Back to Journals » OncoTargets and Therapy » Volume 9
Lead identification for the K-Ras protein: virtual screening and combinatorial fragment-based approaches
Authors Pathan AAK, Panthi B, Khan Z, Koppula PR ![]() , Alanazi M, Sachchidanand S, Parine NR, Chourasia M
, Alanazi M, Sachchidanand S, Parine NR, Chourasia M
Received 2 November 2015
Accepted for publication 18 February 2016
Published 2 May 2016 Volume 2016:9 Pages 2575—2584
DOI https://doi.org/10.2147/OTT.S99671
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Akbar Ali Khan Pathan,1,2,* Bhavana Panthi,3,* Zahid Khan,1 Purushotham Reddy Koppula,4–6 Mohammed Saud Alanazi,1 Sachchidanand,3 Narasimha Reddy Parine,1 Mukesh Chourasia3,*
1Genome Research Chair (GRC), Department of Biochemistry, College of Science, King Saud University, 2Integrated Gulf Biosystems, Riyadh, Kingdom of Saudi Arabia; 3Department of Pharmacoinformatics, National Institute of Pharmaceutical Education and Research, Hajipur, India; 4Department of Internal Medicine, School of Medicine, 5Harry S. Truman Memorial Veterans Affairs Hospital, 6Department of Radiology, School of Medicine, Columbia, MO, USA
*These authors contributed equally to this work
Objective: Kirsten rat sarcoma (K-Ras) protein is a member of Ras family belonging to the small guanosine triphosphatases superfamily. The members of this family share a conserved structure and biochemical properties, acting as binary molecular switches. The guanosine triphosphate-bound active K-Ras interacts with a range of effectors, resulting in the stimulation of downstream signaling pathways regulating cell proliferation, differentiation, and apoptosis. Efforts to target K-Ras have been unsuccessful until now, placing it among high-value molecules against which developing a therapy would have an enormous impact. K-Ras transduces signals when it binds to guanosine triphosphate by directly binding to downstream effector proteins, but in case of guanosine diphosphate-bound conformation, these interactions get disrupted.
Methods: In the present study, we targeted the nucleotide-binding site in the “on” and “off” state conformations of the K-Ras protein to find out suitable lead compounds. A structure-based virtual screening approach has been used to screen compounds from different databases, followed by a combinatorial fragment-based approach to design the apposite lead for the K-Ras protein.
Results: Interestingly, the designed compounds exhibit a binding preference for the “off” state over “on” state conformation of K-Ras protein. Moreover, the designed compounds’ interactions are similar to guanosine diphosphate and, thus, could presumably act as a potential lead for K-Ras. The predicted drug-likeness properties of these compounds suggest that these compounds follow the Lipinski’s rule of five and have tolerable absorption, distribution, metabolism, excretion and toxicity values.
Conclusion: Thus, through the current study, we propose targeting only “off” state conformations as a promising strategy for the design of reversible inhibitors to pharmacologically inhibit distinct conformations of K-Ras protein.
Keywords: antitumor agent, K-Ras, molecular docking, molecular modeling, virtual screening
Corrigendum for this paper has been published
Introduction
Guanosine triphosphatases (GTPases) are a class of proteins that include the Ras proteins. These related proteins are mainly involved in signal transduction, cell growth, motility, differentiation, and cell death.1 During the signaling process, Ras proteins act as molecular switches and bind to guanosine triphosphate (GTP) in the active state and revert to inactive form upon hydrolysis of GTP to guanosine diphosphate (GDP).2,3 Two flexible regions, switch-I that involves residues 32–38 and switch-II that involves residues 60–75, mediate the transition from one form to another.4 This change is catalyzed by guanine nucleotide exchange factors and GTPase-activating proteins (GAPs). In the last decade, numerous mutations, which reflect the oncogenic potential of Ras, have been reported in these two regions; most notably, the regions involving Gly12 and Gly13 in the G1 box and Gln61 in the G3 box are important. Point mutation in these coding regions hinders the activity of GTPase and makes Ras insensitive to GAP action. This initiates multiple signaling pathways with various downstream effectors, such as the Raf kinase and phosphatidylinositol 3-kinase-related protein families.5,6 Mutations in these regions are reported in almost all types of human cancer. Of these, 60%–90% are in pancreatic cancer and 30%–50% in colorectal cancers, with Kirsten rat sarcoma (K-Ras) being the most frequently mutated RAS gene.7–10 Weinstein reported that survival of cells depends on the activated RAS oncogene function and inhibition of activated RAS would lead to tumor regression and cell death.11,12 The guanine nucleotide (GN)-binding pocket dictates the switch conformation; therefore, developing compounds binding to this region will have a likelihood of modulating K-Ras signaling. Small molecules targeting the GN-binding site have been disregarded due to the pM affinity of GTP and GDP to K-Ras and their high intracellular concentration. SML-8-73-1 was shown to covalently bind K-Ras G12C mutant at the “off” state conformation, even in the presence of 1 mM concentration of GTP and GDP.13,14 K-Ras protein plays a key role in the signal transduction cascade. Mutation in Gly12, Gly13, and Gln61 residue of K-Ras leads to permanent “on” state results in tumorigenesis due to failed interaction with the GAPs. Three crucial residues, Tyr32, Thr35, and Gly60, show interaction with γ-phosphate of GTP. The inactive GDP-bound structure of K-Ras reveals the absence of these key interactions and increased distance between these residues and position of the γ-phosphate coinciding with large conformational change in both switch regions15–17 (Figure 1).
