Back to Journals » Clinical Interventions in Aging » Volume 10
Link between type 2 diabetes and Alzheimer’s disease: from epidemiology to mechanism and treatment
Received 10 September 2014
Accepted for publication 14 November 2014
Published 10 March 2015 Volume 2015:10 Pages 549—560
DOI https://doi.org/10.2147/CIA.S74042
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Dr Richard Walker
Video abstract presented by Dalin Song
Views: 639
Xiaohua Li,1 Dalin Song,2 Sean X Leng3
1Dalian Medical University, Dalian, 2Department of Geriatrics, Qingdao Municipal Hospital, Qingdao, People’s Republic of China; 3Division of Geriatric Medicine and Gerontology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Abstract: The aim of this paper is to provide a comprehensive review of the epidemiological evidence linking type 2 diabetes mellitus and its related conditions, including obesity, hyperinsulinemia, and metabolic syndrome, to Alzheimer’s disease (AD). Several mechanisms could help to explain this proposed link; however, our focus is on insulin resistance and deficiency. Studies have shown that insulin resistance and deficiency can interact with amyloid-ß protein and tau protein phosphorylation, each leading to the onset and development of AD. Based on those epidemiological data and basic research, it was recently proposed that AD can be considered as “type 3 diabetes”. Special attention has been paid to determining whether antidiabetic agents might be effective in treating AD. There has been much research both experimental and clinical on this topic. We mainly discuss the clinical trials on insulin, metformin, thiazolidinediones, glucagon-like peptide-1 receptor agonists, and dipeptidyl peptidase-4 inhibitors in the treatment of AD. Although the results of these trials seem to be contradictory, this approach is also full of promise. It is worth mentioning that the therapeutic effects of these drugs are influenced by the apolipoprotein E (APOE)-ε4 genotype. Patients without the APOE-ε4 allele showed better treatment effects than those with this allele.
Keywords: type 2 diabetes mellitus, Alzheimer’s disease, insulin
A Letter to Editor has been received and published for this article
Introduction
Type 2 diabetes mellitus (T2DM) is currently extremely common due to the prevalence of obesity, as well as the aging of the population.1 Prevention and treatment strategies for the classical macrovascular and microvascular complications of diabetes mellitus have significantly improved. Therefore, people are living longer with diabetes mellitus, which might lead to the emergence of new complications. Dementia is one example of these emerging new complications.2 Compared with the general population, the increased risk of dementia is 50%–150% in people with T2DM.3–5 Prince et al6 predicted that people living with dementia worldwide would increase from 35.6 million in 2010 to 115.4 million in 2050. If current studies have correctly predicted the association between dementia and T2DM, then the future burden of dementia, eg, Alzheimer’s disease (AD) and vascular dementia, might be even greater than that estimated as the prevalence of diabetes mellitus continues to rise.7 AD is the most common form and cause of dementia, accounting for 60%–80% of all cases.8
Over the past three decades, numerous epidemiological studies have shown a clear association between T2DM and an increased risk of developing AD. In addition, T2DM-related conditions, including obesity,9 hyperinsulinemia,10 and metabolic syndrome, may also be risk factors for AD. The exact mechanisms with clinical relevance are unclear. Several mechanisms have been proposed, including insulin resistance and deficiency, impaired insulin receptor and impaired insulin growth factor (IGF) signaling, glucose toxicity, problems due to advanced glycation end products and their receptors, cerebrovascular injury, vascular inflammation, and others.11–13 In this review, we discuss insulin resistance and deficiency. Currently, the drugs available are able to slow worsening of symptoms for 6–12 months but are effective in only about half of the treated population.14 Also, no effective drugs are expected to be approved soon, given that several promising new agents have failed in Phase III clinical trials.15,16 Therefore, it is important to accurately define the role of T2DM in the development of AD for preventing and treating the disease. In this review, we discuss the clinical trials on antidiabetic agents, ie, insulin, metformin, thiazolidinediones, glucagon-like peptide-1 receptor (GLP-1R) agonists, and dipeptidyl peptidase (DPP)-4 inhibitors, in the treatment of AD.
Alzheimer’s disease
Clinically, AD is manifested by progressive memory loss and a gradual decline in cognitive function, eventually leading to premature death of the individual, that occurs typically 3–9 years after diagnosis.17 The neuropathological features associated with the disease include the presence of extracellular senile plaques containing amyloid-β (Aβ) protein, neurofibrillary tangles that consist mainly of intracellular and abnormally phosphorylated tau protein, and a dramatic loss of neurons and synapses, especially in the hippocampus and cortex.18–21 Considering these pathological changes, the “amyloid cascade hypothesis” is certainly the most popular current view. This hypothesis proposes that accumulation of Aβ, as either a consequence of increased production or decreased removal of Aβ, instigates all other downstream AD-associated phenomena and ultimately the disease itself.22–24 Despite the indistinguishable clinical symptoms of dementia, there are two different types of origin-based AD. In a small proportion (familial early-onset AD), the disease has a genetic origin and is caused by missense mutations in three genes, ie, Aβ protein precursor, presenilin-1, and presenilin-2.25 These genes affect less than 5% of cases of AD, that usually happen in middle age.26 The great majority of AD cases are sporadic in origin, with older age, being female, vascular disease, head trauma, family history of dementia, and genetic factors (eg, apolipoprotein E [APOE] ε4 allele) as the mainly immutable risk factors. Aside from these factors, there are several controllable risk factors. In a recent study, international experts reached an agreement that more than half of sporadic or late-onset AD cases were related to seven controllable risk factors, ie, depression, diabetes, smoking, and obesity in middle age, high blood pressure in midlife, lack of exercise, and a lower level of education.27
T2DM and its related conditions
When considering the links between T2DM and AD, it is important to consider the natural history that leads to T2DM. There are two underlying mechanisms involved, ie, insulin resistance and inadequate insulin secretion from pancreatic β-cells.28 Initially, pancreatic β-cells increase insulin secretion in response to insulin resistance, causing hyperinsulinemia, and are able to effectively maintain glucose levels below the T2DM range. When β-cell function begins to decline, insulin production is inadequate to overcome insulin resistance, and blood glucose levels rise, resulting in prediabetes and T2DM. Being overweight or obese is the major reason for insulin resistance.29 This natural history is also part of the metabolic syndrome, which includes hypertension, dyslipidemia, and elevated systemic inflammation.30 From a mechanistic standpoint, it is difficult to discern whether the main mechanism linking T2DM to AD is glycemia, hypertension, insulin resistance, or factors specifically related to adipose tissue. Because they are related sequentially and often occur simultaneously, understanding this relationship is fundamental to the study of the role of adiposity, hyperinsulinemia, metabolic syndrome, and diabetes in AD.
