Back to Journals » Clinical Ophthalmology » Volume 16
Late-Onset Retinal Degeneration: Clinical Perspectives
Received 3 August 2022
Accepted for publication 13 September 2022
Published 30 September 2022 Volume 2022:16 Pages 3225—3246
DOI https://doi.org/10.2147/OPTH.S362691
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Leonardo Lando,1,2 Shyamanga Borooah1
1Shiley Eye Institute, University of California San Diego, La Jolla, CA, USA; 2Department of Ophthalmology and Vision Sciences, University of Toronto, Toronto, ON, Canada
Correspondence: Shyamanga Borooah, Shiley Eye Institute, University of California San Diego, 9415 Campus Point Drive, La Jolla, CA, 92093, USA, Email [email protected]
Abstract: Late-onset retinal degeneration (L-ORD) is a type of retinal dystrophy marked by nyctalopia and subretinal pigment epithelium deposits, which eventually promote retinal atrophy with final visual compromise. L-ORD may also present with changes in the anterior segment, notably long anterior zonules and iris atrophy, distinguishing it from other inherited eye conditions. Although it can clinically simulate age-related macular degeneration, L-ORD has a different course of progression and prognosis, requiring adequate diagnosis for patient counseling. This review summarizes the main clinical, genetic, pathophysiological, diagnostic, and therapeutic aspects of L-ORD to help ophthalmologists identify and manage this rare ocular disease.
Keywords: retinal dystrophy, retinal degeneration, macular dystrophy, late-onset retinal degeneration, C1QTNF5, CTRP5
Introduction
Late-onset retinal degeneration (L-ORD) is a fully penetrant autosomal dominant inherited retinal disease (IRD) that primarily affects the retinal pigment epithelium (RPE) but also later involves the choroid and retina. Clinically, L-ORD results in marked retinal and RPE atrophy, which eventually extends to the whole retina. On histopathology, L-ORD is marked by sub-RPE deposits and occasionally other pathology, including choroidal neovascularization (CNV).1–26 In addition to the posterior segment findings, L-ORD can also present with changes in the anterior segment, particularly long anterior zonules (LAZ), which may be found before fundus compromise.3,8,20,21 A typical case of L-ORD with anterior and posterior segment involvement is illustrated in Figure 1. Of particular interest to general ophthalmologists and retinal specialists, L-ORD recapitulates many features of more common retinal degenerative entities, including age-related macular degeneration (AMD).1,3,5,14,17,26–28 As a result, distinguishing L-ORD from other differential diagnoses is essential for both prognosis and family genetic counseling.
 |
Figure 1 Photo-mosaic demonstrates findings in a 62-year-old man with nyctalopia and a molecular diagnosis of late-onset retinal degeneration (Ser163Arg). In the anterior segment (A and B), the patient presents with bilateral peripupillary iris ruff atrophy (arrowheads) in association with long, anteriorly inserted zonules (arrows), mostly visible under transillumination. In addition, color fundus photos of both eyes (C and D) show subtle subretinal yellow deposits (pseudodrusen) and pigmentary changes starting inferotemporally in correspondence to an increased reticular pattern (hyperautofluorescence) on scanning laser ophthalmoscopy (E and F). |
Recently, clinical and basic science studies have expanded the body of knowledge on L-ORD by identifying new mutations and by better phenotyping of clinical findings. In addition, insights have also been made into disease pathophysiology. Yet, there continues to be a gap in knowledge of the exact underlying mechanisms leading to subretinal deposit formation. In this review, we summarize the clinical, genetic, pathophysiological, diagnostic, and therapeutic aspects of L-ORD to provide clinicians with an overview of this rare disease (Table 1).
 |
Table 1 Key Clinical Findings in Late-Onset Retinal Degeneration |
Methodology
The literature consulted for this review consisted of all articles involving humans and published in the English language retrieved from the MEDLINE database until June 2022. The following search terms were used: late-onset retinal degeneration, LORD, L-ORD, autosomal dominant hemorrhagic macular dystrophy, late-onset macular degeneration, choroidal dystrophy, retinal dystrophy, inherited retinal disease, inherited eye disease, macular dystrophy, inherited macular degeneration. Major clinical studies on L-ORD and used in this review are summarized in Table 2. Other studies on L-ORD, including ones involving animal models, were cited throughout the text for a comprehensive coverage of the topics.