 | Figure 1 Superposition of GDP (green) and GTP (magenta)-bound conformation of K-Ras protein. |
Despite many attempts, there has not been much success in finding drugs for K-Ras. Therefore, it still remains a very challenging therapeutic target for the modelers to design inhibitors. In an attempt to identify new leads to inhibit K-Ras, we looked at the interaction18 of the tertiary structures of K-Ras with existing US Food and Drug Administration (FDA)-approved drugs and compounds from different databases by using structure-based virtual screening (VS). We utilized fragment-based approach and combinatorial library design strategy from screened library of different small molecule databases to find out better interacting molecules with favorable interaction followed by absorption, distribution, metabolism, excretion and toxicity property prediction to ensure the pharmacokinetic and toxicity profile of designed molecules. We further performed induced fit docking calculation in order to confirm protein ligand interactions in the binding site.
Materials and methods
Dataset collection and preparation
Compounds were selected from six different databases (Asinex, BindingDB, DrugBank, National Cancer Institute [NCI], PubChem, and Chembridge database) of varying size (numbers of molecules) and type (divers scaffolds) as a starting point for VS.19 In this study, 1 13,962 diverse small molecules from Asinex platinum collection, 1,602 FDA-approved small molecules drugs from DrugBank,20–22 408 Ras protein inhibitors from BindingDB,23–26 4,000 lead-like small molecules from KinaCore library and 10,000 drug-like compounds from KinaSet library of Chembridge database,27 1,990 compounds of NCI diversity set from NCI database,28 155 kinase inhibitors from PubChem compound databases,29 and 267 active molecules from PubChem bioassay (AID-759) entitled HTS were taken to identify specific small molecule inhibitors of Ras and Ras-related GTPases, specifically Ras wild type. The entire set of molecules was subjected to ligand preparation. Ligands were submitted to the LigPrep module of Schrödinger (Schrödinger Release 2015-1; Schrödinger, LLC, NY, USA) to generate ionization states populated at crystallized condition of K-Ras protein pH 8.5 by using epik, enumerating isomers and tautomers and generating three-dimensional conformations.
Protein preparation
The crystal structure of K-Ras protein complex with GDP (Protein Data Bank [PDB] ID: 4OBE) and GTP (PDB ID: 4DSO) was downloaded from PDB.30 4OBE is wild-type GDP-bound structure of K-Ras having a resolution 1.24 Å, and 4DSO is a G12D GTP-bound structure of K-Ras having a resolution 1.85 Å. 4DSO mutant is converted into wild-type structure by mutated Asp12 to Gly12. The mutation was incorporated into the structure by homology modeling of K-Ras sequence, for which 4DSO was used as a template. Both proteins were prepared by using protein preparation wizard panel of Schrödinger Suites 2015.1 (Schrödinger Release 2015-1). Hydrogens were added to its corresponding carbon atom and crystallized water molecules were removed from the structures. Protonation states of the titratable residues were assigned at 8.5 pH. The structures were minimized by using Optimized Potentials for Liquid Simulations 2005 force field. The grid was generated by considering cocrystallized GDP/GTP molecule as a grid center using glide for docking calculations.31
VS of chemical database
VS approach has been widely used in drug discovery process for lead identification and optimization.32 It provides an inexpensive and fast alternative to high-throughput screening for discovering new drugs.33 There are two broad categories of computational techniques for VS: 1) ligand-based drug design (eg, ligand similarity); and 2) structure-based drug design (ligand docking) methods. In this study, structure-based drug design (ligand docking) method was used.34–36 VS was performed by using glide module of Schrödinger suites 2015.1. In VS, prepared ligands from different databases were used as a ligand input and filter criterion based on Lipinski’s rule of five and removal of reactive functional groups were applied to the dataset and docked to the protein by using high-throughput VS.37–39 Molecules obtained from high-throughput VS were docked using the standard precision method, and the standard precision docked poses were further docked by using extra precision method.40,41 The top 100 molecules were finally selected out of 7,894 screened molecules from 113,962 molecules of Asinex platinum library, 16 hits out of 408 molecules obtained from the BindingDB database, 1,320 hits out of 4,000 molecules from KinaCore library and 1,006 hits from total 10,000 molecules from KinaSet of Chembridge database, 83 hits out of 1,480 FDA-approved drugs from Drug Bank database, 150 hits from 1,990 total molecules of NCI diversity set of NCI, and eight hits from 155 kinase inhibitors of PubChem Compound database and 26 hits out of 267 molecules from PubChem Bioassay of H-Ras in VS. Top-ranked molecules based on docking score and poses from each database were analyzed and selected for further study (Figure 2).