Epidemiological studies linking T2DM, obesity, hyperinsulinemia, and metabolic syndrome to AD
T2DM
In the 1990s, the Rotterdam Study, aiming to determine the influence of T2DM on the risk of dementia and AD, found that T2DM almost doubled the risk of dementia and AD.31 In a longitudinal study of 1,138 subjects, they explored the relationship between the aggregation of vascular risk factors (hypertension, heart disease, current smoking) and AD and showed that diabetes and smoking were the strongest risk factors and the risk of AD associated with diabetes was stronger than previously reported (relative risk 3.8), independent of other vascular conditions.32 So far, numerous prospective epidemiological studies have explored the relationship between diabetes and AD, and most have identified diabetes as a risk factor for AD33 (Table 1). Studies specifically assessing the incidence of dementia in people with diabetes mellitus, adjusting for glycemic control, microvascular complications, and comorbidity (eg, hypertension and stroke), have also demonstrated an increased risk; eight of 13 longitudinal population-based studies were reviewed and an excess risk for AD in adults with diabetes was found, ranging from 50% to 100%.3 In a comprehensive meta-analysis, with a total of 6,184 people with diabetes and 38,530 without diabetes, the aggregate relative risk of AD for people with diabetes was 1.5 (95% confidence interval [CI] 1.2–1.8).34
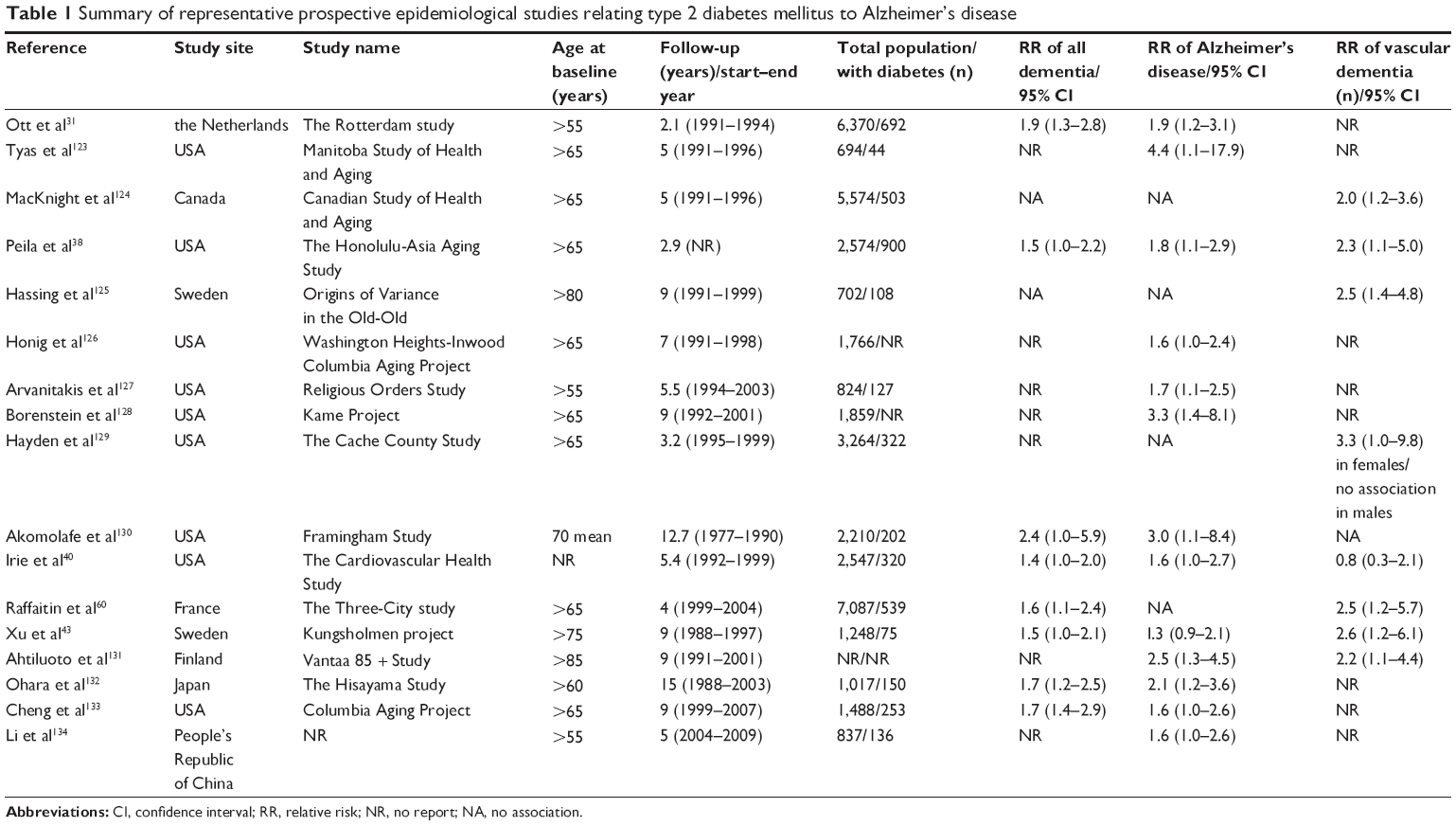 | Table 1 Summary of representative prospective epidemiological studies relating type 2 diabetes mellitus to Alzheimer’s disease |
While the relationship between diabetes and the major types of dementia is controversial, T2DM is a complex metabolic disorder that is closely associated with other identified risk factors for dementia, including atherosclerotic vascular disease, the APOE-ε4 allele, and some indicators of diabetes, such as diabetes duration, high glucose level, and insulin treatment, may also be risk factors of dementia. A study in Japanese Americans found no association between diabetes in middle age and dementia;35 however, both Whitmer et al36 and Schnaider et al37 found that diabetes in middle age increased the risk of dementia in the elderly when considering prospective studies with a large sample size. The causes of such differences are still not known. The Honolulu-Asia Aging Study also found that diabetes in old age was related to a higher risk of AD (relative risk 1.8) and AD pathology on autopsy, particularly in subjects with the APOE-ε4 allele.38 Several other studies have shown that diabetes with the APOE-ε4 allele was associated with increased risk of dementia.39–41 One study in postmenopausal women found that the risk of mild cognitive impairment and dementia increased with each 1% elevation in glycosylated hemoglobin, a marker of glucose control, even in women without T2DM.42 One study from Sweden analyzed 1,301 community dwellers aged 75 years and older and found that diabetes had no significant relationship with AD, but was related to vascular dementia.43 This association was stronger in subjects with diabetes who reported treatment with insulin. In a meta-analysis, the aggregate relative risk of AD for people with diabetes was 1.5 (95% CI 1.2–1.8), while for vascular dementia the relative risk was 2.5 (95% CI 2.1–3.0).34 Another meta-analysis found a similar result, ie, the relative risk of developing AD was 1.6 (95% CI 1.4–1.7) and vascular dementia was 2.3 (95% CI 2.0–2.7).33 In general, it is clear that T2DM has a higher dementia risk, but the associations are stronger for vascular dementia compared with AD. The association is also influenced by many factors, including ethnicity, the APOE-ε4 allele, diabetes duration, glycemic level, and insulin treatment. Determining what factors and what percentages of these factors contribute to this link is also quite important.