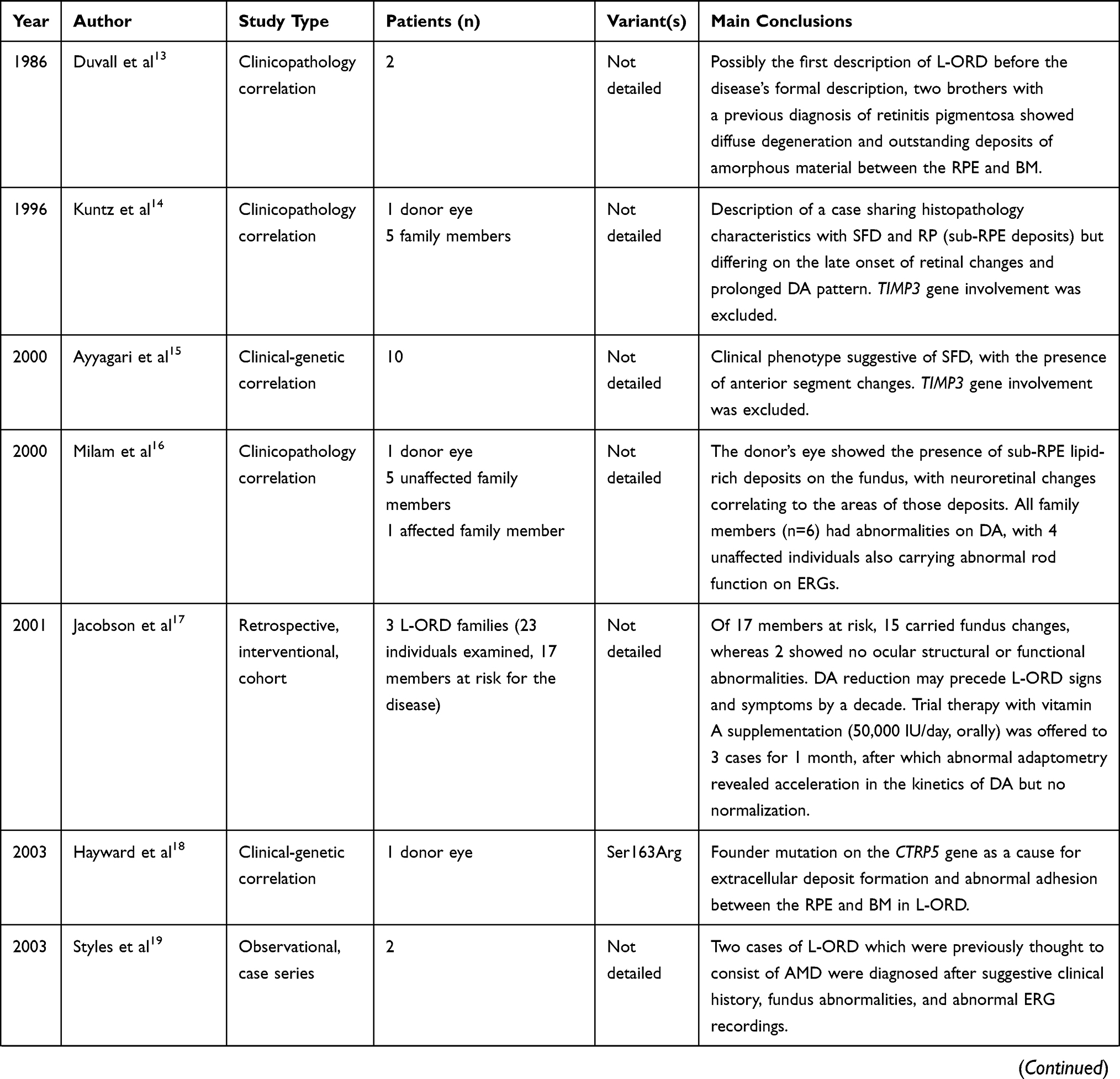 |  |  |
Table 2 Summary of Major Clinical Studies in Late-Onset Retinal Degeneration |
Historical Landmarks
L-ORD is a relatively new term, with the first use approximately 25 years ago. Despite this, cases of L-ORD have previously been described in the literature, with other individuals later found in members of families with genetically confirmed L-ORD. In 1986, Duvall et al published a clinical–pathological study of two brothers, aged 62 and 74, manifesting a progressive condition starting with night blindness around the 50s, symmetrical pigmentary disturbance from the equator to the posterior pole, areas of depigmentation, and extensive sub-RPE deposits.13 These cases were diagnosed as “dominant retinitis pigmentosa” as they presented at a relatively late stage but probably corresponded to the earliest cases of L-ORD in the literature, although they were not recognized as such at the time.
