Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
“Denervation” of autonomous nervous system in idiopathic pulmonary arterial hypertension by low-dose radiation: a case report with an unexpected outcome
Authors Hohenforst-Schmidt W, Zarogoulidis P, Oezkan F, Mahnkopf C, Grabenbauer G, Kreczy A, Bartunek R, Darwiche K, Freitag L, Li Q, Huang H, Vogl T ![]() , LePlvert P, Tsiouda T, Tsakiridis K, Zarogoulidis K, Brachmann J
, LePlvert P, Tsiouda T, Tsakiridis K, Zarogoulidis K, Brachmann J
Received 4 December 2013
Accepted for publication 29 January 2014
Published 27 March 2014 Volume 2014:10 Pages 207—215
DOI https://doi.org/10.2147/TCRM.S58705
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Wolfgang Hohenforst-Schmidt,1 Paul Zarogoulidis,2 Filiz Oezkan,3 Christian Mahnkopf,4 Gerhard Grabenbauer,5 Alfons Kreczy,6 Rolf Bartunek,7 Kaid Darwiche,3 Lutz Freitag,3 Qiang Li,8 Haidong Huang,8 Thomas Vogl,9 Patrick LePilvert,10 Theodora Tsiouda,11 Kosmas Tsakiridis,12 Konstantinos Zarogoulidis,2 Johannes Brachmann1
1II Medical Clinic, Coburg Clinic, University of Würzburg, Coburg, Germany; 2Pulmonary Department-Oncology Unit, G Papanikolaou General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece; 3Department of Interventional Pneumology, Ruhrlandklinik, West German Lung Center, University Hospital, University of Duisburg-Essen, Essen, Germany; 4II Medizinische Klinik, Klinik für Kardiologie, Angiologie, Pneumologie, Klinikum Coburg, 5Department of Radiotherapy, 6Department of Pathology, Cytology and Molecular Diagnostics, 7Institute of Diagnostic and Interventional Radiology, Coburg Clinic, University of Wüerzburg, Coburg, Germany; 8Department of Respiratory Diseases, Changhai Hospital/First Affiliated Hospital of the Second Military Medical University, Shanghai, People’s Republic of China; 9Department of Diagnostic and Interventional Radiology, Goethe University of Frankfurt, Frankfurt, Germany; 10Interventional Drug Delivery Systems and Strategies (ID2S2), Medical Cryogenics, Lakeland Court Jupiter, FL, USA; 11Internal Medicine Unit, Theagenio Cancer Hospital, Thessaloniki, 12Cardiothoracic Surgery Department, Saint Luke Private Hospital, Thessaloniki, Greece
Abstract: Vasointestinal peptide metabolism plays a key physiological role in multimodular levels of vasodilatory, smooth muscle cell proliferative, parenchymal, and inflammatory lung reactions. In animal studies, vasointestinal peptide relaxes isolated pulmonary arterial segments from several mammalian species in vitro and neutralizes the pulmonary vasoconstrictor effect of endothelin. In some animal models, it reduces pulmonary vascular resistance in vivo and in monocrotaline-induced pulmonary hypertension. A 58-year-old woman presented with dyspnea and mild edema of the lower extremities. A bronchoscopy was performed without any suspicious findings suggesting a central tumor or other infiltrative disease. Endobronchial ultrasound revealed enlarged pulmonary arteries containing thrombi, a few enlarged lymph nodes, and enlarged mediastinal tissue anatomy with suspicion for mediastinal infiltration of a malignant process. We estimated that less than 10% of the peripheral vascular bed of the lung was involved in direct consolidated fibrosis as demonstrated in the left upper lobe apex. Further, direct involvement of fibrosis around the main stems of the pulmonary arteries was assumed to be low from positron emission tomography and magnetic resonance imaging scans. Assuming a positive influence of low-dose radiation, it was not expected that this could have reduced pulmonary vascular resistance by over two thirds of the initial result. However; it was noted that this patient had idiopathic pulmonary arterial hypertension mixed with “acute” (mediastinal) fibrosis which could have contributed to the unexpected success of reduction of pulmonary vascular resistance. To the best of our knowledge, this is the first report of successful treatment of idiopathic pulmonary arterial hypertension, probably as a result of low-dose radiation to the pulmonary arterial main stems. The patient continues to have no specific complaints concerning her idiopathic pulmonary arterial hypertension.
