s Republic of China
Abstract: Dysregulated lipid metabolism plays crucial roles in various diseases, including diabetes mellitus, cancer, and neurodegeneration. Recent studies suggest that alterations in major lipid metabolic pathways contribute to pathogenesis of lung diseases, including chronic obstructive pulmonary disease (COPD). These changes allow lung tissue to meet the energy needs and trigger anabolic pathways that initiate the synthesis of active molecules directly involved in the inflammation. In this review, we summarize the changes of catabolism and anabolism of lipids, lipid molecules including lipid mediators, lipid synthesis transcription factors, cholesterol, and phospholipids, and how those lipid molecules participate in the initiation and resolution of inflammation in COPD.
Keywords: COPD, lipid metabolism, airway inflammation
Introduction
Lipids are hydrophobic molecules with three general functions, which include energy storage, membrane building, and signal transduction.1 Lipids can be divided into different classes, including triglycerides, cholesterol, phospholipids, and glycolipids. Metabolism of lipids includes anabolism and catabolism, which involves the synthesis of new lipids from smaller molecules and oxidation of lipids to supply energy or generate other lipids mediators, respectively.2
Lipid metabolism has been implicated in diabetes mellitus, cancer, neurodegeneration, and cardiovascular diseases.3,4 Lipid metabolism contributes to the pathogenesis of diabetes mellitus by modulating the insulin resistance and inflammation.5 As highly proliferating cells, tumor cells need lipids for synthesis of membranes and signaling molecules.6,7 Recent evidence suggests that lipid metabolism in lung disease might also be altered.8–10
Once considered as static metabolic energy reserves, lipids have emerged as important components of cellular signal transduction pathways. Their roles in modulating host inflammatory responses, either promotion or resolution, are of clinical interests.11 At present, there are rapidly expanding insights into novel lipid mediators that function to regulate inflammation.
Chronic obstructive pulmonary disease (COPD) is a pulmonary dysfunction characterized by irreversible and progressive airflow obstruction, chronic airway inflammation, and systemic effects or comorbidities. Several studies have suggested that COPD is a disease associated with derangement of lipids. A study conducted in the Warsaw Ghetto during World War II showed that a high percentage of people who died of starvation had emphysema.12 Furthermore, rats with restricted caloric intake for a few weeks showed changes in pulmonary mechanics and lung structure that were described as “emphysema-like”.13–18 Besides, there are increasing evidence suggesting that obesity is associated with declined lung function and increased morbidity in moderate to severe COPD.19,20
More recent studies have suggested that alterations in major metabolic pathways contribute to pathogenesis of COPD.21,22 The lung is a complex organ composing a large number of cell types. Same lipid molecules might be differentially modulated in various cells in COPD, and they may exert differential effects on the eventual COPD pathogenesis. Thus, studying the cell-specific alterations and functions of lipid molecules in COPD is of great importance. In this review, we will discuss the role of lipid metabolism, focusing on the alterations of various lipids molecules, and the consequences of these changes in COPD.
Lipid catabolism in COPD
As a mechanism for the utilization of fatty acids, fatty acid oxidation (FAO) (Figure 1) is a major bioenergetic pathway that is upregulated under conditions of prolonged fasting, exercise, or metabolic stress.23 The mitochondrial membrane is impermeable to fatty acids. They must be conjugated to carnitine to enter mitochondria. Carnitine forms a high-energy ester bond with fatty acids by the action of carnitine palmitoyl transferase 1 (CPT1), located in the inner aspect of the outer mitochondrial membrane, with the formation of acylcarnitines. Carnitine acylcarnitine translocase (CACT) exchanges acylcarnitines and carnitine between outer and inner membranes of mitochondrial and finally acylcarnitines are converted back into acyl-CoAs for oxidation by CPT2.24,25 Fatty acids are an important source of energy, as FAO produces 2.5 times as much ATP per mole as oxidation of glucose.26
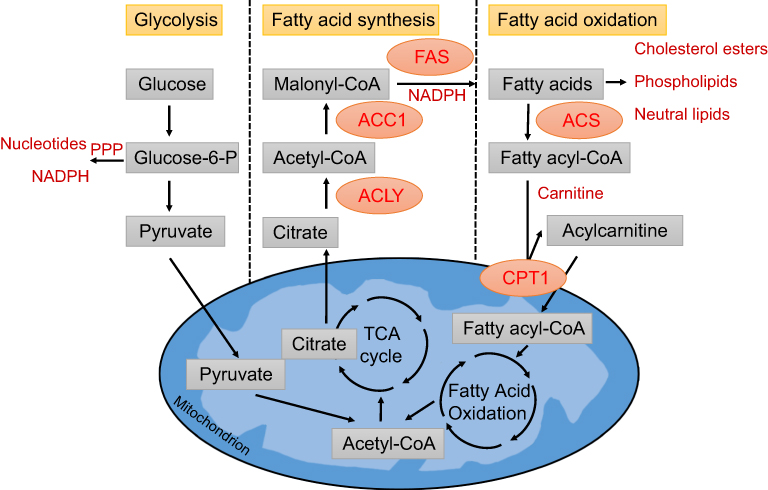 | Figure 1 Metabolic pathway of glycolysis, FAS and FAO. Glycolysis converts glucose into pyruvate, which can enter the tricarboxylic acid (TCA) cycle. Glycolysis also feeds the pentose phosphate pathway (PPP), which generates ribose for nucleotides and NADPH. Citrate can be fully oxidized to generate ATP or transported to the cytoplasm where it is converted back to acetyl-CoA by ATP citrate lyase (ACLY). A portion of the acetyl-CoA is carboxylated to malonyl-CoA by acetyl-CoA carboxylase 1 (ACC1). Fatty acid synthase (FAS) performs the condensation of acetyl-CoA and malonyl-CoA to produce the 16-carbon saturated fatty acid palmitate and other saturated long-chain FAs. In the cytosol, fatty acy-CoA synthases (ACS)activate fatty acids by converting them to fatty acyl-CoA. Fatty acyl-CoA is converted to acylcarnitine by CPT1 on ther outer mitochondrial membrane and transported to the mitochondrial matrix. In the mitochondrial matrix, fatty acyl-CoA is oxidized to acetyl-CoA through fatty acid oxidation. Abbreviations: ACC1, acetyl-CoA carboxylase α; ACLY, ATP citrate lyase; ACS, acyl-CoA synthase; CPT1, carnitine palmitoyltransferase 1; FAS, fatty acid synthase; TCA cycle, tricarboxylic acid cycle. |