T2DM-related conditions
Obesity
Many studies have explored the association between obesity and AD. Obesity, especially obesity in middle age, usually assessed by body mass index (BMI) or waist circumference, has a strong and independent association with an increased risk for AD. The Baltimore Longitudinal Study of Aging showed that the incidence of AD increased in men who gained weight between the ages of 30 and 45 years and in women with a BMI >30 at ages 30, 40, and 45 years.9 A meta-analysis reported an increased risk (95% CI 1.59–2.62) of AD with obesity (BMI ≥30), and people with the APOE-ε4 allele had a higher level of risk.44 The Honolulu-Asia Aging Study reported that groups with and without dementia showed no differences in weight from midlife to late life.31 The explanation for this difference may be ethnicity, ie, Asians may be more susceptible to the effects of obesity compared with Europeans.45 A meta-analysis of 16 papers found that many types of abnormal body weight in midlife, including underweight, overweight, and obesity, increase the risk of dementia.46
The results of studies of late-life obesity with AD have been conflicting, and the following four conditions have been suggested: an increased risk, a reduced risk, no relationship, and a U-shaped relationship, with both high and low BMI related to an increased risk of AD. Higher BMI at ages 70, 75, and 79 years predicts a higher dementia risk;47 however, an analysis of the Cardiovascular Health Study of subjects ≥65 years of age found a 40% decreased risk with BMI >30 compared with subjects of normal weight.48 Whitmer49 reported a decreased risk of dementia with increasing BMI in subjects ≥76 years of age, but a U-shaped association in subjects <76 years of age. They also found that a higher waist circumference is related to a higher AD risk in the younger elderly, but not in the oldest population. The causes for this paradox remain unclear. There may be weight decreases with aging and frailty.50 It has been suggested that changes in body composition with age make BMI a poor measure of obesity in the elderly;49 survival bias related to high obesity may be an important factor, and midlife studies had a much longer period of follow-up prior to diagnosis of dementia. The average length of follow-up for the late-life studies was 7.13 years, which would include the prodromal phase of AD in particular.51 Further data are required from studies with longer durations of follow-up in late life, and more suitable weight indicators are needed for the elderly.
Hyperinsulinemia
Recent attention has turned to the question of whether hyperinsulinemia may directly increase the risk of cognitive decline. Two longitudinal studies, one in elderly Japanese Americans in Hawaii10 and another in persons aged 65 years and older from northern Manhattan,52 found that the risk of incident AD was higher in persons with hyperinsulinemia. These studies also found that the risk of AD related to hyperinsulinemia was higher among persons with the APOE-ε4 allele. Another study found that higher c-peptide levels, a measure of insulin secretion, may be related to worse cognition, even among those without diabetes.53 The Nurses’ Health Study found that “young-old” women (mean age 64 years) without diabetes but with higher c-peptide levels showed cognitive decline approximately 10 years later.54 In the Physicians’ Health Study II, older men aged 60–92 (mean 71.3) years showed similar results.55 Therefore, hyperinsulinemia may be the reason for T2DM being associated with an increased risk for AD.
Metabolic syndrome
Metabolic syndrome, first described about 40 years ago,56 has a cluster of risk factors including abdominal obesity, hypertension, lipid abnormalities, and impaired metabolism of glucose and insulin. Studies addressing the association between metabolic syndrome and AD are limited and the results are mixed. One study in 2,632 elderly black and white people found that the metabolic syndrome measured using National Cholesterol Education Program guidelines was associated with a higher risk of cognitive decline, particularly among those with high levels of inflammatory markers.57 A population-based study of 980 elderly subjects aged 69–78 years found that the metabolic syndrome is significantly associated with AD.58 However, this suggested association between metabolic syndrome and AD was not confirmed by four large, longitudinal, population-based studies, including the Honolulu-Asia Aging Study,59 the Three-City Study,60 the Italian Longitudinal Aging Study,61 and a multiethnic elderly cohort in the USA.62 However, the prevalence of metabolic syndrome depends on the studied population and the definition, except for the most commonly used definition, ie, the Third Adults Treatment Panel of the National Cholesterol Education Program criteria.63 At least two other more recent sets of clinical criteria were also presented, ie, the National Heart, Lung and Blood Institute/American Heart Association criteria64 and the International Diabetes Federation criteria.65 It is still unclear if one or two separate components of metabolic syndrome can drive the relationship with cognitive decline, or whether the individual components are additive or interact in some way.66 Individual components of the metabolic syndrome should not be evaluated in isolation, and careful methodological approaches are needed to understand the timing and non-linear relationships between these components over time.67
Insulin resistance and deficiency: potential mechanisms linking T2DM and its related conditions to AD
A large number of studies have shown that insulin resistance and deficiency, a marker of T2DM, play an important role in AD pathology. The first molecular clue as to how the brain might become insulin-resistant in AD came from studies demonstrating that Aβ oligomers bind to hippocampal neurons and trigger the removal of dendritic insulin receptor substrates (IRs) from the plasma membrane,68 which was subsequently demonstrated in AD brains.69 Lower levels and sensitivity of insulin, IGF, and IRs were observed in AD neuropathology.70–72 Greatly increased biomarkers of peripheral insulin resistance in the hippocampus of non-diabetic AD patients further implicated insulin resistance in AD.73 In T2DM, tumor necrosis factor (TNF)-α signaling activates c-Jun N-terminal kinase,74 resulting in IRs-1 serine phosphorylation and peripheral insulin resistance.68 Similarly, Aβ oligomers cause abnormal activation of the TNF-α/c-Jun N-terminal kinase pathway and inhibition of IRs-1 in cultured hippocampal neurons.75 Recently, it was even proposed that AD can be an “insulin-resistant brain state” or even a “type 3 diabetes”.76 Insulin was found to modulate Aβ protein precursor expression and processing both in vivo and in vitro. Insulin and IGF-1 inhibited Aβ production through Akt-mediated phosphorylation/inactivation of glycogen synthase kinase-3β77 and prevented abnormal intracellular accumulation of Aβ by increasing its extracellular secretion in the brain and accelerating its trafficking from the Golgi and trans-Golgi network to the plasma membrane.69,78 Insulin and IGF-1 also prevented accumulation of Aβ by promoting the transport of Aβ-binding carrier proteins, including transthyretin and albumin, into the brain.79–81 Devi et al82 demonstrated that streptozotocin-induced insulin-deficient diabetes accelerates Aβ accumulation via the translational upregulation of the β-secretase enzyme, BACE1, and its substrate, amyloid precursor protein, in a transgenic mouse model of AD. Another potential mechanism could be the interference of insulin with extracellular proteolytic Aβ degradation occurs via the insulin-degrading enzyme, a metalloprotease that also catabolizes insulin and IGF-1.69,79,80,83,84 Under insulin resistance conditions, insulin may competitively inhibit the insulin-degrading enzyme, thus impairing degradation of Aβ, increasing its neurotoxicity and promoting AD.69,79,80,83,84 Besides Aβ, insulin resistance and deficiency also increases tau protein phosphorylation through activation of glycogen synthase kinase-3β.85,86
Considering the above, neurons in the T2DM brain could be more vulnerable to the toxicity of Aβ due to insulin resistance and deficiency.87 Conversely, insulin resistance and deficiency could lead to increased production of Aβ and Aβ-induced oxidative damage at the mitochondria.88 Therefore, the current hypothesis regarding insulin resistance and deficiency may represent a critical contributing factor in the acceleration of Aβ production during the progression of sporadic AD (Figures 1 and 2), and thus insulin resistance and deficiency may be an important therapeutic target in patients with AD.