Keywords: radiation, pulmonary hypertension, denervatio
Background
In general, radiation therapy when used in lymphoma or breast cancer1 is associated with long-term deterioration of the functional capacity of the cardiopulmonary system, with pulmonary hypertension in addition to other adverse cardiopulmonary effects.2–4 However, in such cases, the typical radiation exposure is 20–35 Gy, which is significantly higher than the low dose of 4 Gy used in the patient described here. Radiation therapy to the abdominal lymph nodes involved in seminoma has been reported to damage peripheral nerves,5 but again the radiation doses applied were higher than 35 Gy. Significant damage to the sciatic nerves was found at one year post-radiation in dogs exposed to intraoperative abdominal irradiation with 28 Gy.6
The scant literature on the cellular effects of experimental (low-dose) radiation is discussed here in support of our hypothesis that low-dose radiation can have a positive hemodynamic effect. A study by Al-Jahdari et al7 demonstrated a significant adverse effect of X-rays at doses of 3 Gy and higher on formation of the growth cone in the sympathetic chain ganglion of chick embryos in vitro. In that study, the radiosensitivity of autonomic nerve fibers was rated the same as that of peripheral nerve fibers. Tan et al8 demonstrated a 50% reduction of neuronal progenitors in the hippocampi of adult male rats after local irradiation with doses as low as 0.78 Gy, which was partially reversible over time. However, at doses over 5 Gy, there was almost complete and irreversible block of neurogenesis without compensatory proliferation. This paper discusses how radiation doses as low as 0.1 Gy may induce damage in the neurogenic regions of the hippocampus. It has to be mentioned that central neuronal structures are generally considered to be more radiosensitive than peripheral structures. Höckerfelt et al9 reported a significant more than three-fold increase in vasoactive intestinal peptide (VIP) content in the rat parotid gland at 10 days following cessation of a 5-day treatment with a total radiation dose of 30–40 Gy. In a study by Otterson et al,10 a significant two-fold increase in intestine VIP content was reported in dogs after a total dose of 1,500 Gy in 250 Gy fractions, although accompanied with some regional variations. Steensma et al11 reported a clinically relevant decrease in pulmonary hypertension in three of four patients with myelofibrosis (including myeloid metaplasia) 72 hours after application of a single 100 Gy dose of whole lung radiation. However, the mechanistic understanding is rather a radiation-induced reduction of stem cell proliferation in the pulmonary vascular bed obstructing pulmonary circulation than an effect by VIP or others.
New concepts in the treatment of pulmonary arterial hypertension (PAH) are discussed here in regard to our case. The concept of treating elevated levels of sympathetic or adrenal neurohormonal levels using beta-blockers has been proposed for right ventricular dysfunction in PAH in the same way as for left heart disease;12–15 this treatment strategy has already been used safely in patients with PAH.16 A genetic right ventricular failure program could be partly reversed by beta-blockers in an animal model.14 In conclusion, the level of activity of the sympathetic autonomic nervous system seems to be directly linked to the development of PAH, and therefore represents a novel target for treatment of PAH.15
Successful treatment of idiopathic PAH by direct inhibition of the autonomic nervous system around the pulmonary arterial main stems in humans was demonstrated for the first time in the PADN-1 (first-in-man Pulmonary Artery DeNervation for treatment of pulmonary artery hypertension) study17 in patients with pure idiopathic PAH who were on specific (at least dual) therapy but still showing resistant unacceptable hemodynamics. This study was conducted using an ablation device similar to that used for renal artery denervation in the treatment of resistant systemic arterial hypertension. Using this type of ablation approach, endoluminal thermal energy damages the adjacent but abluminal autonomous nerve fibers. In our study, there was a highly significant reduction of more than 50% in mean pulmonary arterial resistance (PAR) from a preablative value of 1,883 dyn · s · cm−5 as early as 24 hours following ablation. Additionally, the PAR decreased in the next 3 months by an average of more than 60% compared with the preablation value. This is 2–3-fold higher than the PAR reduction of >20% that is considered significant when using medical therapy in patients with idiopathic PAH.
Hypotheses
From our point of view two hypotheses can be generated by this case. First, metabolism of VIP plays a key physiological role in multimodular levels of vasodilatory, smooth muscle cell proliferative, parenchymal, and inflammatory lung reactions.18 Second, VIP relaxes isolated pulmonary arterial segments in several mammalian species in vitro and neutralizes the pulmonary vasoconstrictor effect of endothelin.18 In some animal models, VIP reduces pulmonary vascular resistance in vivo and in monocrotaline-induced pulmonary hypertension.18 Applied on initiation in a monocrotaline animal model, VIP prevented the animal converting into PAH hemodynamics, and in doing so was more effective than bosentan.19 VIP attenuates hypoxic pulmonary vasoconstriction in newborn lambs and in Fawn-Hooded rats, which are known to have an exaggerated pulmonary pressure response to alveolar hypoxia.18 Together with nitric oxide, VIP is a likely mediator of the nonadrenergic, noncholinergic relaxation system in the pulmonary vasculature.18 The combined activation of the guanylate cyclase and adenylate cyclase systems, respectively, by nitric oxide and VIP, maximizes smooth muscle relaxation.18
In human beings, VIP inhibits the in vitro proliferation of pulmonary vascular smooth muscle cells from patients with idiopathic PAH.18 Given for 3 months by daily inhalation, VIP markedly improved exercise capacity and pulmonary hemodynamics in patients with idiopathic PAH.18 VIP-containing nerves, normally plentiful in the walls of the pulmonary arteries, are absent in patients with idiopathic PAH.18 Moreover, serum VIP levels in patients with idiopathic PAH have been found to be low (13.16±8.5 pg/mL versus 42.4±17.1 pg/mL in healthy controls).20
Low-dose radiation could lead to elevated VIP levels in some tissues.10 Therefore, it is possible that low-dose irradiation was linked to elevated VIP levels in this woman with idiopathic PAH, thereby contributing to decreased pulmonary vascular resistance. Unfortunately we did not measure her VIP concentrations.