Although several factors, including genetic or environmental ones, may contribute to the development of COPD, cigarette smoking (CS) is still the highest risk factor.27 Glucose metabolism decreased in lung alveolar cells after subchronic cigarette smoke exposure (8 weeks). This impairment was compensated by enhanced FAO to maintain cellular energy homeostasis, accompanied by an increased expression of CPT1. Increased utilization of palmitate (most likely from dipalmitoyl-phosphatidylcholine) was found in alveolar type 2 cells.21,28 Consistently, cigarette smoke exposure promoted FAO and increased mitochondrial respiration in human bronchial epithelial cells.29
This switch might be important to maintain adenosine triphosphate (ATP) levels and delay CS-induced cellular damage. Metabolomics screening showed that reduced L-carnitine, a metabolite critical for transporting long-chain fatty acids into the mitochondrial for their subsequent β-oxidation, was associated with impaired lung function.30
However, sustained elevation in FAO may also aggravate CS-induced ROS accumulation, mitochondrial damage, and cell death.29 As phosphatidylcholine represents approximately 80% of the surfactant phospholipid, continuously increased FAO may lead to surfactant deficiency, which had been observed in smokers and patients with COPD.31,32
The FAO level might vary according to cell types as showed by reduced FAO capacity in airway smooth muscle cell (ASMC) of COPD patients. Changes in fatty acid metabolism may contribute to increased biosynthesis, supporting ASMC hypertrophy and/or hyperplasia.33 Future studies are required to fully clarify the metabolic responses of other cells, including those immune cells.
Catabolic lipid mediators in COPD
Eicosanoids are a family of very potent biological signaling molecules that act as short-range messengers, affecting tissues near the cells that produce them. In response to inflammation, phospholipase A2 attacks membrane phospholipids, releasing arachidonic acid (AA) from the middle carbon of glycerol. Arachidonic acid, a 20-carbon fatty acid containing four double bonds, is a primary target of regulated PLA2 hydrolysis, and is the precursor to a vast array of lipid signaling molecules.34 Cyclooxygenases (COX) 1/2 oxidize AA to generate the prostaglandin family of oxidized fatty acids. In general, these lipid species can be proinflammatory, propagating their signaling actions via receptor-mediated mechanisms, and are responsible for many of the phenomena of inflammation.35,36
Prostanoids are a family of metabolites of the fatty acid arachidonic acid, which include PGD2, PGE2, PGF2, and PGI2, as well as thromboxane A.37 PGE2 levels were increased in respiratory secretions from patients with COPD, which could contribute to airway inflammation and the impaired lung repair.38–40
Leukotrienes are a family of eicosanoid inflammatory mediators, which are synthesized in the cell from arachidonic acid by arachidonate 5-liopxygenase. In COPD, activated macrophages, neutrophils, and epithelial cells produce cysteinyl leukotrienes (LTC4, LTD4, and LTE4) and Leukotriene B4 (LTB4).41 Leukotriene B4 (LTB4), a neutrophil and T-cell chemoattractant, is a potent inflammatory mediator. LTB4 concentrations were increased in induced sputum and were further increased in sputum and exhaled breath condensate during acute exacerbations.42,43
The role of oxidized lipids as proinflammatory mediators is well established. However, in the past decades several classes of lipid mediators have been discovered whose signaling actions act to resolve rather than to promote inflammation. These bioactive lipid mediators are called pro-resolving lipid mediators (SPMs).44 SPMs are mainly biosynthesized from omega-3 essential fatty acids, including eicosapentaenoic and docosahexaenoic acids, and are generally categorized into five families: lipoxins, D-series resolvins, E-series resolvins, protectins, and maresins. These molecules are currently the subject of intense research, as they represent an emerging generation of investigational new drugs to address inflammatory diseases.
Pro-resolving pathways were disrupted in lung tissue from patients with COPD. Supplementation with resolvin D1 was associated with a reduced development of cigarette-smoke-induced increase of neutrophils and total cell counts, emphysema and airspace enlargement, with concurrent reductions in inflammation, oxidative stress, and cell death. D-series resolvins decreased inflammatory cytokines and enhanced phagocytosis in human alveolar macrophages from COPD patients. Thus, these results suggest that supplementation with SPMs might reduce the development of emphysema by controlling chronic inflammation.45–47
Nitrated fatty acids (NFAs), endogenous products of nonenzymatic reactions of NO-derived reactive nitrogen species with unsaturated fatty acids, exhibit substantial anti-inflammatory activities. NFA treatment downregulated expression and activity of the inflammatory transcription factor NF-κB while upregulated PPAR-γ. It also downregulated production of inflammatory cytokines and chemokines and of the protease cathepsin S (Cat S), a key mediator of emphysematous septal destruction.48
The progression of COPD is often accelerated by its exacerbation.49 A primary goal of treatment is to reduce the frequency of exacerbation. Thus, a specific biomarker for detection and diagnosis of exacerbations is needed. Considering the role of those lipid mediators in COPD, whether they could become a biomarker of exacerbations need further investigation.
It is now clear that the lipid mediators play an important role in regulating generation and resolution of inflammation by modulating the balance between proinflammatory and anti-inflammatory mediators; however, it remains mysterious how lung shift from the secretion of proinflammatory to anti-inflammatory molecules. More studies are needed to directly assess the impact of lipid metabolic genes on COPD development, which are important for production of various lipid mediators.