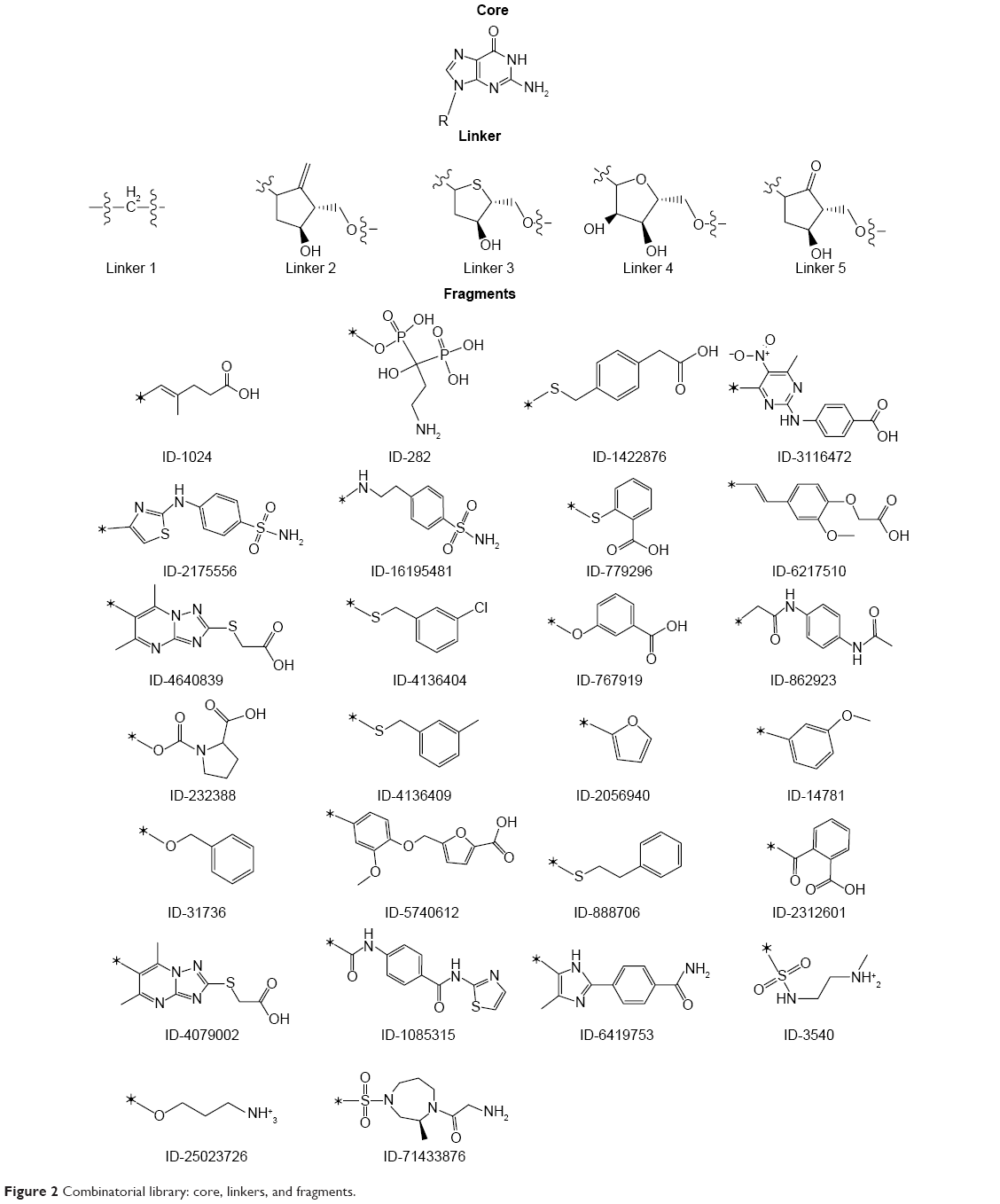 | Figure 2 Combinatorial library: core, linkers, and fragments. |
Generation of fragment library
New lead molecules for K-Ras were designed from the screened molecules by analyzing their interactions with the binding site residues.42,43 The binding-site occupancy of screened molecules from VS, particularly FDA-approved drugs, Ras inhibitors from BindingDB, kinase and H-Ras inhibitors from PubChem database, have been used as a starting point to formulate a dictionary of keys. To optimize the chemotype requirements for the GDP/GTP-bound conformations, fragment library was generated by pruning screened molecules from these databases on the basis of binding interactions and were further suggested according to the (or) part of the pocket occupied.44
Design of combinatorial library
Combinatorial library was generated by using CombiGlide workflow of Schrödinger. These workflows can be used either as a prelude to combinatorial libraries’ design that may be screened for leads and identifying novel scaffolds, or generate focused libraries in support of lead discovery and optimization process. To design lead molecule for the K-Ras GN-binding pocket, combinatorial library design strategy was utilized. The fragments were substituted into a common template core (guanine) at a defined attachment point to graft structural features that are present in different structures (Figure 3). All possible combinations of fragments and linker with the core were explored. The core, linker, and fragments were assembled into about 260 molecules.