Case report
A 58-year-old woman presented to the Coburg Clinic in February 2013 with dyspnea, loss of energy, slight edema of the lower extremities, and a 4 kg weight loss in the 4 weeks before presentation. A 3/6 systolic murmur with a left apical maximum point was noted on physical examination. Except for an appendectomy more than 30 years earlier, the patient had never been hospitalized or on any medication. She had never smoked. Family history was unremarkable, except for a sister who had experienced uncomplicated tuberculosis. She had worked as a seamstress until giving birth to a healthy daughter in 1973. From 1976 to 1980 and then again from 1985 to 1997 she reported having worked in the ceramics industry, but her job was not associated with significant exposure to ceramic dust. During that time, living in the former German Democratic Republic, she had had annual chest X-rays without any abnormalities. During a second examination, she reported having felt dyspnea on exercise since 2003, but did not complain of loss of energy, and had been able to perform regular work. The electrocardiogram performed on admission showed sinus tachycardia at a rate of 109 beats per minute, incomplete right bundle branch block, and preterminal inverted T-waves in II, III, aVF, and V1–V6.
Body plethysmography showed slight restriction with forced expiratory volume in one second being 68% predicted, inspiratory vital capacity 70% predicted, and a Tiffeneau index of 101%. Her diffusion capacity was reduced at 60%. Her respiratory insufficiency was managed with oxygen substitution therapy (pO2 60 mmHg and pCO2 33 mmHg under oxygen 3 L per minute).
An initial transthoracic echocardiogram on February 1, 2013 demonstrated dilatation of the right ventricle to 38 mm and dilatation of the left atrium to 43 mm. Sclerosis of the aortic valve, minimal aortic insufficiency, first-degree mitral insufficiency, obvious tricuspid valve insufficiency with a diastolic gradient of 67 mmHg, and diastolic dysfunction of the left ventricle with a slightly pathological E/A ratio could also be visualized. Left ventricular ejection fraction was normal at 65%.
The woman presented for the first time to the pulmonary consultant late on a Friday afternoon with dyspnea and spontaneous oxygen saturation of 83% on the bronchoscopy table (pO2 38 mmHg, pCO2 31 mmHg). She complained of rapid deterioration in the previous 2 days in comparison with her status on admission. Her weight was 58 kg, but she complained a 19 kg weight loss in the previous 2 months. Her height was 158 cm (body mass index 23.1 kg/m2). At that time, only a standard chest X-ray taken on admission was available, and showed a prominent bihilar region with a suspicious mass in the left apex of the lung (Figure 1). Standard serum analysis showed slightly elevated lactate dehydrogenase and uric acid levels, with a mild anemia. On fluoroscopy, the right hilum appeared to be enlarged compared with the X-ray taken on admission. No result suspicious for a central tumor or other infiltrative disease was seen on bronchoscopy. Endobronchial ultrasound revealed enlarged pulmonary arteries containing multiple thrombi, a few enlarged lymph nodes, and abnormal mediastinal tissue anatomy suspicious for mediastinal infiltration of a malignant process. We did not perform transbronchial needle aspiration before the weekend due to her dyspnea. She was put on low-dose low molecular weight heparin. The decision was taken to start her on radiotherapy over the weekend due to the suspicion of rapid progression of small cell lung cancer or lymphoma in the mediastinum. Computed tomography planning was done in the radiotherapy department on the following day and she received radiation with 4 Gy to the mediastinum including both hila over the weekend (2 Gy per day).