Lipid anabolism in COPD
Fatty acid synthesis (FAS) (Figure 1) uses products derived from several other metabolic pathways, notably glycolysis, the TCA cycle, and pentose phosphate pathway. These metabolic pathways cross-talk with FAS by providing precursors and NADPH. Glycolysis feeds the pentose phosphate pathway, which generates ribose for nucleotides and NADPH.50 TCA cycle-derived citrate may be exported from the mitochondria into the cytosol via the citrate carrier, where ATP citrate lyase converts it into acetyl-CoA. FAS takes place in the cytoplasm of the cell, commencing with the ATP-dependent carboxylation of acetyl-CoA to malonyl-CoA. This committed step of FAS is catalyzed by acetyl-CoA carboxylase 1 (ACC1).51 The next step is performed by FA synthase (FAS), a key multifunctional enzyme that acts in an NADPH-dependent manner to catalyze the condensation of acetyl-CoA and malonyl-CoA to produce the 16-carbon saturated FA palmitate and other saturated long-chain FAs (LCFAs).52 Saturated LCFAs can be further modified by elongases or desaturases to form more complex lipids, which are then used for the synthesis of phospholipids, triglycerides, and cholesterol esters.
Viral infections can provoke acute worsening of disease in patients with COPD.53 Study showed that the activity of FAS was enhanced when infected with rhinovirus in bronchial epithelial cells, and that inhibition of FAS reduced the rhinoviral infections.54 Therefore, FAS might be a potential target for exacerbation of COPD.
Prolonged cigarette smoke may lead to different metabolic changes. Warburg effect is the phenomenon that tumor cells consume glucose at a surprisingly high rate compared to normal cells and secrete most of the glucose-derived carbon as lactate rather than oxidizing it completely. Similar to the role of “Warburg effect” in cancer cells, glucose consumption and lactate production were increased in human bronchial epithelial BEAS2B cells treated with cigarette smoke condensate for 7 months. Besides, lipid biosynthetic capacity and net reductive carboxylation were enhanced, suggesting a profound anabolic reprogramming in the airway epithelium.55,56 Although speculative, lipid metabolism changes may help us understand why COPD patients tend to get lung cancer.57
Lipid synthesis transcription factors in COPD
Transcriptional mechanisms regulating lipid synthesis are known to be dependent on a number of transcription factors, including peroxisome proliferator activated receptors (PPARs), liver X receptor (LXR), and SREBPs.58–61 Although transcriptional networks regulating lipid homeostasis have been extensively studied in other cell types, including hepatocytes and adipocytes, less is known regarding the transcriptional control of lipid homeostasis in the respiratory system. Those transcription factors might be pathophysiologically or therapeutically relevant to COPD if their potential roles in COPD are well characterized.
PPAR-γ is a member of the PPAR subfamily of nuclear hormone receptors that function as transcription factors, modulating glucose and lipid metabolism, inflammation, and cell proliferation. PPAR-γ forms heterodimers with retinoid X receptors, which bind to PPAR response elements and regulate transcription of target genes.61 PPAR-γ was down-regulated in lung tissue, epithelial cells, and myeloid dendritic cells in COPD patients.62,63 Mice lacking PPAR specifically in airway epithelial cells displayed increased susceptibility to chronic CS-induced emphysema, with excessive macrophages accumulation.64 Conditional knock-out of PPAR-γ in antigen-presenting cells (APC) led to spontaneous lung inflammation and emphysema that resembled the phenotype of smoke-exposed mice.63 Treating epithelial cells with synthetic (rosiglitazone) or endogenous PPAR-γ agonists strongly up-regulated PPAR-γ expression and activity, suppressed CSE-induced production and secretion of inflammatory cytokines.62 Later study showed that PPAR-γ ligands reduced proinflammatory cytokine production, including tumor necrosis factor-α and CCL5 in COPD alveolar macrophages. Rosiglitazone, one of PPAR-γ agonist, increased the levels of M2 genes that are involved in anti-inflammatory effects and tissue repair and enhanced the clearance of apoptotic neutrophils. Moreover, Rosiglitazone attenuated airway neutrophilia in a corticosteroid-resistant mouse model of pulmonary inflammation, without leading to an increase in the pulmonary bacterial burden, unlike dexamethasone.65,66 These data suggest that activation of PPAR-γ in local airways might be an effective therapeutic strategy for COPD.
LXR is a sensor of cellular cholesterol load, which regulates the transcription of genes involved in cholesterol efflux and low-density lipoprotein receptor degradation.60 The levels of LXR were significantly increased in whole lung tissue extracts of COPD patients and smokers, and those increases were located in small airway and alveolar epithelium.67
Sterol-response element-binding proteins (SREBPs) are transcription factors that regulate the expression of genes encoding enzymes required for synthesis of cholesterol and unsaturated fatty acids.5 Conditionally deleting the SREBP cleavage-activating protein gene, Scap, in alveloar epithelial cells decreased the phospholipid synthesis while lipid storage, synthesis, and transfer by lung lipofibroblasts were increased.68 Second-hand smoke stimulates lipid accumulation in the liver by modulating AMPK and SREBP1.69 Further studies are needed to investigate the role of SREBPs in lung diseases.
Cholesterol in COPD
Cholesterol is an essential component of cellular membranes that is therefore required for cell proliferation.70 Several intermediates of the cholesterol biosynthetic pathway, as well as oxidized cholesterol products such as oxysterols, modulate immune cell functions by affecting signaling molecules or by binding to cellular receptors.71 Cholesterol is produced from acetyl-CoA via cytosolic HMG-CoA reductase (HMGCR) pathway. The activity of this enzyme is regulated by the levels of intracellular cholesterol by means of feedback inhibition, control of gene expression via SREBPs, and cholesterol-induced polyubiquitination and proteasomal degradation.72 In addition, the activity of HMGCR is regulated through phosphorylation by AMP-activated protein kinase (AMPK).
