Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Leukocyte Function in COPD: Clinical Relevance and Potential for Drug Therapy
Authors Baker JR, Donnelly LE
Received 8 April 2021
Accepted for publication 19 July 2021
Published 30 July 2021 Volume 2021:16 Pages 2227—2242
DOI https://doi.org/10.2147/COPD.S266394
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Jonathan R Baker, Louise E Donnelly
Airway Disease, National Heart and Lung Institute, Imperial College London, London, UK
Correspondence: Louise E Donnelly
Airway Disease, National Heart and Lung Institute, Imperial College London, Dovehouse Street, London, SW3 6LY, UK
Tel +44 207 594 7895
Email [email protected]
Abstract: Chronic obstructive pulmonary disease (COPD) is a progressive lung condition affecting 10% of the global population over 45 years. Currently, there are no disease-modifying treatments, with current therapies treating only the symptoms of the disease. COPD is an inflammatory disease, with a high infiltration of leukocytes being found within the lung of COPD patients. These leukocytes, if not kept in check, damage the lung, leading to the pathophysiology associated with the disease. In this review, we focus on the main leukocytes found within the COPD lung, describing how the release of chemokines from the damaged epithelial lining recruits these cells into the lung. Once present, these cells become active and may be driven towards a more pro-inflammatory phenotype. These cells release their own subtypes of inflammatory mediators, growth factors and proteases which can all lead to airway remodeling, mucus hypersecretion and emphysema. Finally, we describe some of the current therapies and potential new targets that could be utilized to target aberrant leukocyte function in the COPD lung. Here, we focus on old therapies such as statins and corticosteroids, but also look at the emerging field of biologics describing those which have been tested in COPD already and potential new monoclonal antibodies which are under review.
Keywords: neutrophil, macrophage, eosinophil, CD8+ T cell, mast cell
Introduction
Patients with chronic obstructive pulmonary disease (COPD) often present with shortness of breath on exertion. This is due to several structural changes in the lung including destruction of the alveoli leading to airspace enlargement and stenosis of the small airways. Some patients also exhibit chronic bronchitis, with this excess mucus production that also contributes to occlusion of the airways. At present, treatment for this debilitating condition is purely symptomatic with no pharmaceutical intervention able to reduce disease progression. COPD is a chronic inflammatory condition, and as such is associated with increased leukocyte infiltration in the lung. This influx is thought to be mediated by epithelial damage by exogenous factors such as cigarette smoke leading to release of chemoattractants that recruit activated leukocytes. Once present in the lung, these cells may then release further mediators such as growth factors and proteases that contribute to lung remodeling and disease pathophysiology. Understanding the mechanisms underlying these inflammatory and structural changes in the lung is fundamental to the design of new therapeutic interventions. This review will examine the different leukocyte populations associated with COPD and the possibility of targeting these responses as a therapeutic option in this condition.
Leukocytes in COPD
Macrophages
In health, macrophages are the most abundant leukocyte in the lung and have a vital role in maintaining lung homeostasis.1 They are one of the major innate immune defense mechanisms against inhaled particulates and pathogens. In the COPD lung, macrophage numbers increase by at least 20-fold,2 but at least half of COPD patients are chronically colonized with bacteria3 suggesting a defect in their innate immune functioning. The origin of macrophages in the lung is a current area of research; macrophages may derive from circulating monocytes that are recruited to the lung and subsequently differentiate in the tissue.4–6 However, in the lung, there appears to be a population of tissue-resident macrophages that may originate from the yolk sac7 and fetal liver8 during development with the capacity to self-renew. Within the alveolar compartment, alveolar macrophages have <5% proliferation capacity,9 but whether this activity is sufficient to account for the increased numbers of macrophages in COPD is unknown.10 In IPF there is accumulation of monocyte-derived macrophages (MDM) that are highly pro-inflammatory,11,12 and it is likely that MDM contribute to much of the observed macrophage population found in COPD.13 It is clear that the macrophage in COPD is dysfunctional and releases high levels of inflammatory mediators including CXCL8 and granulocyte macrophage-colony stimlating factor (GM-CSF); the latter is important in the context of macrophage development as it drives macrophage differentiation.14 Furthermore, these cells release high levels of activated proteases including matrix metalloproteinase (MMP)-2 and MMP-915,16 that contribute to destruction of the lung tissue and emphysema. The role of the macrophage is to remove any pathogen or particle, and both alveolar macrophages and MDM from patients with COPD are poor at clearing bacteria,17–19 apoptotic cells20 and fungal spores.21 Exactly why macrophages from COPD patients differ from those from healthy controls is not clear, although environmental exposures such as cigarette smoke or viral challenges appear to alter their function.22–24 This defect is associated with dysfunctional mitochondrial responses25,26 and therefore may be due to reduced availability of ATP in these cells. A distinct, COPD macrophage phenotype has been described that is glucocorticosteroid-insensitive and highly inflammatory,27 although again whether this is driven by local environment, systemic changes or is an epigenetic response is not clear. There are differences in circulating monocytes in COPD patients, which may account for differences in the lung macrophage population. Although chemokine receptor expression appears similar to those of control cells,28,29 the chemotactic response of these cells to specific chemokines, namely CXCL1 and CXCL5, shows an enhanced migratory response,28 thus suggesting alterations in the circulating macrophage progenitors. Given the highly inflammatory and dysfunctional profile of macrophages in COPD, these cells could be responsible for many of the pathophysiologies observed in this disease, and targeting these cells could be a therapeutic option.
Neutrophils
Neutrophils have long been considered a key inflammatory cell in COPD contributing to tissue destruction via the release of highly activated proteases including neutrophil elastase, cathepsin G and proteinase 3.30 Their numbers are increased in the airways of patients with COPD, and sputum neutrophilia is considered a hallmark of inflammation in COPD.31 Neutrophils are released into the circulation from the bone marrow and are relatively short-lived with a half-life of ~19 h.32 They have a characteristic three-lobed nucleus and contain pre-formed granules,33 azurophilic granules, specific granules and gelatinase granules. Neutrophils are recruited to sites of inflammation by the release of chemotactic molecules including CXCL8 and leukotriene (LT)B4 as well as bacterial cell wall fragments such as formyl-methionine-leucine-phenylalanine (f-MLP).34 As such, neutrophils are key components of the innate immune response. However, in COPD, the accumulation of these cells appears to be detrimental.35 There are alterations to neutrophil responses in COPD including aberrant migration.36,37 More detailed analysis of neutrophil migration in COPD has suggested that these cells have lost direction,38 leading to an increased presence of these cells in tissue with increased opportunity to destroy extracellular matrix and hence contribute to emphysema.39 Neutrophil elastase is also a potent stimulator of mucus hypersecretion and thus contributes to chronic bronchitis.
Neutrophils play a crucial role in the innate immune response.32 They have the capacity to phagocytose pathogens and produce reactive oxygen species (ROS) to kill these species. In addition, neutrophils can control infection via the production of neutrophil extracellular traps (NETS) and degranulation and release of proteases.39 In addition to the defect in migration observed in neutrophils from COPD patients, other functions attributed to these cells may also be aberrant. There are studies that suggest phagocytosis of bacteria is reduced in neutrophils from COPD patients40 as well as alterations in NET formation,41 although further studies are required. However, it is clear that neutrophilia is observed in the airways of patients with COPD, and this is associated with increased expression of chemokines including CXCL1 and CXCL842,43 as well as lipid mediators such as leukotriene (LT)B4 that recruit these cells into the lung where they increase oxidant stress and propagate tissue destruction, leading to disease pathophysiology.
Eosinophils
Eosinophils are granulocytes that are formed in the bone marrow from a CD34+ precursor and require IL-5 for maturation and then are released into the circulation.44,45 In healthy people, the level of circulating eosinophils is low, in the range 0.05–0.5×109 cells/L,46 but blood eosinophilia is associated with atopy and is associated with an allergic asthma phenotype.47 Eosinophils accumulate in the lung tissue of asthmatics and are associated with bronchial hyperresponsiveness as, upon activation, these cells release numerous bronchoconstrictors including cysteinyl leukotrienes.48 The presence of eosinophils has been noted in COPD with approximately one-third of patients exhibiting sputum eosinophils,49 and there is a suggestion that this may be associated with increased exacerbation frequency;50 however, their abundance is reduced compared with asthma.51 More recently, the presence of eosinophils in the blood of a subgroup of COPD patients has garnered interest.52 In particular, the presence of eosinophils in blood of 100–300 cells/μL appears to be a biomarker for glucocorticosteroid responsiveness.52,53 However, there has been debate as to the threshold of eosinophil numbers needed to assign a COPD patient eosinophilic.54 High blood eosinophil levels are considered >2% of total leukocytes.55 In some COPD patients, there is an increase in eosinophils during exacerbations that are associated with a distinct inflammatory profile.56 Whether these patients could represent an asthma–COPD overlap remains a subject of debate; they remain a significant COPD population.