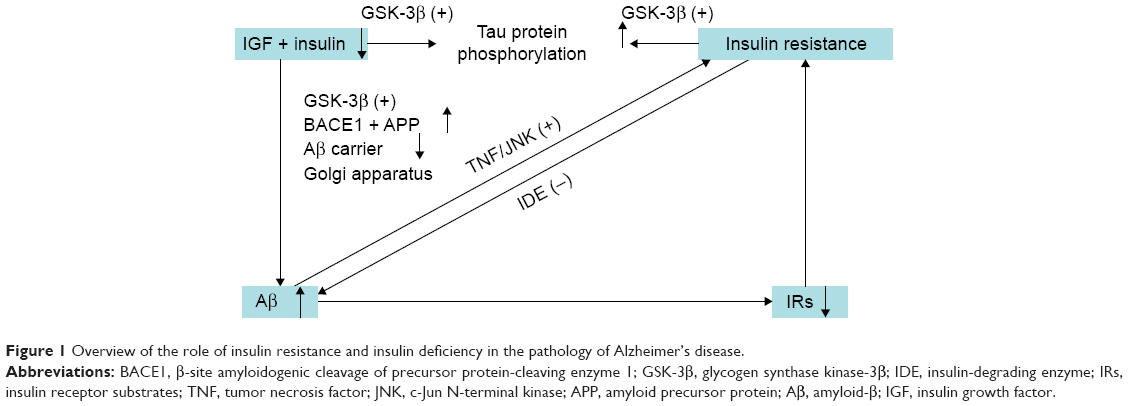 | Figure 1 Overview of the role of insulin resistance and insulin deficiency in the pathology of Alzheimer’s disease. |
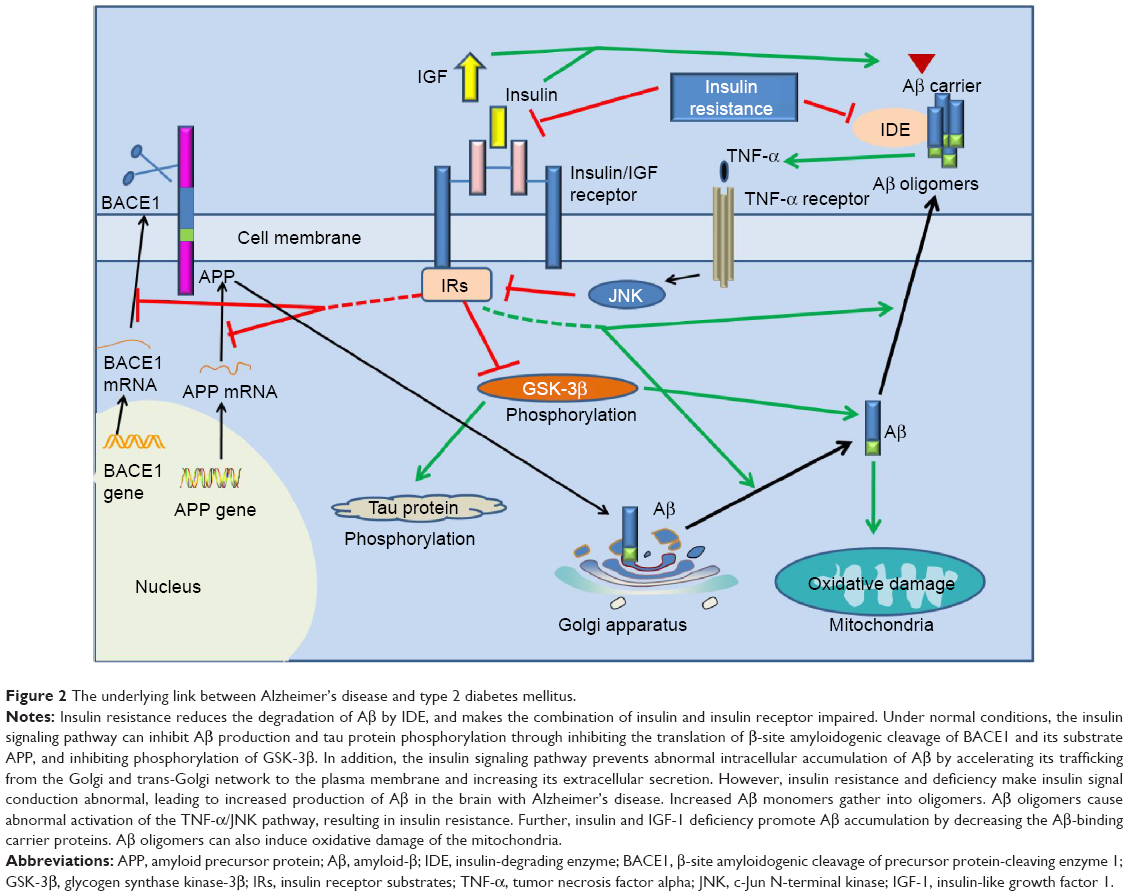 | Figure 2 The underlying link between Alzheimer’s disease and type 2 diabetes mellitus. |
Implications for treatment of AD with or without T2DM
Given the role of insulin resistance and deficiency in the pathogenesis of AD, it could be possible that a drug currently approved for T2DM may also be useful for AD (Figure 3). We now summarize some of the relevant clinical trials.