Patients with very severe COPD exhibited higher level of cholesterol and serum Apolipoprotein M in blood.73,74 Oxidized cholesterol products 27-hydroxycholesterol (27-HC) and 25-hydroxycholesterol (25-HC) were elevated in the airways of COPD patients compared with those in healthy subjects.75,76 Inhibition of HMGCR by statin attenuated key genes involved in inflammatory processes, immune response, and leukocyte activation in lung tissues of COPD patients.77 Studies in animal models revealed that treatment with statin prior to and continued throughout smoke exposure reduced the total influx of leukocytes, neutrophils, and macrophages into the lung and airways.78 Treatment with the concentration of 27-HC detected in COPD airways significantly augmented expression of senescence-associated proteins and senescence-associated β-galactosidase activity, and delayed cell growth through the prostaglandin E2-reactive nitrogen species pathway.79 Moreover, 25-HC in airway epithelial cells regulated CS-induced B-cell migration and inducible bronchus-associated lymphoid tissue (iBALT) formation, a tertiary lymphoid organ that develops in the lungs during infection, autoimmunity, or chronic inflammation. Mice deficient with 25-hydroxylase, the key enzyme for 25-HC production, or the oxysterol receptor Epstein–Barr virus-induced gene 2 exhibited decreased iBALT and subsequently attenuated CS-induced emphysema.76
The upregulation of cholesterol can be interpreted as smoking has an adverse impact on the lipoprotein. Cholesterol is insoluble in water, which is carried in blood plasma as plasma lipoproteins. Much of the cholesterol synthesis takes place in the liver. Low-density lipoprotein (LDL) carries cholesterol to extrahepatic tissues, and high-density lipoprotein (HDL) transports cholesterol to the liver. Smoking is associated with an increase in circulating LDL, and a decrease in plasma HDL levels.80,81 Apolipoprotein A1 (ApoA1) is the major protein in HDL that plays an important role in reverse cholesterol transport by extracting cholesterol and phospholipid from various cell types, including lung cells, and transferring them to the liver. ApoA1 levels were significantly decreased in the lungs of patients with COPD and in the lungs of mice exposed to CS. ApoA1 overexpression attenuated lung inflammation, oxidative stress, metalloprotease activation, and apoptosis in CS-exposed mouse lungs. Mechanistically, ApoA1 prevented CSE-induced translocation of Fas and downstream death-inducing signaling complex into lipid rafts, thereby inhibiting Fas-mediated apoptosis.82
Observational studies suggested that statin use was associated with reduced morbidity and/or mortality in COPD, whereas a large randomized controlled trial concluded that simvastatin did not reduce exacerbations in patients with COPD that had no cardiovascular indication for statin treatment.83 Therefore, it is possible that a subgroup of COPD patients with cardiovascular indications and/or systemic inflammation may obtain clinical benefit from statin treatment.
Phospholipid in COPD
Phospholipids are a class of lipids that are major component of all cell membranes, composed of sphingomyelin and glycerophospholipids. Sphingolipids were significantly higher expressed in smokers with COPD than in smokers without COPD in sputum using untargeted lipidomic analysis.84 A recent study found that sphingomyelins were strongly associated with emphysema and glycosphingolipids were associated with COPD exacerbations.85
Ceramide is a component of sphingomyelin, which is found in the cell membrane and is associated with a wide variety of cellular functions, including growth, differentiation, and apoptosis. Ceramide is de novo generated by serine palmitoyl transferase, ceramide synthase, or via membrane sphingomyelin hydrolysis by sphingomyelinase. It can be directly phosphorylated by ceramide kinase to ceramide-1-phosphate (C1P).86