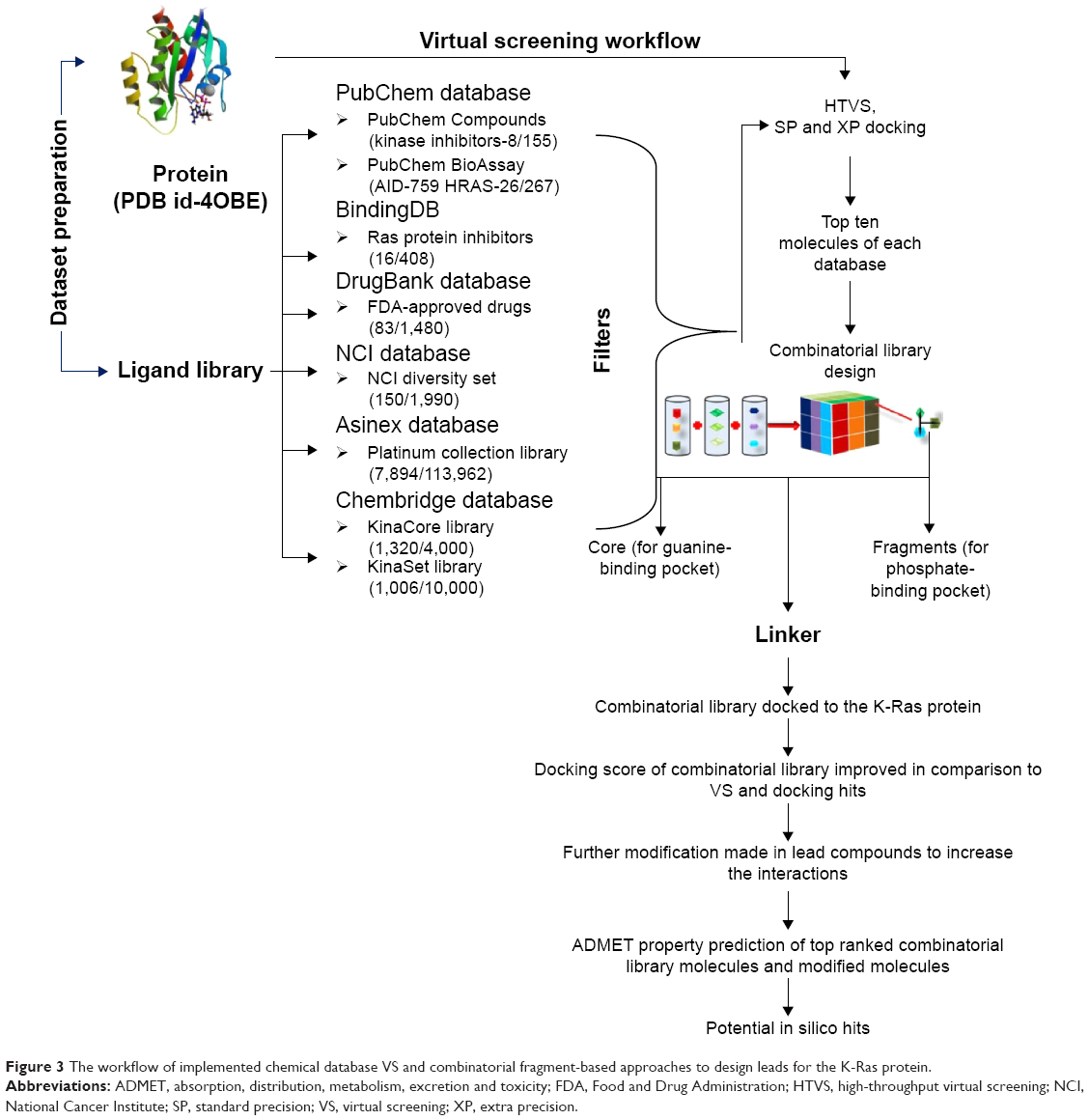 | Figure 3 The workflow of implemented chemical database VS and combinatorial fragment-based approaches to design leads for the K-Ras protein. |
Results
Active site analysis
The catalytic GN-binding site in the Ras family is conserved in spite of the variations in protein function and cellular localizations.45 The conformational changes are witnessed in the switch-I and switch-II loop region of GN-binding site on the conversion of GTP to GDP, which facilitates to address the specificity issue in the design of lead in the reactive and product two-state conformations. The binding pockets of different Ras are similar in sequence and size, whereas they vary in the conformation of protein in the “on” state and “off” state.
The Ras inhibitors developed over the years are mainly for the “on” state and failed to address the selectivity issue. The binding site and inhibitor surfaces are complementary to each other; therefore, it is essential to comprehend and investigate the properties of the binding pockets to add cognizance to the lead design. The active site of Ras has been divided into three subpockets, namely, guanine-binding pocket (GBP), ribose-binding pocket (RBP), and phosphate-binding pocket (PBP). In the GDP-bound conformation, one extended cavity was also observed (Figure 4). This cavity was formed by the residues Tyr32, Thr35, Gly60, which interact with the γ-phosphate in the presence of GTP. These interacting residues are exposed to the surface after phosphate hydrolysis, which creates a cavity. The backbone of Asp33 and Ile36 residues orients and exposes itself inside this cavity, which were inaccessible at GTP-bound conformation. Such conformational rearrangement in the “off” state allowed us to gaze extended cavity for the inhibitor design aspects which was not explored earlier. Its relatively big volume and varying nature of the inner lining in “off” state conformation enabled us to use different combinations of fragments that were selective for the subpocket.
