")
Back to Journals » Hepatic Medicine: Evidence and Research » Volume 11
“Let my liver rather heat with wine” - a review of hepatic fibrosis pathophysiology and emerging therapeutics
Authors Moscoso CG , Steer CJ
Received 25 April 2019
Accepted for publication 17 August 2019
Published 2 September 2019 Volume 2019:11 Pages 109—129
DOI https://doi.org/10.2147/HMER.S213397
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Carlos G Moscoso,1 Clifford J Steer1,2
1Department of Medicine, Division of Gastroenterology, Hepatology and Nutrition; 2Department of Genetics, Cell Biology and Development, University of Minnesota Medical School, Minneapolis, MN 55455, USA
Correspondence: Carlos G Moscoso; Clifford J Steer
Department of Medicine, Division of Gastroenterology, Hepatology and Nutrition, University of Minnesota Medical School, Phillips-Wangensteen Building, 516 Delaware Street, S.E., Minneapolis, MN 55455, USA
Tel +1 612 625 8999
Fax +1 612 625 5620
Email [email protected]
[email protected]
Abstract: Cirrhosis is characterized by extensive hepatic fibrosis, and it is the 14th leading cause of death worldwide. Numerous contributing conditions have been implicated in its development, including infectious etiologies, medication overdose or adverse effects, ingestible toxins, autoimmunity, hemochromatosis, Wilson’s disease and primary biliary cholangitis to list a few. It is associated with portal hypertension and its stigmata (varices, ascites, hepatic encephalopathy, combined coagulopathy and thrombophilia), and it is a major risk factor for hepatocellular carcinoma. Currently, orthotopic liver transplantation has been the only curative modality to treat cirrhosis, and the scarcity of donors results in many people waiting years for a transplant. Identification of novel targets for pharmacologic therapy through elucidation of key mechanistic components to induce fibrosis reversal is the subject of intense research. Development of robust models of hepatic fibrosis to faithfully characterize the interplay between activated hepatic stellate cells (the principal fibrogenic contributor to fibrosis initiation and perpetuation), hepatocytes and extracellular matrix components has the potential to identify critical components and mechanisms that can be exploited for targeted treatment. In this review, we will highlight key cellular pathways involved in the pathophysiology of fibrosis from extracellular ligands, effectors and receptors, to nuclear receptors, epigenetic mechanisms, energy homeostasis and cytokines. Further, molecular pathways of hepatic stellate cell deactivation are discussed, including apoptosis, senescence and reversal or transdifferentiation to an inactivated state resembling quiescence. Lastly, clinical evidence of fibrosis reversal induced by biologics and small molecules is summarized, current compounds under clinical trials are described and efforts for treatment of hepatic fibrosis with mesenchymal stem cells are highlighted. An enhanced understanding of the rich tapestry of cellular processes identified in the initiation, perpetuation and resolution of hepatic fibrosis, driven principally through phenotypic switching of hepatic stellate cells, should lead to a breakthrough in potential therapeutic modalities.
Keywords: fibrosis, cirrhosis, hepatic stellate cell, reversal, transdifferentiation, senescence, apoptosis
Introduction
The role of the liver in human physiology has long fascinated physicians and philosophers. Interestingly, its regenerative properties were alluded to in the Greek myth of Prometheus and the Roman myth of Tityus, in which birds of prey feasted on the livers of the respective protagonists, only to have their livers regenerate overnight. In the 5th century BCE, Hippocrates noted associations between jaundice and behavioral anomalies in his “Prognostics and Prorrhetics of Hippocrates.”1 The Roman physician Galen saw the liver as the predominant organ from which all other organs originated, conjecturing that it was “the principal instrument of sanguification,” the site from which blood was derived.2 Indeed, given the contemporaneous theory of four bodily humors, the liver, spleen and gallbladder were thought to be the sites of production and storage of three of the four humors. However, the liver was not uniformly ascertained to play such a principal role by all philosophers and physicians; Aristotle ascribed a central role to the heart,3 a view shared by the Islamic medical scholar Avicenna, who nonetheless noted that the liver was “the seat of the nutritive or vegetative faculties” and the “seat of manufacture of the dense part of the humors.”4
In 1685, an English surgeon named John Browne described a “liver appearing glandulous,” which is thought to be one of the first surgical descriptions of cirrhosis.5 The term cirrhosis was initially used by Rene Laennec in 1819 in his seminal work on auscultation after his invention of the stethoscope.6 The term was not mentioned in the medical literature again until William Osler used it as the eponym Laennec’s cirrhosis in his essential medical textbook “Principles and Practice of Medicine,” first published in 1901.7 The prolific and renowned British hepatologist Sheila Sherlock published the seminal “Diseases of the Liver and Biliary System” in 1955,8 along with over 600 articles on hepatic pathology, with momentous contributions to the field of hepatology, including the first serologic test for primary biliary cholangitis.9
Cirrhosis epidemiology, diagnosis and treatment
Estimates of the epidemiology of the various etiologies of cirrhosis vary significantly. Overall, the prevalence and incidence of cirrhosis is likely underestimated given its asymptomatic initial stages. Alcoholic liver disease is thought to account for almost half of liver-related deaths worldwide.10 Currently, alcohol use disorder affects nearly 1 in 6 Americans, an increase of ~50% between 2001 and 2012.11 Of the viral etiologies, there are approximately 2.2 million US adults with HBV,12,13 and more than 2.5 million US adults with HCV, a significant proportion of whom are unaware of their infection.12–15 The US prevalence of non-alcoholic fatty liver disease (NAFLD) has a broad range of between 29 and 113 million; of these, between 3.2 and 9.7 million have non-alcoholic steatohepatitis (NASH).16–18 Inherited metabolic disorders as etiologies of cirrhosis have a lower prevalence with α-1 antitrypsin (α1AT) deficiency present in 20 per 60,000 to 100,000 patients,19–21 hemochromatosis in 300 per 100,000 patients22–24 and Wilson’s disease occurring in 3 per 100,000 patients.25 Cholestatic etiologies such as primary sclerosing cholangitis and primary biliary cholangitis are prevalent in up to 40.2 per 100,000 patients.26 Autoimmune hepatitis is seen in 10 per 50,000 to 100,000 patients.27 The prevalence and/or incidence of vascular etiologies such as Budd-Chiari have not been determined.
The impact on the health care system is profound, as cirrhosis is the 14th leading cause of death in the world28 and the 10th leading cause of death in men,29 resulting in the yearly deaths of over 1 million persons globally, and over 33,000 persons in the US in 2015.30 Treatment and management of its complications cost ~$9.5 billion dollars in one 2018 study,31 of which $5.3 billion was due to alcoholic cirrhosis alone. Combined direct and indirect costs for care of cirrhosis and its complications exceed $12 billion in the US yearly.32 From 2006 to 2011, over 3 million emergency department visits in the US were due to cirrhosis and its complications,33 while in 2010, about 100,000 US inpatient hospitalizations were due to cirrhosis and chronic liver disease.34 Between 2002 and 2014, there were 1.24 million inpatient admissions for cirrhosis and its complications in the US, with an aggregate cost of $28 billion, rising to $2.8 billion in 2014 alone.35 Further, patients affected by cirrhosis tend to be in lower socioeconomic strata, have adverse effects on employment, and are negatively affected by medical expenditures.36 Additionally, these patients report low health-related quality of life.37
The initial suspicion for hepatic pathology is usually borne out of symptoms, signs, or during routine evaluation with a panel of liver function tests. Work-up for acute or chronic liver damage typically includes imaging, serologies to look for viral or autoimmune etiologies, evidence of ingestion, levels of ceruloplasmin and α1AT serologies, among other tests. There are several well-validated combinations of non-invasive markers of cirrhosis in clinical use. While the gold standard for diagnosis remains a liver biopsy, imaging modalities including ultrasound-based transient elastography have emerged as validated alternatives for diagnosis of cirrhosis.38 Staging of fibrosis, from stage I (early) to IV (previously thought to be irreversible cirrhosis) can be attained from either invasive or imaging-based non-invasive modalities to risk-stratify patients. The well-known model for end-stage liver disease (MELD) score39 relies on multiple serologic measurements to produce a score that is used as the principal ranking parameter on transplantation lists. Mortality is quite high when cirrhosis decompensates, as the portal hypertension driven by the diffuse fibrotic tissue in the hepatic parenchyma has multiple downstream repercussions that often involve other tissues such as lung (portopulmonary hypertension, hepatopulmonary syndrome), kidney (hepatorenal syndrome) and heart (cirrhotic cardiomyopathy, portopulmonary hypertension, hepatopulmonary syndrome), not to mention splenomegaly, portal vein and splenic vein thromboses, ascites, hepatic encephalopathy and varices. Sequelae of portal hypertension can be recognized on physical exam by presence of hepatosplenomegaly, spider angiomas, palmar erythema, Dupuytren’s contractures, and Terry’s nails.
Treatment of cirrhosis is complicated by its previously assumed irreversibility, which has been challenged by multiple in vivo and in vitro lines of evidence showing reversibility in various settings. Typically, cessation of the offending agent, if ingestion is the etiology, is of paramount importance. If of a viral etiology, treatment with antiviral therapy can result in cessation of progression, as outlined below. The only curative treatment for cirrhosis remains orthotopic liver transplantation. Mortality is estimated based on Child-Pugh and MELD scores. Child-Pugh provides information about life expectancy and perioperative mortality for abdominal surgeries,40 while MELD provides estimated 3-month mortality rates based on score ranges.39 Management of decompensated cirrhosis is largely aimed at prophylaxis and surveillance of variceal bleeding, mitigation of hepatic encephalopathy, therapeutic paracenteses for excessive ascites and prophylaxis of spontaneous bacterial peritonitis. Preventative strategies are limited to screening for esophageal varices by endoscopy and radiologic screening for hepatocellular carcinoma.
Pathophysiology of cirrhosis
A recent drive toward alternative treatment modalities for cirrhosis has focused on the pathophysiologic mechanisms undergirding the development and exacerbation of fibrosis. Much has been gleaned from study of isolated hepatocytes in addition to various in vivo systems of induced cirrhosis in mice and rats, using carbon tetrachloride (CCl4), bile duct ligation (BDL) and thioacetamide (TAA) treatment. Numerous studies in mice have examined accelerated or attenuated cirrhosis induced by CCl4 in transgenic mice with various modifications, including dietary components. Additionally, in vitro models of liver fibrosis, while challenging to develop, have nevertheless been bolstered recently by co-culture of human hepatocyte lines (HepaRG) with hepatic stellate cell-like cells derived from induced pluripotent stem cells (iPSC-HSCs).41 These “3D cultures” have recently been validated42 by the composition of cells within the culture, as well as by the response of constituent cells to various factors, including activation of hepatic stellate cells (HSCs) with subsequent fibrogenic activity. Such advancements in development of in vitro models of cirrhosis will yield further insights into the mechanisms of fibrosis reversal.
The liver parenchyma is primarily composed of epithelial hepatocytes, making up 60–80% of the total liver mass and arranged in lobules composed of a portal triad at the periphery and a central vein. Other cell types include Kupffer cells, which are tissue macrophages that reside in the lumen of sinusoids and are members of the reticuloendothelial system; HSCs of mesenchymal lineage that comprise 10–15% of the resident liver cells and which reside in the subendothelial space of Disse; liver sinusoidal endothelial cells with prominent fenestrae that line the sinusoids; bile ductular cells, also known as cholangiocytes, into which bile canaliculi empty; and the hepatic and portal vasculature.43 As the disease process advances to frank cirrhosis (Figure 1), the liver undergoes wide-spread fibrogenesis with formation of fibrotic septa surrounding areas of diffuse nodular regeneration. As these changes progress, they ultimately lead to parenchymal extinction with subsequent necroinflammation, as well as distortion and collapse of vascular architecture.44 There is resultant increased resistance to portal blood flow, resulting in portal hypertension and its stigmata, as well as decreased hepatic synthetic function. This process is principally driven by HSCs, which are activated or transdifferentiated into contractile, migratory, proliferative and fibrogenic myofibroblast-like cells that secrete copious extracellular matrix components into the space of Disse.