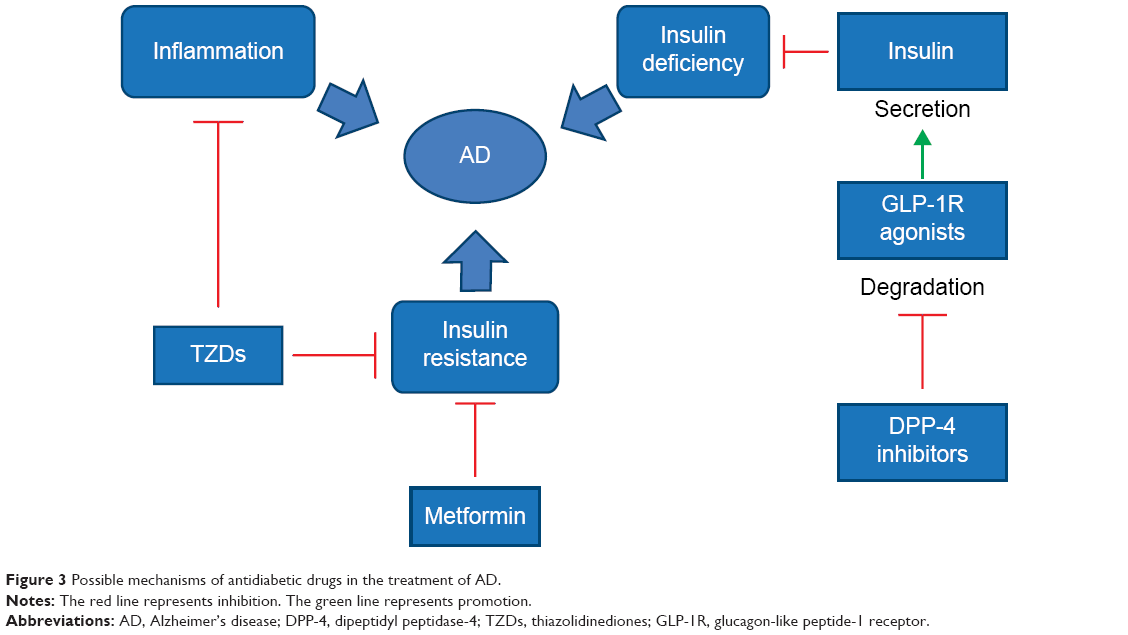 | Figure 3 Possible mechanisms of antidiabetic drugs in the treatment of AD. |
Intranasal insulin
Insulin has also been studied in cognitively impaired patients, because intranasal administration can quickly deliver insulin to the central nervous system across olfactory and trigeminal perivascular channels and axonal pathways,89 and there are fewer potential side effects, such as hypoglycemia, when compared with intravenous insulin infusions. Thus, intranasal delivery of insulin is a viable long-term therapy for AD. A 2008 study reported that intranasal insulin (20 IU, twice a day) for 21 days improved story recall, attention, and caregiver-rated functional status in cognitively impaired subjects or individuals with AD.90 In another study, 40 individuals with AD and 64 with mild cognitive impairment received either placebo, 20 IU of insulin, or 40 IU of insulin administered via nasal spray over the course of 4 months. The between-subject comparison showed that intranasal insulin improved delayed memory and cognitive function. Based on positron emission tomography findings, the authors also provided evidence that both doses of insulin had a higher 18F fluorodeoxyglucose uptake in the parietotemporal, frontal, precuneus, and cuneus regions of the central nervous system following administration of intranasal insulin compared with placebo, linking the enhancement in function of those brain areas.91 Given what is known about AD and diabetes, it is not surprising that intranasal insulin could have different effects depending on the APOE-ε4 genotype. In a study by Reger et al92 the participants were further subdivided based on APOE-ε4 genotype (APOE-ε4 negative or positive allele), and cognitive function was measured 45 minutes after administration of intranasal treatment. Participants with the APOE-ε4 negative allele displayed significant cognitive improvement on the 20 IU and 40 IU doses when compared with the placebo group and the APOE-ε4 positive group, and patients in the APOE-ε4 negative group taking 40 IU of insulin performed better than patients in the other groups. In the APOE-ε4 positive group, cognitive function (working and verbal memory) was reduced with treatment. Currently, the sample size of clinical trials exploring insulin administration in AD patients seems to be relatively low, with a relatively short treatment period. Therefore, longer trials are needed, as well as APOE-ε4 allele related differences in insulin metabolism.
Metformin
Metformin is an orally active biguanide that lowers blood glucose levels by suppressing hepatic glucose output, increasing insulin-mediated glucose disposal, increasing intestinal glucose use, and decreasing fatty acid oxidation. It also reduces insulin levels,93 inflammation and thrombosis,94 and the risks of metabolic syndrome95 and diabetes96 in persons without diabetes. Long-term use of metformin is also associated with a lower risk of certain cancers.97,98 While the mechanisms of action are not completely understood for metformin, studies have shown that patients with T2DM and AD, and receiving antidiabetic drugs including metformin, have a lower rate of cognitive impairment than in untreated patients.99 This result suggests that diabetic medication might somehow affect neuronal networks in the brain, leading to functional preservation or benefit in AD patients. In a large epidemiological study comparing individuals with T2DM, either taking antidiabetic drugs or not, metformin and sulfonylureas decreased the risk of dementia by 35% over 8 years in patients with T2DM.100 Notably, another large epidemiological trial, based on the UK-based General Practice Research Database, including 7,086 individuals aged 65 years and older with an incident diagnosis of AD and the same number of matched controls without dementia, showed that patients with T2DM who were long-term users of metformin, had a slightly higher risk of AD than those who did not receive the drug.101 The conflicting results of these studies point to more research being needed.
Thiazolidinediones
Thiazolidinediones are peroxisome proliferator-activated receptor-γ (PPARγ) agonists and potent insulin sensitizers.102 Their mechanism involves stimulation of the action of PPARγ in response to changes in insulin, thereby triggering a drop in serum glucose.103 The best characterized PPARγ agonists are pioglitazone and rosiglitazone. Thiazolidinediones also have potent anti-inflammatory properties.104 Given the role of insulin resistance and inflammation in the pathogenesis of AD,105,106 these agents are being studied as a potential treatment for AD. One small study showed that persons receiving rosiglitazone 4 mg daily had improved memory and selective attention.107 In a larger trial, more than 500 patients with mild to moderate AD were randomized to 6 months of treatment with placebo or rosiglitazone 2, 4, or 8 mg, resulting in significant improvement on the Alzheimer’s Disease Assessment Scale-cognitive subscale in APOE-ε4-negative patients on 8 mg rosiglitazone, while persons with the APOE-ε4 allele showed no benefit.108 However, a Phase III trial of rosiglitazone (NCT00428090) in mild to moderate AD found no benefit.109 Pioglitazone and rosiglitazone seem to have similar results. Researchers in one study reported improvement of cognition with pioglitazone in patients with both T2DM and AD, whereas another study showed no effect.110 The major limitation of thiazolidinediones in the prevention of dementia is the side effects of edema and congestive heart failure. In the interests of safety, the USA and Europe have either partially or completely restricted the use of rosiglitazone for treatment of T2DM.111 Therefore, solving these side effect issues is very important for the future application of thiazolidinediones.