Studies have shown that ceramide triggered apoptosis of pulmonary epithelial cells and was involved in the destruction of alveolar structure in emphysema.87,88 Cigarette smoke exposure could increase the level of C16-Cer, and C16-Cer accumulation contributed to mitochondrial damage and PINK1-mediated necroptosis.89 While C1P reduced CS-induced acute and chronic lung inflammation and development of emphysema in mice, which was associated with a reduction in neutral sphingomyelinase (nSMase) and NF-κB activity in the lungs, nSMase can hydrolyze the phosphodiester bond of sphingomyelin to release the ceramide, whose activity in human serum was correlated negatively with lung function decline.90
Conclusion remarks
Though current literature have addressed certain parts of lipid metabolism in COPD pathogenesis (summarized in Table 1), future studies are encouraged to address the following concerns.
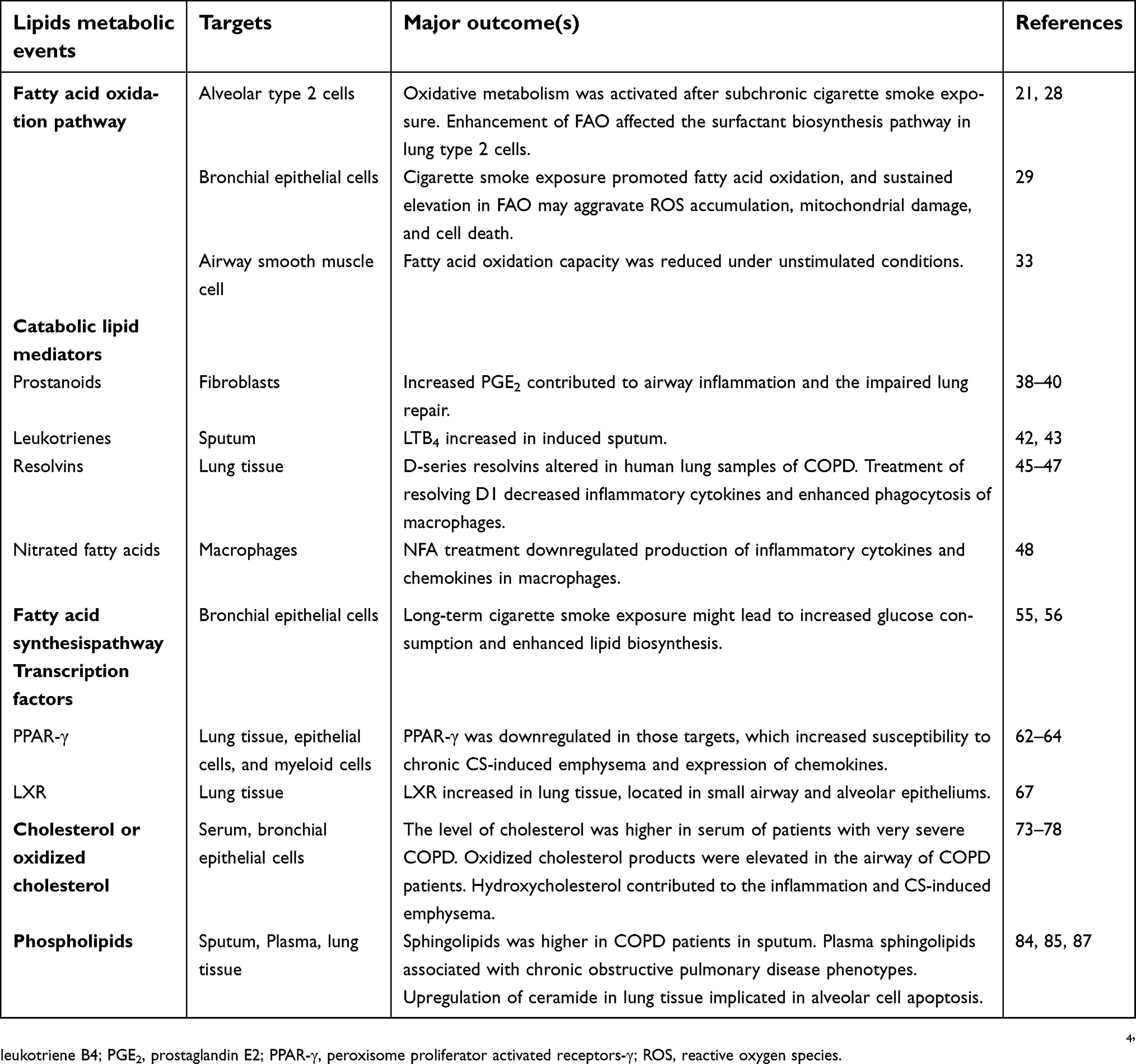 | Table 1 Lipids alterations in COPD |
Although COPD is a lung disease, it is associated with systemic manifestations and comorbid conditions, including skeletal muscle wasting, cachexia, and lung cancer.91,92 It will be interesting to determine whether lipid metabolism alteration in organs like liver or adipose tissue is consistent with changes in lung. The detailed effects and mechanisms of cigarette smoke putting on lipid metabolism in those organs will enhance our understanding of the relations between COPD and comorbidities.
Most of the currently available studies about energy metabolism in COPD focused on epithelial cells and ASMCs, while few advances have been made to unveil the metabolic changes in those immune cells. Macrophages function in host defense, tissue homeostasis and repair, pathology, and development. To accommodate this function, macrophages usually undergo a metabolism reprogramming to adopt an activation state.93 Alveolar macrophages play a key role in sensing and eliminating microbial agents early in the course of an infection, while cigarette smoke compromises the ability of alveolar macrophages to phagocytose bacteria.94,95 Activation of PPAR-γ is able to reduce cigarette-smoke-induced inflammation and to decrease the magnitude of bacterial exacerbations.66 Thus, it can be estimated that macrophages might have a dysregulated lipid metabolism that have a compromised function to phagocytose bacteria. Targeting those metabolism pathways in macrophages might be an effective therapeutic approach to improve COPD management.
Little is known about the changes of FAS in COPD, as balance of FAS and FAO is a key molecular integrator of energy homeostasis. Understanding how the balance between FAS and FAO is regulated under conditions of inflammation, and examining the external and intracellular signaling pathways regulating this balance, are also challenges in COPD research. In addition, it will be necessary to address the role FAS and coordinating transcription regulators in COPD using conditional knock-out mice. Pulmonary surfactant consists of lipids and related surfactant protein, and it is also not clear whether dysregulated lipid metabolism will affect the homeostasis of pulmonary surfactant and eventually contribute to the development of emphysema.
Lipidomics should be applied to the study in COPD. Lipidomics is an emerging field of biomedical research which includes complex lipidome analysis. Lipidomics involves system-level identification and quantitation of thousands of pathways and networks of cellular lipid molecular species.96–98 Pilot study employing lipidomic analysis have uncovered that sphingolipid pathways are highly expressed in smokers with COPD in sputum.84 Nevertheless, such lipidomic analyses need to be extensively employed in COPD research.