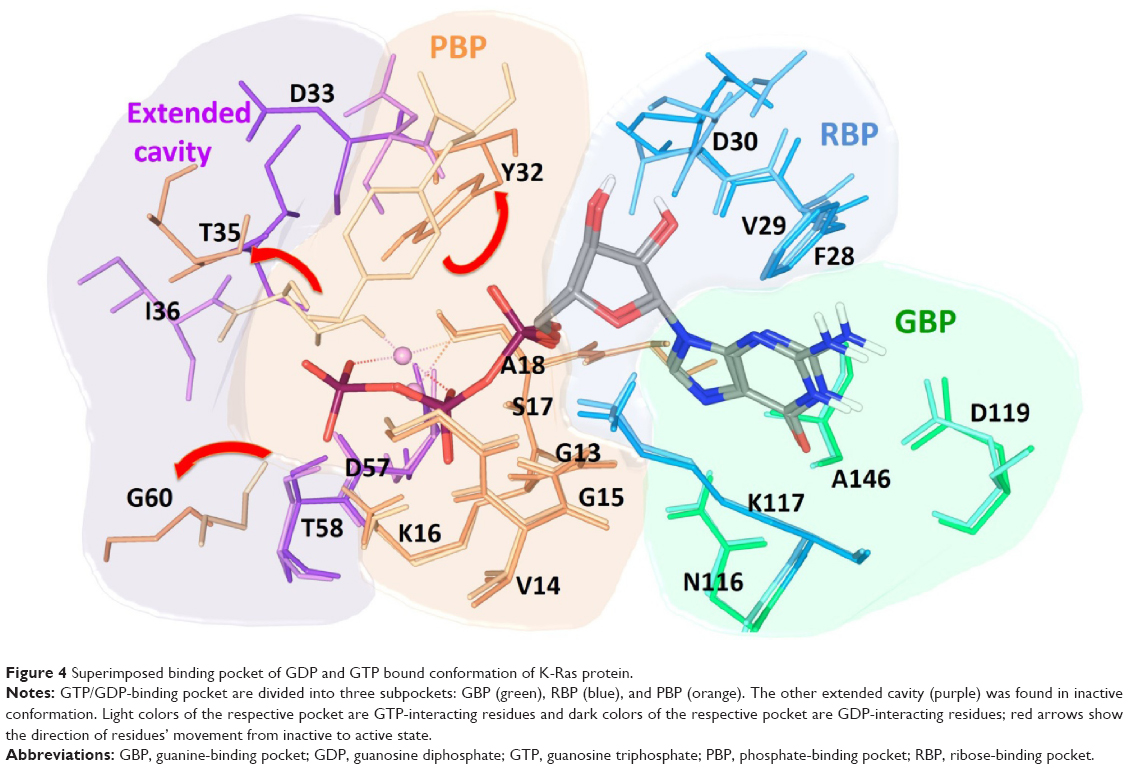 | Figure 4 Superimposed binding pocket of GDP and GTP bound conformation of K-Ras protein. |
The tightly packed guanine base at the GBP accommodates and forms hydrogen bonds (H-bonds) with active site residues Asn116, Lys117, Asp119, and Ala146. The residues Phe28 and Lys117 are positioned in such a way that it can form additional π–π and cation–π interactions, respectively, with guanine base. Asp30 shows H-bonds with ribose sugar. Residues Gly13, Val14, Gly15, Lys16, Ser17, and Ala18 form very strong H-bond network with phosphates in PBP. The superimposed GTP-bound conformation over GDP-bound conformation was showing considerably significant conformational changes in switch-I and -II regions of active site. GTP and GDP share common interaction with the GBP residues, but due to the presence of γ-phosphate in GTP, it forms an additional H-bond interaction with Val29, Tyr32, Thr35, and Gly60.
Database screening
VS methods are central to many problems of chemoinformatics in design, selection, and analysis of small molecules. VS is a useful technique to rapidly assess a large library of compounds in order to identify those molecules that most likely bind to a drug target. The docking protocol for VS has been validated by redocking cocrystallized GDP molecule to its own receptor-binding site. The lowest energy-docked conformation corroborated well with the cocrystallized GDP in the protein showing root mean square deviation of 0.4185 Å, which indicates that this docking protocol is predicting the pose correctly. The results were analyzed on the basis of poses, interactions, and binding score obtained after screening the datasets. Based on our interests, we sorted out the top ten molecules from each database after screening. NCI database molecules showed better binding interaction energy with K-Ras, when compared with other databases (Table S1). However, these molecules were not able to explore the entire binding cavity for the interaction. The top-ranked molecule (NSC_1945) from NCI diversity set database had −9.215 docking score, and was found to interact with PBP residues Gly13, Gly15, Lys16, Val29, and Tyr32 and was unable to approach the other two pockets. The compound (Code: 70014) from Asinex Platinum collection database showing −8.663 docking score occupies GBP and PBP partially and interacts with Gly13, Gly15, Lys16, Ala146, and Lys147. The interactions of other database molecules are not mentioned here because of their insignificant contributions in the binding.
Binding-site occupancy and interactions with the screened compounds
The molecules from different datasets were found to be poor binders on their own, so combinatorial and knowledge-based approach of drug design has been employed. The 260 designed molecules have been docked on the “on” and “off” state conformations of K-Ras protein. The molecules having >10 kcal/mol interaction docked energy were selected and discussed as potential lead molecules. Molecules 13 and 18 (Figure S1) were found to be within the cut-off energy of the “off” and “on” state conformations, respectively, and their corresponding docking score and energy decomposition are given in Tables S2 and S3.