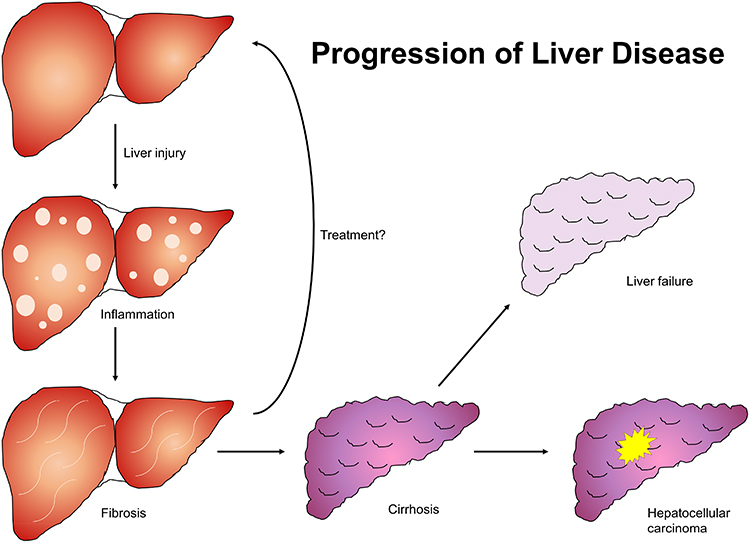 |
Figure 1 Natural progression of liver disease. Liver insult of any etiology results in inflammatory changes in hepatic parenchyma which progress to fibrosis and ultimately cirrhosis if unaddressed. Cirrhosis culminates in liver failure and is a principal risk factor for hepatocellular carcinoma. |
The pathophysiology of portal hypertension (recently reviewed by Gracia-Sancho and colleagues)45 can be mostly attributed to increased hepatic resistance, though splanchnic vasodilation and the formation of portosystemic collaterals including varices also contribute. Increased hepatic resistance to portal blood flow has two primary contributors, namely anatomic aberrations and functional abnormalities. As mentioned earlier, the altered anatomy of hepatic parenchyma accounts for about 70% of the hepatic vascular resistance, with loss of endothelial fenestration resulting in sinusoidal capillarization, as well as fibrogenesis and angiogenesis, leading to formation of intrahepatic shunts. Functional abnormalities in the form of endothelial dysfunction, increased response to vasoconstrictors and increased hepatic vascular tone account for about 30% of the total portal pressure. Splanchnic vasodilation is primarily an adaptive response to altered intrahepatic hemodynamics and opposes the increased hepatic vascular tone. The resultant increased blood flow into the portal system exacerbates portal pressure. Not infrequently, it can become so pronounced that in advanced cirrhosis it causes a hyperdynamic splanchnic and systemic circulation, contributing to ascites and to hepatorenal syndrome.46 Systemic vasodilation exacerbated by hyperdynamic circulation also leads to pulmonary ventilation/perfusion mismatch, which in advanced cases can lead to hepatopulmonary syndrome with arterial hypoxemia.47 In this setting, portopulmonary hypertension due to pulmonary vasoconstriction can also develop; the underlying pathophysiology is thought to be due to pulmonary vasculature endothelial dysfunction.48 Further, development of varices due to portosystemic collateral formation can lead to hematemesis in the case of esophageal varices, as well as portal hypertensive gastropathy and bleeding in the setting of dilated gastric mucosal vasculature.49 Hepatic encephalopathy can ensue via portosystemic collaterals shunting blood from the portal circulation into the systemic circulation.50 Lastly, intrahepatic shunting adversely impacts hepatocyte perfusion, a significant contributor to hepatic failure.
Evaluation of liver fibrosis and inflammation
The gold standard for evaluation of grade (degree of inflammation, corresponding to active liver injury) and stage (established fibrosis) of liver pathology is a liver biopsy. The most common histologic scoring systems to grade and stage liver pathology include METAVIR,51 the International Association for Study of the Liver (IASL),52 and Batts and Ludwig.53 Other systems, such as Ishak54 and Knodell,55 are more complex and are usually used in research settings, including in clinical trials. Liver biopsies can be obtained via percutaneous, transjugular and laparoscopic routes. Given their invasive nature, liver biopsies are not without complications; further, sampling error and interobserver variability can lead to incorrect staging of fibrosis. As such, there are various noninvasive modalities to predict fibrosis, including direct and indirect laboratory markers, and radiologic modalities. Direct laboratory markers include procollagens I, III and IV, matrix metalloproteinases (MMPs), cytokines and chemokines. The FIBROSpect II assay combines hyaluronic acid, tissue inhibitor of metalloproteinase-1 (TIMP-1) and α2-macroglobulin to predict fibrosis at stages F2-F4.56 There are various indirect markers of fibrosis, including platelet count, prothrombin time, albumin, total bilirubin, ALT and AST, as well as hyaluronic acid and α2-macroglobulin. Many algorithms exist that use these markers to predict fibrosis with varying degrees of sensitivity and specificity, including AST to platelet ratio index (APRI),57 FIB-4,58 FibroIndex,59 Forns Index,60 HepaScore,61 FibroTest and ActiTest,62 Lastly, radiologic modalities that can estimate fibrosis include liver ultrasound, transient elastography (FibroScan), shear wave elastography (used less often than transient elastography in the US) and magnetic resonance elastography.38
Cellular mechanisms of hepatic fibrosis
A common feature to the chronic fibro-proliferative disorders affecting liver, kidney and lung is the presence of the myofibroblast. These are cells of mesenchymal lineage which have contractile, proliferative, secretory and migratory properties. In liver, quiescent HSCs are activated by a variety of intracellular, microenvironment, extracellular and extrahepatic processes, resulting in transdifferentiation into myofibroblast-like cells that exhibit pro-fibrotic transcriptional and secretory properties. These activated HSCs secrete excess extracellular matrix (ECM) components into the space of Disse, resulting in their accumulation as well as loss of endothelial fenestration.
There are other potential sources of myofibroblasts, including bone marrow-derived fibrocytes and mesenchymal stem cells. Resident epithelial cells, including hepatocytes and cholangiocytes, are capable of differentiating into a mesenchymal phenotype by losing epithelial markers and gaining mesenchymal markers. This well-characterized process is known as epithelial-to-mesenchymal transition (EMT)63 and likely represents only a minor source of myofibroblasts. Similarly, endothelial cells are capable of a phenotypic switch to mesenchymal cells in a process known as endothelial-to-mesenchymal transition (EndMT),63 although the extent of contribution via this mechanism to the myofibroblast pool is unclear. Mesothelial cells, which make up ~15% of the resident liver cells and which express Wt1 (Wilms’ tumor 1), are another potential source of myofibroblasts through mesothelial-to-mesenchymal transition (MMT).64 In mice, chlorhexidine gluconate-induced liver fibrosis has been shown to trigger MMT.65 Animal studies have suggested that HSC transdifferentiation is the primary source of myofibroblasts involved in fibrogenesis. A cell fate tracing study in rats, in which HSCs were genetically labeled to express fluorescent Cre reporter proteins under the control of the lecithin-retinol acyltransferase (LRAT) promoter, found that 82–96% of the myofibroblasts originated from HSCs in CCl4, BDL and TAA models of cirrhosis.66 A murine study found that in CCl4-induced cirrhosis, HSCs were the predominant source of myofibroblasts, while in cholestatic BDL-induced cirrhosis, portal fibroblasts were the major source of myofibroblasts.67 The data thus far suggest that HSCs are the predominant source of myofibroblasts; however, these rodent studies have not yet been shown to recapitulate the human condition(s).
There are various mechanisms whereby HSCs become activated, then initiate and perpetuate hepatic fibrosis. A variety of extracellular and intracellular events contribute to HSC activation, encompassing a wide range of cellular processes. Histologically, a prominent feature of quiescent HSCs is the presence of retinoid droplets in the cytoplasm, which are lost during transdifferentiation.68,69 Many different marker transcripts and proteins specific for HSCs have been identified over the past decade. Together, they have advanced research into histologic detection, cell fate tracing, genetic targeting, imaging and ultimately therapeutic targeting through identification of relevant mechanisms. The paradigm of fibrogenesis and its perpetuation encompass the hallmarks of HSC activity, notwithstanding its initial description ~20 years ago.70 Initiation refers to an initial phenotypic switch favoring contractility and fibrogenicity, transcription and translation of growth factor receptors, and modulation of growth factor signaling. Perpetuation encompasses processes that amplify the phenotypic switch, including paracrine, autocrine, juxtacrine and matricrine interactions. Lastly, clearance of HSCs includes pathways such as apoptosis, necroinflammation and reversion to a quiescent state.
Extracellular mechanisms of HSC activation
There are numerous events occurring extracellularly that contribute to activation of HSCs (Figure 2). Parenchymal damage to hepatocytes due to processes such as NASH and viral hepatitis can result in the release of various ligands and intracellular proteins, nucleic acids and molecules that are able to elicit a non-infectious “sterile” inflammatory and profibrotic milieu. Damage-associated molecular patterns (DAMPs), such as mitochondrial and nuclear DNA, ATP, heat shock proteins and S100 proteins, bind to pattern-recognition receptors such as Toll-like receptors (TLRs) including TLR9, TLR4 and purine P2X7 receptors.71 Murine models with constitutively active inflammasome components (NLRP3) exhibited increased rates of hepatocyte caspase-1-dependent pyroptosis and HSC activation,72 underscoring the role of cellular death in activation of HSCs. IL-33 is a cytokine released by hepatocytes during liver injury, which activates innate lymphoid cells. These cells are known to trigger HSCs and promote liver fibrosis.73 Phagocytosis of apoptotic bodies by HSCs also promotes HSC activation.74,75 These hepatocyte-mediated mechanisms of HSC activation highlight the role of released intracellular components and apoptotic bodies in enabling a pro-inflammatory microenvironment.
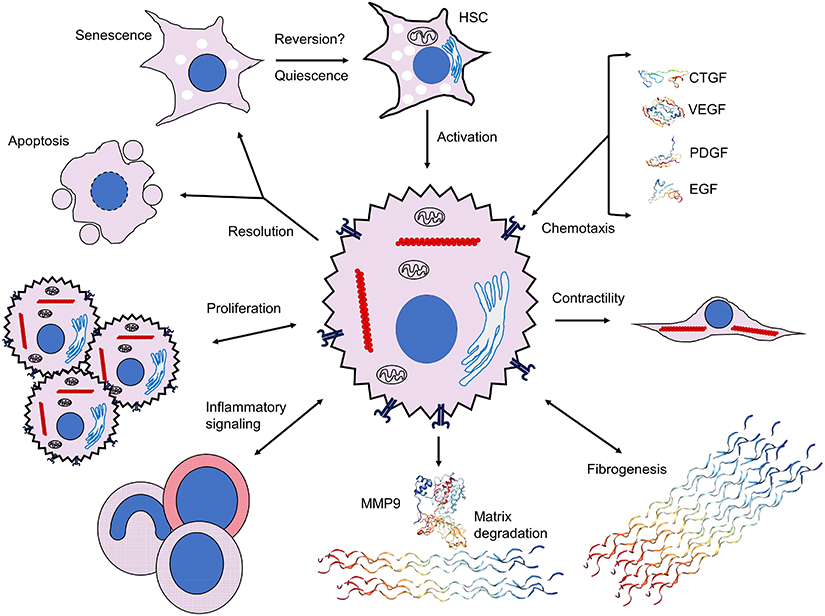 |
Figure 2 Extracellular signaling mechanisms initiated by and contributing to hepatic stellate cell activation. Activation of HSCs and initiation of fibrogenic, contractile, proliferative and chemotactile processes facilitate perpetuation of fibrogenesis. Chemotaxis through synthesis and secretion of various growth factors, such as CTGF (SWISS-MODEL accession P29279), VEGF (PDB ID 1TZH),226 PDGF (PDB ID 4QCI)227 and EGF (PDB ID 2KV4),228 promote proliferation, secretion of ECM components and maintenance of a profibrotic milieu via juxtacrine, paracrine and autocrine interactions. Upregulation of fibrogenic genes, including αSMA (red chains) to enable contractility and collagen III (PDB ID 6A0A),229 leads to expansion of ECM and fibrotic septa, and integrin-dependent matricrine interactions facilitate perpetuation of the activated state. Dysregulation of matrix degradation through differential expression of various MMPs, including MMP9 (PDB ID 1L6J),230 enables accumulation of ECM components and sustenance of a profibrotic microenvironment. Inflammatory signaling recruits various WBCs and macrophages with profibrotic downstream effectors through Th1 and Th17 juxtacrine and paracrine signaling. Activated HSCs proliferate via TGF-β-, Ras- and Hedgehog-dependent pathways. Resolution of HSC activation proceeds via apoptosis, senescence or reversal to a quiescent state; it is unclear whether reversal to the deactivated state fully occurs. Crystal structures rendered with NGL Viewer.231 Abbreviations: αSMA, alpha-smooth muscle actin; CTGF, connective tissue growth factor; ECM, extracellular matrix; EGF, epidermal growth factor; HSC, hepatic stellate cell; MMP, matrix metalloproteinase; PDB, Protein Data Bank; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor beta; VEGF, vascular endothelial growth factor; WBC, white blood cell. |