In 1996, the first reference to a late-onset retinal degeneration was made by Kuntz et al in the form of “dominant late-onset retinal degeneration”.14 The authors, based on the histopathology study of a post-mortem donor retina from an 80-year-old patient, found widespread, thick sub-RPE deposits distributed between the Bruch’s membrane (BM) and the RPE from the optic nerve to the ora serrata which disrupted almost the entire photoreceptor layer.14 Although the findings were noted to somewhat overlap with features of Sorsby fundus dystrophy (SFD), dominant drusen (DD), and retinitis pigmentosa (RP), they were also felt to differ in terms of deposits pattern and quantity.14
In 2003, a founder mutation (Ser163Arg) in the short-chain collagen gene known as either C1QTNF5 or CTRP5 was identified and associated with a late-onset retinal degeneration phenotype in gene linkage studies.15 Hayward et al also used the phrase “late-onset retinal degeneration” for the disease.18 In the subsequent decades, several other families were identified worldwide through familial and genetic studies, which traced a common ancestor who probably lived in southeast Scotland.3,18,20 In 2009, a classification by Borooah et al was designed to assist in categorizing these patients based on disease severity.3 However, variabilities in the clinical and imaging findings noted in the more recently published cases have limited the applicability of this generalized staging system to all cases. More recently, new pathogenic variants of C1QTNF5 have been reported. Findings from the clinical and basic science work associated with these mutations will be discussed further below.29,30
Terminology
The name late-onset retinal degeneration (OMIM: 605670, 608752) has been employed in the literature with certain consensus by the majority of ocular geneticists and authors since the identification of the C1QTNF5 mutation.18 The “late-onset” terminology was used because of the relatively later onset for Mendelian inherited retinal degeneration. The term has been considered adequate for alluding to the disease’s typical presentation, which usually starts in older patients compared to other retinal dystrophies, in which retinal dysfunction usually starts in the first or second decades of life.14,31,32 Furthermore, the designation retinal degeneration points to L-ORD’s natural course until the late stage in which atrophy of the entire retina is encountered.
However, the term “late-onset” has caused some confusion, as the onset of macular degeneration is earlier than in traditional age-related macular degeneration, one of its primary differential diagnoses. While original studies have referred to L-ORD as “autosomal dominant hemorrhagic macular dystrophy” or “late-onset macular degeneration”, these names became historically less frequently used.3,15,20 Additionally, some studies have looked to highlight the relatively early macular involvement and used the term “late-onset macular degeneration”.3,20 None of the past or present synonyms of L-ORD have captured the anterior segment findings of the phenotype.
Demographics
Prevalence
The exact prevalence of L-ORD is not known. Reports in the literature and our clinical experience from a database generated from some European patients estimate that there are now over a hundred patients with clinically or genetically confirmed disease. Since the mapping of C1QTNF5,18 families in the United States,4,6,9,14,15,20 United Kingdom,1,3,5,7,8,10,12,17–19,21,23–25 Canada,11 France,26 and Belgium2 have been studied. L-ORD cases are likely underreported due to misdiagnosis for AMD and the limited availability of genetic testing in many countries worldwide.27,28,32 However, there must be some caution as the number of cases listed in some studies may describe the patients from the same family who are reporting to different centers.