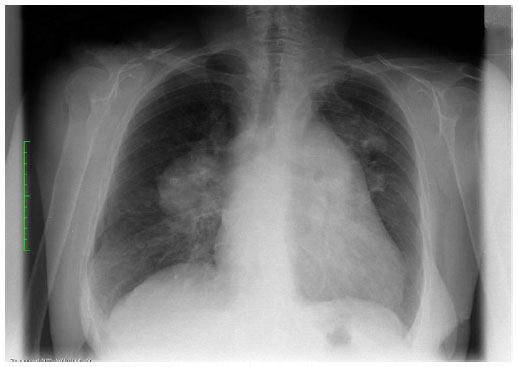 | Figure 1 Standard chest X-ray on admission. |
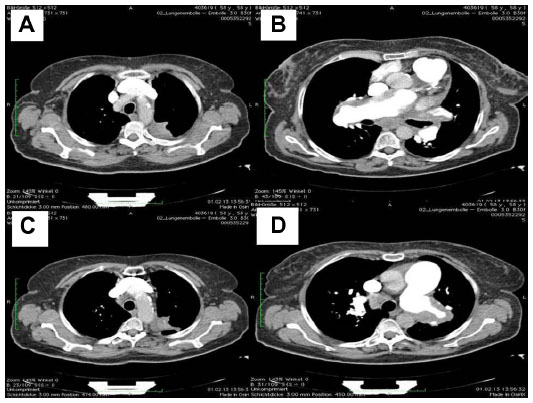
After the weekend, we observed a mediastinal mass, abnormally enlarged central pulmonary arteries with tortuous peripheral branches, and a consolidation in the left upper lobe on the patient’s computed tomography scan (Figures 2 and 3). At that point we stopped radiation therapy because it was clear that we were not dealing with a typical case of suspected small cell lung cancer or lymphoma. Repeat bronchoscopy and endobronchial ultrasound demonstrated partial compression of the central vessels by a mass located in the left mediastinum, and partial infiltration of the walls by this mass was suspected. Given that the lymph nodes in position 10 R (right) and 7 had a diameter of 15–20 mm, we took several biopsies from these sites in addition to biopsies of the mediastinal mass. Histologically, there was no suggestion of malignancy, but a strong suspicion for fibrosis was raised on evaluation of the magnetic resonance imaging (MRI) scans (Figure 4) but could not be proven.
 | Figure 2 Esophageal ultrasound. |
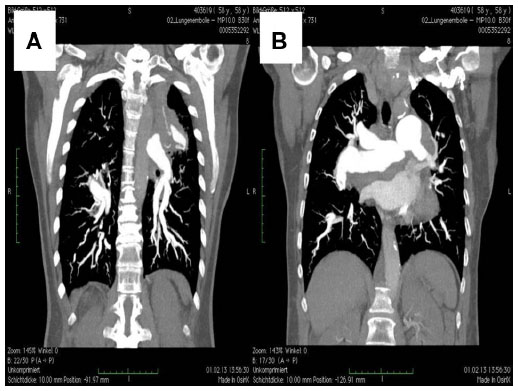 | Figure 3 Maximum intensity projection view. |
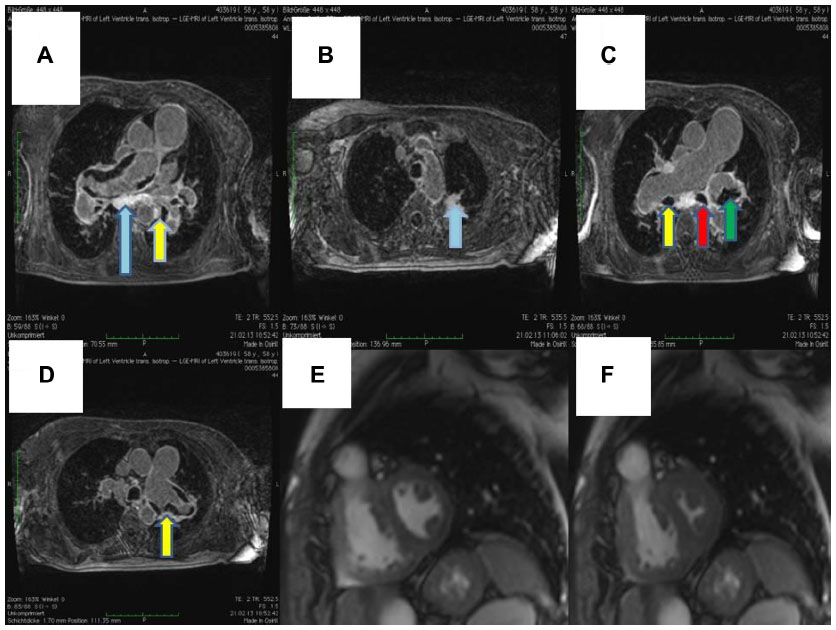 | Figure 4 Magnetic resonance imaging. |
Two weeks later, an endoesophageal ultrasound was performed with a thicker needle in lymph node position 7. Again there was only a hint of fibrosis. A transthoracic lung puncture of the mass in the left upper lobe revealed thickened pulmonary arterial walls, activated pneumocytes, and fibrosis (Figures 5–7). Several endoscopic ultrasound consultants and endobronchial ultrasound examinations reported massive “smoke” (indicating prethrombotic formations) in the central pulmonary arterial vessels. Tuberculosis was ruled out by a negative interferon gamma release assay, several negative bronchial specimens examined by microscopy, polymerase chain reaction, and cultures for tuberculosis collected by bronchoscopy. Nontuberculous mycobacteria was ruled out by negative polymerase chain reaction, microscopy, and culture results, as was sarcoidosis, given the patient’s normal serum soluble interleukin-2 receptor and angiotensin-converting enzyme levels. We could not find any histological suggestion of histoplasmosis,21 but this diagnosis was not ruled out by serum analysis as the patient appeared to be immunocompetent with normal immunoelectrophoresis and a negative human immunodeficiency virus test result.