About 80% of all macrophages in the body reside in the liver.76 Liver macrophages, including Kupffer cells, monocyte-derived macrophages and resident macrophages, activate HSCs during hepatic injury through production of cytokines and chemokines, including TGFβ, PDGF, TNF, IL-1β, MCP1, CCL3 and CCL5. There are subsets of macrophage populations that are profibrotic and pro-inflammatory (M1 polarization) and associated with Th1 responses, as well as some that down-modulate inflammation, promote resolution of inflammation and fibrosis (M2 polarization) and are associated with Th2 responses. Expression of fibrolytic MMPs including MMP12 and MMP13 is a key component of fibrosis resolution.77,78 A specific population of murine macrophages has been identified (CD11bhi/F4/80int/LY6Clow) that is derived from a phenotypic switch of pro-inflammatory and profibrogenic LY6Chi macrophages.79 This population is restorative, with enhanced MMP expression and upregulation of CX3CR1, a chemokine receptor whose interaction with its ligand CX3CL1 inhibits pro-inflammatory properties in Kupffer cells and macrophages, resulting in decreased hepatic fibrosis in a murine CCl4 model.80
The subendothelial ECM, composed primarily of type IV collagen, heparan sulfate proteoglycan and laminin, appears to be crucial in maintaining the function of all types of resident liver cells. As hepatic fibrosis progresses, both the amount and type of ECM changes; both collagenous and non-collagenous ECM components increase 3- to 5-fold, and type IV collagen is replaced by fibril-forming type I and III collagens.81,82 These changes result in increased density and stiffness of ECM and serve as mechanical stimuli to further activate HSCs in part via integrin signaling pathways, resulting in a positive feedback loop.83,84 HSCs express integrins as well as discoid domain-containing tyrosine kinase receptors (DDRs) that bind to triple helical collagen on HSCs and promote further fibrosis.82,85 ECM that has expanded during liver injury can also function as a reservoir for various growth factors, including platelet-derived growth factor (PDGF), epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF) and vascular endothelial growth factor (VEGF). Such growth factors promote expansion of HSCs.86,87
Other cell types are also either directly or indirectly involved in HSC activation. Platelets promote fibrosis in various hepatic disorders, including hepatitis B and C, alcoholic liver disease (ALD) and NASH, through their release of platelet-derived growth factor beta (PDGFβ) and transforming growth factor (TGFβ).88,89 Natural killer (NK) cells target activated HSCs for apoptosis via NK-derived interferon-γ (IFNγ)90 and death receptor-induced apoptosis.91 NK cells also promoted resolution of fibrosis by targeting senescent activated HSCs.92 Liver sinusoidal endothelial cells (LSECs) in their differentiated, fenestrated phenotype were stimulated by VEGF to produce nitric oxide (NO), which prevented HSC activation and also promoted reversal of HSC activation, resulting in quiescence.93 However, LSECs can act in opposing fashion depending on whether there is acute or chronic liver injury through action of pro-regenerative CXCR7 and profibrotic CXCR4 receptors.94 Lastly, HSCs appear to influence innate B cell activity via retinoic acid- and MyD88-dependent B cell recruitment of dendritic cells and monocytes in mice.95 A study with B cell-deficient mice showed decreased fibrosis induced by CCl4, suggesting a role for B cells in the perpetuation of hepatic fibrosis.96
Metabolic and molecular dysregulation of hepatic stellate cells
Membrane receptors
The HSC intracellular processes regulating initiation and perpetuation of fibrosis are complex and multifactorial, involving various and disparate signaling pathways and cellular biologic mechanisms (Figure 3). Multiple transcription factors are implicated in modulation of HSC activation, including ERAS, GIV, YAP, SOX9, GATA4, Kruppel-like factors, AhR, MRTF-A and NR4A1/2. Various membrane receptor-signaling pathways have been implicated. TGFβ is secreted in a latent form by activated HSCs and promotes hepatic fibrosis via an autocrine positive feedback loop through the action of SMAD2/SMAD3 transcription factors.97,98 PDGF is a chemoattractant to HSCs and myofibroblasts,68 and expression of its receptor, PDGFRβ, was upregulated in mice with HSC activation.99,100 Connective tissue growth factor (CTGF) knockdown in mice resulted in decreased CCl4-induced fibrosis.101 EGF receptor antagonism was associated with reduced hepatic fibrosis and HCC in rodent models,102 while transgenic mice lacking macrophage EGF receptor showed a decrease in HCC.103 As noted previously, integrins play a key role in driving fibrogenesis through interactions with ECM and the activation of latent TGFβ.68,86 Of note, deletion of αv integrin in activated HSCs reduced CCl4-induced fibrosis.84 The Hedgehog pathway is a well-characterized signal transduction pathway implicated in development, stem cell maintenance and carcinogenesis. Rodent hepatocyte Hedgehog ligand expression induced HSC activation, resulting in fibrogenesis and HCC.104,105 Inhibition of this pathway by forskolin reduced CCl4-induced fibrosis in rats,106 and by vismodegib in both BDL-induced and NASH-induced fibrosis.107 Cannabinoid receptors CB1 and CB2 are neuromodulatory and immunomodulatory GPCRs; CB1 has pro-fibrogenic function, and CB2 displayed hepatoprotective properties.108 Agonism of CB2 induced HSC quiescence and/or apoptosis, and also reduced IL-17 production by Th17 cells, leading to a reduction in hepatic fibrosis.109
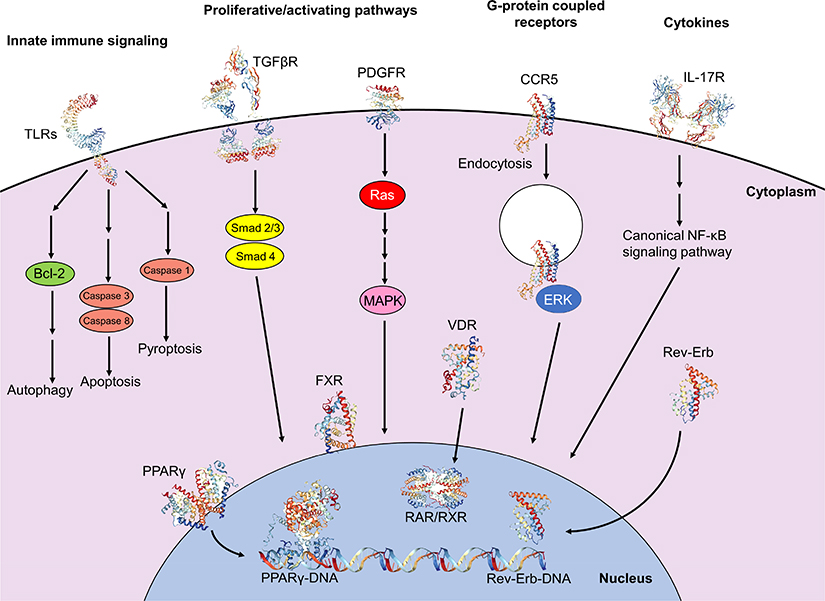 |
Figure 3 Intracellular signaling pathways involved in initiation and perpetuation of hepatic stellate cell activation. Initiation of the activated state involves engagement of proliferative and activating pathways, including TGFβR (PDB IDs 1KTZ, 3TZM, 5E8U, 5E8V)232–234 binding and downstream nuclear translocation of Smad2/3 and Smad 4 proteins, resulting in transcription and translation of profibrotic genes. Binding of PDGF to its receptor PDGFR (PDB ID 5GRN)235 results in activation of cytoplasmic Ras and MAPK effectors, with subsequent nuclear translocation and expression of proliferative genes. Engagement of the GPCR CCR5 (PDB ID 6AKX)236 results in clathrin-mediated endocytosis, activation and nuclear translocation of ERK1/2, and expression of proliferative genes, also allowing for migratory properties. Cytokines also have significant modulatory properties on fibrogenesis; for example, binding of IL-17 to its receptor IL-17R (PDB ID 4HSA)237 results in activation of the canonical NF-κB pathway, resulting in pro-inflammatory gene expression. Programmed cell death can be facilitated via innate immune signaling; engagement of TLRs (PDB IDs 6NIH, 2MKA, 2J67)238–240 with various TLR ligands propagates downstream signaling culminating in autophagy, apoptosis or pyroptosis. Nuclear membrane receptors also have large roles in modulating profibrogenic properties; engagement of ligands, including fatty acids and thiazolidinediones, with receptors of the PPAR family, including PPARγ (PDB ID 2Q8S),241 results in nuclear translocation, heterodimerization with RXR and binding with DNA (PDB ID 3DZU),242 with subsequent expression of antifibrogenic genes. Binding of obeticholic acid to FXR and of vitamin D to VDR results in receptor nuclear translocation, heterodimerization with RXR and expression of antifibrogenic genes. Meanwhile, binding of heme to Rev-Erb upregulates Rev-Erb expression, accumulation of Rev-Erb in the cytoplasm and promotion of a profibrogenic, contractile HSC phenotype. Crystal structures rendered with NGL Viewer.231 Abbreviations: CB1, cannabinoid receptor type 1; CCR5, C-C chemokine receptor type 5; CX3CR1, C-X3-C motif chemokine receptor 1; DNA, deoxyribonucleic acid; EKR1/2, extracellular signal-regulated protein kinases 1 and 2; FXR, farsenoid X receptor; GPCR, G-protein coupled receptor; HSC, hepatic stellate cell; IL-17, interleukin 17; IL-17R, IL-17 receptor; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PDGF, platelet-derived growth factor; PDGFR, PDGF receptor; PPARγ, peroxisome proliferator-activated receptor gamma; RXR, retinoid X receptor; TGFβR, transforming growth factor beta receptor; TLR, Toll-like receptor; VDR, vitamin D receptor. |
Nuclear receptors
Various nuclear receptors are implicated in the activation of HSCs and perpetuation of fibrosis (Figure 3). Peroxisome proliferating activated receptors (PPARs), which recognize fatty acids and various derivatives, are involved in regulation of lipid homeostasis.110 In HSCs, PPARγ suppresses PDGFRβ signaling and TGFβ1 by virtue of the β-catenin pathway.111,112 Various PPAR agonists are currently under investigation in clinical trials (ClinicalTrials.gov identifier NCT02704403, Clinical Trial Registry India CTRI/2015/10/006236).113,114 Signaling through the farsenoid X receptor (FXR), which is activated by bile acids, increased sensitivity to insulin and enhances β-oxidation of fatty acids in hepatocytes.115 A phase II clinical trial of a synthetic FXR agonist (obeticholic acid) demonstrated reduced liver fibrosis in NASH patients without cirrhosis,116 and this agent is also under investigation in a phase III trial of NASH patients with fibrosis (REGENERATE, ClinicalTrials.gov identifier NCT02548351). Vitamin D receptor (VDR) activation reduced TGFβ and SMAD3 target gene transcription, thereby inhibiting fibrosis.117 TGFβ1 rearranged VDR binding sites throughout the genome in HSCs at SMAD3 target genes via chromatin remodeling. VDR signaling promoted by p62/SQSTM1 suppressed HSC activation, decreasing fibrogenesis and HCC. The effect was offset by VDR knockout in a murine model.117 Retinoid receptors, including retinoic acid receptors (RARs) and retinoid X receptors (RXRs), participate in various metabolic processes. Through interaction with all-trans retinoic acid (ATRA), RARs inhibited expression of various genes, including α-smooth muscle actin (αSMA, a marker of HSC activation), TGFβ, IL-6, procollagen I, III and IV, fibronectin and laminin, with a resultant inhibitory effect on HSC activation.118 RXRs heterodimerize with VDR, thereby transducing cytoplasmic signals to the nucleus in HSCs with profibrogenic, pro-inflammatory and carcinogenic effects.119 Other nuclear receptors, including Rev-Erb and liver X receptors, also modulated HSC activation.120,121 Retinoids activated latent TGFβ in rat HSCs, with the predicted profibrotic effects.122
Epigenetic dysregulation of hepatic stellate cell activation
Activation of HSCs results in global epigenomic and local epigenetic dysregulation, promoting fibrogenesis.123 DNA methyltransferase (DNMT) expression appears to play a role in HSC activation, and suppression of DNMTs dampened HSC activation.124–126 DNA hypermethylation by DNMT1 of phosphatase and tensin homolog (PTEN) protein resulted in HSC activation.127 Inheritable adaptation might also play a role, where family history of liver damage in rats was associated with epigenetic modifications resulting in increased expression of PPARγ, reduced TGFβ expression and fewer myofibroblasts.128 Dysregulation of various micro-RNAs (miRNAs) and other non-coding RNAs has been implicated in HSC activation and inactivation, via post-translational modifications. Profiling of miRNAs using high-throughput methods has identified profibrogenic miRNAs that are upregulated in activated HSCs, as well as anti-fibrotic miRNAs.129–131 For example, miR-378 appears to inhibit HSC activation through suppression of Gli3, a downstream effector and transcription factor in the Hedgehog pathway.132 Overexpression of miR-200a suppressed translation of αSMA and inhibited TGFβ by various mechanisms associated with reduced fibrogenesis.133–135 There is also evidence that ethanol induces lysine methyltransferase 2A (KMT2A), resulting in transcriptional activation of elastin and other ECM genes,136 underscoring the role of toxins in epigenetic regulation of fibrogenesis.