Visual inspection of the docked poses of these molecules has shown similar binding as GDP and GTP when bound to their corresponding crystal structures of K-Ras protein, which may indicate that designed molecules were forming favorable interactions with the receptor (Tables S4 and S5). The guanine moiety of all designed molecules has shown similar binding mode and interaction at GBP as in the case of GDP (Figure S2) and GTP (Figure S3). The top-ranked molecule (M3) from the series of “off” state compound has shown similar docking score (−13.30) as its own GDP ligand (−13.33). However, the top-ranked molecule (M12) of “on” state has a lower docking score (−17.6) than GTP (−21.47). Such discrepancy in the energy between designed and GDP/GTP molecules arises mainly from the electrostatic component because GDP/GTP was docked in the presence of Mg2+ ion which adds more electrostatic component in the docking score, but the designed molecules were docked in the absence of Mg2+ ion. Therefore, it becomes very difficult to compensate such a large electrostatic component and replace GDP/GTP from its binding site. The cyclopentyl group of M3 (Figure 5A, B) molecule in “off” conformation is placed like ribose sugar at RBP. The 4-hydroxy and 2-oxo form H-bond interaction with Val29 and Lys117, respectively. The 3-ammonio-1-hydroxy-1-(hydroxyphosphinato) propyl phosphonate group is positioned in such a way that it can go deep into the extended cavity and form strong H-bond interaction with the side chain of Lys16, Ser17, and backbone carbonyl oxygen of Val14, Asp33, and Ile36. In addition to H-bond interaction, a salt bridge interaction was also observed between phosphonate and carboxylate of Asp57. The lipophilic term derived from hydrophobic grid potential and fraction of the total protein ligand vdW energy (LipophilicEvdW) has shown less contribution in M3 docking score. The electrostatic energy term in the docking score of GDP has been compensated by ‘reward for hydrophobically packed correlated H-bond’ (PhobEnPairHB) and H-bond term in designed M3 molecule. The top-ranked docked molecule (M12) in “on” state is also found to occupy similar GTP conformation in the binding cavity. The interaction networks between the “on” conformation and M12 (Figure 5C, D) are made up of a closely knit circuit of noncovalent interactions, such as electrostatic and hydrophobic, in addition to H-bonds to achieve proper binding. The phenyl ring in M12 is perfectly positioned between RBP and PBP to form π–π interaction with Tyr32. Also, 2-(ammoniomethyl)-3-oxopropanoate occupies PBP, where it forms H-bonds with Gly13, Asp57, Thr58, and Gly60 and a salt bridge with Lys16. The electrostatic energy term in the docking score of GTP has been partially compensated by PhobEnPairHB and LipophilicEvdW terms in designed M12 molecule. Per residue H-bonding, vdW and Coulomb contributions in the binding of molecules have also been calculated in both the conformations and mentioned in the supporting information (Figures S4–S9).
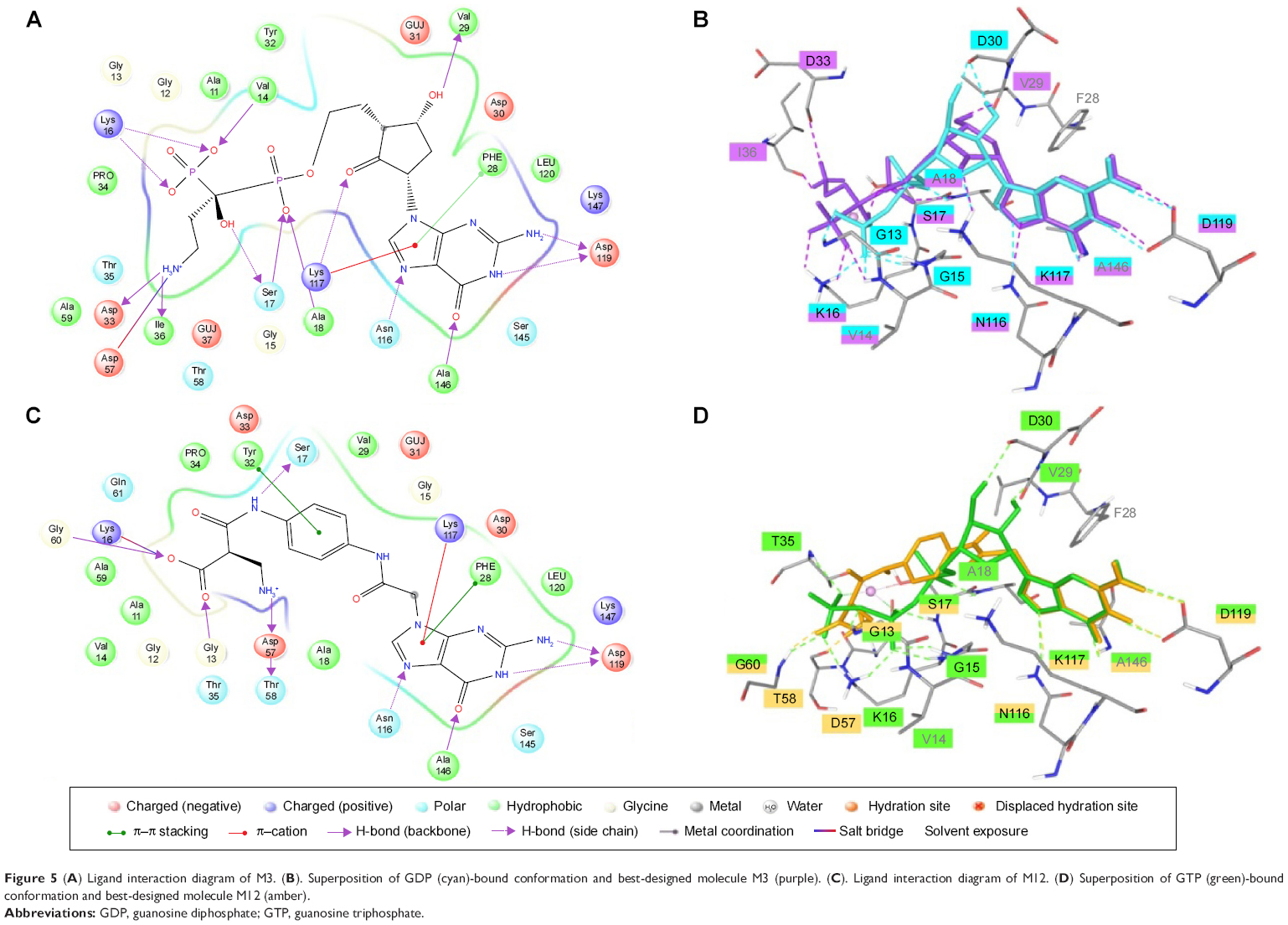 | Figure 5 (A) Ligand interaction diagram of M3. (B). Superposition of GDP (cyan)-bound conformation and best-designed molecule M3 (purple). (C). Ligand interaction diagram of M12. (D) Superposition of GTP (green)-bound conformation and best-designed molecule M12 (amber). |
Validation
The top-rank designed molecules were evaluated on the basis of their binding. The influence of induced conformation changes on docking results becomes apparent when the ligand is docked to a receptor cocrystallized with another inhibitor. Docking scores of molecules were compared with induced-fit docking score (Figure S10). The result of flexible docking corroborates the docking score obtained from Glide, but with a change in the ranking order, which suggests that small variations in the structure of the binding pocket also have a large impact on the interaction energy.