GLP-1R agonists and DPP-IV inhibitors
GLP-1 is a gut-derived incretin hormone that enhances glucose-stimulated insulin secretion and suppresses glucagon secretion.112 GLP-1 is rapidly degraded by DPP-4; however, administration of specific DPP-4 inhibitors can increase the half-life of endogenous GLP-1 and hence prolong the activation of GLP-1R in different cell types.113,114 Currently, GLP-1R agonists and DPP-4 inhibitors are routinely used to treat T2DM.112,114,115 In agreement with the proposed role of insulin signaling declining with the development of AD, GLP-1R agonists are an attractive option because they activate pathways common to bypassing IRs and boost insulin-related signaling pathways through G protein-dependent signaling.116 In fact, animal studies have revealed that GLP-1R plays an important role in the control of synaptic plasticity and in some forms of memory formation.117,118 Exendin-4 and liraglutide, two types of GLP-1R agonists, also restored impaired insulin signaling, exerting neuroprotective effects on neurons and synapses, improving cognition, and decreasing Aβ accumulation in the brain in a transgenic mouse model of AD.75,119 As recently suggested, an agent that chronically decreases Aβ levels should be beneficial in APOE-ε4 allele carriers.120 If the beneficial effect of GLP-1R agonists is found to translate to primates, APOE-ε4 allele carriers may possibly benefit from the use of GLP-1R agonists. Similarly, the DPP-4 inhibitors, sitagliptin and vildagliptin, have been found to have beneficial effects on learning and memory in animal models.121,122 Unfortunately, no relevant clinical data are available. We are awaiting the results of two clinical trials, ie, a study of exendin-4 in 230 patients with mild cognitive impairment/early-stage AD (NCT01255163) and a large-scale Phase II clinical trial assessing the safety and efficacy of liraglutide in 206 patients with mild cognitive impairment (NCT01843075).
Summary
T2DM and AD have traditionally been treated as independent disorders. With extensive and indepth research on T2DM and AD, epidemiological associations and some common pathophysiological mechanisms have been found. If demonstrated to be true, common pharmacotherapy should be effective, and clinical trials testing the effectiveness of antidiabetic drugs in AD patients should be initiated. The results will not only be important for the treatment of AD patients, but will also be key to understanding the connection between these serious but seemingly unrelated disorders.
Acknowledgment
We thank LetPub (www.letpub.com) for its assistance with English language during preparation of this manuscript.
Disclosure
The author reports no conflicts of interest in this work.
References
Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes estimates for the year 2000 and projections for 2030. Diabetes Care.. 2004;27(5):1047–1053. | ||
Strachan MW, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7(2):108–114. | ||
Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5(1):64–74. | ||
Cukierman T, Gerstein H, Williamson J. Cognitive decline and dementia in diabetes – systematic overview of prospective observational studies. Diabetologia. 2005;48(12):2460–2469. | ||
Strachan MW, Deary IJ, Ewing FM, Frier BM. Is type II diabetes associated with an increased risk of cognitive dysfunction? A critical review of published studies. Diabetes Care. 1997;20(3):438–445. | ||
Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and meta-analysis. Alzheimers Dement. 2013;9(1):63–75. | ||
Strachan MW, Price JF, Frier BM. Diabetes, cognitive impairment, and dementia. BMJ. 2008;336(7634):6. | ||
Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10(2):e47–e92. | ||
Beydoun MA, Lhotsky A, Wang Y, et al. Association of adiposity status and changes in early to mid-adulthood with incidence of Alzheimer’s disease. Am J Epidemiol. 2008;168(10):1179–1189. | ||
Peila R, Rodriguez BL, White LR, Launer LJ. Fasting insulin and incident dementia in an elderly population of Japanese–American men. Neurology. 2004;63(2):228–233. | ||
Sjoholm A, Nystrom T. Inflammation and the etiology of type 2 diabetes. Diabetes Metab Res Rev. 2006;22(1):4–10. | ||
Luchsinger JA. Type 2 diabetes and cognitive impairment: linking mechanisms. J Alzheimer’s Dis. 2012;30 Suppl 2:S185–S198. | ||
de la Monte SM. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012;72(1):49–66. | ||
Lanctot KL, Rajaram RD, Herrmann N. Therapy for Alzheimer’s disease: how effective are current treatments? Ther Adv Neurol Disord. 2009;2(3):163–180. | ||
Green RC, Schneider LS, Amato DA, et al. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA. 2009;302(23):2557–2564. | ||
Quinn JF, Raman R, Thomas RG, et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA. 2010;304(17):1903–1911. | ||
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–344. | ||
Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. 2001;3(1):75–80. | ||
Moreira PI, Santos MS, Seica R, Oliveira CR. Brain mitochondrial dysfunction as a link between Alzheimer’s disease and diabetes. J Neurol Sci. 2007;257(1–2):206–214. | ||
Moreira PI, Honda K, Zhu X, et al. Brain and brawn: parallels in oxidative strength. Neurology. 2006;66(2 Suppl 1):S97–S101. | ||
Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314(5800):777–781. | ||
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. | ||
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. | ||
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–185. | ||
Rocchi A, Pellegrini S, Siciliano G, Murri L. Causative and susceptibility genes for Alzheimer’s disease: a review. Brain Res Bull. 2003;61(1):1–24. | ||
Cummings JL. Alzheimer’s disease. N Engl J Med. 2004;351(1):56–67. | ||
Smith AD, Yaffe K. Dementia (including Alzheimer’s disease) can be prevented: statement supported by international experts. J Alzheimers Disease. 2014;38(4):699–703. | ||
DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37(6):667–687. | ||
Stefan N, Kantartzis K, Machann J, et al. Identification and characterization of metabolically benign obesity in humans. Arch Intern Med. 2008;168(15):1609–1616. | ||
Luchsinger JA, Gustafson DR. Adiposity, type 2 diabetes, and Alzheimer’s disease. J Alzheimers Disease. 2009;16(4):693–704. | ||
Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology. 1999;53(9):1937–1942. | ||
Luchsinger J, Reitz C, Honig LS, Tang M-X, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65(4):545–551. | ||
Gudala K, Bansal D, Schifano F, Bhansali A. Diabetes mellitus and risk of dementia: a meta-analysis of prospective observational studies. J Diabetes Investig. 2013;4(6):640–650. | ||
Cheng G, Huang C, Deng H, Wang H. Diabetes as a risk factor for dementia and mild cognitive impairment: a meta-analysis of longitudinal studies. Intern Med J. 2012;42(5):484–491. | ||
Curb JD, Rodriguez BL, Abbott RD, et al. Longitudinal association of vascular and Alzheimer’s dementias, diabetes, and glucose tolerance. Neurology. 1999;52(5):971–975. | ||
Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005;64(2):277–281. | ||