HSC activation results in dysregulation of cellular energy homeostasis, as the energy required for various intracellular processes is shunted toward the transcriptional and translational demands of HSC transdifferentiation. Autophagy in HSCs is induced by endoplasmic reticulum (ER) and oxidative stress in response to chronic liver injury137 and provides ATP to fuel HSC activation.138 Specifically, the knockdown of essential autophagy genes Atg5 and Atg7 not only inhibited autophagy but also suppressed HSC activation.139 Another ER stress effector pathway, the unfolded protein response (UPR), is implicated in HSC activation through stimulation of the TGFβ pathway in protein kinase R-like ER kinase (PERK)-mediated UPR.140–142 Chaperone proteins involved in protein folding are also implicated in ER stress and HSC activation; deletion of collagen-specific heat shock protein 47 (Hsp47), which mediates folding of collagen I, resulted in collagen I accumulation, induction of autophagy and reduced levels of apoptosis.143 Various other pathways, effectors and receptors, such as advanced glycation end products (AGE), inflammasome (NLRP3), Rev-Erb nuclear receptors, LXR, PPARγ and hypoxia-inducible factor-1α (HIF1α) have been implicated in the regulation of autophagy, ER stress and HSC activation. Further, retinoids appeared to activate latent TGFβ in rat HSCs, exacerbating hepatic fibrosis.122 Alcohol dehydrogenase 3-mediated oxidation of retinol to retinoic acid promoted expression of TGFβ and collagen in HSCs, while retinol secreted by HSCs promoted HSC survival via suppression of NK cells.144 Lastly, protein ubiquitination was inhibited during HSC activation through upregulation of ubiquitin C-terminal hydrolase 1 (UCHL1), a deubiquitinase, resulting in proliferation of activated HSCs in CCl4 and BDL mice.145 Many pathways are currently under investigation, including YAP1 (Hippo pathway), endosialin, BRD4, galectin 3, GATA4, BMP6, GAS6 and AXL.
Cytokines also play an important role in HSC activation. IL-17, secreted by Th17 cells, activated the STAT3 signaling pathway, inducing synthesis of type I collagen and promoting fibrosis.146 IL-20 is a pro-inflammatory cytokine that promotes TGFβ expression and activates HSCs, and antibody neutralization of either IL-20 or its receptor inhibited expression of TGFβ, activation of HSCs and hepatic fibrosis.147 Meanwhile, IL-15 and IL-15 receptor α (IL-15Rα) have a hepatoprotective role by downmodulating progression of hepatic fibrosis via inhibition of collagen expression in HSCs, as well as by promoting NK cell homeostasis.148 IL-22 has been shown to mitigate hepatic fibrosis via induction of HSC senescence. An in vivo study of increased hepatic IL-22 expression in transgenic mice resulted in accelerated fibrosis resolution following liver injury, through reduced αSMA expression and increased numbers of senescent HSCs.149 Murine administration of IL-22 improved BDL-induced fibrosis,146 while deletion of IL-22 worsened fibrosis.149
Deactivation pathways of hepatic stellate cells
Cessation of chronic liver injury can lead to fibrosis reversal, even at the cirrhotic stage, whereas previously it was thought to be an end-stage and irreversible condition. There are three predominant mechanisms that contribute to clearance of activated HSCs and resolution of fibrosis, namely induction of HSC apoptosis, senescence and reversion/transdifferentiation to an inactivated state, all ostensibly culminating in a non-fibrogenic state (Figure 3). There is evidence that antiviral treatment for HBV and HCV results in some degree of fibrosis reversal, as detailed below.
HSC apoptosis
Induction of apoptosis serves to decrease the number of activated HSCs, leading in part to resolution of fibrosis. In the activated state, HSCs express various “death” receptors, including those for TNF receptor 1 (TNFR1), first apoptosis signal (FAS), p75 neutrophin (p75NTR) and TNF-related apoptosis-inducing ligand (TRAIL).150 In a transgenic mouse model lacking the p75NTR ligand-binding domain, there was decreased resolution of fibrosis and decreased apoptosis of myofibroblasts.151 Exogenous pegylated TRAIL improved CCl4-induced fibrosis and induced apoptosis of activated HSCs in a rat model.152 Notwithstanding this seemingly increased susceptibility to apoptosis, activated HSCs do display increased resistance to apoptosis. There are various pathways of programmed cell death (PCD); apoptosis, autophagy, oncosis leading to necrosis, and pyroptosis.153 PCD pathways, including apoptosis, are suppressed by NF-κB, which induces transcription of various antiapoptotic genes, such as Bcl-2 family members like Bcl-XL and Bfl-1,154 as well as TRAF1, TRAF2, XIAP, FLIP and c-IAP.155,156 TNF and IL-1β activate NF-κB and promote resistance to HSC apoptosis.157 In rats, inhibition of NF-κB by the small molecule BAY 11-7082 reduces hepatic fibrosis in vivo.158 Further, proteasome inhibition by small molecules bortezomib and MG132 induced apoptosis via three mechanisms that included inhibition of (i) nuclear translocation of RelA/p65 protein; (ii) I-κBα degradation (preventing NF-κB nuclear translocation)in and (iii) NF-κB DNA binding activity, thus preventing expression of Bcl-2 family genes involved in apoptosis.158 Additionally, TIMP1 and TGFβ have been shown to have anti-apoptotic action and to promote survival of activated HSCs.159 Inhibition of histone deacetylation by the small molecule nilotinib was shown to induce apoptosis and autophagy, exemplifying epigenetic regulation of PCD.160 Lastly, the Akt anti-apoptotic pathway, when activated, suppressed apoptosis mediated by Jun N-terminal kinase (JNK).161 Inhibition of several tyrosine and Raf kinases, including Akt pathway kinases, induced both apoptosis and autophagy in HSCs.162
Senescence
Cellular senescence is typically associated with some type of cell damage, as well as with developmental differentiation. There are various damage-induced senescence mechanisms, through which cells lose proliferative markers, express tumor suppression gene products, DNA damage markers, secrete various signaling molecules and express cell cycle inhibitors.163 Replicative senescence is the result of telomere shortening due to consecutive cycles of cell division, which is detected as a form of DNA damage. Induction of senescence is achieved through multiple signaling pathways, most of which activate p53, that ultimately converge on activation of cyclin-dependent kinase (CDK) inhibitors p16, p15, p21 and p27. Such activation results in proliferative arrest, mostly mediated by hypophosphorylated retinoblastoma (RB) protein.164 Stress-induced senescence is mediated through increased levels of reactive oxygen species (ROS). As an example, the RAS-RAF-MEK-ERK cascade, which can lead to high intracellular ROS levels, activates p38 MAPK, which upregulates p21 and increased transcription of p53.165 Activation of oncogenes can also induce senescence, an observation first made when an oncogenic isoform of RAS was overexpressed in human fibroblasts.166 Other oncogenes that can result in induction of senescence include von Hippel-Lindau disease tumor suppressor (VHL), neurofibromin (NF1) and PTEN.167 Murine activation of the p53/p21 pathway inhibits activated HSC proliferation and attenuates hepatic fibrosis.92,168 Senescence results in a pro-inflammatory phenotype, the senescence-associated secretory phenotype (SASP)169–172 that upregulates TGFβ, IL-6, IL-8, various chemokines and macrophage inflammatory proteins, proteases, growth factors and granulocyte-macrophage colony-stimulating factor (GM-CSF).173,174 Such an environment is conducive to recruitment of phagocytic macrophages and NK cells, ultimately leading to clearance of senescent activated HSCs.
Reversal/transdifferentiation to an inactivated state
In rodents, there is some evidence that activated HSCs have the capacity to undergo reversion into an inactivated phenotype not unlike the quiescent, pre-activation phenotype. It was further observed that these reverted HSCs have a lowered threshold for reactivation. While profibrogenic genes such as Col1a1, Acta2, Timp1 and Tgfbr1 are downregulated, genes associated with quiescence, including Gfap, Adfp and Adipor1, are not expressed.175,176 A murine study with genetically labeled activated HSCs showed that after stopping CCl4 administration, expression of labeled genes decreased significantly, with some apoptosis and regression to the inactivated but primed state.175 Similarly, single-cell PCR and genetic tracing in mice found that upon withdrawal of the offending agent (CCl4), HSCs reverted back to a quiescent-like phenotype, yet remained “semi-activated” and primed to reactivation.176 In vitro, human fetal limb fibroblasts were induced to transdifferentiate to hepatocyte-like cells using lentiviral expression of FOXA3, HNF1A and GATA4.177 This study was replicated in vivo via lentiviral induction of fibroblasts into hepatocyte-like cells via ectopic expression of FOXA3, GATA4, HNF1A and HNF4A.178 Though advances have been made in identifying relevant mechanisms in this partial reversal to a non-fibrogenic, inactivated state, much work remains to be done to identify targets amenable to therapeutic intervention.
Clinical evidence to reverse fibrosis
Reversal of HBV fibrosis following antiviral treatment
There is growing evidence that antiviral treatment of HBV is associated with hepatic fibrosis reversal. The standard of care prior to the newer generation of antivirals, including nucleoside/nucleotide analog reverse transcriptase inhibitors, was pegylated IFNα (PEG-IFNα), which has been shown to inhibit TGFβ, reduce HSC activation and stimulate HSC apoptosis in vitro.179 A case series of 110 patients with chronic HBV (CHB) treated with PEG-IFNα showed a 27% histologic improvement in hepatic fibrosis at 16 months.180 In a small case study of patients with CHB, treatment with the nucleoside analog lamivudine was associated with fibrosis reversal in 68% of the patients, including complete regression of fibrosis or cirrhosis in 21% of the patients.181 An earlier case series of CHB patients treated with lamivudine demonstrated histologic cirrhosis reversal in 73% of the patients at 36 months.182 A larger case study of 348 CHB patients treated with the reverse transcriptase inhibitor tenofovir showed a 51% improvement in fibrosis at 5 years’ follow-up.183–185 A cohort of 57 patients with CHB were treated with entecavir, another reverse transcriptase inhibitor, and showed a remarkable 88% histologic improvement in fibrosis.186,187 A randomized controlled trial (RCT) of 125 patients with CHB treated with adefovir, a nucleotide analog of adenosine, showed histologic fibrosis improvement in 71% of the patients at 240 weeks (Table 1).188 A study of 57 patients with CHB treated with the thymidine nucleoside analog telbivudine demonstrated significant improvements in Ishak fibrosis scores after long-term treatment (mean duration 261 weeks).189 Improvement in histologic fibrosis stage has been associated with, and perhaps dependent on a reduction in HBV viral load.190,191
 |
Table 1 Summary of published and ongoing phase II/III randomized controlled trials (RCTs) of therapeutic agents for hepatic fibrosis reversal |
Reversal of HCV-induced fibrosis via DAA treatment
Patients with chronic hepatitis C (CHC) differentially benefited from PEG-IFNα treatment based on genotype, displaying viral eradication rates from 40% with genotype 1 to 70% with genotypes 2 or 3. A study of 60 patients with CHC treated with PEG-IFNα and ribavirin showed an improvement in fibrosis of 82% at 60-month follow-up.192 The newer generation of oral direct-acting antivirals (DAA) for HCV has shown excellent efficacy in curing HCV. Indirect measurements of fibrosis and cirrhosis made by transient elastography indicated that patients that achieved a sustained virologic response (SVR) following therapy with DAA exhibited lower FibroScan scores, as well as indirect biochemical markers of fibrosis.193 More recently, short-term evaluation of 51 patients with CHC treated with DAA and who underwent SVR showed moderate improvement in necroinflammation, though no histologic improvement in fibrosis was observed at a mean of 41 weeks after completion of DAA treatment.194 In a recent prospective study of 260 patients with CHC treated with DAA, 94.6% of whom achieved SVR, 40% underwent significant regression in fibrosis measured indirectly via transient elastography. Interestingly, the change was more pronounced in patients with higher baseline fibrosis scores.195 The use of angiotensin receptor blockers such as losartan, thought to inhibit NADPH oxidase-induced oxidative stress and resultant fibrogenesis, has also been shown to reduce hepatic fibrosis. In a small case study of 14 patients with CHC treated with losartan, 50% displayed histologic improvement.196 Lastly, an RCT of 109 patients with CHC treated with the PPARγ agonist farglitazar did not show any improvement in hepatic fibrosis at 18-month follow-up (Table 1).197
Other etiologies of hepatic fibrosis/cirrhosis