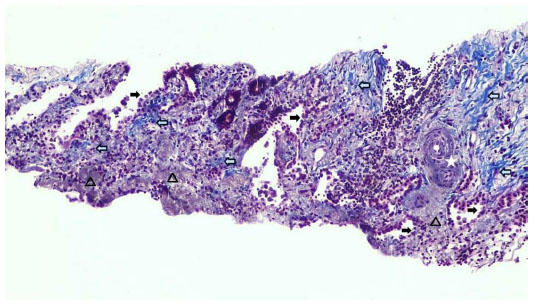 | Figure 5 Lung biopsy showing increased interstitial collagen ( blue), elastic fibers ( gray), activated pneumocytes (), and pathological vessels with thickened walls. 100×. |
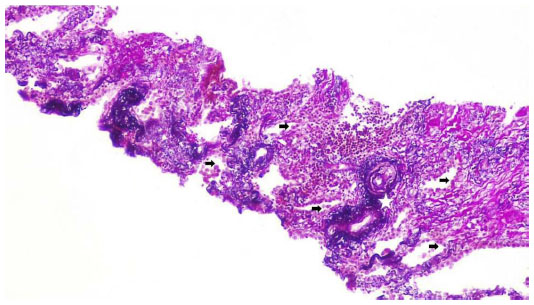 | Figure 6 Lung biopsy with increased interstitial collagen (red) and elastic fibers (blue), Activated pneumocytes, Pathologic vessels with thick walls. evaluation of vessel invasion, 100×. |
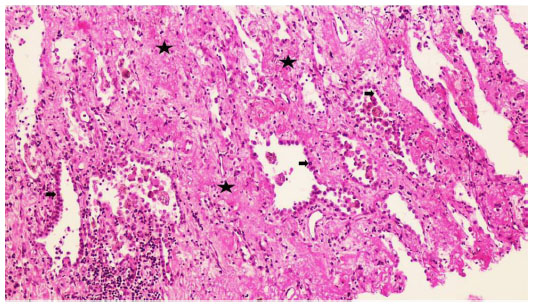 | Figure 7 Lung biopsy with interstitial fibrosis and activated pneumocytes. Hematoxylin and eosin, 100×. |
Cardiac MRI showed reduced right ventricular function, dilatation of the right ventricle and a hypertrophic wall, a flattened septum with paradoxical movement (Figure 4), and dilated pulmonary artery trunks. There was no suggestion of intravascular thrombus on pulmonary angiography during cardiac MRI. A sudden change in diameter of the pulmonary arteries could be seen in the peripheral regions. Small tortuous peripheral pulmonary arteries were seen on cardiac MRI and computed tomography (Figure 3). The aforementioned abnormal mediastinal mass and left upper lobe mass were also observed.
Lung embolism was excluded using ventilation and perfusion scintigraphy. Combined ventilation and perfusion defects in the left upper and middle lobes were associated with the mediastinal mass.
Precapillary PAH with a PAR of 1,360 dyn · s · cm−5 and completely normal wedge pressure was seen on right heart Swan-Ganz catheterization, coronary angiography, and dextrography (Figure 8). There were no definite criteria for reactive PAH on inhalation of iloprost. The right ventricle was hypokinetic especially in its anterior portion (Figure 8C and D) with an enlarged wall which, on echocardiography, was 8 mm thick (ie, thickness of right ventricular anterior wall in end diastole). The median right atrial pressure was 8 mmHg and borderline for right heart decompensation. The pulmonary arteries were shown to be dilated on angiography, but there was no indication of peripheral obstruction or chronic thromboembolic pulmonary hypertension. The left marginal artery and diagonal branch showed an angiographic stenosis of 70% (Figure 8A and B), and left ventricular ejection fraction was 70%. There was endothelial dysfunction with low flow phenomenon in different coronary arteries, besides the partly abnormal coronary anatomy with dilated cardiomyopathy (Figure 8A and B). There was no suggestion of vasculitis, coagulopathy, a thyroid or hepatic disorder, or collagenosis on serum analysis. No other predominant cardiopulmonary disease directly involved in the initially highly elevated PAH hemodynamics was found after working through the algorithm for PAH recommended by the European Society of Cardiology.