Mast Cells
Mast cells have a well-defined role in IgE-mediated airway disease such as allergic asthma,57 but their role in COPD is much less understood. Although considered tissue-resident cells, mast cells are derived from CD34+/KIT+ hematopoietic stem cells in the bone marrow.58,59 These cells then migrate from the bone marrow to various organs including the lung. However, the possibility of a local, self-renewing mast stem cell population has derived from murine studies where progenitors have been identified in the yolk sac.60 These progenitor cells require exposure to the ligand for cKIT, stem cell factor (SCF), for differentiation into a mast cell committed progenitor with IL-4 driving subsequent maturation, although other cytokines may also have a role. Mast cells exist as two separate subtypes in the lung that can be classified based on expression of tryptase or chymase.57,59 Mast cells located predominately in the bronchiolar epithelium lack chymase expression, whereas those in the interstitium express both chymase and tryptase.61 Mast cells accumulate in the lung tissue of asthmatics62,63 and have an important role in driving airway inflammation in allergic lung disease; they can mediate bronchoconstriction via the release of mediators including histamine and prostaglandin (PG)D2.64 Classically, this activation pathway can occur by IgE-mediated events, but mast cells can also be activated by IgE-independent pathways including activation via pathogen-associated molecular patterns (PAMPs) including lipopolysaccharide (LPS).65
The role of mast cells in COPD remains unclear. In vivo models have suggested a role for mast cell-derived tryptase in the early development of COPD,66 but this has not been translated into human studies of early COPD at present. Mast cells have been examined in lung tissue from COPD patients, and such studies indicate an increase in mast cell density with more severe disease,67,68 but not in stable disease.69 Ballarin et al examined lung tissue from patients with panlobular emphysema and centrilobular emphysema and compared these tissues to those from non-smokers and smokers without disease.68 They showed that there were increased mast cell numbers in the lung tissue of patients with centrilobular emphysema and that this was associated with increased airway smooth muscle deposition. This suggests that mast cells can discriminate between emphysema phenotypes and may have a role in pathophysiology.
Lymphocytes
Th1/Th2 CD4+ Cells
CD4+ T cells are key components of a healthy immune system and modulate both the innate and adaptive immune systems via the regulation of macrophage microbe killing, the recruitment of immune cells and assisting B cells in antibody production.70 CD4+ T cell function is dictated by environment, with naive CD4+ T cells differentiating into mature CD4+ T cells via environmental cues. Th1 and Th2 cells are the most characterized and classically described CD4+ T cell phenotypes. Pro-inflammatory/anti-inflammatory environments induce activated Th1 (IL-12 and interferon-γ (IFNγ)) and Th2 (IL-2 and IL-4) cells via activation of the transcription factors STAT4/T-Bet and GATA3, respectively.71 These two T cell subsets have distinct cytokine production and functions, with Th1 cells being associated with the host defense against intracellular viral and bacterial pathogens, whereas Th2 cells respond against extracellular pathogens and parasites. Th1 cells predominantly produce IFNγ, IL-2 and TNFβ, whilst Th2 cells predominantly release IL-4, IL-5 and IL-13.
Within the COPD lung, there appears to be a more dominant Th1 phenotype with elevated numbers of these cells as well as increased expression of Th1 cytokines,72 although some conflicting studies show the heterogeneity of COPD as a disease.73 However, in the lung of smokers with COPD, CD4+ T cells express increased levels of the tissue-specific chemokine receptor CXCR3, along with the epithelium expressing increased levels of its ligand, CXCL10,74 with these cells showing a Th1 phenotype. CD4+ T cells in the lungs of smokers with COPD also express STAT4 and IFNγ suggesting an active Th1-mediated inflammatory process, with these levels correlating with the degree of airflow obstruction.75 Overactivation or increase numbers of T cells within the COPD lung may therefore drive COPD progression via the over-recruitment/activation of other immune cells via the aberrant section of inflammatory cytokines and chemokines.
Treg Cells
Another set of CD4+ T cells are Tregs. Tregs suppress the activation, proliferation and cytokine production of CD4+ and CD8+ T cells, via the release of TGF-β, IL-10 and adenosine, along with direct cell contact.76 The numbers of CD4+CD25+FOXP3+ regulatory T cells are found to be reduced in the sputum of COPD patients, whilst there is an increase in CD4+CD25+ regulatory T cells in the BAL of smokers compared to non-smokers, but this does not occur in COPD patients.73,77 Loss of Tregs may therefore lead to overactivated and uncontrolled T cell activation and inflammation, leading to COPD pathogenesis.
Th17 Cells
A more recently described subset of CD4+ T cells is known as Th17 cells. Th17 cells are differentiated from naïve CD4+ T cells when stimulated with antigen-presenting cells (APCs) in the presence of IL-6 and IL-1β in humans,78 with IL-23 secondarily promoting complete differentiation. Th17 cells uniquely release IL-17, a highly inflammatory cytokine which acts mainly upon stromal cells in many tissues including the lung.78 IL-17 activates bronchial epithelial cells to release neutrophil chemokines such as CXCL8 and GM-CSF, as well as cytokines such as IL-6, along with antimicrobial peptides such as β-defensins and S100 proteins.79 However, dysregulated Th17 cytokines may lead to disease pathophysiology. Increased Th17 cytokines are found in the bronchial mucosa of COPD patients80 and are associated with driving emphysema via MMP-9 production in COPD, neutrophilia and cytokine release in vivo.81
CD8+ T Cells
CD8+ T cells are important for immune defense against intracellular pathogens. Upon antigen detection, CD8+ T cells become activated and utilize three major mechanisms to kill infected cells.82 The first is secretion of cytokines, primarily TNFα and IFNγ, which have anti-viral effects. The second major function is the production and release of cytotoxic granules. These consist of perforin and granzymes. Perforin proteins form a pore in the membrane of a target cell, allowing entry of granzymes into the cell. Granzymes are a type of serine proteases which cleave intracellular proteins, resulting in apoptosis. CD8+ T cells can also destroy infected cells via Fas/Fas ligand (FasL) interactions. Upon activation CD8+ T cells begin to express FasL on their surface, and this can bind to Fas on the surface of the infected cell. Binding induces activation of caspase cascade, ultimately resulting in apoptosis of the target cell.83
Dendritic cells (DCs) are the predominant APCs for the priming of naïve CD8+ T cells, with this occurring mainly by MHC class I antigen presentation to the T cell receptor. Antigen presentation leads to proliferation and differentiation of the T cell. In the lung, under viral challenge, DCs which have taken up a viral antigen stimulate the activation of naive CD8+ T cells in the draining lymph nodes.84 As patients with COPD have a known history of viral infection and these are known to play a crucial role in COPD exacerbations, increased numbers/overactive CD8+ T cells may play a role in this disease.
CD8+ T cells appear to be the largest subset of T cells found in the COPD lung, and numbers are found to correlate with lung function.85,86 Current smokers with COPD and chronic airflow limitation have an increased number of CD8+ T-lymphocytes,85 and these have increased activation markers.87,88 To enter the lung CD8+ T cells utilize chemokine receptors to exit the bloodstream/lymph nodes, with CD8+ T cells recruited into the lung via chemokine/chemokine receptor gradients. In COPD, expression of the chemokine receptors CCR5, CXCR3 and CXCR6 were increased on CD8+ T cells, and this correlated with severity of COPD89 showing an altered subset of CD8+ T cells in COPD. A recent systematic review of papers published on CD8+ T cells in the last 15 years showed that not only were the number of CD8+ T cells increased in COPD, but these cells had an increased capacity to exert effector functions such as increased inflammatory mediator release and increased expression of cytotoxic mediators such as granzyme and/or perforin.90 Immunosenescence is also evident in COPD patients, with increased numbers of T cells that do not express CD28; these cells release increased amounts of inflammatory cytokines as well as elevated levels of perforins and granzyme B.91 The mechanisms by which these elevated numbers of CD8+ T cells may destroy lung parenchyma are not clear, but they may be able to lyse target cells, via the release of cytotoxic proteins, such as perforin or granzyme, or by inducing apoptosis via the Fas ligand-Fas pathway.92
The comparative roles of the innate and adaptive immune systems in COPD remains an area of contention, with suggestions that activation of the innate immune system drives early disease. Evidence of this is taken from in vivo mice models, where removal of T cells does not change the levels of emphysema seen in mice exposed to cigarette smoke for 7 months.93 However, there is evidence to suggest that both the innate and adaptive immune system may be important in disease progression and pathophysiology.
B Cells
The presence of lymphoid follicles, of which B cells are the major cell constituent, in the tissue surrounding the small airways of COPD patients suggests that these cells may be important in small airways disease and progression as the number of B cells identified is significantly higher in those patients with severe disease.94 The number of B cells in the sub-epithelium of the bronchial airways of COPD patients was also found to be significantly higher than in controls,95 and this is also observed within the lung parenchyma,96 suggesting this to be a global phenomenon within the epithelial airway lining. The role these B cells play in the progression of the disease is still not fully understood, but the secretion and levels of autoantibodies in COPD has been extensively studied. Autoantibodies can drive disease and have pathogenic effects by increasing sensitivity of cell surface receptors to their agonist and therefore causing overactivation of these receptors. They may also cause neutrophil activation and induce inflammation at the site of auto-antibody binding.97 In COPD, elevated levels of auto-antibodies against elastin are detected in the blood of patients with emphysema, with low-grade elastin breakdown caused by cigarette exposure thought to cause this effect, but how these autoantibodies drive the disease is not fully understood.98 There are also increased levels of antinuclear antibodies (ANA) and anti-tissue (AT) antibodies in stable-state COPD patients, further suggesting an autoimmune component to COPD.99 However, there is conflicting data on the role of these auto-antibodies and whether they play a role in disease progression or are a consequence of the disease.