Interventions for various other etiologies of hepatic fibrosis and/or cirrhosis have been assessed for their potential to reverse fibrosis (Table 1). In alcohol-related cirrhosis, abstinence of alcohol was shown to have a survival benefit, although no fibrosis follow-up data were reported.198 An RCT of 85 patients with alcohol-related cirrhosis treated with the ACE inhibitor candesartan showed a 33% improvement in fibrosis, compared to 12% with placebo.199 Ursodeoxycholic acid (UDCA), a naturally occurring hydrophilic bile acid, has been used for treatment of primary biliary cholangitis (PBC) and other cholestatic liver disorders. UDCA works to protect cholangiocytes and hepatocytes against the cytotoxicity of static bile acids, as well as by stimulation of hepatobiliary secretion and subsequent anticholestatic effects.200 An RCT including 146 patients with PBC treated with UDCA showed slowing of fibrosis progression but no improvement in stage,201 while 103 PBC patients treated with UDCA in a separate study also showed a lower rate of fibrogenesis.202 The use of immunosuppression in autoimmune hepatitis (AIH) has also been evaluated for improvement in fibrosis. In a study of 87 patients with AIH treated with corticosteroids, 53% showed improvement in histologic fibrosis scores and there was no further progression in 26% of the patients. In addition, the frequency of cirrhosis decreased from 16% to 11% over a mean treatment period of 62 months.203 A cohort of 19 patients with AIH treated with cyclosporine A versus prednisolone for 6 months, and then maintained on azathioprine, showed a significant decrease in fibrosis stage in paired liver biopsies, as well as a decrease in inflammatory grade and non-invasive biochemical markers of fibrosis.204 There has been one recent study of 36 patients with hereditary hemochromatosis and severe fibrosis or cirrhosis (METAVIR scores F3 or F4), in which liver fibrosis or cirrhosis was assayed histologically following phlebotomy in C282Y homozygotes. Fibrosis regressed at least 2 METAVIR units in 69% of the patients with stage F3 fibrosis at baseline, and in 35% of the patients with stage F4 cirrhosis at baseline.205
There are currently no FDA-approved treatments for non-alcoholic fatty liver disease (NAFLD) or for non-alcoholic steatohepatitis (NASH), other than addressing risk factors, including weight loss through lifestyle modification. The NAFLD Activity Score (NAS) is a scoring system to histologically assess the features observed; typically, a score equal to or greater than 5 is used for a histologic diagnosis of NASH.206 An RCT assessing weight loss in 31 patients with NASH over a 48-week period found significant decreases in NAS after an average weight loss of 9.3% but did not show improvement in fibrosis.207 Various other agents are under active investigation for treatment of NASH (Table 1). An RCT of 74 patients with NASH treated with the PPARγ agonist pioglitazone versus placebo for 12 months found that fibrosis and hepatocellular injury were significantly reduced in the treatment arm.208 An RCT comparing pioglitazone versus vitamin E versus placebo in 247 patients with NASH found that vitamin E was superior to placebo in decreasing NAS, but showed no improvement in fibrosis. Pioglitazone therapy did not demonstrate any advantage over placebo in the primary outcome (improved NAS), though there was benefit in several secondary outcomes.113 Another PPARγ agonist, rosiglitazone, was not found to have any effect on improvement of fibrosis in an RCT of 53 patients with NASH.209 The phosphodiesterase inhibitor pentoxifylline was compared to placebo in an RCT of 55 patients with NASH showing a significant decrease in NAS, as well as a significant improvement in fibrosis.210 In a much anticipated study, the farsenoid X receptor agonist obeticholic acid showed a statistically significant improvement in fibrosis (45% vs 21%, P=0.002) at 24-weeks versus placebo in 141 patients with NASH.116
Biologic and small molecule therapeutics under investigation to reverse fibrosis
There are no FDA-approved therapies for fibrosis reversal, though many promising agents are being actively investigated (Table 1). Hepatic fibrosis can be achieved via a variety of mechanisms, including suppression of HSC activation, reversal or transdifferentiation of activated HSCs, immune clearance of HSCs, induction of HSC apoptosis and promotion of HSC senescence. A few biologic therapies have also been investigated. A human monoclonal antibody targeting CTGF, pamrevlumab (FG-3019) was initially tested for hepatic fibrosis and is currently on fast-track approval by the FDA for treatment of idiopathic pulmonary fibrosis.211 Targeting ECM genes such as lysyl oxidase-like 2 (LOXL2) to down-modulate fibrogenesis has been found to be an attractive target. Two phase IIb RCTs of simtuzumab, a humanized monoclonal antibody to LOXL2, failed to decrease hepatic collagen content or hepatic venous pressure gradient.212
In addition to the molecules described in various areas earlier, in vivo inhibition of TGFβ1 activation via integrin αvβ6 antagonism with EMD527040 in a BDL model of cirrhosis was found to significantly decrease bile duct proliferation and peribiliary collagen deposition by 40–50%.213 Cenicriviroc is an inhibitor to both CCR2 and CCR5 that leads to decreased recruitment of inflammatory monocytes and macrophages via CCR2 and decreased recruitment of HSCs and lymphocytes via CCR5. Cenicriviroc demonstrated significant improvement in NAS after 1 year in a phase IIb RCT of 289 patients with NASH214 and is currently being studied in a phase III clinical trial (AURORA trial, ClinicalTrials.gov identifier NCT03028740). An RCT of 86 cirrhotic patients treated with emricasan, a potent irreversible pan-caspase inhibitor, found significant reductions in mean MELD and Child-Pugh scores after 3 months of treatment, an effect that was sustained or improved at 6 months.215 Elafibranor, a PPARα and PPARδ agonist, was evaluated in a phase IIb RCT of 276 patients with NASH and found to significantly reduce fibrosis.114 Inhibition of apoptosis signal-regulating kinase 1 (ASK1) by selonsertib with and without simtuzumab led to improvement in liver fibrosis, measured both directly and indirectly, in a phase II RCT of 72 patients with NASH.216 These promising results are encouraging signs that effective therapies for NASH and potentially other hepatic fibrosis etiologies are on the horizon (Table 1).
Mesenchymal stem cell transplantation
Lastly, there have been efforts at utilizing mesenchymal stem cells (MSCs) derived from umbilical cord and from bone marrow to treat liver fibrosis. However, such efforts have been hampered by the inability to monitor the cells in the transplant recipient and by a lack of standardized protocols governing timing, dose and route of administration. Additionally, concerns have been raised regarding potential tumorigenicity in vivo, while other data suggested that there is an antitumor effect and down-modulation of signaling pathways associated with tumor growth and cell division. Nevertheless, the small case series thus far showed a clear trend toward improved MELD and liver function; and there is only one study that showed histologic improvement of fibrosis.217 Several reports using intravenous administration of umbilical cord-derived MSCs showed various improvement in biochemical markers, including improvement in MELD and liver function,218,219 reduced ascites,218,220 and decreased serum alkaline phosphatase and γ-glutamyltransferase levels.220 Six studies of autologous bone marrow-derived MSCs administered via various routes showed improvements in MELD score,221–224 Child-Pugh score,217 liver function222,224 and improvement in various liver function tests.217,223,225 It should be noted that none of these studies have been performed in the US, most likely given the paucity of data regarding tumorigenicity; hence, additional in vitro and in vivo studies are required to better characterize the risk potential of autologous MSC transplantation.
Conclusion
The wealth of knowledge that has accumulated over the past 15 years illuminating the multifaceted intricacies of HSC activation, apoptosis, immune recruitment and senescence have paved the way for translational therapeutics with the potential to reverse fibrosis. As we continue to discover and elucidate the relevant cellular pathways, it is hoped that other therapeutic targets will be identified to develop new modalities of treatment for this all-too-common and increasing ailment. Promising agents currently underway in clinical trials will help to design and implement additional novel approaches with improved efficacy. Lastly, a greater understanding of the fundamental mechanisms of fibrosis and its reversal could and will lead to therapeutic agents for other organs, such as kidney, lung and heart.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Moffat J. Pre-1801 imprint collection (Library of Congress), Joseph Meredith Toner Collection (Library of Congress). In: The Prognostics and Prorrhetics of Hippocrates. London: Printed by T. Bensley for C. Elliot, T. Kay, and Co., opposite Somerset-House, in the Strand; 1788.
2. Galen MMT. Galen on the Usefulness of the Parts of the Body. Peri Chreias Moriōn [romanized Form] De Usu Partium. Ithaca, N.Y.: Cornell University Press; 1968.
3. Ross WD. The Works of Aristotle Translated into English. Oxford: Clarendon Press; 1928.
4. Avicenna GOC. A Treatise on the Canon of Medicine of Avicenna, Incorporating A Translation of the First Book. New York: AMS Press; 1973.
5. Brown J. A remarkable account of a liver, appearing glandulous to the eye; communicated by Mr. John Brown, Chirurgeon of St. Thomas’s Hospitall in Southwark; in a letter to one of the Secretarys of the Royal Society. Philos Trans R Soc London. 1685;15(178):1266–1268. doi:10.1098/rstl.1685.0087
6. Laennec RTH, Forbes J, Fisher JD, Andral G, Harvard Medical S. A Treatise on the Diseases of the Chest, and on Mediate Auscultation. New York: Samuel S. and William Wood; 1838.
7. Osler W. The Principles and Practice of Medicine, Designed for the Use of Practitioners and Students of Medicine. New York: D. Appleton and Company; 1901.
8. Sherlock S. Diseases of the Liver and Biliary System. Oxford: Blackwell Scientific Publications; 1958.
9. Walker JG, Doniach D, Roitt IM, Sherlock S. Serological tests in diagnosis of primary biliary cirrhosis. Lancet. 1965;1(7390):827–831. doi:10.1016/s0140-6736(65)91372-3
10. Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol. 2013;59(1):160–168. doi:10.1016/j.jhep.2013.03.007
11. Grant BF, Chou SP, Saha TD, et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: results from the National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry. 2017;74(9):911–923. doi:10.1001/jamapsychiatry.2017.2161
12. Sarpel D, Baichoo E, Dieterich DT. Chronic hepatitis B and C infection in the United States: a review of current guidelines, disease burden and cost effectiveness of screening. Expert Rev Anti Infect Ther. 2016;14(5):511–521. doi:10.1586/14787210.2016.1174066
13. Gish RG, Cohen CA, Block JM, et al. Data supporting updating estimates of the prevalence of chronic hepatitis B and C in the United States. Hepatology. 2015;62(5):1339–1341. doi:10.1002/hep.28026
14. Edlin BR, Eckhardt BJ, Shu MA, Holmberg SD, Swan T. Toward a more accurate estimate of the prevalence of hepatitis C in the United States. Hepatology. 2015;62(5):1353–1363. doi:10.1002/hep.27978
15. Udompap P, Mannalithara A, Heo NY, Kim D, Kim WR. Increasing prevalence of cirrhosis among U.S. adults aware or unaware of their chronic hepatitis C virus infection. J Hepatol. 2016;64(5):1027–1032. doi:10.1016/j.jhep.2016.01.009
16. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011;34(3):274–285. doi:10.1111/j.1365-2036.2011.04724.x
17. Lazo M, Hernaez R, Eberhardt MS, et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol. 2013;178(1):38–45. doi:10.1093/aje/kws448
18. Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the United States and the rest of the world. Clin Liver Dis. 2016;20(2):205–214. doi:10.1016/j.cld.2015.10.001
19. Colp C, Pappas J, Moran D, Lieberman J. Variants of α 1-antitrypsin in Puerto Rican children with asthma. Chest. 1993;103(3):812–815. doi:10.1378/chest.103.3.812
20. Silverman EK, Miletich JP, Pierce JA, et al. α-1-antitrypsin deficiency. High prevalence in the St. Louis area determined by direct population screening. Am Rev Respir Dis. 1989;140(4):961–966. doi:10.1164/ajrccm/140.4.961
21. O’Brien ML, Buist NR, Murphey WH. Neonatal screening for α1-antitrypsin deficiency. J Pediatr. 1978;92(6):1006–1010. doi:10.1016/s0022-3476(78)80388-6
22. Distante S, Robson KJ, Graham-Campbell J, Arnaiz-Villena A, Brissot P, Worwood M. The origin and spread of the HFE-C282Y haemochromatosis mutation. Hum Genet. 2004;115(4):269–279. doi:10.1007/s00439-004-1152-4
23. Allen KJ, Gurrin LC, Constantine CC, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358(3):221–230. doi:10.1056/NEJMoa073286
24. McLaren GD, Gordeuk VR. Hereditary hemochromatosis: insights from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Hematology Am Soc Hematol Educ Program. 2009;195–206. doi:10.1182/asheducation-2009.1.195
25. Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47(6):2089–2111. doi:10.1002/hep.22261
26. Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol. 2012;56(5):1181–1188. doi:10.1016/j.jhep.2011.10.025
27. Fallatah HI, Akbar HO. Autoimmune hepatitis as a unique form of an autoimmune liver disease: immunological aspects and clinical overview. Autoimmune Dis. 2012;2012:312817.
28. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet. 2012;380(9859):2095–2128. doi:10.1016/S0140-6736(12)61728-0
29. National Center for Health Statistics. Health, United States, 2016: With Chartbook on Long-term Trends in Health. DHHS Pub No. (PHS) 2017-1232. Hyattsville, MD.
30. Hoyert DL, Xu J. Deaths: preliminary data for 2011. Natl Vital Stat Rep. 2012;61(6):1–51.
31. Mellinger JL, Shedden K, Winder GS, et al. The high burden of alcoholic cirrhosis in privately insured persons in the United States. Hepatology. 2018;68(3):872–882. doi:10.1002/hep.29887
32. Ge PS, Runyon BA. Treatment of patients with cirrhosis. N Engl J Med. 2016;375(21):2104–2105. doi:10.1056/NEJMc1612334
33. Pant C, Olyaee M, Gilroy R, et al. Emergency department visits related to cirrhosis: a retrospective study of the nationwide emergency department sample 2006 to 2011. Medicine (Baltimore). 2015;94(1):e308. doi:10.1097/MD.0000000000000874
34. National Center for Health Statistics, Center for Disease Control and Prevention, United States Census Bureau. United States National Hospital Discharge Survey: 2007 Summary. Hyattsville, MD: National Health Statistics Reports No. 29.