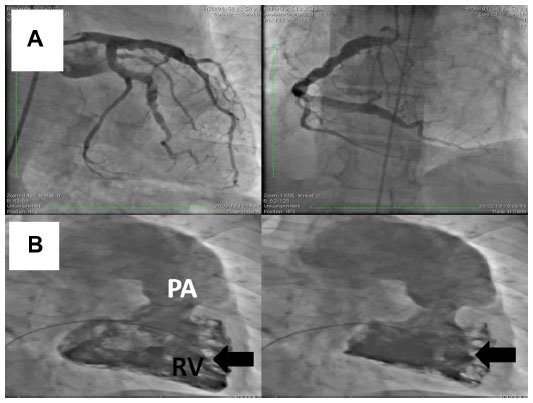 | Figure 8 Coronary angiography. |
A dobutamine MRI stress test was performed a week later without any suspicion for ischemia, so stenting of the abovementioned stenosis observed on angiography was not performed. Because thrombus was expected in the pulmonary arterial walls, transesophageal echocardiography was performed and supported the presumption of thrombus in the pulmonary arterial walls, including prethrombotic “smoke” in the central vessels. No shunt was observed. Phenprocoumon was started as oral anticoagulation therapy. Thoracic surgeons at the nearby Kutzenberg Hospital refused to do mediastinoscopy because of the diagnosis of PAH.
Positron emission tomography at the University of Erlangen showed no suspicion of tumor or lymphoma in the mediastinum or elsewhere, and mediastinal fibrosis was again diagnosed. Lymphoma was ruled out in an oncologic ambulatory setting via peripheral lymph node biopsies from the groin, serum electrophoresis, urinalysis, and bone marrow puncture. Serum beta2-microglobulin was negative.
At the end of April, the patient was presented for the first time at the National Pulmonary Hypertension Center of Excellence in Bad Nauheim/Giessen and a second time in May 2013 (Figure 9) with a differential diagnosis of idiopathic PAH. This was done because the referring cardiopulmonary consultant at Coburg Clinic, who is also an expert in the treatment of PAH and a member of the national guideline group, was surprised by this patient’s abnormal anatomy and the massive extent of central smoke without any peripheral suggestion of embolism. Repeat pulmonary angiography showed no thrombus (Figure 9), and chronic thromboembolic pulmonary hypertension was excluded on ventilation/perfusion (V/Q) scintigraphy. Right heart catheterization revealed precapillary pulmonary hypertension with a PAR of 600 dyn · s · cm−5 and no reactive properties, which was quite striking given that our patient had not received any specific treatment in the preceding weeks. Cardiac MRI revealed the same pathologies in the pulmonary arterial walls as before, and functional and anatomic abnormalities of the right ventricle and mediastinal fibrosis were again reported. The diagnosis of idiopathic PAH was confirmed at several conferences with the team led by Ghofrani and Mayer (Professor Hossein A Ghofrani, Pulmonary Department, University Hospital Giessen, Giessen, Germany; and Professor Eckhard Mayer, Thoracic Surgery Department, Bad Nauheim, Germany). During discussion between the experts at both institutions, the question was raised as to whether a spontaneous dissection of the pulmonary arterial walls with secondary thrombus formation in the walls was possible, and a diagnosis of mediastinal fibrosis was accepted.
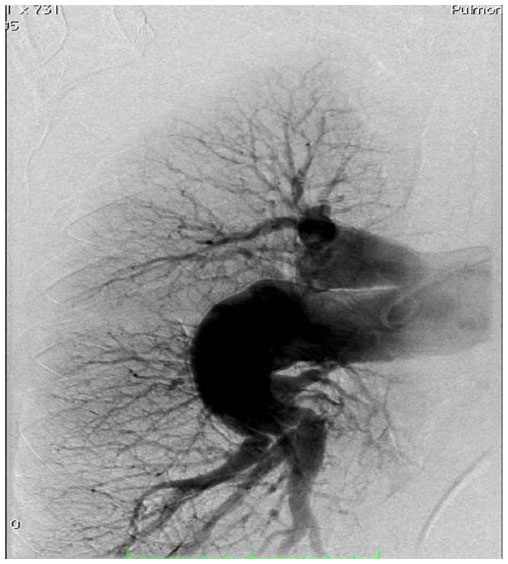 | Figure 9 Right sided pulmonary artery angiography performed at Giessen indicating chronic thromboembolic pulmonary hypertension which was also demonstrated by two ventilation/perfusion (V/Q) mismatches. |
Four months after her first admission, the patient was electively rehospitalized to initiate specific treatment in June 2013. In view of her coronary artery disease without evidence of ischemia, the decision was made to start treatment with bosentan instead of sildenafil. On this admission, an echocardiogram showed no signs of pulmonary hypertension, and no enlargement of the right ventricle or paradoxical septal shift was detectable by an experienced examiner. The patient had not had any complaints for at least 2 months, and was started on bosentan without any problems.