Natural Killer (NK) Cells
Natural killer (NK) cells are defined as effector lymphocytes belonging to the innate immune system which have both cytotoxic and cytokine producing functions. NK cells are major regulators of tumor cells and microbial infections, forming a first line of defense.100 NK cells require priming and can be activated by direct cell contact, with tumor or infected cells, or via other leukocytes such as dendritic cells and macrophages.101 Cytokines also play a role in their activation, including type 1 interferons and IL-12/-18, especially when released from dendritic cells.102 IL-15 is also known to be an unconventional activator of NK cells.103
In humans, NK cells can be defined as CD3−CD56+, with NK cells being further defined into two subsets dependent on function, maturation and homing properties.100 CD56dim CD16+ NK cells are cytotoxic and produce IFNγ and perforin, whilst CD56Bright CD16− NK cells are thought of as being less mature, do not express perforin and their primary role is cytokine secretion.100 Within the lung, most NK cells are CD56dim CD16+.104
NK cells are believed to be in a less active state in the lung, compared to those from peripheral blood, with this being accredited to suppression via pulmonary macrophages.105 NK cell activity is also suggested to be further suppressed by smoking, with release of superoxides by macrophages being the suggested mechanism.106 However, conflicting data are found in the COPD lung; NK cell numbers are elevated in sputum and BAL fluid of COPD patients and are more cytotoxic compared to smokers.107,108 Finch et al also showed dendritic cells to be a major activator of NK cells in COPD, via the release of IL-15, with increased cytotoxic effects of NK cells on airway epithelial cells being greater in COPD patients than smoker controls.109 This may be due to changes within the lung environment in COPD patients.
Localization of Immune Cells May Regulate Their Pathogenic Function
The importance of the localization of immune cells in the COPD lung may be crucial in their ability to drive pathogenesis and the different etiologies of the disease. Recently, interstitial macrophages have been found to locate mainly in the tissue of the alveolar septa, along with lesser numbers being found surrounding the small airways and vessels. The authors show increased numbers within smokers and hypothesize that the interstitial macrophages located in the septa may play a role in driving emphysema in COPD patients.110 Using data from in vivo mouse models in which different strains of mice develop distinct COPD phenotypes when exposed to cigarette smoke, it was shown that distinct macrophage phenotypes are found in these mice and that these may play a role in driving that form of disease pathophysiology.111 Neutrophil location and site of de-granularization may also be important in COPD, with dysregulation of directional migration leading to tissue damage of the parenchyma, whilst de-granularization in the alveolar space or airway drives chronic bronchitis or small airway disease. NK cells are known to be able to destroy the lung parenchyma104 as well as CD8 T cells. Th17 cells on the other hand are found in the bronchial mucosa and therefore may drive airway inflammation.80 All of these data show the importance of the type of sampling technique used when examining the role each immune cell may play in the pathogenesis of COPD, with careful decisions regarding which technique should be used to assess the role of a specific leukocyte and its contribution to disease pathophysiology. However, the increase in inflammatory cells in the lungs of COPD patients, together with skewing of many cell functions towards a destructive rather than a protective pathology (Table 1), suggests that targeting these cells is a possible therapeutic option (Figure 1).
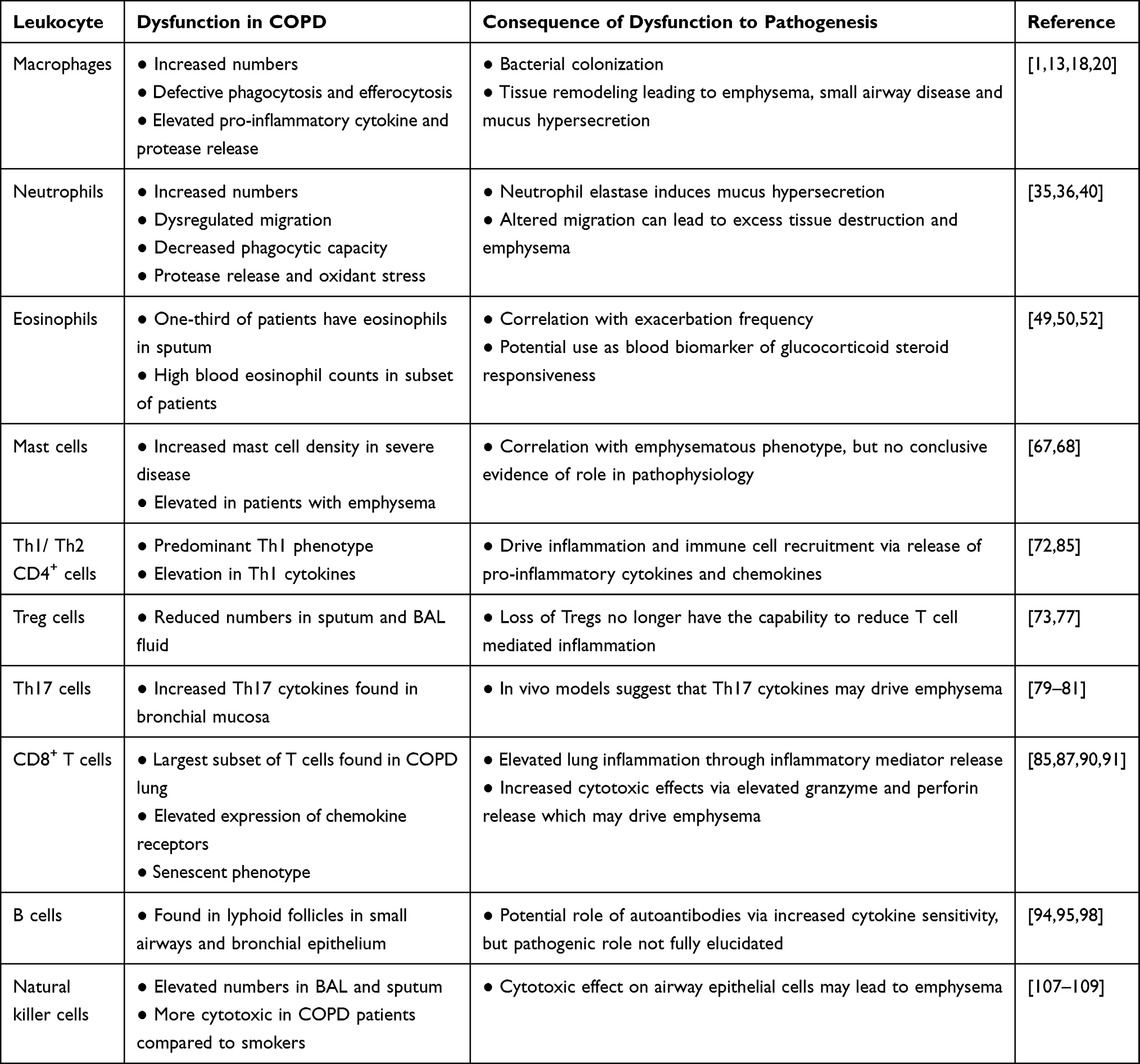 |
Table 1 Contribution of Different Leukocytes to COPD Pathophysiology |
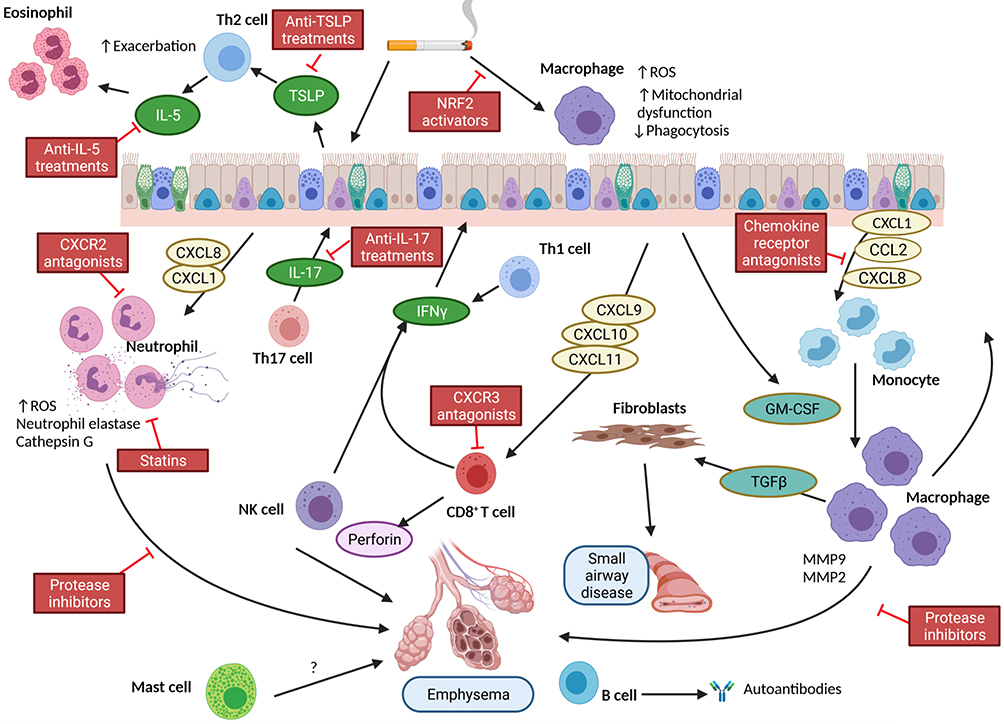 |
Figure 1 Simplified diagram of the role of leukocytes in the pathophysiology of COPD and respective sites of therapeutic interventions. Notes: Exposure of the airway to cigarette smoke leads to expression of cytokine and chemokines that recruit inflammatory cells into the lung. Most notably are neutrophils and monocytes/macrophages. These cells are activated and release proteases that contribute to emphysema. In addition, there is release of TGFβ that activates fibroblasts to synthesize collagen and contribute to small airways fibrosis. There is an additional contribution from Th1 and Th2 cells. The former together with Natural Killer (NK) cells produce IFNγ which stimulates the epithelium to produce CXCR3 ligands and recruit CD8+ cells. NK and CD8+ cells produce enzymes including perforin which drives apoptosis of alveolar cells contributing to emphysema. The role of mast cells is not clear, but their numbers increase in emphysema. Similarly, there is an increase in eosinophils in some patients at exacerbation. The role of increased B cells in more severe disease also remains unclear but may contribute to autoantibody expression. From this schematic, there are a number of points of potential therapeutic interventions highlighted in red boxes. Created with Biorender.com. |
Implications for Therapy
The presence of increased numbers of inflammatory cells in the lung offers a potential mechanism for intervention to return these numbers to those seen in a healthy lung (Figure 1). In addition, the activation of leukocytes in COPD is also a potential target, such that even if there are increased numbers, they are more of a phenotype that supports resolution of inflammation rather than propagating damage. At present, much of therapy for treatment of COPD addresses symptoms, and patients are offered bronchodilators including β2–agonists and muscarinic receptor antagonist as well as anti-inflammatory agents including glucocorticosteroids; however, there are no therapies that address disease progression.