35. Barritt AS, Jiang Y, Schmidt M, Hayashi PH, Bataller R. Charges for alcoholic cirrhosis exceed all other etiologies of cirrhosis combined: a national and state inpatient survey analysis. Dig Dis Sci. 2019;64(6):1460–1469. doi:10.1007/s10620-019-5471-7
36. Bajaj JS, Wade JB, Gibson DP, et al. The multi-dimensional burden of cirrhosis and hepatic encephalopathy on patients and caregivers. Am J Gastroenterol. 2011;106(9):1646–1653. doi:10.1038/ajg.2011.157
37. Thiele M, Askgaard G, Timm HB, Hamberg O, Gluud LL. Predictors of health-related quality of life in outpatients with cirrhosis: results from a prospective cohort. Hepat Res Treat. 2013;2013:479639.
38. Chin JL, Pavlides M, Moolla A, Ryan JD. Non-invasive markers of liver fibrosis: adjuncts or alternatives to liver biopsy? Front Pharmacol. 2016;7:159. doi:10.3389/fphar.2016.00323
39. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464–470. doi:10.1053/jhep.2001.22172
40. Child CG, Turcotte JG. Surgery and portal hypertension. Major Probl Clin Surg. 1964;1:1–85.
41. Coll M, Perea L, Boon R, et al. Generation of hepatic stellate cells from human pluripotent stem cells enables in vitro modeling of liver fibrosis. Cell Stem Cell. 2018;23(1):101–113. doi:10.1016/j.stem.2018.05.027
42. Leite SB, Roosens T, El Taghdouini A, et al. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials. 2016;78:1–10. doi:10.1016/j.biomaterials.2015.11.026
43. Rodés J. Textbook of Hepatology: From Basic Science to Clinical Practice.
44. Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371(9615):838–851. doi:10.1016/S0140-6736(08)60383-9
45. Gracia-Sancho J, Marrone G, Fernández-Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol. 2019;16(4):221–234. doi:10.1038/s41575-018-0097-3
46. Ginès P, Solà E, Angeli P, Wong F, Nadim MK, Kamath PS. Hepatorenal syndrome. Nat Rev Dis Primers. 2018;4(1):23. doi:10.1038/s41572-018-0022-7
47. Rodríguez-Roisin R, Krowka MJ. Hepatopulmonary syndrome–a liver-induced lung vascular disorder. N Engl J Med. 2008;358(22):2378–2387. doi:10.1056/NEJMra0707185
48. Porres-Aguilar M, Mukherjee D. Portopulmonary hypertension: an update. Respirology. 2015;20(2):235–242. doi:10.1111/resp.12455
49. Garcia-Tsao G, Bosch J. Management of varices and variceal hemorrhage in cirrhosis. N Engl J Med. 2010;362(9):823–832. doi:10.1056/NEJMra0901512
50. Wijdicks EF. Hepatic Encephalopathy. N Engl J Med. 2016;375(17):1660–1670. doi:10.1056/NEJMra1600561
51. Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR cooperative study group. Hepatology. 1996;24(2):289–293. doi:10.1002/hep.510240201
52. Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. 1994;19(6):1513–1520.
53. Batts KP, Ludwig J. Chronic hepatitis. An update on terminology and reporting. Am J Surg Pathol. 1995;19(12):1409–1417.
54. Ishak K, Baptista A, Bianchi L, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22(6):696–699. doi:10.1016/0168-8278(95)80226-6
55. Knodell RG, Ishak KG, Black WC, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1(5):431–435.
56. Poordad FF. FIBROSpect II: a potential noninvasive test to assess hepatic fibrosis. Expert Rev Mol Diagn. 2004;4(5):593–597. doi:10.1586/14737159.4.5.593
57. Wai CT, Greenson JK, Fontana RJ, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology. 2003;38(2):518–526. doi:10.1053/jhep.2003.50346
58. Sterling RK, Lissen E, Clumeck N, et al. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology. 2006;43(6):1317–1325. doi:10.1002/hep.21178
59. Koda M, Matunaga Y, Kawakami M, Kishimoto Y, Suou T, Murawaki Y. FibroIndex, a practical index for predicting significant fibrosis in patients with chronic hepatitis C. Hepatology. 2007;45(2):297–306. doi:10.1002/hep.21520
60. Forns X, Ampurdanès S, Llovet JM, et al. Identification of chronic hepatitis C patients without hepatic fibrosis by a simple predictive model. Hepatology. 2002;36(4 Pt 1):986–992. doi:10.1053/jhep.2002.36128
61. Adams LA, Bulsara M, Rossi E, et al. Hepascore: an accurate validated predictor of liver fibrosis in chronic hepatitis C infection. Clin Chem. 2005;51(10):1867–1873. doi:10.1373/clinchem.2005.048389
62. Poynard T, Imbert-Bismut F, Munteanu M, et al. Overview of the diagnostic value of biochemical markers of liver fibrosis (FibroTest, HCV FibroSure) and necrosis (ActiTest) in patients with chronic hepatitis C. Comp Hepatol. 2004;3(1):8. doi:10.1186/1476-5926-3-8
63. Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. doi:10.1172/JCI20530
64. Mutsaers SE, Birnie K, Lansley S, Herrick SE, Lim CB, Prêle CM. Mesothelial cells in tissue repair and fibrosis. Front Pharmacol. 2015;6:113. doi:10.3389/fphar.2015.00113
65. Lua I, Li Y, Pappoe LS, Asahina K. Myofibroblastic conversion and regeneration of mesothelial cells in peritoneal and liver fibrosis. Am J Pathol. 2015;185(12):3258–3273. doi:10.1016/j.ajpath.2015.08.009
66. Mederacke I, Hsu CC, Troeger JS, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823. doi:10.1038/ncomms3823
67. Iwaisako K, Jiang C, Zhang M, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci USA. 2014;111(32):E3297–E3305. doi:10.1073/pnas.1400062111
68. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88(1):125–172.
69. D’Ambrosio DN, Walewski JL, Clugston RD, Berk PD, Rippe RA, Blaner WS. Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS One. 2011;6(9):e24993. doi:10.1371/journal.pone.0024993
70. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275(4):2247–2250. doi:10.1074/jbc.275.4.2247
71. Mihm S. Danger-associated molecular patterns (DAMPs): molecular triggers for sterile inflammation in the liver. Int J Mol Sci. 2018;19:10. doi:10.3390/ijms19103104
72. Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi:10.1002/hep.26592
73. McHedlidze T, Waldner M, Zopf S, et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39(2):357–371. doi:10.1016/j.immuni.2013.07.018
74. Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83(5):655–663.
75. Zhan SS, Jiang JX, Wu J, et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology. 2006;43(3):435–443. doi:10.1002/hep.21093
76. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60(5):1090–1096. doi:10.1016/j.jhep.2013.12.025
77. Fallowfield JA, Mizuno M, Kendall TJ, et al. Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol. 2007;178(8):5288–5295. doi:10.4049/jimmunol.178.8.5288
78. Pellicoro A, Aucott RL, Ramachandran P, et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology. 2012;55(6):1965–1975. doi:10.1002/hep.25567
79. Ramachandran P, Pellicoro A, Vernon MA, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(46):E3186–E3195. doi:10.1073/pnas.1119964109
80. Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52(4):1390–1400. doi:10.1002/hep.23795
81. Schuppan D, Ruehl M, Somasundaram R, Hahn EG. Matrix as a modulator of hepatic fibrogenesis. Semin Liver Dis. 2001;21(3):351–372. doi:10.1055/s-2001-17556
82. Olaso E, Ikeda K, Eng FJ, et al. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest. 2001;108(9):1369–1378. doi:10.1172/JCI12373
83. Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47(4):1394–1400. doi:10.1002/hep.22193
84. Henderson NC, Arnold TD, Katamura Y, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19(12):1617–1624. doi:10.1038/nm.3282
85. Ikeda K, Wang LH, Torres R, et al. Discoidin domain receptor 2 interacts with Src and Shc following its activation by type I collagen. J Biol Chem. 2002;277(21):19206–19212. doi:10.1074/jbc.M201078200
86. Friedman SL, Sheppard D, Duffield JS, Violette S. Therapy for fibrotic diseases: nearing the starting line. Sci Transl Med. 2013;5(167):167sr161. doi:10.1126/scitranslmed.3004700
87. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786–801. doi:10.1038/nrm3904
88. Kurokawa T, Zheng YW, Ohkohchi N. Novel functions of platelets in the liver. J Gastroenterol Hepatol. 2016;31(4):745–751. doi:10.1111/jgh.13244
89. Nowatari T, Murata S, Fukunaga K, Ohkohchi N. Role of platelets in chronic liver disease and acute liver injury. Hepatol Res. 2014;44(2):165–172. doi:10.1111/hepr.12205
90. Jeong WI, Park O, Suh YG, et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology. 2011;53(4):1342–1351. doi:10.1002/hep.24190
91. Glässner A, Eisenhardt M, Krämer B, et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest. 2012;92(7):967–977. doi:10.1038/labinvest.2012.54
92. Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. doi:10.1016/j.cell.2008.06.049
93. Deleve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology. 2008;48(3):920–930. doi:10.1002/hep.22351
94. Ding BS, Cao Z, Lis R, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505(7481):97–102. doi:10.1038/nature12681
95. Thapa M, Chinnadurai R, Velazquez VM, et al. Liver fibrosis occurs through dysregulation of MyD88-dependent innate B-cell activity. Hepatology. 2015;61(6):2067–2079. doi:10.1002/hep.27761
96. Novobrantseva TI, Majeau GR, Amatucci A, et al. Attenuated liver fibrosis in the absence of B cells. J Clin Invest. 2005;115(11):3072–3082. doi:10.1172/JCI24798
97. Shi M, Zhu J, Wang R, et al. Latent TGF-β structure and activation. Nature. 2011;474(7351):343–349. doi:10.1038/nature10152
98. Dooley S, Ten Dijke P. TGF-β in progression of liver disease. Cell Tissue Res. 2012;347(1):245–256. doi:10.1007/s00441-011-1246-y
99. Kocabayoglu P, Lade A, Lee YA, et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J Hepatol. 2015;63(1):141–147. doi:10.1016/j.jhep.2015.01.036
100. Czochra P, Klopcic B, Meyer E, et al. Liver fibrosis induced by hepatic overexpression of PDGF-B in transgenic mice. J Hepatol. 2006;45(3):419–428. doi:10.1016/j.jhep.2006.04.010
101. Hao C, Xie Y, Peng M, et al. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation in vitro and prevents liver fibrosis in vivo. Clin Exp Med. 2014;14(2):141–150. doi:10.1007/s10238-013-0229-6
102. Fuchs BC, Hoshida Y, Fujii T, et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology. 2014;59(4):1577–1590. doi:10.1002/hep.26898
103. Lanaya H, Natarajan A, Komposch K, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol. 2014;16(10):972–977. doi:10.1038/ncb3031
104. Chung SI, Moon H, Ju HL, et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J Hepatol. 2016;64(3):618–627. doi:10.1016/j.jhep.2015.10.007
105. Chen Y, Choi SS, Michelotti GA, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143(5):1319–1329. doi:10.1053/j.gastro.2012.07.115
106. El-Agroudy NN, El-Naga RN, El-Razeq RA, El-Demerdash E. Forskolin, a hedgehog signalling inhibitor, attenuates carbon tetrachloride-induced liver fibrosis in rats. Br J Pharmacol. 2016;173(22):3248–3260. doi:10.1111/bph.13611
107. Pratap A, Singh S, Mundra V, et al. Attenuation of early liver fibrosis by pharmacological inhibition of smoothened receptor signaling. J Drug Target. 2012;20(9):770–782. doi:10.3109/1061186X.2012.719900
108. Mallat A, Teixeira-Clerc F, Lotersztajn S. Cannabinoid signaling and liver therapeutics. J Hepatol. 2013;59(4):891–896. doi:10.1016/j.jhep.2013.03.032
109. Guillot A, Hamdaoui N, Bizy A, et al. Cannabinoid receptor 2 counteracts interleukin-17-induced immune and fibrogenic responses in mouse liver. Hepatology. 2014;59(1):296–306. doi:10.1002/hep.26598
110. Gross B, Pawlak M, Lefebvre P, Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol. 2017;13(1):36–49. doi:10.1038/nrendo.2016.135
111. Zhang F, Kong D, Chen L, et al. Peroxisome proliferator-activated receptor-γ interrupts angiogenic signal transduction by transrepression of platelet-derived growth factor-β receptor in hepatic stellate cells. J Cell Sci. 2014;127(Pt 2):305–314. doi:10.1242/jcs.128306
112. Qian J, Niu M, Zhai X, Zhou Q, Zhou Y. β-Catenin pathway is required for TGF-β1 inhibition of PPARγ expression in cultured hepatic stellate cells. Pharmacol Res. 2012;66(3):219–225. doi:10.1016/j.phrs.2012.06.003
113. Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–1685. doi:10.1056/NEJMoa0907929
114. Ratziu V, Harrison SA, Francque S, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150(5):1147–1159. doi:10.1053/j.gastro.2016.01.038
115. Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut. 2017;66(1):180–190. doi:10.1136/gutjnl-2016-312431
116. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi:10.1016/S0140-6736(14)61933-4
117. Ding N, RT Y, Subramaniam N, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153(3):601–613. doi:10.1016/j.cell.2013.03.028
118. Hellemans K, Grinko I, Rombouts K, Schuppan D, Geerts A. All-trans and 9-cis retinoic acid alter rat hepatic stellate cell phenotype differentially. Gut. 1999;45(1):134–142. doi:10.1136/gut.45.1.134
119. Duran A, Hernandez ED, Reina-Campos M, et al. p62/SQSTM1 by binding to vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell. 2016;30(4):595–609. doi:10.1016/j.ccell.2016.09.004
120. Beaven SW, Wroblewski K, Wang J, et al. Liver X receptor signaling is a determinant of stellate cell activation and susceptibility to fibrotic liver disease. Gastroenterology. 2011;140(3):1052–1062. doi:10.1053/j.gastro.2010.11.053
121. Li T, Eheim AL, Klein S, et al. Novel role of nuclear receptor Rev-erbα in hepatic stellate cell activation: potential therapeutic target for liver injury. Hepatology. 2014;59(6):2383–2396. doi:10.1002/hep.27049
122. Okuno M, Moriwaki H, Imai S, et al. Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-β in liver stellate cells. Hepatology. 1997;26(4):913–921. doi:10.1053/jhep.1997.v26.pm0009328313
123. Hardy T, Mann DA. Epigenetics in liver disease: from biology to therapeutics. Gut. 2016;65(11):1895–1905. doi:10.1136/gutjnl-2015-311292
124. Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138(2):
125. Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 2007;14(2):275–285. doi:10.1038/sj.cdd.4401979
126. El Taghdouini A, Sørensen AL, Reiner AH, et al. Genome-wide analysis of DNA methylation and gene expression patterns in purified, uncultured human liver cells and activated hepatic stellate cells. Oncotarget. 2015;6(29):26729–26745. doi:10.18632/oncotarget.4925
127. Bian EB, Huang C, Ma TT, et al. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol. 2012;264(1):13–22. doi:10.1016/j.taap.2012.06.022
128. Zeybel M, Hardy T, Wong YK, et al. Multigenerational epigenetic adaptation of the hepatic wound-healing response. Nat Med. 2012;18(9):1369–1377. doi:10.1038/nm.2893
129. Kitano M, Bloomston PM. Hepatic stellate cells and microRNAs in pathogenesis of liver fibrosis. J Clin Med. 2016;5:3. doi:10.3390/jcm5030038
130. Yu F, Lu Z, Huang K, et al. MicroRNA-17-5p-activated Wnt/β-catenin pathway contributes to the progression of liver fibrosis. Oncotarget. 2016;7(1):81–93. doi:10.18632/oncotarget.6447
131. Coll M, El Taghdouini A, Perea L, et al. Integrative miRNA and gene expression profiling analysis of human quiescent hepatic stellate cells. Sci Rep. 2015;5:11549. doi:10.1038/srep11549
132. Hyun J, Wang S, Kim J, et al. MicroRNA-378 limits activation of hepatic stellate cells and liver fibrosis by suppressing Gli3 expression. Nat Commun. 2016;7:10993. doi:10.1038/ncomms10993
133. Sun X, He Y, Ma TT, Huang C, Zhang L, Li J. Participation of miR-200a in TGF-β1-mediated hepatic stellate cell activation. Mol Cell Biochem. 2014;388(1–2):11–23. doi:10.1007/s11010-013-1895-0
134. Yang JJ, Tao H, Hu W, et al. MicroRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell Signal. 2014;26(11):2381–2389. doi:10.1016/j.cellsig.2014.07.016
135. Yang JJ, Tao H, Liu LP, Hu W, Deng ZY, Li J. miR-200a controls hepatic stellate cell activation and fibrosis via SIRT1/Notch1 signal pathway. Inflamm Res. 2017;66(4):341–352. doi:10.1007/s00011-016-1020-4
136. Page A, Paoli PP, Hill SJ, et al. Alcohol directly stimulates epigenetic modifications in hepatic stellate cells. J Hepatol. 2015;62(2):388–397. doi:10.1016/j.jhep.2014.09.033
137. Hernández-Gea V, Hilscher M, Rozenfeld R, et al. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol. 2013;59(1):98–104. doi:10.1016/j.jhep.2013.02.016
138. Thoen LF, Guimarães EL, Dollé L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55(6):1353–1360. doi:10.1016/j.jhep.2011.07.010
139. Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946. doi:10.1053/j.gastro.2011.12.044
140. Koo JH, Lee HJ, Kim W, Kim SG. Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK-mediated degradation of HNRNPA1 and up-regulation of SMAD2. Gastroenterology. 2016;150(1):181–193. doi:10.1053/j.gastro.2015.09.039
141. Kim RS, Hasegawa D, Goossens N, et al. The XBP1 arm of the unfolded protein response induces fibrogenic activity in hepatic stellate cells through autophagy. Sci Rep. 2016;6:39342.
142. Heindryckx F, Binet F, Ponticos M, et al. Endoplasmic reticulum stress enhances fibrosis through IRE1α-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol Med. 2016;8(7):729–744. doi:10.15252/emmm.201505925
143. Kawasaki K, Ushioda R, Ito S, Ikeda K, Masago Y, Nagata K. Deletion of the collagen-specific molecular chaperone Hsp47 causes endoplasmic reticulum stress-mediated apoptosis of hepatic stellate cells. J Biol Chem. 2015;290(6):3639–3646. doi:10.1074/jbc.M114.592139
144. Yi HS, Lee YS, Byun JS, et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer cells in mice. Hepatology. 2014;60(3):1044–1053. doi:10.1002/hep.27137
145. Wilson CL, Murphy LB, Leslie J, et al. Ubiquitin C-terminal hydrolase 1: a novel functional marker for liver myofibroblasts and a therapeutic target in chronic liver disease. J Hepatol. 2015;63(6):1421–1428. doi:10.1016/j.jhep.2015.07.034
146. Meng F, Wang K, Aoyama T, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143(3):765–776. doi:10.1053/j.gastro.2012.05.049
147. Chiu YS, Wei CC, Lin YJ, Hsu YH, Chang MS. IL-20 and IL-20R1 antibodies protect against liver fibrosis. Hepatology. 2014;60(3):1003–1014. doi:10.1002/hep.27189
148. Jiao J, Ooka K, Fey H, et al. Interleukin-15 receptor α on hepatic stellate cells regulates hepatic fibrogenesis in mice. J Hepatol. 2016;65(2):344–353. doi:10.1016/j.jhep.2016.04.020
149. Kong X, Feng D, Wang H, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56(3):1150–1159. doi:10.1002/hep.25744
150. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14(3):181–194. doi:10.1038/nri3623
151. Kendall TJ, Hennedige S, Aucott RL, et al. p75 Neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology. 2009;49(3):901–910. doi:10.1002/hep.22701
152. Oh Y, Park O, Swierczewska M, et al. Systemic PEGylated TRAIL treatment ameliorates liver cirrhosis in rats by eliminating activated hepatic stellate cells. Hepatology. 2016;64(1):209–223. doi:10.1002/hep.28432
153. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–1916. doi:10.1128/IAI.73.4.1907-1916.2005
154. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64(5):830–841. doi:10.1136/gutjnl-2014-306842
155. Karin M, Lin A. NF-κB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. doi:10.1038/ni0302-221
156. Kucharczak J, Simmons MJ, Fan Y, Gélinas C. To be, or not to be: NF-κB is the answer–role of Rel/NF-κB in the regulation of apoptosis. Oncogene. 2003;22(56):8961–8982. doi:10.1038/sj.onc.1207230
157. Pradere JP, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–1473. doi:10.1002/hep.26429
158. Anan A, Baskin-Bey ES, Bronk SF, Werneburg NW, Shah VH, Gores GJ. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology. 2006;43(2):335–344. doi:10.1002/hep.21036
159. Murphy FR, Issa R, Zhou X, et al. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition: implications for reversibility of liver fibrosis. J Biol Chem. 2002;277(13):11069–11076. doi:10.1074/jbc.M111490200
160. Shaker ME, Ghani A, Shiha GE, Ibrahim TM, Mehal WZ. Nilotinib induces apoptosis and autophagic cell death of activated hepatic stellate cells via inhibition of histone deacetylases. Biochim Biophys Acta. 2013;1833(8):1992–2003. doi:10.1016/j.bbamcr.2013.02.033
161. Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–6251. doi:10.1038/onc.2008.301
162. Hao H, Zhang D, Shi J, et al. Sorafenib induces autophagic cell death and apoptosis in hepatic stellate cell through the JNK and Akt signaling pathways. Anticancer Drugs. 2016;27(3):192–203. doi:10.1097/CAD.0000000000000316
163. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–496. doi:10.1038/nrm3823
164. Chicas A, Wang X, Zhang C, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17(4):376–387. doi:10.1016/j.ccr.2010.01.023
165. Sun P, Yoshizuka N, New L, et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128(2):295–308. doi:10.1016/j.cell.2006.11.050
166. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi:10.1016/s0092-8674(00)81902-9
167. Alimonti A, Nardella C, Chen Z, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest. 2010;120(3):681–693. doi:10.1172/JCI40535
168. Schnabl B, Purbeck CA, Choi YH, Hagedorn CH, Brenner D. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology. 2003;37(3):653–664. doi:10.1053/jhep.2003.50097
169. Evan GI, d’Adda Di Fagagna F. Cellular senescence: hot or what? Curr Opin Genet Dev. 2009;19(1):25–31. doi:10.1016/j.gde.2008.11.009
170. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi:10.1146/annurev-physiol-030212-183653
171. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
172. Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9(2):81–94. doi:10.1038/nrc2560
173. Acosta JC, O’Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–1018. doi:10.1016/j.cell.2008.03.038
174. Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi:10.1016/j.cell.2008.03.039
175. Kisseleva T, Cong M, Paik Y, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(24):9448–9453. doi:10.1073/pnas.1201840109
176. Troeger JS, Mederacke I, Gwak GY, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143(4):1073–1083. doi:10.1053/j.gastro.2012.06.036
177. Huang P, Zhang L, Gao Y, et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell Stem Cell. 2014;14(3):370–384. doi:10.1016/j.stem.2014.01.003
178. Song G, Pacher M, Balakrishnan A, et al. Direct reprogramming of hepatic myofibroblasts into hepatocytes in vivo attenuates liver fibrosis. Cell Stem Cell. 2016;18(6):797–808. doi:10.1016/j.stem.2016.01.010
179. Chang ML, Yeh CT, Chang PY, Chen JC. Comparison of murine cirrhosis models induced by hepatotoxin administration and common bile duct ligation. World J Gastroenterol. 2005;11(27):4167–4172. doi:10.3748/wjg.v11.i27.4167
180. van Zonneveld M, Zondervan PE, Cakaloglu Y, et al. Peg-interferon improves liver histology in patients with HBeAg-positive chronic hepatitis B: no additional benefit of combination with lamivudine. Liver Int. 2006;26(4):399–405. doi:10.1111/j.1478-3231.2006.01257.x
181. Xu B, Lin L, Xu G, et al. Long-term lamivudine treatment achieves regression of advanced liver fibrosis/cirrhosis in patients with chronic hepatitis B. J Gastroenterol Hepatol. 2015;30(2):372–378. doi:10.1111/jgh.12718
182. Dienstag JL, Goldin RD, Heathcote EJ, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124(1):105–117. doi:10.1053/gast.2003.50013
183. Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet. 2013;381(9865):468–475. doi:10.1016/S0140-6736(12)61425-1
184. Buti M, Fung S, Gane E, et al. Long-term clinical outcomes in cirrhotic chronic hepatitis B patients treated with tenofovir disoproxil fumarate for up to 5 years. Hepatol Int. 2015;9(2):243–250. doi:10.1007/s12072-015-9614-4
185. Tsai NC, Marcellin P, Buti M, et al. Viral suppression and cirrhosis regression with tenofovir disoproxil fumarate in Asians with chronic hepatitis B. Dig Dis Sci. 2015;60(1):260–268. doi:10.1007/s10620-014-3336-7
186. Chang TT, Liaw YF, Wu SS, et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology. 2010;52(3):886–893. doi:10.1002/hep.23785