Schnaider Beeri M, Goldbourt U, Silverman JM, et al. Diabetes mellitus in midlife and the risk of dementia three decades later. Neurology. 2004;63(10):1902–1907. | ||
Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies The Honolulu-Asia Aging Study. Diabetes. 2002;51(4):1256–1262. | ||
Takeda M, Martinez R, Kudo T, et al. Apolipoprotein E and central nervous system disorders: reviews of clinical findings. Psychiatry Clin Neurosci. 2010;64(6):592–607. | ||
Irie F, Fitzpatrick AL, Lopez OL, et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch Neurol. 2008;65(1):89–93. | ||
Dore GA, Elias MF, Robbins MA, Elias PK, Nagy Z. Presence of the APOE epsilon4 allele modifies the relationship between type 2 diabetes and cognitive performance: the Maine-Syracuse Study. Diabetologia. 2009;52(12):2551–2560. | ||
Yaffe K, Blackwell T, Whitmer RA, Krueger K, Barrett Connor E. Glycosylated hemoglobin level and development of mild cognitive impairment or dementia in older women. J Nutr Health Aging. 2006;10(4):293–295. | ||
Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology. 2004;63(7):1181–1186. | ||
Profenno LA, Porsteinsson AP, Faraone SV. Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry. 2010;67(6):505–512. | ||
Reaven GM, Laws A, editors. Insulin Resistance: The Metabolic Syndrome X. Clifton, NJ, USA: Human Press; 1999. | ||
Anstey K, Cherbuin N, Budge M, Young J. Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev. 2011;12(5):e426–e437. | ||
Gustafson D, Rothenberg E, Blennow K, Steen B, Skoog I. An 18-year follow-up of overweight and risk of Alzheimer disease. Arch Intern Med. 2003;163(13):1524–1528. | ||
Fitzpatrick AL, Kuller LH, Lopez OL, et al. Midlife and late-life obesity and the risk of dementia: cardiovascular health study. Arch Neurol. 2009;66(3):336–342. | ||
Whitmer RA. The epidemiology of adiposity and dementia. Curr Alzheimer Res. 2007;4(2):117–122. | ||
Morley JE. Anorexia, sarcopenia, and aging. Nutrition. 2001;17(7):660–663. | ||
Johnson DK, Wilkins CH, Morris JC. Accelerated weight loss may precede diagnosis in Alzheimer disease. Arch Neurol. 2006;63(9):1312–1317. | ||
Luchsinger JA, Tang M-X, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63(7):1187–1192. | ||
Okereke O, Hankinson SE, Hu FB, Grodstein F. Plasma C peptide level and cognitive function among older women without diabetes mellitus. Arch Intern Med. 2005;165(14):1651–1656. | ||
Okereke OI, Pollak MN, Hu FB, Hankinson SE, Selkoe DJ, Grodstein F. Plasma C-peptide levels and rates of cognitive decline in older, community-dwelling women without diabetes. Psychoneuroendocrinology. 2008;33(4):455–461. | ||
Okereke OI, Kurth T, Pollak MN, Gaziano JM, Grodstein F. Fasting plasma insulin, c-peptide and cognitive change in older men without diabetes: results from the Physicians’ Health Study II. Neuroepidemiology. 2010;34(4):200–207. | ||
Avogaro P, Crepaldi G, Enzi G, Tiengo A. Association of hyperlipemia, diabetes mellitus and mild obesity. Acta Diabetol Lat. 1967;4:572–590. | ||
Yaffe K, Kanaya A, Lindquist K, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA. 2004;292(18):2237–2242. | ||
Vanhanen M, Koivisto K, Moilanen L, et al. Association of metabolic syndrome with Alzheimer disease. A population-based study. Neurology. 2006;67(5):843–847. | ||
Kalmijn S, Foley D, White L, et al. Metabolic cardiovascular syndrome and risk of dementia in Japanese-American elderly men: the Honolulu-Asia Aging Study. Arterioscler Thromb Vasc Biol. 2000;20(10):2255–2260. | ||
Raffaitin C, Gin H, Empana J-P, et al. Metabolic syndrome and risk for incident Alzheimer’s disease or vascular dementia: the Three-City Study. Diabetes Care. 2009;32(1):169–174. | ||
Solfrizzi V, Scafato E, Capurso C, et al. Metabolic syndrome and the risk of vascular dementia: the Italian Longitudinal Study on Ageing. J Neurol Neurosurg Psychiatry. 2010;81(4):433–440. | ||
Muller M, Tang M-X, Schupf N, Manly JJ, Mayeux R, Luchsinger JA. Metabolic syndrome and dementia risk in a multiethnic elderly cohort. Dement Geriatr Cogn Disord. 2007;24(3):185–192. | ||
Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on Detection, Evaluation, and Treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA. 2001;285(19):2486–2497. | ||
Grundy SM, Cleeman JI, Daniels SR, et al. Diagnosis and management of the metabolic syndrome an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Circulation. 2005;112(17):2735–2752. | ||
Alberti KG, Zimmet P, Shaw J; IDF Epidemiology Task Force Consensus Group. The metabolic syndrome – a new worldwide definition. Lancet. 2005;366(9491):1059–1062. | ||
Crichton GE, Elias MF, Buckley JD, Murphy KJ, Bryan J, Frisardi V. Metabolic syndrome, cognitive performance, and dementia. J Alzheimers Dis. 2012;30:S77–S87. | ||
Watts AS, Loskutova N, Burns JM, Johnson DK. Metabolic syndrome and cognitive decline in early Alzheimer’s disease and healthy older adults. J Alzheimers Dis. 2013;35(2):253–265. | ||
Zhao W-Q, De Felice FG, Fernandez S, et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22(1):246–260. | ||
Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31(2):224–243. | ||
Steen E, Terry BM, Rivera EJ, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease-is this type 3 diabetes? J Alzheimers Dis. 2005;7(1):63–80. | ||
Hoyer S, Nitsch R. Cerebral excess release of neurotransmitter amino acids subsequent to reduced cerebral glucose metabolism in early-onset dementia of Alzheimer type. J Neural Transm. 1989;75(3):227–232. | ||
Craft S. Alzheimer disease: insulin resistance and AD – extending the translational path. Nat Rev Neurol. 2012;8(7):360–362. | ||
Talbot K, Wang H-Y, Kazi H, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122(4):1316–1338. | ||
Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. | ||
Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J Clin Invest. 2012;122(4):1339–1353. | ||
Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8(3):247–268. | ||
Phiel CJ, Wilson CA, Lee VM-Y, Klein PS. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature. 2003;423(6938):435–439. | ||
Wada A, Yokoo H, Yanagita T, Kobayashi H. New twist on neuronal insulin receptor signaling in health, disease, and therapeutics. J Pharmacol Sci. 2005;99(2):128–143. | ||
Carro E, Trejo JL, Spuch C, Bohl D, Heard JM, Torres-Aleman I. Blockade of the insulin-like growth factor I receptor in the choroid plexus originates Alzheimer’s-like neuropathology in rodents: new cues into the human disease? Neurobiol Aging. 2006;27(11):1618–1631. | ||
Carro E, Torres-Aleman I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur J Pharmacol. 2004;490(1):127–133. | ||
Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol. 2011;68(1):51–57. | ||
Devi L, Alldred MJ, Ginsberg SD, Ohno M. Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer’s disease. PLoS One. 2012;7(3):e32792. | ||
Plum L, Schubert M, Brüning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab. 2005;16(2):59–65. | ||
Gasparini L, Netzer WJ, Greengard P, Xu H. Does insulin dysfunction play a role in Alzheimer’s disease? Trends Pharmacol Sci. 2002;23(6):288–293. | ||