Current Therapies
The effect of bronchodilators on leukocytes in COPD has been examined, in particular the effect of β2-adrenoceptor agonists as anti-inflammatory properties.112 Long-acting β2-adrenoceptor agonists are anti-inflammatory in monocyte-derived macrophages via increased cAMP,113 but their effects have been reported to occur, in part, due to an up-regulation of the glucocorticoid receptor in a number of cell types including sputum macrophages from patients with COPD.114 Furthermore, this interaction may also support prolonged neutrophil survival in COPD.115 More recently, a novel bifunctional anti-muscarinic-β2-adrenoceptor agonist, AZD8999, reduced lipopolysaccharide-induced inflammatory mediator release from neutrophils from patients with COPD and enhanced the effect of the glucocorticosteroid, fluticasone propionate.116
Roflumilast is a phosphodiesterase (PDE)-4 inhibitor that has been shown to have efficacy in COPD patients with chronic bronchitis, leading to reduced exacerbations and sputum neutrophilia.117 It appears to exert effects on leukocytes directly and is reported to improve the phagocytic response of circulating granulocytes in patients with COPD.118 This effect appears to be mostly in neutrophils, and roflumilast has also been shown to modify other neutrophil functions in COPD. The enhanced neutrophil chemotactic response seen in this disease could be attenuated when the cells were incubated in the presence of roflumilast-N-oxide.36 This effect was mediated by inhibition of phosphodiesterase (PDE)-4 and appeared to act via elevation of cAMP and activation of exchange protein directly activated by cAMP (ePAC)1, suggesting that this could be a novel mechanism to reduce neutrophilic inflammation and that ePAC1 activators could be used to avoid the unwanted side effects that are associated with PDE4 inhibition. In addition, roflumilast has also been shown to reverse glucocorticosteroid insensitivity in COPD neutrophils, although whether this occurs via a similar mechanism is not known.119
The effects of current therapies on defective phagocytosis of macrophages has been addressed. However, using data from in vitro studies, glucocorticosteroids, muscarinic receptor antagonists or β2-adrenoceptor agonists have been shown to have no effect on phagocytosis.1,18,120 There is evidence suggesting that macrolide antibiotics such as azithromycin can improve both efferocytosis and phagocytosis in COPD macrophages,121–124 but others have not seen this effect.18 Furthermore, there is evidence that glucocorticosteroids in the presence of TLR ligands induce expression of TLR on alveolar macrophages,125 which could lead to an enhanced response of these cells to TLR ligands which would be abundant when patients were colonized with bacteria.
Glucocorticosteroids are often prescribed to COPD patients particularly during exacerbation.126 However, inflammatory mediator release from macrophages are glucocorticosteroid-insensitive in COPD,11,27,127 and this is associated with reduced expression of histone deacetylase (HDAC)2 in these cells.127 There is no effect of glucocorticosteroids on phagocytic function in macrophages,120 although others report an improvement in efferocytosis.128 Glucocorticosteroid resistance is not limited to macrophages in COPD, but is also observed in CD8+ CD28 null NKT-like pro-inflammatory cytotoxic cells, although the mechanism underlying this remains unknown.129
Drug Development
Chemokine Receptor Antagonists
The presence of increased numbers of leukocytes in the lungs of COPD patients strongly suggests that there is an infiltration of many of these cells from the circulation. Cell migration is complex but is driven primarily by the process of chemotaxis. This suggests that suppression of chemotaxis could be one option to reduce this leukocyte influx. To this end, there have been several studies aimed at inhibiting specific chemokine receptors to reduce cell recruitment. An attractive target has been CXCR2,130 which is expressed highly on neutrophils but also monocytes and has been shown to drive enhanced chemotaxis of these cells in COPD.28,37 Several pharmaceutical companies have developed molecules to block this receptor and have performed clinical trials in COPD patients. One of the earliest investigated in patients was navarixin (MK723/SCH 571213). In a trial of COPD patients, sputum neutrophilia was reduced, but the effects on lung function were only seen in current smoking patients and not in those who no longer smoked.131 Other CXCR2 antagonists have been investigated, including AZD5069 which appeared to be safe132 but reduced circulating neutrophils.133 More recently, there have been reports of another CXCR2 antagonist, danirixin (GSK1325756). A trial in 93 COPD patients initially suggested some benefit to reduce symptoms but could not see any difference between current smokers and non-smokers.134 However, further investigation of this drug demonstrated an increase in the incidence of exacerbations in patients with chronic mucus secretion.135 These observations have now led to the suggestion that such therapies targeting neutrophils will be of benefit in a subset of patients,136 but identification of these is not clear.
Other chemokine receptor antagonists have been examined in patients with COPD, including AZD2423, a CCR2 antagonist that would inhibit monocyte recruitment to the lung. To date, the only study in COPD patients has been to examine safety, and no effect on clinical outcomes has been reported.137 CXCR3 appears to be a promising target in COPD as it would reduce the increased migratory response of monocytes29 as well as suppressing CD8+ cells, but although such drugs exist, such as AMG487,138 to date there have been no trials in COPD.
Statins
Statins, or 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors, are mainly used to lower cholesterol but may be beneficial during periods of infection. This is because statins may have anti-inflammatory properties due to their ability to disrupt protein–protein interactions and prevent anchoring of signaling molecules to the cell membrane.139 Little is known about the role of statins in COPD, although many patients take statins due to the high prevalence of comorbidities in these patients. However, altered neutrophil migratory function and increased NET release is a key phenotype of COPD neutrophils and can lead to disease pathology. In vitro studies suggest that treatment with the statin simvastatin improves neutrophil migratory accuracy of COPD neutrophils.140 Simvastatin has also been shown to improve chemotaxis and reduce NETosis in a pilot randomized controlled trial in sepsis patients, suggesting that there are implications in treating COPD patients.141 Statins can also have anti-inflammatory effects on macrophages, reducing MMP-9 activity142 and release of inflammatory mediators.143 These data suggest that statin treatment of COPD patients may improve the function and reduce the inflammation by the two main phagocytes found within the COPD lung.
Biologics
Anti-IL-5
Blood eosinophil numbers, and the role they play in COPD, is an area of much contention, with eosinophil counts being assessed as markers of patients who frequently exacerbate and their responsiveness to corticosteroid treatment.144,145 However, patients with eosinophilic COPD have increased IL-5 concentrations in their sputum, suggesting that targeting IL-5 may reduce eosinophilic inflammation and may be beneficial in these selected patients with COPD.31 Currently there are three anti-IL-5 therapies available: mepolizumab, benralizumab and reslizumab. Mepolizumab is an anti-interleukin-5 (anti-IL-5) monoclonal antibody, whereas benralizumab and reslizumab target and block the IL-5 receptor. Trials of anti-IL-5 therapy have been disappointing in COPD. The phase 3, METREX trial of mepolizumab in COPD patients did show a reduced annual rate exacerbation compared to placebo in patients with COPD with an eosinophilic phenotype, cutting them by 18% compared to placebo. However, in a second study, METREO, mepolizumab failed to meet its primary outcome.146 Also, two clinical trials of benralizumab, named the GALATHEA and TERRANOVA studies, were unable to show a benefit compared to placebo in reducing exacerbations.147 These findings appear inconclusive for the effectiveness of these treatments in COPD and may be due to the definition of patients with eosinophilic COPD and the numbers of eosinophils a patient may need for these drugs to be effective.