ADMET property calculation
The shortlisted ligands were then screened for their ADME and toxicity properties using QikProp module of Schrödinger software.46,47 It predicts physically significant descriptor and pharmaceutically relevant properties of organic molecules. All compounds were found to be within the limit of approved drug parameter range (Tables S6 and S7).
Discussion
Our objective was to screen all kinase inhibitor libraries as well as some generalized libraries and identify the compounds that compete with cytosolic GDP/GTP, inhibit the interaction of Ras, and ultimately arrest the Ras signaling pathway. We extracted molecules from different databases, screened out molecules, and compared the poses of top-ranked molecules from each database with the GDP/GTP-bound K-Ras protein. We analyzed the pose of the molecules and found that molecules screened from databases were showing a poor docking score. Some of their docking scores were too low, resulting in unfavorable interactions, and in many others, the stereochemical orientation was very poor. Also, some of the molecules were found to bind to GBP and others were found to bind to PBP, and none of them was able to occupy the GDP/GTP-binding pocket completely. Therefore, this could be the possible reason for the poor binding or docking score, but some small molecular fragments from Ras-focused dataset were showing good interactions. To improve the interaction and docking score, we utilized the fragment-based combinatorial library design strategies. We selected molecules that showed interactions, like GDP/GTP in PBP and RBP, and fragmented and divided them on the basis of RBP and PBP interacting and noninteracting part. We utilized the interacting parts of the molecules that can bind to both RBP and PBP. Guanine-containing molecules from the database showed the best docking score and interaction. Thus, we fixed guanine as a core molecule, as few FDA drugs also contain guanine,48,49 and selected the fragments that can bind to RBP and PBP, which were further used to design the combinatorial library.
Targeting RAS gene is not uncommon. There have been several reports showing small molecules getting bound to GDP and inhibiting the son of sevenless-mediated nucleotide exchange. We selected molecules from FDA-approved drugs, PubChem bioassay compounds that are active against H-Ras protein (isoform of K-Ras), kinase inhibitors from PubChem compounds, and Ras inhibitors from BindingDB for the generation of fragment library. The reason for selecting these molecules is the availability of in vitro activity data, which would have less possibility of toxicity. The docking results of these molecules in “on” and “off” states of K-Ras were encouraging, and we found that these molecules were able to occupy the entire nucleotide-binding cavity with a knitting of noncovalent and H-bonding interactions. Our findings of designed molecular interactions with K-Ras might open new avenues in investigating its use in cancer therapy, where Ras pathway activation plays an important role. The predicted molecules from the studies may be synthesized and tested through in vitro and in vivo experiments and can also be further modified to achieve specificity.
Conclusion
Individual molecules from different datasets on its own has not been that salient, but blending of its fragments has significantly improved binding in the active site of K-Ras. The present study was conducted to identify small molecules as potent K-Ras inhibitors for the treatment of different cancers using in silico tools and techniques. The interactions between K-Ras and designed ligands were studied by using Glide docking protocol. Based on the docking score and interactions, the docking results were analyzed. The results were compared with GDP/GTP to find out the best ligand that can inhibit GTPase activity of Ras. The shortlisted molecules in our study that bind at the GTP/GDP-binding pocket of K-Ras might have the potential of inhibiting its activity, which will further strengthen this concept. While the docking scores for all compounds with K-Ras were comparable to GDP/GTP conformations, all the compounds exhibited significantly higher binding with K-Ras. With this study, we are trying to bring Ras back into focus as a drug target.
Acknowledgments
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award no. 12-MED2622-02. BP and MC thank NIPER, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers for providing the fellowship and facility.
Disclosure
The authors report no conflicts of interest in this work.