187. Schiff ER, Lee SS, Chao YC, et al. Long-term treatment with entecavir induces reversal of advanced fibrosis or cirrhosis in patients with chronic hepatitis B. Clin Gastroenterol Hepatol. 2011;9(3):274–276. doi:10.1016/j.cgh.2010.11.040
188. Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B for up to 5 years. Gastroenterology. 2006;131(6):1743–1751. doi:10.1053/j.gastro.2006.09.020
189. Hou JL, Xu D, Shi G, et al. Long-term telbivudine treatment results in resolution of liver inflammation and fibrosis in patients with chronic hepatitis B. Adv Ther. 2015;32(8):727–741. doi:10.1007/s12325-015-0232-2
190. Lok AS, Everhart JE, Chung RT, et al. Evolution of hepatic steatosis in patients with advanced hepatitis C: results from the hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) trial. Hepatology. 2009;49(6):1828–1837. doi:10.1002/hep.22865
191. Chen CJ, Yang HI, Iloeje UH, Group R-HS. Hepatitis B virus DNA levels and outcomes in chronic hepatitis B. Hepatology. 2009;49(5 Suppl):S72–S84. doi:10.1002/hep.22884
192. George SL, Bacon BR, Brunt EM, Mihindukulasuriya KL, Hoffmann J, Di Bisceglie AM. Clinical, virologic, histologic, and biochemical outcomes after successful HCV therapy: a 5-year follow-up of 150 patients. Hepatology. 2009;49(3):729–738. doi:10.1002/hep.22694
193. Bachofner JA, Valli PV, Kröger A, et al. Direct antiviral agent treatment of chronic hepatitis C results in rapid regression of transient elastography and fibrosis markers fibrosis-4 score and aspartate aminotransferase-platelet ratio index. Liver Int. 2017;37(3):369–376. doi:10.1111/liv.13256
194. Enomoto M, Ikura Y, Tamori A, et al. Short-term histological evaluations after achieving a sustained virologic response to direct-acting antiviral treatment for chronic hepatitis C. United Eur Gastroenterol J. 2018;6(9):1391–1400. doi:10.1177/2050640618791053
195. Lledó GM, Carrasco I, Benítez-Gutiérrez LM, et al. Regression of liver fibrosis after curing chronic hepatitis C with oral antivirals in patients with and without HIV coinfection. AIDS. 2018;32(16):2347–2352. doi:10.1097/QAD.0000000000001966
196. Colmenero J, Bataller R, Sancho-Bru P, et al. Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am J Physiol Gastrointest Liver Physiol. 2009;297(4):G726–G734. doi:10.1152/ajpgi.00162.2009
197. McHutchison J, Goodman Z, Patel K, et al. Farglitazar lacks antifibrotic activity in patients with chronic hepatitis C infection. Gastroenterology. 2010;138(4):
198. Verrill C, Markham H, Templeton A, Carr NJ, Sheron N. Alcohol-related cirrhosis–early abstinence is a key factor in prognosis, even in the most severe cases. Addiction. 2009;104(5):768–774. doi:10.1111/j.1360-0443.2009.02521.x
199. Kim MY, Cho MY, Baik SK, et al. Beneficial effects of candesartan, an angiotensin-blocking agent, on compensated alcoholic liver fibrosis - a randomized open-label controlled study. Liver Int. 2012;32(6):977–987. doi:10.1111/j.1478-3231.2012.02774.x
200. Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36(3):525–531. doi:10.1053/jhep.2002.36088
201. Poupon RE, Poupon R, Balkau B. Ursodiol for the long-term treatment of primary biliary cirrhosis. The UDCA-PBC study group. N Engl J Med. 1994;330(19):1342–1347. doi:10.1056/NEJM199405123301903
202. Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology. 2000;32(6):1196–1199. doi:10.1053/jhep.2000.20240
203. Czaja AJ, Carpenter HA. Decreased fibrosis during corticosteroid therapy of autoimmune hepatitis. J Hepatol. 2004;40(4):646–652. doi:10.1016/j.jhep.2004.01.009
204. Mohamadnejad M, Malekzadeh R, Nasseri-Moghaddam S, et al. Impact of immunosuppressive treatment on liver fibrosis in autoimmune hepatitis. Dig Dis Sci. 2005;50(3):547–551. doi:10.1007/s10620-005-2472-5
205. Falize L, Guillygomarc’h A, Perrin M, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: a study of 36 cases. Hepatology. 2006;44(2):472–477. doi:10.1002/hep.21260
206. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander-Tetri BA; (CRN) NCRN. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology. 2011;53(3):810–820. doi:10.1002/hep.24127
207. Promrat K, Kleiner DE, Niemeier HM, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51(1):121–129. doi:10.1002/hep.23276
208. Aithal GP, Thomas JA, Kaye PV, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135(4):1176–1184. doi:10.1053/j.gastro.2008.06.047
209. Ratziu V, Charlotte F, Bernhardt C, et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51(2):445–453. doi:10.1002/hep.23270
210. Zein CO, Yerian LM, Gogate P, et al. Pentoxifylline improves nonalcoholic steatohepatitis: a randomized placebo-controlled trial. Hepatology. 2011;54(5):1610–1619. doi:10.1002/hep.24544
211. Gorina E, Richeldi L, Raghu G, et al. PRAISE, a randomized, placebo-controlled, double-blind Phase 2 clinical trial of pamrevlumab (FG-3019) in IPF patients. Eur Respir J. 2017;50(suppl61):OA3400. doi:10.1183/13993003.00711-2017
212. Harrison SA, Abdelmalek MF, Caldwell S, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. 2018;155(4):1140–1153. doi:10.1053/j.gastro.2018.07.006
213. Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin αvβ6 on cholangiocytes blocks transforming growth factor-β activation and retards biliary fibrosis progression. Gastroenterology. 2008;135(2):660–670. doi:10.1053/j.gastro.2008.04.009
214. Friedman SL, Ratziu V, Harrison SA, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67(5):1754–1767. doi:10.1002/hep.29477
215. Frenette CT, Morelli G, Shiffman ML, et al. Emricasan improves liver function in patients with cirrhosis and high Model for End-Stage Liver Disease scores compared with placebo. Clin Gastroenterol Hepatol. 2019;17(4):774–783. doi:10.1016/j.cgh.2018.06.012
216. Loomba R, Lawitz E, Mantry PS, et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology. 2018;67(2):549–559. doi:10.1002/hep.29514.
217. Jang YO, Kim YJ, Baik SK, et al. Histological improvement following administration of autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: a pilot study. Liver Int. 2014;34(1):33–41. doi:10.1111/liv.12218
218. Zhang Z, Lin H, Shi M, et al. Human umbilical cord mesenchymal stem cells improve liver function and ascites in decompensated liver cirrhosis patients. J Gastroenterol Hepatol. 2012;27(Suppl 2):112–120. doi:10.1111/j.1440-1746.2011.07024.x
219. Shi M, Zhang Z, Xu R, et al. Human mesenchymal stem cell transfusion is safe and improves liver function in acute-on-chronic liver failure patients. Stem Cells Transl Med. 2012;1(10):725–731. doi:10.5966/sctm.2012-0034
220. Wang L, Li J, Liu H, et al. Pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J Gastroenterol Hepatol. 2013;28(Suppl 1):85–92. doi:10.1111/jgh.12029
221. Mohamadnejad M, Alimoghaddam K, Mohyeddin-Bonab M, et al. Phase 1 trial of autologous bone marrow mesenchymal stem cell transplantation in patients with decompensated liver cirrhosis. Arch Iran Med. 2007;10(4):459–466.
222. Kharaziha P, Hellström PM, Noorinayer B, et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: a phase I-II clinical trial. Eur J Gastroenterol Hepatol. 2009;21(10):1199–1205. doi:10.1097/MEG.0b013e32832a1f6c
223. Peng L, Xie DY, Lin BL, et al. Autologous bone marrow mesenchymal stem cell transplantation in liver failure patients caused by hepatitis B: short-term and long-term outcomes. Hepatology. 2011;54(3):820–828. doi:10.1002/hep.24434
224. El-Ansary M, Abdel-Aziz I, Mogawer S, et al. Phase II trial: undifferentiated versus differentiated autologous mesenchymal stem cells transplantation in Egyptian patients with HCV induced liver cirrhosis. Stem Cell Rev. 2012;8(3):972–981. doi:10.1007/s12015-011-9322-y
225. Amin MA, Sabry D, Rashed LA, et al. Short-term evaluation of autologous transplantation of bone marrow-derived mesenchymal stem cells in patients with cirrhosis: Egyptian study. Clin Transplant. 2013;27(4):607–612. doi:10.1111/ctr.12179
226. Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proc Natl Acad Sci U S A. 2004;101(34):12467–12472. doi:10.1073/pnas.0401786101
227. Kuai J, Mosyak L, Brooks J, et al. Characterization of binding mode of action of a blocking anti-platelet-derived growth factor (PDGF)-B monoclonal antibody, MOR8457, reveals conformational flexibility and avidity needed for PDGF-BB to bind PDGF receptor-β. Biochemistry. 2015;54(10):1918–1929. doi:10.1021/bi5015425
228. Huang HW, Mohan SK, Yu C. The NMR solution structure of human epidermal growth factor (hEGF) at physiological pH and its interactions with suramin. Biochem Biophys Res Commun. 2010;402(4):705–710. doi:10.1016/j.bbrc.2010.10.089
229. Hua C, Zhu Y, Xu W, et al. Characterization by high-resolution crystal structure analysis of a triple-helix region of human collagen type III with potent cell adhesion activity. Biochem Biophys Res Commun. 2019;508(4):1018–1023. doi:10.1016/j.bbrc.2018.12.018
230. Elkins PA, Ho YS, Smith WW, et al. Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 7):1182–1192. doi:10.1107/s0907444902007849
231. Rose AS, Bradley AR, Valasatava Y, Duarte JM, Prlic A, Rose PW. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018;34(21):3755–3758. doi:10.1093/bioinformatics/bty419
232. Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, Hinck AP. Crystal structure of the human TβR2 ectodomain–TGF-β3 complex. Nat Struct Biol. 2002;9(3):203–208. doi:10.1038/nsb766
233. Ogunjimi AA, Zeqiraj E, Ceccarelli DF, Sicheri F, Wrana JL, David L. Structural basis for specificity of TGFβ family receptor small molecule inhibitors. Cell Signal. 2012;24(2):476–483. doi:10.1016/j.cellsig.2011.09.027
234. Tebben AJ, Ruzanov M, Gao M, et al. Crystal structures of apo and inhibitor-bound TGFβR2 kinase domain: insights into TGFβR isoform selectivity. Acta Crystallogr D Struct Biol. 2016;72(Pt 5):658–674. doi:10.1107/S2059798316003624
235. Wang Q, Liu F, Qi S, et al. Discovery of 4-((N-(2-(dimethylamino)ethyl)acrylamido)methyl)-N-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)phenyl)benzamide (CHMFL-PDGFR-159) as a highly selective type II PDGFRα kinase inhibitor for PDGFRα driving chronic eosinophilic leukemia. Eur J Med Chem. 2018;150:366–384. doi:10.1016/j.ejmech.2018.03.003
236. Peng P, Chen H, Zhu Y, et al. Structure-based design of 1-heteroaryl-1,3-propanediamine derivatives as a novel series of CC-chemokine receptor 5 antagonists. J Med Chem. 2018;61(21):9621–9636. doi:10.1021/acs.jmedchem.8b01077
237. Liu S, Song X, Chrunyk BA, et al. Crystal structures of interleukin 17A and its complex with IL-17 receptor A. Nat Commun. 2013;4:1888. doi:10.1038/ncomms2880
238. Su L, Wang Y, Wang J, et al. Structural basis of TLR2/TLR1 activation by the synthetic agonist diprovocim. J Med Chem. 2019;62(6):2938–2949. doi:10.1021/acs.jmedchem.8b01583
239. Mineev KS, Goncharuk SA, Arseniev AS. Toll-like receptor 3 transmembrane domain is able to perform various homotypic interactions: an NMR structural study. FEBS Lett. 2014;588(21):3802–3807. doi:10.1016/j.febslet.2014.08.031
240. Nyman T, Stenmark P, Flodin S, Johansson I, Hammarström M, Nordlund P. The crystal structure of the human toll-like receptor 10 cytoplasmic domain reveals a putative signaling dimer. J Biol Chem. 2008;283(18):11861–11865. doi:10.1074/jbc.C800001200
241. Casimiro-Garcia A, Bigge CF, Davis JA, et al. Effects of modifications of the linker in a series of phenylpropanoic acid derivatives: synthesis, evaluation as PPARα/γ dual agonists, and X-ray crystallographic studies. Bioorg Med Chem. 2008;16(9):4883–4907. doi:10.1016/j.bmc.2008.03.043
242. Chandra V, Huang P, Hamuro Y, et al. Structure of the intact PPAR-γ-RXR- nuclear receptor complex on DNA. Nature. 2008;456(7220):350–356. doi:10.1038/nature07413
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.