Anti-IL-17
Targeting IL-17 in COPD has not been well studied, but mechanistic data suggest this to be a potential therapeutic avenue. Secukinumab, a monoclonal antibody against IL-17A, and brodalumab, a monoclonal antibody against IL-17 receptor A, have been tested in asthma, but not in patients with COPD. A phase 2 clinical trial of an IL-17A-neutralizing antibody, CNTO 6785, has been tested in COPD patients but failed to meet the primary endpoints.148 There are also concerns that targeting the IL-17 pathway may result in further exacerbation of bacterial infection in COPD. However, in a AECOPD mouse model, mice treated with an IL-17-neutralizing antibody were protected against enhanced pulmonary neutrophilia.149 Targeting IL-17 in COPD may therefore be a novel therapeutic target, with further clinical trials and the use of precision medicine, potentially by targeting well characterized neutrophilic patients and potentially using nasal sampling to detect patients with high IL-17 levels, to identify COPD patients who may respond well to these drugs and benefit.
Anti-TSLP
Thymic stromal lymphopoietin (TSLP) is a member of the IL-17 family and an alarmin released from epithelial cells. TSLP can promote the differentiation of Th2 T cells and release of chemokine which can attract them. TSLP is seen to be elevated in the BAL of COPD patients compared to non-smokers and may therefore be a target in COPD.150 Currently a phase 2 study, utilizing tezepelumab, a blocking antibody to TSLP, is underway and is looking at the effects of this drug during exacerbations in patients with moderate to severe COPD and are receiving triple inhaled maintenance therapy, and have had two or more exacerbations in the 12 months prior to visit 1.151 There are numerous other biologics in development to target specific inflammatory mediators, and many of these are under investigation for COPD. These include antibodies targeting IL-6, IL-18, IL-23, IL-33 and granulocyte-macrophage colony-stimulating factor (GM-CSF).
Other Anti-Inflammatory Pathway Inhibitors
Several p38 inhibitors have been used in clinical trials in COPD, although none have shown long-term efficacy. A novel p38 kinase inhibitor, RV568, has recently been shown to reduce inflammation in COPD and reduce neutrophil counts in an LPS-treated mouse model. The drug was shown to have synergistic effects when used in combination with corticosteroids. A small clinical trial of this compound showed a small improvement in pre-bronchodilator FEV1 after 14 days of treatment.152 Losmapimod (GW856553) is an oral p38 inhibitor; a six-month trial failed to see any effect on exercise tolerance or lung function,153 and a shorter study (12 weeks) failed to see any change in sputum neutrophils but did report reduced circulating fibrinogen.154 However, when patients were stratified based on eosinophil count, those with <2% eosinophils did show some efficacy with losmapimod with respect to time to next exacerbation,155 suggesting that stratifying patients based on eosinophil count might be one way of identifying responsive patients. AZD7624 is an inhaled p38 inhibitor; in a trial to prevent exacerbations in patients with COPD, it was not effective in increasing days to first exacerbation,156 but there was a reduction in sputum neutrophils in these patients, demonstrating an effect on inflammatory indices. The effect of acumapimod (BCT197) was recently evaluated in acute exacerbations of COPD but did not improve outcomes, and no measurements of changes in leukocyte accumulation were reported.157 Overall, the effects of p38 inhibitors in clinical trials for COPD have not been successful, and alternative strategies are needed.
Other kinase pathways under investigation include the PI3kinase pathway and at present CHF6523, an inhaled drug, is currently undergoing safety and tolerability tests in COPD patients.158 Nemiralisib (GSK2269557) is an inhibitor of PI3Kδ and is an inhaled drug that did not show improvement in patients with asthma159 but is also currently under investigation with safety studies in smokers160 and COPD patients.161
There has been little investigation into restoring defective innate immune responses in COPD. However, sulforaphane has been shown to improve phagocytosis by COPD macrophages162,163 acting via Nrf2, although a four-week clinical trial of this drug in patients with COPD failed to show an improvement in expression of Nrf2 or any clinical parameters.164 This may be due to the lack of stability of the compound, therefore discovery of a more stable analogue of sulforaphane may allow further studies.165
Although we are now learning more about the inflammatory profile observed in the lungs of patients with COPD, the role of increased leukocyte accumulation is still not fully understood. Environmental changes to cellular activation states or phenotypes may derive from structural cells or exposure to inhaled pollutants such as cigarette smoke. Further understanding is needed to differentiate a “normal” physiological response to these insults compared to a more pathological response of leukocytes. Similarly, understanding as to whether there are epigenetic changes in precursor cells is also required. Nevertheless, targeting the changes in leukocyte populations in COPD with pharmacological interventions (Table 2) could offer a strategy to normalize the immune response in these patients and reduce disease progression and improve quality of life.
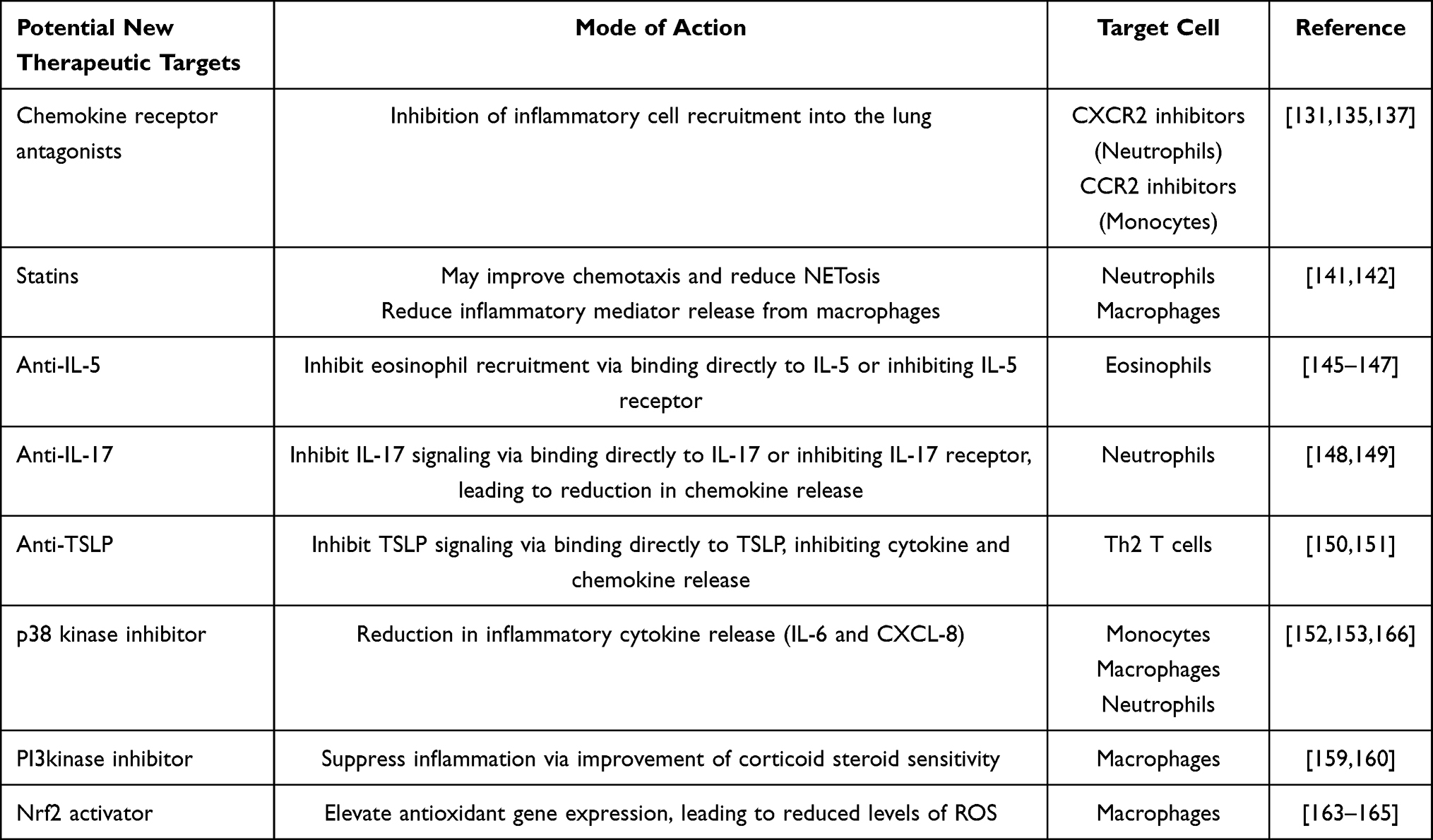 |
Table 2 Potential Therapeutic Interventions in COPD and Their Leukocyte Target |
Disclosure
LED has received unrestricted research grants from AstraZeneca and Boehringer-Ingelheim, outside the submitted work. The authors report no conflicts of interest in this work.
References
1. Belchamber KBR, Donnelly LE. Macrophage dysfunction in respiratory disease. Results Probl Cell Differ. 2017;62:299–313.
2. Hill AT, Campbell EJ, Hill SL, Bayley DL, Stockley RA. Association between airway bacterial load and markers of airway inflammation in patients with stable chronic bronchitis. Am J Med. 2000;109(4):288–295.
3. Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57(10):847–852.
4. Cai Y, Sugimoto C, Arainga M, Alvarez X, Didier ES, Kuroda MJ. In vivo characterization of alveolar and interstitial lung macrophages in rhesus macaques: implications for understanding lung disease in humans. J Immunol. 2014;192(6):2821–2829.
5. Perez-Rial S, Del Puerto-Nevado L, Terron-Exposito R, Giron-Martinez A, Gonzalez-Mangado N, Peces-Barba G. Role Oo recently migrated monocytes in cigarette smoke-induced lung inflammation in different strains of mice. PLoS One. 2013;8(9):e72975.