Kremer A, Louis JV, Jaworski T, Van Leuven F. GSK3 and Alzheimer’s disease: facts and fiction. Front Mol Neurosci. 2011;4:17. | ||
Li X, Lu F, Tian Q, Yang Y, Wang Q, Wang JZ. Activation of glycogen synthase kinase-3 induces Alzheimer-like tau hyperphosphorylation in rat hippocampus slices in culture. J Neural Transm. 2006;113(1):93–102. | ||
Picone P, Giacomazza D, Vetri V, et al. Insulin-activated Akt rescues Aβ oxidative stress-induced cell death by orchestrating molecular trafficking. Aging Cell. 2011;10(5):832–843. | ||
Suzanne M. Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs. 2012;72(1):49–66. | ||
Thorne R, Pronk G, Padmanabhan V, Frey WH 2nd. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience. 2004;127(2):481–496. | ||
Reger M, Watson G, Green P, et al. Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology. 2008;70(6):440–448. | ||
Craft S, Baker LD, Montine TJ, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69(1):29–38. | ||
Reger M, Watson G, Frey WH 2nd, et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging. 2006;27(3):451–458. | ||
Kitabchi AE, Temprosa M, Knowler WC, et al. Diabetes Prevention Program Research Group. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the diabetes prevention program: effects of lifestyle intervention and metformin. Diabetes. 2005;54(8):2404–2414. | ||
Goldberg R, Horton E, Marcovina S, et al. Intensive lifestyle intervention or metformin on inflammation and coagulation in participants with impaired glucose tolerance. Diabetes. 2005;54(5):1566–1572. | ||
Orchard TJ, Temprosa M, Goldberg R, et al. The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: the Diabetes Prevention Program randomized trial. Ann Intern Med. 2005;142(8):611–619. | ||
Knowler WC, Barrett-Connor E, Fowler SE, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. | ||
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. | ||
Currie C, Poole C, Gale E. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52(9):1766–1777. | ||
Domínguez RO, Marschoff ER, González SE, Repetto MG, Serra JA. Type 2 diabetes and/or its treatment leads to less cognitive impairment in Alzheimer’s disease patients. Diabetes Res Clin Pract. 2012;98(1):68–74. | ||
Hsu C-C, Wahlqvist ML, Lee M-S, Tsai H-N. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis. 2011;24(3):485–493. | ||
Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: a population-based case-control study. J Am Geriatr Soc. 2012;60(5):916–921. | ||
Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351(11):1106–1118. | ||
Malinowski JM, Bolesta S. Rosiglitazone in the treatment of type 2 diabetes mellitus: a critical review. Clin Ther. 2000;22(10):1151–1168. | ||
Nesto R. C-reactive protein, its role in inflammation, type 2 diabetes and cardiovascular disease, and the effects of insulin-sensitizing treatment with thiazolidinediones. Diabet Med. 2004;21(8):810–817. | ||
Tuppo EE, Arias HR. The role of inflammation in Alzheimer’s disease. Int J Biochem Cell Biol. 2005;37(2):289–305. | ||
Craft S. Insulin resistance and Alzheimers disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007;4(2):147–152. | ||
Watson G, Cholerton BA, Reger MA, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13(11):950–958. | ||
Risner M, Saunders A, Altman J, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6(4):246–254. | ||
Gold M, Alderton C, Zvartau-Hind M, et al. Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord. 2010;30(2):131–146. | ||
Miller BW, Willett KC, Desilets AR. Rosiglitazone and pioglitazone for the treatment of Alzheimer’s disease. Ann Pharmacother. 2011;45(11):1416–1424. | ||
Cheung BM. Behind the rosiglitazone controversy. Expert Rev Clin Pharmacol. 2010;3(6):723–725. | ||
Deacon CF, Ahrén B. Physiology of incretins in health and disease. Rev Diabet Stud. 2011;8(3):293. | ||
Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. | ||
Deacon C. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: a comparative review. Diabetes Obes Metab. 2011;13(1):7–18. | ||
Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5(5):262–269. | ||
Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Both GLP-1 and GIP are insulinotropic at basal and postprandial glucose levels and contribute nearly equally to the incretin effect of a meal in healthy subjects. Regul Pept. 2003;114(2):115–121. | ||
During MJ, Cao L, Zuzga DS, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9(9):1173–1179. | ||
Abbas T, Faivre E, Hölscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: interaction between type 2 diabetes and Alzheimer’s disease. Behav Brain Res. 2009;205(1):265–271. | ||
McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31(17):6587–6594. | ||
Selkoe DJ. Preventing Alzheimer’s disease. Science. 2012;337(6101):1488–1492. | ||
Pipatpiboon N, Pintana H, Pratchayasakul W, Chattipakorn N, Chattipakorn SC. DPP4-inhibitor improves neuronal insulin receptor function, brain mitochondrial function and cognitive function in rats with insulin resistance induced by high-fat diet consumption. Eur J Neurosci. 2013;37(5):839–849. | ||
Pintana H, Apaijai N, Chattipakorn N, Chattipakorn SC. DPP-4 inhibitors improve cognition and brain mitochondrial function of insulin-resistant rats. J Endocrinol. 2013;218(1):1–11. | ||
Tyas SL, Manfreda J, Strain LA, Montgomery PR. Risk factors for Alzheimer’s disease: a population-based, longitudinal study in Manitoba, Canada. Int J Epidemiol. 2001;30(3):590–597. | ||
MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord. 2002;14(2):77–83. | ||
Hassing LB, Johansson B, Nilsson SE, et al. Diabetes mellitus is a risk factor for vascular dementia, but not for Alzheimer’s disease: a population-based study of the oldest old. Int Psychogeriatr. 2002;14(3):239–248. | ||
Honig LS, Tang MX, Albert S, et al. Stroke and the risk of Alzheimer disease. Arch Neurol. 2003;60(12):1707–1712. | ||
Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61(5):661–666. | ||
Borenstein AR, Wu Y, Mortimer JA, et al. Developmental and vascular risk factors for Alzheimer’s disease. Neurobiol Aging. 2005;26(3):325–334. | ||
Hayden KM, Zandi PP, Lyketsos CG, et al. Vascular risk factors for incident Alzheimer disease and vascular dementia: the Cache County study. Alzheimer Dis Assoc Disord. 2006;20(2):93–100. | ||
Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risk of developing Alzheimer disease: results from the Framingham Study. Arch Neurol. 2006;63(11):1551–1555. | ||
Ahtiluoto S, Polvikoski T, Peltonen M, et al. Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology. 2010;75(13):1195–1202. | ||
Ohara T, Doi Y, Ninomiya T, et al. Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology. 2011;77(12):1126–1134. | ||
Cheng D, Noble J, Tang MX, Schupf N, Mayeux R, Luchsinger JA. Type 2 diabetes and late-onset Alzheimer’s disease. Dement Geriatr Cogn Disord. 2011;31(6):424–430. | ||
Li J, Wang YJ, Zhang M, et al. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology. 2011;76(17):1485–1491. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.