References
Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9(7):517–531. | ||
Hall BE, Bar-Sagi D, Nassar N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc Natl Acad Sci U S A. 2002;99(19):12138–12142. | ||
Milburn MV, Tong L, deVos AM, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247(4945):939–945. | ||
Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81(1):153–208. | ||
Fiegen D, Dvorsky R, Ahmadian MR. Structural principles of Ras interaction with regulators and effectors. In: Der C, editor. RAS Family GTPases. Vol 4. Amsterdam, Springer; 2006:45–66. | ||
Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2013;3(1):112–123. | ||
Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400(6743):468–472. | ||
Macaluso M, Russo G, Cinti C, Bazan V, Gebbia N, Russo A. Ras family genes: an interesting link between cell cycle and cancer. J Cell Physiol. 2002;192(2):125–130. | ||
Knijn N, Mekenkamp LJ, Klomp M, et al. KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br J Cancer. 2011;104(6):1020–1026. | ||
Zorde Khvalevsky E, Gabai R, Rachmut IH, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(51):20723–20728. | ||
Weinstein IB. Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science. 2002;297(5578):63–64. | ||
Podsypanina K, Politi K, Beverly LJ, Varmus HE. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci U S A. 2008;105(13):5242–5247. | ||
Hunter JC, Gurbani D, Ficarro SB, et al. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci U S A. 2014;111(24):8895–8900. | ||
Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KA. K-Ras(G12C) inhibitor allosterically control GTP affinity and effector interactions. Nature. 2013;503(7477):548–551. | ||
Shima F, Yoshikawa Y, Ye M, et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci. 2013;110(20):8182–8187. | ||
Muraoka S, Shima F, Araki M, et al. Crystal structures of the state 1 conformations of the GTP-bound H-Ras protein and its oncogenic G12V and Q61L mutants. FEBS Lett. 2012;586(12):1715–1718. | ||
Burns MC, Sun Q, Daniels RN, et al. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc Natl Acad Sci. 2014;111(9):3401–3406. | ||
Chourasia M, Sastry GM, Sastry GN. Aromatic-Aromatic Interactions Database, A(2)ID: an analysis of aromatic pi-networks in proteins. Int J Biol Macromol. 2011;48(4):540–552. | ||
Mukherjee P, Desai P, Ross L, White EL, Avery MA. Structure-based virtual screening against SARS-3CL(pro) to identify novel non-peptidic hits. Bioorg Med Chem. 2008;16(7):4138–4149. | ||
Wishart DS, Knox C, Guo AC, et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34(Database issue):D668–D672. | ||
Wishart DS, Knox C, Guo AC, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36(Database issue):D901–D906. | ||
Knox C, Law V, Jewison T, et al. DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 2011;39(Database issue):D1035–D1041. | ||
Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35(Database issue):D198–D201. | ||
Chen X, Lin Y, Gilson MK. The binding database: overview and user’s guide. Biopolymers. 2001;61(2):127–141. | ||
Chen X, Lin Y, Liu M, Gilson MK. The binding database: data management and interface design. Bioinformatics. 2002;18(1):130–139. | ||
Chen X, Liu M, Gilson MK. Binding DB: A web-accessible molecular recognition database. Comb Chem High-Throughput Screen. 2001;4:719–725. | ||
Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr B. 2002;58(Pt 3 Pt 1):380–388. | ||
Milne GWA, Nicklaus MC, Driscoll JS, Wang S, Zaharevitz D. The NCI drug information system 3D database. J Chem Inf Comput Sci. 1994;34:1219–1224. | ||
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37(Web Server issue):W623–W633. | ||
Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. | ||
Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. | ||
Badrinarayan P, Sastry GN. Virtual high throughput screening in new lead identification. Comb Chem High Throughput Screen. 2011;14:840–860. | ||
Badrinarayan P, Choudhury C, Sastry GN. Molecular modeling. In: Singh V, Dhar PK, editors. Systems and Synthetic Biology. Dordrecht: Springer; 2014. | ||
Onodera K, Satou K, Hirota H. Evaluations of molecular docking programs for virtual screening. J Chem Inf Model. 2007;47(4):1609–1618. | ||
McGaughey GB, Sheridan RP, Bayly CI, et al. Comparison of topological, shape, and docking methods in virtual screening. J Chem Inf Model. 2007;47(4):1504–1519. | ||
Cross JB, Thompson DC, Rai BK, et al. Comparison of several molecular docking programs: pose prediction and virtual screening accuracy. J Chem Inf Model. 2009;49:1455–1474. | ||
Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47(7):1750–1759. | ||
Kim R, Skolnick J. Assessment of programs for ligand binding affinity prediction. J Comput Chem. 2008;29(8):1316–1331. | ||
Huang SY, Grinter SZ, Zou X. Scoring functions and their evaluation methods for protein-ligand docking: recent advances and future directions. Phys Chem Chem Phys. 2010;12(40):12899–12908. | ||
Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: successes and challenges. Curr Pharm Des. 2005;11(3):323–333. | ||
Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49(21):6177–6196. | ||
Badrinarayan P, Sastry GN. Specificity rendering ‘hot-spots’ for aurora kinase inhibitor design: the role of non-covalent interactions and conformational transitions. PLoS One. 2014;9(12):e113773. | ||
Badrinarayan P, Sastry GN. Sequence, structure, and active site analyses of p38 MAP kinase: exploiting DFG-out conformation as a strategy to design new type II leads. J Chem Inf Model. 2011;51:115–119. | ||
Badrinarayan P, Sastry GN. Virtual screening filters for the design of type II p38 MAP kinase inhibitors: a fragment based library generation approach. J Mol Graph Model. 2012;34:89–100. | ||
Quinlan MP, Settleman J. Isoform-specific ras functions in development and cancer. Future Oncol. 2009;5(1):105–116. | ||
Colmenarejo G, Alvarez-Pedraglio A, Lavandera JL. Cheminformatic models to predict binding affinities to human serum albumin. J Med Chem. 2001;44(25):4370–4378. | ||
Cheng A, Dixon SL. In silico models for the prediction of dose-dependent human hepatotoxicity. J Comput Aided Mol Des. 2003;17(12):811–823. | ||
Walsh AW, Langley DR, Colonno RJ, Tenney DJ. Mechanistic characterization and molecular modeling of hepatitis B virus polymerase resistance to entecavir. PLoS One. 2010;5(2):e9195. | ||
Sims KA, Woodland AM. Entecavir: a new nucleoside analog for the treatment of chronic hepatitis B infection. Pharmacotherapy. 2006;26(12):1745–1757. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.