6. Evren E, Ringqvist E, Tripathi KP, et al. Distinct developmental pathways from blood monocytes generate human lung macrophage diversity. Immunity. 2020;54(2):259–275
7. Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–551.
8. Tan SY, Krasnow MA. Developmental origin of lung macrophage diversity. Development. 2016;143(8):1318–1327.
9. Tomita K, Caramori G, Lim S, et al. Increased p21(CIP1/WAF1) and B cell lymphoma leukemia-x(L) expression and reduced apoptosis in alveolar macrophages from smokers. Am J Respir Crit Care Med. 2002;166(5):724–731.
10. Kulikauskaite J, Wack A. Teaching old dogs new tricks? The plasticity of lung alveolar macrophage subsets. Trends Immunol. 2020;41(10):864–877.
11. Culpitt SV, Rogers DF, Shah P, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167(1):24–31.
12. Russell RE, Culpitt SV, DeMatos C, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26(5):602–609.
13. Davies LC, Rice CM, Palmieri EM, Taylor PR, Kuhns DB, McVicar DW. Peritoneal tissue-resident macrophages are metabolically poised to engage microbes using tissue-niche fuels. Nat Commun. 2017;8(1):2074.
14. Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol. 2016;100(3):481–489.
15. Finlay GA, O’Driscoll LR, Russell KJ, et al. Matrix metalloproteinase expression and production by alveolar macrophages in emphysema. Am J Respir Crit Care Med. 1997;156(1):240–247.
16. Russell RE, Thorley A, Culpitt SV, et al. Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol. 2002;283(4):L867–L873.
17. Berenson CS, Garlipp MA, Grove LJ, Maloney J, Sethi S. Impaired phagocytosis of nontypeable haemophilus influenzae by human alveolar macrophages in chronic obstructive pulmonary disease. J Infect Dis. 2006;194(10):1375–1384.
18. Taylor AE, Finney-Hayward TK, Quint JK, et al. Defective macrophage phagocytosis of bacteria in COPD. Eur Respir J. 2010;35(5):1039–1047.
19. Bewley MA, Belchamber KB, Chana KK, et al. Differential effects of p38, MAPK, PI3K or Rho Kinase Inhibitors on bacterial phagocytosis and efferocytosis by macrophages in COPD. PLoS One. 2016;11(9):e0163139.
20. Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81(4):289–296.
21. Wrench C, Belchamber KBR, Bercusson A, et al. Reduced clearance of fungal spores by chronic obstructive pulmonary disease GM-CSF- and M-CSF-derived macrophages. Am J Respir Cell Mol Biol. 2018;58(2):271–273.
22. Birrell MA, Wong S, Catley MC, Belvisi MG. Impact of tobacco-smoke on key signaling pathways in the innate immune response in lung macrophages. J Cell Physiol. 2008;214(1):27–37.
23. Oliver BG, Lim S, Wark P, et al. Rhinovirus exposure impairs immune responses to bacterial products in human alveolar macrophages. Thorax. 2008;63(6):519–525.
24. Finney LJ, Belchamber KBR, Fenwick PS, et al. Human rhinovirus impairs the innate immune response to bacteria in alveolar macrophages in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2019;199(12):1496–1507.
25. Belchamber KBR, Singh R, Batista CM, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. 2019;54:4.
26. Bewley MA, Preston JA, Mohasin M, et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am J Respir Crit Care Med. 2017;196(7):845–855.
27. Chana KK, Fenwick P, Nicholson AG, Barnes PJ, Donnelly LE. Identification of a distinct glucocorticoid-insensitive pulmonary macrophage phenotype in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2014;133:1.
28. Traves SL, Smith SJ, Barnes PJ, Donnelly LE. Specific CXC but not CC chemokines cause elevated monocyte migration in COPD: a role for CXCR2. J Leukoc Biol. 2004;76:441–450.
29. Costa C, Traves SL, Tudhope SJ, et al. Enhanced monocyte migration to CXCR3 and CCR5 chemokines in COPD. Eur Respir J. 2016;47(4):1093–1102.
30. Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5(14):1317–1327.
31. Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74(7):1249–1256.
32. Patel AA, Ginhoux F, Yona S. Monocytes, macrophages, dendritic cells and neutrophils: an update on lifespan kinetics in health and disease. Immunology. 2021;163(3):250–261.
33. Sheshachalam A, Srivastava N, Mitchell T, Lacy P, Eitzen G. Granule protein processing and regulated secretion in neutrophils. Front Immunol. 2014;5:448.
34. Metzemaekers M, Gouwy M, Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords. Cell Mol Immunol. 2020;17(5):433–450.
35. Hughes MJ, Sapey E, Stockley R. Neutrophil phenotypes in chronic lung disease. Expert Rev Respir Med. 2019;13(10):951–967.
36. Dunne AE, Kawamatawong T, Fenwick PS, et al. Direct inhibitory effect of the PDE4 inhibitor roflumilast on neutrophil migration in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2019;60(4):445–453.
37. Burnett D, Chamba A, Hill SL, Stockley RA. Neutrophils from subjects with chronic obstructive lung disease show enhanced chemotaxis and extracellular proteolysis. Lancet. 1987;2(8567):1043–1046.
38. Sapey E, Stockley JA, Greenwood H, et al. Behavioral and structural differences in migrating peripheral neutrophils from patients with COPD. Am J Respir Crit Care Med. 2011;183(9):176–186.
39. Butler A, Walton GM, Sapey E. Neutrophilic inflammation in the pathogenesis of chronic obstructive pulmonary disease. COPD. 2018;15(4):392–404.
40. Shanmugam L, Ravinder S, Johnson P, Padmavathi R, Rajagopalan B, Kindo A. Assessment of phagocytic activity of neutrophils in chronic obstructive pulmonary disease. Lung India. 2015;32(5):437–440.
41. Uddin M, Watz H, Malmgren A, Pedersen F. NETopathic inflammation in chronic obstructive pulmonary disease and severe asthma. Front Immunol. 2019;10:47.
42. Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med. 1996;153(2):530–534.
43. Traves SL, Culpitt SV, Russell RE, Barnes PJ, Donnelly LE. Increased levels of the chemokines GROα and MCP-1 in sputum samples from patients with COPD. Thorax. 2002;57(7):590–595.
44. Mori Y, Iwasaki H, Kohno K, et al. Identification of the human eosinophil lineage-committed progenitor: revision of phenotypic definition of the human common myeloid progenitor. J Exp Med. 2009;206(1):183–193.
45. Olsson A, Salomonis N, Singh H, Grimes HL. Identification of the origin of eosinophils. Blood. 2015;126(23):886.
46. Vedel-Krogh S. The search for the “healthy” blood eosinophil count. Eur Respir J. 2020;55(5):2000473.
47. Pavord ID. Eosinophilic phenotypes of airway disease. Ann Am Thorac Soc. 2013;10(Suppl):S143–149.
48. Thompson-Souza GA, Gropillo I, Neves JS. Cysteinyl leukotrienes in eosinophil biology: functional roles and therapeutic perspectives in eosinophilic disorders. Front Med (Lausanne). 2017;4:106.
49. Leigh R, Pizzichini MMM, Morris MM, Maltais F, Hargreave FE, Pizzichini E. Stable COPD: predicting benefit from high-dose inhaled corticosteroid treatment. Eur Respir J. 2006;27(5):964–971.
50. Felton JM, Lucas CD, Rossi AG, Dransfield I. Eosinophils in the lung – modulating apoptosis and efferocytosis in airway inflammation. Front Immunol. 2014;5:302.
51. Jeffery PK. Comparison of the structural and inflammatory features of COPD and asthma. Giles F. Filley Lecture. Chest. 2000;117(5Suppl 1):251S–260S.
52. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. 2020;76(2):188–195.
53. Bafadhel M, McKenna S, Terry S, et al. Blood eosinophils to direct corticosteroid treatment of exacerbations of chronic obstructive pulmonary disease: a randomized placebo-controlled trial. Am J Respir Crit Care Med. 2012;186(1):48–55.
54. Tashkin DP, Wechsler ME. Role of eosinophils in airway inflammation of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2018;13:335–349.
55. Littner MR, Ilowite JS, Tashkin DP, et al. Long-acting bronchodilation with once-daily dosing of tiotropium (Spiriva) in stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1136–1142.
56. Bafadhel M, McKenna S, Terry S, et al. Acute exacerbations of chronic obstructive pulmonary disease identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184(6):662–671.
57. Méndez-Enríquez E, Hallgren J. Mast cells and their progenitors in allergic asthma. Front Immunol. 2019;10:821.
58. Födinger M, Fritsch G, Winkler K, et al. Origin of human mast cells: development from transplanted hematopoietic stem cells after allogeneic bone marrow transplantation. Blood. 1994;84(9):2954–2959.
59. Valent P, Akin C, Hartmann K, et al. Mast cells as a unique hematopoietic lineage and cell system: from Paul Ehrlich’s visions to precision medicine concepts. Theranostics. 2020;10(23):10743–10768.
60. Sonoda T, Hayashi C, Kitamura Y. Presence of mast cell precursors in the yolk sac of mice. Dev Biol. 1983;97(1):89–94.
61. Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A. 1986;83(12):4464–4468.
62. Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346(22):1699–1705.
63. Carroll NG, Mutavdzic S, James AL. Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur Respir J. 2002;19(5):879–885.
64. Raible DG, Schulman ES, DiMuzio J, Cardillo R, Post TJ. Mast cell mediators prostaglandin-D2 and histamine activate human eosinophils. J Immunol. 1992;148(11):3536–3542.