The patient was evaluated 2 months later by Swan-Ganz catheterization of the right heart on full-dose bosentan 125 mg twice daily for 4 weeks. Again, precapillary pulmonary hypertension was reported and her PAR had dropped to 450 dyn · s · cm−5. Meanwhile, her spontaneous pO2 at rest had increased to 76 mmHg and her 6-minute walking distance was 360 m (Figure 10).
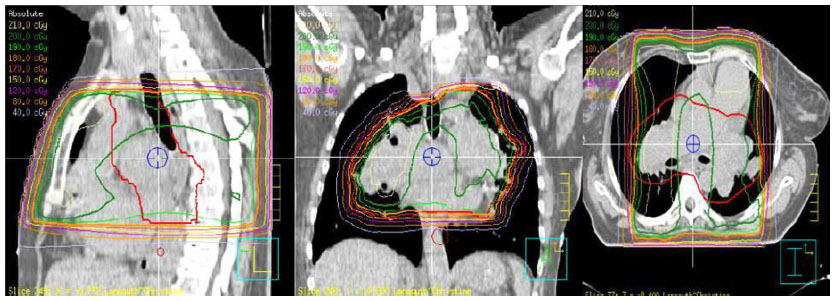 | Figure 10 Isodoses clearly showing the main branch of the pulmonary artery with the first segmentation included. |
As a proof-of-concept of our hypotheses, we interrupted her full-dose bosentan therapy for 2 weeks and again obtained invasive hemodynamic measurements via Swan-Ganz catheterization of the right heart. The results seemed to be only slightly different and obvious precapillary pulmonary hypertension was again demonstrated but, interestingly, her PAR was 400 dyn · s · cm−5, and therefore not elevated compared with the last measurement taken when the patient was on bosentan 125 mg twice daily.
Seven and a half months after the initial diagnosis of idiopathic PAH with a PAR of 1,360 dyn · s · cm−5, we observed a reduction in PAR of at least two thirds without having established any specific treatment, except for low-dose radiation to the main stems of the pulmonary arteries (Figure 10).
Discussion
To the best of our knowledge, this is the first report of successful treatment of idiopathic PAH by low-dose irradiation of the main stems of the pulmonary arteries. Seven months after a mediastinal radiation dose of 4 Gy, the patient still has no specific complaints related to her idiopathic PAH and is in very good health. This case report raises several questions and issues.
First, the pulmonary consultants of the Coburg Clinic and Bad Nauheim/University of Giessen belong to the national pulmonary hypertension guideline group and have extensive experience with regard to diagnostic and therapeutic approaches in pulmonary hypertension. A discussed spontaneous dissection of the pulmonary main stems without urgent clinical presentation could not be assumed. However, there was no change in the local anatomy of the central pulmonary vessels seen on timely MRI scans. Therefore, we believe that neither this etiology nor any congenital malformation played a role in this case. Thoracic X-rays over two decades had not revealed any evidence of malformation. Serum analysis revealed no signs of vasculitis, coagulopathy, thyroid or hepatic disorder, or collagenosis. There was no other predominant cardiopulmonary disease directly involved in the initially highly elevated PAH hemodynamics, and the European Society of Cardiology algorithm for PAH was worked through thoroughly. Mistakes in measurement or misdiagnosis can be ruled out of the discussion from our point of view.
Second, to the best of our knowledge, spontaneous healing of an idiopathic PAH has not been reported in the literature. Further, the patient did not show reactivity on an iloprost inhalation test.
Third, the histological evidence of activated pneumocytes in the left upper lobe mass gave a clue towards an acute inflammatory pathobiology of mediastinal and pulmonary localized fibrosis. It is conceivable that low-dose radiation could have reduced this active inflammatory process and consequently the PAR in these areas. On the other hand, the areas involved were not very large, and we estimate that less than 10% of the peripheral vascular bed of the entire lung was involved in direct consolidated fibrosis as shown in the left upper lobe apex. Further, the direct involvement of fibrosis around the main stems of the pulmonary arteries was assumed to be rather low on positron emission tomography and MRI scans. Assuming a positive influence of low-dose radiation, it was unlikely that this could have reduced PAR by more than two thirds of the initial value. However, it is notable that this patient did not have pure idiopathic PAH, but was mixed with some “acute” (mediastinal) fibrosis which could have contributed to the unexpected success of PAR reduction.
Fourth, in regard to anatomic considerations, the only treatment given was low-dose (4 Gy) radiation to the hila and therefore the main stems of the pulmonary arteries on both sides. We therefore hypothesize that this low-dose irradiation positively influenced the idiopathic PAH and at least in part contributed to the reduced PAR. The sympathetic autonomic nervous system is generally accepted to play a key role in the development of PAH, and it is notable that low-dose radiation seems to disturb the growth and repair capacity of the autonomic nerve system.1 Therefore, we hypothesize that the reduction of sympathetic efferent signals by 4 Gy radiation could have contributed to the reduction in pulmonary vascular resistance seen over time in the case described here. However, because this clinical effect was unexpected, we did not perform any measurements of autonomic nervous system function.