In conclusion, lipid metabolic pathways and molecules are clearly altered in COPD, and these alterations may in turn modulate the functions of specific cells, such as production of inflammatory mediators, immune regulation, or cell death, eventually contributing to COPD development. However, deeper metabolic studies of different lipid species in various lung cell types are required in vitro as well as in COPD patients and in animal models. Also, cell specifically genetic modification of certain lipid metabolic molecules in mouse lung cells are critical to clarify the cell-type-dependent functions of lipid metabolism in COPD pathogenesis. With such efforts, lipid metabolic signals might represent some novel therapeutic targets for COPD.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant 2016YFA0501602 to ZC) and the Major and General Projects from the National Natural Science Foundation of China (Grant 81670031 to ZC).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015;36(2):81–91. doi:10.1016/j.it.2014.12.005
2. Walther TC, Farese RV
3. Lane-Donovan C, Philips GT, Herz J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron. 2014;83(4):771–787. doi:10.1016/j.neuron.2014.08.005
4. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9(2):162–176. doi:10.1038/nrm2335
5. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology – divergent pathophysiology. Nat Rev Endocrinol. 2017;13(12):710–730. doi:10.1038/nrendo.2017.91
6. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi:10.1016/j.cmet.2007.10.002
7. Currie E, Schulze A, Zechner R, Walther TC, Farese RV
8. Alvarado A, Arce I. Metabolic functions of the lung, disorders and associated pathologies. J Clin Med Res. 2016;8(10):689–700. doi:10.14740/jocmr2668w
9. Lamonaca P, Prinzi G, Kisialiou A, Cardaci V, Fini M, Russo P. Metabolic disorder in Chronic Obstructive Pulmonary Disease (COPD) patients: towards a personalized approach using marine drug derivatives. Mar Drugs. 2017;15(3). doi:10.3390/md15030081
10. Kilk K, Aug A, Ottas A, Soomets U, Altraja S, Altraja A. Phenotyping of chronic obstructive pulmonary disease based on the integration of metabolomes and clinical characteristics. Int J Mol Sci. 2018;19(3):666. doi:10.3390/ijms19030666
11. Yedgar S, Krimsky M, Cohen Y, Flower RJ. Treatment of inflammatory diseases by selective eicosanoid inhibition: a double-edged sword? Trends Pharmacol Sci. 2007;28(9):459–464. doi:10.1016/j.tips.2007.07.005
12. Coxson HO, Chan IHT, Mayo JR, Hlynsky J, Nakano Y, Birmingham CL. Early emphysema in patients with anorexia nervosa. Am J Respir Crit Care Med. 2004;170(7):748–752. doi:10.1164/rccm.200405-651OC
13. Gail DB, Massaro GD, Massaro D. Influence of fasting on the lung. J Appl Physiol. 1977;42(1):88–92. doi:10.1152/jappl.1977.42.1.88
14. D‘Amours R, Clerch L, Massaro D. Food deprivation and surfactant in adult rats. J Appl Physiol. 1983;55(5):1413–1417. doi:10.1152/jappl.1983.55.5.1413
15. Sahebjami H, MacGee J. Effects of starvation on lung mechanics and biochemistry in young and old rats. J Appl Physiol. 1985;58(3):778–784. doi:10.1152/jappl.1985.58.3.778
16. Sahebjami H, Vassallo CL, Wirman JA. Lung mechanics and ultrastructure in prolonged starvation. Am Rev Respir Dis. 1978;117(1):77–83. doi:10.1164/arrd.1978.117.1.77
17. Sahebjami H, Wirman JA. Emphysema-like changes in the lungs of starved rats. Am Rev Respir Dis. 1981;124(5):619–624. doi:10.1164/arrd.1981.124.5.619
18. Kerr JS, Riley DJ, Lanza-Jacoby S, et al. Nutritional emphysema in the rat. Influence of protein depletion and impaired lung growth. Am Rev Respir Dis. 1985;131(4):644–650. doi:10.1164/arrd.1985.131.4.644
19. Lambert AA, Putcha N, Drummond MB, et al. Obesity is associated with increased morbidity in moderate to severe COPD. Chest. 2017;151(1):68–77. doi:10.1016/j.chest.2016.08.1432
20. Hanson C, Rutten EP, Wouters EF, Rennard S. Influence of diet and obesity on COPD development and outcomes. Int J Chron Obstruct Pulmon Dis. 2014;9:723–733. doi:10.2147/COPD.S50111
21. Agarwal AR, Yin F, Cadenas E. Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am J Respir Cell Mol Biol. 2014;51(2):284–293. doi:10.1165/rcmb.2013-0523OC
22. Titz B, Boue S, Phillips B, et al. Effects of cigarette smoke, cessation, and switching to two heat-not-burn tobacco products on lung lipid metabolism in C57BL/6 and Apoe-/- mice – an integrative systems toxicology analysis. Toxicol Sci. 2016;149(2):441–457. doi:10.1093/toxsci/kfv244
23. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227–232. doi:10.1038/nrc3483
24. Qu Q, Zeng F, Liu X, Wang QJ, Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7:e2226. doi:10.1038/cddis.2016.132
25. Longo N, Frigeni M, Pasquali M. Carnitine transport and fatty acid oxidation. Biochim Biophys Acta. 2016;1863(10):2422–2435. doi:10.1016/j.bbamcr.2016.01.023
26. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(1):11–31. doi:10.1038/nrclinonc.2016.60
27. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9
28. Agarwal AR, Zhao LQ, Sancheti H, Sundar IK, Rahman I, Cadenas E. Short-term cigarette smoke exposure induces reversible changes in energy metabolism and cellular redox status independent of inflammatory responses in mouse lungs. Am J Physiol-Lung Cell Mol Physiol. 2012;303(10):L889–L898. doi:10.1152/ajplung.00219.2012
29. Jiang Z, Knudsen NH, Wang G, et al. Genetic control of fatty acid beta-oxidation in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2017;56(6):738–748. doi:10.1165/rcmb.2016-0282OC
30. Conlon TM, Bartel J, Ballweg K, et al. Metabolomics screening identifies reduced L-carnitine to be associated with progressive emphysema. Clin Sci. 2016;130(4):273–287. doi:10.1042/CS20150438
31. More JM, Voelker DR, Silveira LJ, Edwards MG, Chan ED, Bowler RP. Smoking reduces surfactant protein D and phospholipids in patients with and without chronic obstructive pulmonary disease. BMC Pulm Med. 2010;10:53. doi:10.1186/1471-2466-10-53
32. Ohlmeier S, Vuolanto M, Toljamo T, et al. Proteomics of human lung tissue identifies surfactant protein A as a marker of chronic obstructive pulmonary disease. J Proteome Res. 2008;7(12):5125–5132. doi:10.1021/pr800423x
33. Michaeloudes C, Kuo CH, Haji G, et al. Metabolic re-patterning in COPD airway smooth muscle cells. Eur Respir J. 2017;50(5). doi:10.1183/13993003.00711-2017.
34. Leslie CC. Regulation of arachidonic acid availability for eicosanoid production. Biochem Cell Biol. 2004;82(1):1–17. doi:10.1139/o03-080
35. Calder PC. Dietary modification of inflammation with lipids. Proc Nutr Soc. 2002;61(3):345–358.
36. Ferrer R, Moreno JJ. Role of eicosanoids on intestinal epithelial homeostasis. Biochem Pharmacol. 2010;80(4):431–438. doi:10.1016/j.bcp.2010.04.033
37. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875.
38. Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology. 2008;13(7):1014–1021. doi:10.1111/j.1440-1843.2008.01365.x
39. Montuschi P, Kharitonov SA, Ciabattoni G, Barnes PJ. Exhaled leukotrienes and prostaglandins in COPD. Thorax. 2003;58(7):585–588.
40. Dagouassat M, Gagliolo JM, Chrusciel S, et al. The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med. 2013;187(7):703–714. doi:10.1164/rccm.201208-1361OC
41. Drakatos P, Lykouras D, Sampsonas F, Karkoulias K, Spiropoulos K. Targeting leukotrienes for the treatment of COPD? Inflamm Allergy Drug Targets. 2009;8(4):297–306. doi:10.2174/187152809789352177
42. Beeh KM, Kornmann O, Buhl R, Culpitt SV, Giembycz MA, Barnes PJ. Neutrophil chemotactic activity of sputum from patients with COPD: role of interleukin 8 and leukotriene B4. Chest. 2003;123(4):1240–1247.
43. Biernacki WA, Kharitonov SA, Barnes PJ. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax. 2003;58(4):294–298.
44. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi:10.1038/nature13479
45. Hsiao HM, Thatcher TH, Colas RA, Serhan CN, Phipps RP, Sime PJ. Resolvin D1 reduces emphysema and chronic inflammation. Am J Pathol. 2015;185(12):3189–3201. doi:10.1016/j.ajpath.2015.08.008
46. Pena KB, Ramos CO, Soares NP, et al. The administration of a high refined carbohydrate diet promoted an increase in pulmonary inflammation and oxidative stress in mice exposed to cigarette smoke. Int J Chron Obstruct Pulmon Dis. 2016;11:3207–3217. doi:10.2147/COPD.S119485
47. Croasdell A, Thatcher TH, Kottmann RM, et al. Resolvins attenuate inflammation and promote resolution in cigarette smoke-exposed human macrophages. Am J Physiol Lung Cell Mol Physiol. 2015;309(8):L888–L901. doi:10.1152/ajplung.00125.2015
48. Reddy AT, Lakshmi SP, Muchumarri RR, Reddy RC. Nitrated fatty acids reverse cigarette smoke-induced alveolar macrophage activation and inhibit protease activity via electrophilic S-Alkylation. PLoS One. 2016;11(4):e0153336. doi:10.1371/journal.pone.0153336
49. Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60(11):925–931. doi:10.1136/thx.2005.040527
50. O‘Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–565. doi:10.1038/nri.2016.70
51. Kim KH. Regulation of mammalian acetyl-coenzyme A carboxylase. Annu Rev Nutr. 1997;17:77–99. doi:10.1146/annurev.nutr.17.1.77
52. Smith S, Witkowski A, Joshi AK. Structural and functional organization of the animal fatty acid synthase. Prog Lipid Res. 2003;42(4):289–317.
53. Britto CJ, Brady V, Lee S, Dela Cruz CS. Respiratory viral infections in chronic lung diseases. Clin Chest Med. 2017;38(1):87–96. doi:10.1016/j.ccm.2016.11.014
54. Nguyen A, Guedan A, Mousnier A, et al. Host lipidome analysis during rhinovirus replication in HBECs identifies potential therapeutic targets. J Lipid Res. 2018;59(9):1671–1684. doi:10.1194/jlr.M085910
55. Rahman SM, Ji X, Zimmerman LJ, et al. The airway epithelium undergoes metabolic reprogramming in individuals at high risk for lung cancer. JCI Insight. 2016;1(19):e88814. doi:10.1172/jci.insight.88814
56. Solanki HS, Babu N, Jain AP, et al. Cigarette smoke induces mitochondrial metabolic reprogramming in lung cells. Mitochondrion. 2018;40:58–70. doi:10.1016/j.mito.2017.10.002
57. Houghton AM. Mechanistic links between COPD and lung cancer. Nat Rev Cancer. 2013;13(4):233–245. doi:10.1038/nrc3477
58. Nerlov C. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 2007;17(7):318–324. doi:10.1016/j.tcb.2007.07.004
59. DeBose-Boyd RA, Ye J. SREBPs in lipid metabolism, insulin signaling, and beyond. Trends Biochem Sci. 2018;43:358–368. doi:10.1016/j.tibs.2018.01.005
60. Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discovery. 2014;13(6):433–444. doi:10.1038/nrd4280
61. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi:10.1146/annurev.biochem.77.061307.091829
62. Lakshmi SP, Reddy AT, Zhang Y, et al. Down-regulated peroxisome proliferator-activated receptor gamma (PPARgamma) in lung epithelial cells promotes a PPARgamma agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem. 2014;289(10):6383–6393. doi:10.1074/jbc.M113.536805
63. Shan M, You R, Yuan X, et al. Agonistic induction of PPARgamma reverses cigarette smoke-induced emphysema. J Clin Invest. 2014;124(3):1371–1381. doi:10.1172/JCI70587
64. Solleti SK, Simon DM, Srisuma S, et al. Airway epithelial cell PPARgamma modulates cigarette smoke-induced chemokine expression and emphysema susceptibility in mice. Am J Physiol Lung Cell Mol Physiol. 2015;309(3):L293–L304. doi:10.1152/ajplung.00287.2014
65. Lea S, Plumb J, Metcalfe H, et al. The effect of peroxisome proliferator-activated receptor- ligands on in vitro and in vivo models of COPD. Eur Respir J. 2013;43(2):409–420. doi:10.1183/09031936.00187812
66. Morissette MC, Shen P, Thayaparan D, Stampfli MR. Impacts of peroxisome proliferator-activated receptor-gamma activation on cigarette smoke-induced exacerbated response to bacteria. Eur Respir J. 2015;45(1):191–200. doi:10.1183/09031936.00004314
67. Higham A, Lea S, Plumb J, et al. The role of the liver X receptor in chronic obstructive pulmonary disease. Respir Res. 2013;14:106. doi:10.1186/1465-9921-14-19
68. Besnard V, Wert SE, Stahlman MT, et al. Deletion of Scap in alveolar type II cells influences lung lipid homeostasis and identifies a compensatory role for pulmonary lipofibroblasts. J Biol Chem. 2009;284(6):4018–4030. doi:10.1074/jbc.M805388200
69. Yuan H, Shyy JYJ, Martins-Green M. Second-hand smoke stimulates lipid accumulation in the liver by modulating AMPK and SREBP-1. J Hepatol. 2009;51(3):535–547. doi:10.1016/j.jhep.2009.03.026
70. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi:10.1146/annurev.cellbio.22.010305.104656
71. Spann NJ, Glass CK. Sterols and oxysterols in immune cell function. Nat Immunol. 2013;14(9):893–900. doi:10.1038/ni.2681
72. Brown MS, Radhakrishnan A, Goldstein JL. Retrospective on cholesterol homeostasis: the central role of scap. Annu Rev Biochem. 2018;87:783–807.