65. Murakami D, Yamada H, Yajima T, Masuda A, Komune S, Yoshikai Y. Lipopolysaccharide inhalation exacerbates allergic airway inflammation by activating mast cells and promoting Th2 responses. Clin Exp Allergy. 2007;37(3):339–347.
66. Beckett EL, Stevens RL, Jarnicki AG, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immun. 2013;131(3):752–762.
67. Andersson CK, Mori M, Bjermer L, Löfdahl C-G, Erjefält JS. Alterations in lung mast cell populations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(3):206–217.
68. Ballarin A, Bazzan E, Zenteno RH, et al. Mast cell infiltration discriminates between histopathological phenotypes of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(3):233–239.
69. López-Giraldo A, Cruz T, Molins L, et al. Characterization, localization and comparison of c-Kit+ lung cells in never smokers and smokers with and without COPD. BMC Pulm Med. 2018;18(1):123.
70. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol. 2012;12(2):136–148.
71. Kanhere A, Hertweck A, Bhatia U, et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nat Commun. 2012;3:1–2.
72. Hodge G, Nairn J, Holmes M, Reynolds PN, Hodge S. Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin Exp Immunol. 2007;150(1):22–29.
73. Barcelo B, Pons J, Ferrer JM, Sauleda J, Fuster A, Agusti AG. Phenotypic characterisation of T-lymphocytes in COPD: abnormal CD4+CD25+ regulatory T-lymphocyte response to tobacco smoking. Eur Respir J. 2008;31(3):555–562.
74. Saetta M, Mariani M, Panina-Bordignon P, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;165(10):1404–1409.
75. Di Stefano A, Caramori G, Capelli A, et al. STAT4 activation in smokers and patients with chronic obstructive pulmonary disease. Eur Respir J. 2004;24(1):78–85.
76. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. 2012;3:51.
77. Smyth LJ, Starkey C, Vestbo J, Singh D. CD4-regulatory cells in COPD patients. Chest. 2007;132(1):156–163.
78. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113.
79. Le Rouzic O, Pichavant M, Frealle E, Guillon A, Si-Tahar M, Gosset P. Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur Respir J. 2017;50:4.
80. Di Stefano A, Caramori G, Gnemmi I, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157(2):316–324.
81. Prause O, Bozinovski S, Anderson GP, Linden A. Increased matrix metalloproteinase-9 concentration and activity after stimulation with interleukin-17 in mouse airways. Thorax. 2004;59(4):313–317.
82. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35(2):161–168.
83. Suzuki I, Martin S, Boursalian TE, Beers C, Fink PJ. Fas ligand costimulates the in vivo proliferation of CD8+ T cells. J Immunol. 2000;165(10):5537–5543.
84. Schmidt ME, Varga SM. The CD8 T cell response to respiratory virus infections. Front Immunol. 2018;9:678.
85. Saetta M, Di Stefano A, Turato G, et al. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(3):822–826.
86. Barnes PJ. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immun. 2016;138(1):16–27.
87. Roos-Engstrand E, Ekstrand-Hammarstrom B, Pourazar J, Behndig AF, Bucht A, Blomberg A. Influence of smoking cessation on airway T lymphocyte subsets in COPD. Copd. 2009;6(2):112–120.
88. Wang J, Urbanowicz RA, Tighe PJ, Todd I, Corne JM, Fairclough LC. Differential activation of killer cells in the circulation and the lung: a study of current smoking status and chronic obstructive pulmonary disease (COPD). PLoS One. 2013;8(3):e58556.
89. Freeman CM, Curtis JL, Chensue SW. CC chemokine receptor 5 and CXC chemokine receptor 6 expression by lung CD8+ cells correlates with chronic obstructive pulmonary disease severity. Am J Pathol. 2007;171(3):767–776.
90. Williams M, Todd I, Fairclough LC. The role of CD8 + T lymphocytes in chronic obstructive pulmonary disease: a systematic review. Inflamm Res. 2021;70(1):11–18.
91. Hodge G, Mukaro V, Reynolds PN, Hodge S. Role of increased CD8/CD28(null) T cells and alternative co-stimulatory molecules in chronic obstructive pulmonary disease. Clin Exp Immunol. 2011;166(1):94–102.
92. Kagi D, Ledermann B, Burki K, Zinkernagel RM, Hengartner H. Molecular mechanisms of lymphocyte-mediated cytotoxicity and their role in immunological protection and pathogenesis in vivo. Annu Rev Immunol. 1996;14:207–232.
93. De Cunto G, Lunghi B, Bartalesi B, et al. Severe reduction in number and function of peripheral T cells does not afford protection toward emphysema and bronchial remodeling induced in mice by cigarette smoke. Am J Pathol. 2016;186(7):1814–1824.
94. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653.
95. Gosman MM, Willemse BW, Jansen DF, et al. Increased number of B-cells in bronchial biopsies in COPD. Eur Respir J. 2006;27(1):60–64.
96. van der Strate BW, Postma DS, Brandsma CA, et al. Cigarette smoke-induced emphysema: a role for the B cell? Am J Respir Crit Care Med. 2006;173(7):751–758.
97. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, et al. Mechanisms of autoantibody-induced pathology. Front Immunol. 2017;8:603.
98. Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;13(5):567–569.
99. Nunez B, Sauleda J, Anto JM, et al. Anti-tissue antibodies are related to lung function in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;183(8):1025–1031.
100. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510.
101. Culley FJ. Natural killer cells in infection and inflammation of the lung. Immunology. 2009;128(2):151–163.
102. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633–640.
103. Huntington ND. The unconventional expression of IL-15 and its role in NK cell homeostasis. Immunol Cell Biol. 2014;92(3):210–213.
104. Freeman CM, Stolberg VR, Crudgington S, et al. Human CD56+ cytotoxic lung lymphocytes kill autologous lung cells in chronic obstructive pulmonary disease. PLoS One. 2014;9(7):e103840.
105. Robinson BW, Pinkston P, Crystal RG. Natural killer cells are present in the normal human lung but are functionally impotent. J Clin Invest. 1984;74(3):942–950.
106. Takeuchi M, Nagai S, Nakajima A, et al. Inhibition of lung natural killer cell activity by smoking: the role of alveolar macrophages. Respiration. 2001;68(3):262–267.
107. Hodge G, Mukaro V, Holmes M, Reynolds PN, Hodge S. Enhanced cytotoxic function of natural killer and natural killer T-like cells associated with decreased CD94 (Kp43) in the chronic obstructive pulmonary disease airway. Respirology. 2013;18(2):369–376.
108. Urbanowicz RA, Lamb JR, Todd I, Corne JM, Fairclough LC. Enhanced effector function of cytotoxic cells in the induced sputum of COPD patients. Respir Res. 2010;11:76.
109. Finch DK, Stolberg VR, Ferguson J, et al. Lung dendritic cells drive natural killer cytotoxicity in chronic obstructive pulmonary disease via IL-15Ralpha. Am J Respir Crit Care Med. 2018;198(9):1140–1150.
110. Hume P, Gibbings S, Jakubzick CV, et al. Localization of macrophages in the human lung via design-based stereology. Am J Respir Crit Care Med. 2020;201.
111. De Cunto G, Cavarra E, Bartalesi B, Lungarella G, Lucattelli M. Alveolar macrophage phenotype and compartmentalization drive different pulmonary changes in mouse strains exposed to cigarette smoke. COPD. 2020;17(4):429–443.
112. Theron AJ, Steel HC, Tintinger GR, Feldman C, Anderson R. Can the anti-inflammatory activities of β2-agonists be harnessed in the clinical setting? Drug Des Devel Ther. 2013;7:1387–1398.
113. Donnelly LE, Tudhope SJ, Fenwick PS, Barnes PJ. Effects of formoterol and salmeterol on cytokine release from monocyte-derived macrophages. Eur Respir J. 2010;36(1):178–186.
114. Haque R, Hakim A, Moodley T, et al. Inhaled long-acting β2 agonists enhance glucocorticoid receptor nuclear translocation and efficacy in sputum macrophages in COPD. J Allergy Clin Immun. 2013;132(5):1166–1173.
115. Perttunen H, Moilanen E, Zhang X, Barnes PJ, Kankaanranta H. Beta2-agonists potentiate corticosteroid-induced neutrophil survival. Copd. 2008;5(3):163–169.
116. Milara J, Contreras S, de Diego A, et al. In vitro anti-inflammatory effects of AZD8999, a novel bifunctional muscarinic acetylcholine receptor antagonist /β2-adrenoceptor agonist (MABA) compound in neutrophils from COPD patients. PLoS One. 2019;14(1):e0210188.
117. Calverley P, Rabe K, Goehring U, Kristiansen S, Fabbri L, Martinez F. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009;374:685–694.
118. Porpodis K, Domvri K, Zarogoulidis P, et al. Roflumilast, a phosphodiesterase-4 inhibitor, induces phagocytic activity in Greek COPD patients. Int J Chron Obstruct Pulmon Dis. 2015;10:1123–1128.
119. Milara J, Lluch J, Almudever P, Freire J, Xiaozhong Q, Cortijo J. Roflumilast N-oxide reverses corticosteroid resistance in neutrophils from patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2014;134(2):314–322.e319.