Disclosure
The authors report no conflicts of interest in this work.
References
McGale P, Darby SC, Hall P, et al. Incidence of heart disease in 35,000 women treated with radiotherapy for breast cancer in Denmark and Sweden. Radiother Oncol. 2011;100(2):167–175. | |
Heidenreich PA, Hancock SL, Lee BK, Mariscal CS, Schnittger I. Asymptomatic cardiac disease following mediastinal irradiation. J Am Coll Cardiol. 2003;42(4):743–749. | |
Heidenreich PA, Hancock SL, Vagelos RH, Lee BK, Schnittger I. Diastolic dysfunction after mediastinal irradiation. Am Heart J. 2005; 150(5):977–982. | |
Ghobadi G, van der Veen S, Bartelds B, et al. Physiological interaction of heart and lung in thoracic irradiation. Int J Radiat Oncol Biol Phys. 2012;84(5):e639–e646. | |
Ebbeskov Lauritsen L, Meidahl Petersen P, Daugaard G. Neurological adverse effects after radiation therapy for Stage II seminoma. Case Rep Oncol. 2012;5(2):444–448. | |
Vujaskovic Z, Gillette SM, Powers BE, Thurmond DN, Gillette EL, Colacchio TA. Ultrastructural morphometric analysis of peripheral nerves after intraoperative irradiation. Int J Radiat Biol. 1995;68(1):71–76. | |
Al-Jahdari WS, Suzuki Y, Yoshida Y, et al. Growth cone collapse and neurite retractions: an approach to examine X-irradiation affects on neuron cells. J Radiat Res. 2008;49(5):481–489. | |
Tan YF, Rosenzweig S, Jaffray D, Wojtowicz JM. Depletion of new neurons by image guided irradiation. Front Neurosci. 2011;5:59. | |
Hockerfelt U, Kjorell U, Malm V, Henriksson R, Franzen L, Forsgren S. VIP in the rat parotid gland in response to irradiation. Regul Pept. 1997;68(2):83–89. | |
Otterson MF, Koch TR, Zhang Z, Leming SC, Moulder JE. Fractionated irradiation alters enteric neuroendocrine products. Dig Dis Sci. 1995;40(8):1691–1702. | |
Steensma DP, Hook CC, Stafford SL, Tefferi A. Low-dose, single-fraction, whole-lung radiotherapy for pulmonary hypertension associated with myelofibrosis with myeloid metaplasia. Br J Haematol. 2002;118(3):813–816. | |
Bogaard HJ, Natarajan R, Mizuno S, et al. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182(5):652–660. | |
Ishikawa M, Sato N, Asai K, Takano T, Mizuno K. Effects of a pure alpha/beta-adrenergic receptor blocker on monocrotaline-induced pulmonary arterial hypertension with right ventricular hypertrophy in rats. Circ J. 2009;73(12):2337–2341. | |
Drake JI, Bogaard HJ, Mizuno S, et al. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45(6):1239–1247. | |
Handoko ML, de Man FS, Allaart CP, Paulus WJ, Westerhof N, Vonk-Noordegraaf A. Perspectives on novel therapeutic strategies for right heart failure in pulmonary arterial hypertension: lessons from the left heart. Eur Respir Rev. 2010;19(115):72–82. | |
So PP, Davies RA, Chandy G, et al. Usefulness of beta-blocker therapy and outcomes in patients with pulmonary arterial hypertension. Am J Cardiol. 2012;109(10):1504–1509. | |
Chen SL, Zhang FF, Xu J, et al. Pulmonary artery denervation to treat pulmonary arterial hypertension: the single-center, prospective, first-in-man PADN-1 study (first-in-man pulmonary artery denervation for treatment of pulmonary artery hypertension). J Am Coll Cardiol. 2013;62(12):1092–1100. | |
Said SI. Mediators and modulators of pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2006;291(4):L547–L558. | |
Hamidi SA, Lin RZ, Szema AM, Lyubsky S, Jiang YP, Said SI. VIP and endothelin receptor antagonist: an effective combination against experimental pulmonary arterial hypertension. Respir Res. 2011;12:141. | |
Haberl I, Frei K, Ramsebner R, et al. Vasoactive intestinal peptide gene alterations in patients with idiopathic pulmonary arterial hypertension. Eur J Hum Genet. 2007;15(1):18–22. | |
Seferian A, Jais X, Creuze N, et al. Mediastinal fibrosis mimicking proximal chronic thromboembolic disease. Circulation. 2012; 125(16):2045–2047. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.