73. Zafirova-Ivanovska B, Stojkovikj J, Dokikj D, et al. The level of cholesterol in COPD patients with severe and very severe stage of the disease. Open Access Maced J Med Sci. 2016;4(2):277–282. doi:10.3889/oamjms.2016.063
74. Li H, Liu Y, Wang L, et al. High apolipoprotein M serum levels correlate with chronic obstructive pulmonary disease. Lipids Health Dis. 2016;15:59. doi:10.1186/s12944-016-0228-1
75. Kikuchi T, Sugiura H, Koarai A, et al. Increase of 27-hydroxycholesterol in the airways of patients with COPD: possible role of 27-hydroxycholesterol in tissue fibrosis. Chest. 2012;142(2):329–337. doi:10.1378/chest.11-2091
76. Jia J, Conlon TM, Sarker RS, et al. Cholesterol metabolism promotes B-cell positioning during immune pathogenesis of chronic obstructive pulmonary disease. EMBO Mol Med. 2018;10:5. doi:10.15252/emmm.201708349
77. Mroz RM, Lisowski P, Tycinska A, et al. Anti-inflammatory effects of atorvastatin treatment in chronic obstructive pulmonary disease. A controlled pilot study. J Physiol Pharmacol. 2015;66(1):111–128.
78. Davis BB, Zeki AA, Bratt JM, et al. Simvastatin inhibits smoke-induced airway epithelial injury: implications for COPD therapy. Eur Respir J. 2013;42(2):350–361. doi:10.1183/09031936.00042512
79. Hashimoto Y, Sugiura H, Togo S, et al. 27-Hydroxycholesterol accelerates cellular senescence in human lung resident cells. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1028–L1041. doi:10.1152/ajplung.00351.2015
80. Craig WY, Palomaki GE, Haddow JE. Cigarette smoking and serum lipid and lipoprotein concentrations: an analysis of published data. BMJ. 1989;298(6676):784–788.
81. Mjos OD. Lipid effects of smoking. Am Heart J. 1988;115(1 Pt 2):272–275. doi:10.1016/0002-8703(88)90649-7
82. Kim C, Lee J-M, Park S-W, et al. Attenuation of cigarette smoke-induced emphysema in mice by apolipoprotein A-1 overexpression. Am J Respir Cell Mol Biol. 2016;54(1):91–102. doi:10.1165/rcmb.2014-0305OC
83. Thomson NC. Clinical studies of statins in asthma and COPD. Curr Mol Pharmacol. 2017;10(1):60–71. doi:10.2174/1874467209666160112125911
84. Telenga ED, Hoffmann RF, Ruben TK, et al. Untargeted lipidomic analysis in chronic obstructive pulmonary disease. Uncovering sphingolipids. Am J Respir Crit Care Med. 2014;190(2):155–164. doi:10.1164/rccm.201312-2210OC
85. Bowler RP, Jacobson S, Cruickshank C, et al. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am J Respir Crit Care Med. 2015;191(3):275–284. doi:10.1164/rccm.201410-1771OC
86. Chalfant CE, Spiegel S. Sphingosine 1-phosphate and ceramide 1-phosphate: expanding roles in cell signaling. J Cell Sci. 2005;118(Pt 20):4605–4612. doi:10.1242/jcs.02637
87. Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–498. doi:10.1038/nm1238
88. Schweitzer KS, Hatoum H, Brown MB, et al. Mechanisms of lung endothelial barrier disruption induced by cigarette smoke: role of oxidative stress and ceramides. Am J Physiol Lung Cell Mol Physiol. 2011;301(6):L836–L846. doi:10.1152/ajplung.00385.2010
89. Mizumura K, Justice MJ, Schweitzer KS, et al. Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. FASEB J. 2018;32(4):1880–1890. doi:10.1096/fj.201700571R
90. Baudiß K, Ayata CK, Lazar Z, et al. Ceramide-1-phosphate inhibits cigarette smoke-induced airway inflammation. Eur Respir J. 2015;45(6):1669–1680. doi:10.1183/09031936.00080014
91. Decramer M, Janssens W. Chronic obstructive pulmonary disease and comorbidities. Lancet Respir Med. 2013;1(1):73–83. doi:10.1016/S2213-2600(12)70060-7
92. Vanfleteren L, Spruit MA, Wouters EFM, Franssen FME. Management of chronic obstructive pulmonary disease beyond the lungs. Lancet Respir Med. 2016;4(11):911–924. doi:10.1016/S2213-2600(16)00097-7
93. Kelly B, O‘Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–784. doi:10.1038/cr.2015.68
94. King TE
95. Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S. Impaired phagocytosis of nontypeable Haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. 2006;194(10):1375–1384. doi:10.1086/508428
96. Shevchenko A, Simons K. Lipidomics: coming to grips with lipid diversity. Nat Rev Mol Cell Biol. 2010;11(8):593–598. doi:10.1038/nrm2934
97. Wenk MR. Lipidomics: new tools and applications. Cell. 2010;143(6):888–895. doi:10.1016/j.cell.2010.11.033
98. Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat Biotechnol. 2018;36(4):316–320. doi:10.1038/nbt.4101
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.