120. Belchamber KB, Thomas CM, Dunne AE, Barnes PJ, Donnelly LE. Comparison of fluticasone propionate and budesonide on COPD macrophage and neutrophil function. Int J Chron Obstruct Pulmon Dis. 2018;13:2883–2897.
121. Hodge S, Hodge G, Brozyna S, Jersmann H, Holmes M, Reynolds PN. Azithromycin increases phagocytosis of apoptotic bronchial epithelial cells by alveolar macrophages. Eur Respir J. 2006;28(3):486–495.
122. Hodge S, Hodge G, Jersmann H, et al. Azithromycin improves macrophage phagocytic function and expression of mannose receptor in COPD. Am J Respir Crit Care Med. 2008;178(2):139–148.
123. Hodge S, Reynolds PN. Low-dose azithromycin improves phagocytosis of bacteria by both alveolar and monocyte-derived macrophages in chronic obstructive pulmonary disease subjects. Respirology. 2012;17(5):802–807.
124. Hodge S, Tran HB, Hamon R, et al. Nonantibiotic macrolides restore airway macrophage phagocytic function with potential anti-inflammatory effects in chronic lung diseases. Am J Physiol-Lung C. 2017;312(5):L678–L687.
125. Ji J, von Schéele I, Billing B, et al. Effects of budesonide on toll-like receptor expression in alveolar macrophages from smokers with and without COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:1035–1043.
126. GOLD. Executive summary: global strategy for the diagnosis, management, and prevention of COPD; 2020. Available from: http://www.goldcopd.com/Guidelineitem.asp?l1=2&l2=1&intId=996.
127. Cosio BG, Tsaprouni L, Ito K, Jazrawi E, Adcock IM, Barnes PJ. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med. 2004;200(5):689–695.
128. Higham A, Scott T, Li J, et al. Effects of corticosteroids on COPD lung macrophage phenotype and function. Clin Sci (Lond). 2020;134(7):751–763.
129. Hodge G, Hodge S. Steroid resistant CD8(+)CD28(null) NKT-like pro-inflammatory cytotoxic cells in chronic obstructive pulmonary disease. Front Immunol. 2016;7:617.
130. Donnelly LE, Barnes PJ. Chemokine receptors as therapeutic targets in chronic obstructive pulmonary disease. Trends Pharmacol Sci. 2006;27(10):546–553.
131. Rennard SI, Dale DC, Donohue JF, et al. CXCR2 antagonist MK-7123. A Phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2015;191(9):1001–1011.
132. Kirsten AM, Förster K, Radeczky E, et al. The safety and tolerability of oral AZD5069, a selective CXCR2 antagonist, in patients with moderate-to-severe COPD. Pulm Pharmacol Ther. 2015;31:36–41.
133. Jurcevic S, Humfrey C, Uddin M, Warrington S, Larsson B, Keen C. The effect of a selective CXCR2 antagonist (AZD5069) on human blood neutrophil count and innate immune functions. Br J Clin Pharmacol. 2015;80(6):1324–1336.
134. Lazaar AL, Miller BE, Tabberer M, et al. Effect of the CXCR2 antagonist danirixin on symptoms and health status in COPD. Eur Respir J. 2018;52:4.
135. Lazaar AL, Miller BE, Donald AC, et al. CXCR2 antagonist for patients with chronic obstructive pulmonary disease with chronic mucus hypersecretion: a Phase 2b trial. Respir Res. 2020;21(1):149.
136. Keir HR, Richardson H, Fillmore C, et al. CXCL-8-dependent and -independent neutrophil activation in COPD: experiences from a pilot study of the CXCR2 antagonist danirixin. ERJ Open Res. 2020;6:4.
137. AZD2423 safety and tolerability study in patients with moderate and severe Chronic Obstructive Pulmonary Disease(COPD). Available from: https://ClinicalTrials.gov/show/NCT01215279.
138. Liu J, Fu Z, Li AR, et al. Optimization of a series of quinazolinone-derived antagonists of CXCR3. Bioorg Med Chem Lett. 2009;19(17):5114–5118.
139. Greenwood J, Steinman L, Zamvil SS. Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol. 2006;6(5):358–370.
140. Walton GM, Stockley JA, Griffiths D, Sadhra CS, Purvis T, Sapey E. Repurposing treatments to enhance innate immunity can statins improve neutrophil functions and clinical outcomes in COPD? J Clin Med. 2016;5:10.
141. Sapey E, Patel JM, Greenwood H, et al. Simvastatin improves neutrophil function and clinical outcomes in pneumonia. A pilot randomized controlled clinical trial. Am J Respir Crit Care Med. 2019;200(10):1282–1293.
142. Bellosta S, Via D, Canavesi M, et al. HMG-CoA reductase inhibitors reduce MMP-9 secretion by macrophages. Arterioscl Throm Vas. 1998;18(11):1671–1678.
143. Tuomisto TT, Lumivuori H, Kansanen E, et al. Simvastatin has an anti-inflammatory effect on macrophages via upregulation of an atheroprotective transcription factor, Kruppel-like factor 2. Cardiovasc Res. 2008;78(1):175–184.
144. Singh D, Kolsum U, Brightling CE, et al. Eosinophilic inflammation in COPD: prevalence and clinical characteristics. Eur Respir J. 2014;44(6):1697–1700.
145. Wedzicha JA. Eosinophils as biomarkers of chronic obstructive pulmonary disease exacerbation risk maybe just for some? Am J Respir Crit Care Med. 2016;193(9):937–938.
146. Pavord ID, Chanez P, Criner GJ, et al. Mepolizumab for eosinophilic chronic obstructive pulmonary disease. N Engl J Med. 2017;377(17):1613–1629.
147. Criner GJ, Celli BR, Brightling CE, et al. Benralizumab for the prevention of COPD exacerbations. N Engl J Med. 2019;381(11):1023–1034.
148. Eich A, Urban V, Jutel M, et al. A randomized, placebo-controlled Phase 2 trial of CNTO 6785 in Chronic Obstructive Pulmonary Disease. Copd. 2017;14(5):476–483.
149. Yanagisawa H, Hashimoto M, Minagawa S, et al. Role of IL-17A in murine models of COPD airway disease. Am J Physiol Lung Cell Mol Physiol. 2017;312(1):L122–L130.
150. Ying S, O’Connor B, Ratoff J, et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J Immunol. 2008;181(4):2790–2798.
151. Tezepelumab COPD exacerbation study. Available from: https://ClinicalTrials.gov/show/NCT04039113.
152. Charron CE, Russell P, Ito K, et al. RV568, a narrow-spectrum kinase inhibitor with p38 MAPK-alpha and -gamma selectivity, suppresses COPD inflammation. Eur Respir J. 2017;50:4.
153. Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(1):63–72.
154. Lomas DA, Lipson DA, Miller BE, et al. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012;52(3):416–424.
155. Marks-Konczalik J, Costa M, Robertson J, McKie E, Yang S, Pascoe S. A post-hoc subgroup analysis of data from a six month clinical trial comparing the efficacy and safety of losmapimod in moderate-severe COPD patients with ≤2% and >2% blood eosinophils. Respir Med. 2015;109(7):860–869.
156. Patel NR, Cunoosamy DM, Fagerås M, et al. The development of AZD7624 for prevention of exacerbations in COPD: a randomized controlled trial. Int J Chron Obstruct Pulmon Dis. 2018;13:1009–1019.
157. Strâmbu IR, Kobalava ZD, Magnusson BP, MacKinnon A, Parkin JM. Phase II study of single/repeated doses of acumapimod (BCT197) to treat acute exacerbations of COPD. Copd. 2019;16(5–6):344–353.
158. Investigate the Safety, Tolerability and Pharmacokinetics of CHF6523 in Healthy and in COPD Subjects. Available from: https://ClinicalTrials.gov/show/NCT04032535.
159. Khindri S, Cahn A, Begg M, et al. A multicentre, randomized, double-blind, placebo-controlled, crossover study to investigate the efficacy, safety, tolerability, and pharmacokinetics of repeat doses of inhaled nemiralisib in adults with persistent, uncontrolled asthma. J Pharmacol Exp Ther. 2018;367(3):405–413.
160. Begg M, Wilson R, Hamblin JN, et al. Relationship between pharmacokinetics and pharmacodynamic responses in healthy smokers informs a once-daily dosing regimen for nemiralisib. J Pharmacol Exp Ther. 2019;369(3):337–344.
161. Dose finding study of nemiralisib (GSK2269557) in subjects with an acute moderate or severe exacerbation of Chronic Obstructive Pulmonary Disease (COPD). Available from: https://ClinicalTrials.gov/show/NCT03345407.
162. Harvey CJ, Thimmulappa RK, Sethi S, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. 2011;3(78):78ra32.
163. Bewley MA, Budd RC, Ryan E, et al. Opsonic phagocytosis in chronic obstructive pulmonary disease is enhanced by Nrf2 agonists. Am J Respir Crit Care Med. 2018;198(6):739–750.
164. Wise RA, Holbrook JT, Criner G, et al. Lack of effect of oral sulforaphane administration on Nrf2 expression in COPD: a randomized, double-blind, placebo controlled trial. PLoS One. 2016;11(11):e0163716.
165. Boehm J, Davis R, Murar CE, et al. Discovery of a crystalline sulforaphane analog with good solid-state stability and engagement of the Nrf2 pathway in vitro and in vivo. Bioorg Med Chem. 2019;27(4):579–588.
166. Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med. 2011;32(4–6):234–246.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.