Age
Several subtle signs and symptoms can precede degeneration noted on fundoscopy in patients with L-ORD.1,2,15,17,20,21 The age of first symptoms can be variable but generally tends to occur in the 5th decade, starting with dark adaptation (DA) delay.1,2,4,5,7,11,12,17,23 Anterior segment findings, especially LAZ, can be identified before the age of 40, functioning as an important diagnostic marker for L-ORD disease.20,21,33,34 In contrast to L-ORD, AMD generally manifests after the age of 55.27,28 DA delay can be a finding in AMD, similar to L-ORD, especially in cases with the reticular pseudodrusen endophenotype.10,17,24,28
Sex
No comparative data on gender distribution is available for L-ORD. However, the condition is not believed to have any gender preference in the largest cohort descriptions.1,2,7,8,11,24 This understanding is also supported by the fact that the causal C1QTNF5 gene is not found on the sex chromosomes and that the disease appears fully penetrant in both males and females. Comparatively, AMD has equal incidence in men and women. The higher AMD prevalence amongst women is thought to be due to a longer expectancy in women.28
Ethnicity
No ethnic predisposition has been elucidated in L-ORD, given the relatively small number of patients described. In most cohorts, a predominance in Caucasian populations originating in Western Europe has been observed.1,2,7,8,11,24 Similarly, AMD has a vast predominance in Caucasians, who are also at increased risk of disease progression.28 In the Asian population, a high rate of polypoidal choroidal vasculopathy, a clinical variant of neovascular AMD, has been encountered.28,35,36
Genetic Background
L-ORD is a dystrophy showing an autosomal dominant pattern of inheritance with full gene penetrance. Initial genetic studies into L-ORD aimed to exclude known pathogenic variants in coding genes that resulted in diseases with a similar phenotype, including rhodopsin, peripherin/RDS, and TIMP3.15,16 The initial L-ORD pathogenic variant (Ser163Arg) was identified at chromosome 11 in the complement 1Q tumor necrosis factor 5 (C1QTNF5) by gene linkage analysis, with the region 11q23.3. identified as responsible for the phenotypic changes.18,37–40
C1QTNF5 features 3 exons and 7764 bases. The synonymous C1QTNF5 protein, composed of a globular domain, collagen-like fibril domain, and a nitrile terminal domain, carries 243 amino acids.39,41 The mutations all occur in a region of the gene encoding the globular C1q domain. C1QTNF5 protein is thought to be part of the adiponectin superfamily, having a function that is only partially understood. Increasing evidence indicates that the C1QTNF5 plays a part in the cellular energy homeostasis of the RPE and extraocular tissues.40,42
Recently, it was shown that C1QTNF5 also binds to the adiponectin receptor 1 (ADIPOR1) on RPE. The mutant C1QTNF5 constitutively activates the receptor leading to altered AMP-activated protein kinase (AMPK) metabolism as the cell loses its ability to sense AMP/ATP ratio alterations.42 The resultant abnormal protein leads to the aggregation and formation of high-molecular-weight precipitates. These have been found to alter the protein interactions at ocular sites by modifying C1QTNF5 folding and its final forms, misplacing the protein during its secretory routes, and potentially causing resultant abnormal cellular adhesion.20,38–40,42–44 In addition, the mutant protein has been found to interact with HTRA1 (HtrA Serine Peptidase 1), a protein implicated in AMD. Both mutant and wild-type C1QTNF5 bind to HTRA1, being cleaved by HTRA1, although the mutant is more resistant to cleavage, leading to a hypothesis that C1QTNF5 and HTRA1 are found in sub-RPE aggregates. Increased CTRP5 in sub-RPE deposits has been confirmed using a mouse model of L-ORD.43
Since the identification of the most commonly reported mutation, Ser163Arg (c.489C > G), in 2003, five additional missense variants have so far been added to the list – p.(Gly216Cys), p.(Pro188Thr), and p.(Ser163Arg) (c.489C > A) – and two other by whole genome sequencing – p.(Pro186Ser) and p.(Ser190Trp).29,30 In addition, exome sequencing analysis has identified a variant p.(Pro188Ala) as likely pathogenic in one L-ORD individual.45 As a result, when L-ORD is suspected, it is best that whole exome sequencing, polymerase chain reaction, or panel covering the whole region encoding the globular domain be used to identify potentially pathogenic variants. A dominant-negative disease mechanism using in silico and in vitro studies has been proposed.29,30
Histopathology
Histopathological studies of L-ORD eyes have allowed for adequate correlation with the clinical and imaging findings, mainly using optical coherence tomography (OCT), and offering some insights into disease mechanisms.4,5,10 Five major investigations have focused on pathology analysis of L-ORD,5,16,20 two before the disease description.13,14 The common finding from all these studies was that gross pathology of L-ORD retina carried a resemblance to other degenerative and inherited conditions, including AMD and SFD.16,20
On microscopy, L-ORD is marked by the formation of thick deposits between the RPE and BM, causing displacement of the RPE cells and disruption of the photoreceptor layer, with a generalized reduction in photoreceptor density and many structurally abnormal photoreceptors including short outer segments and disorganized disc membranes, or complete loss of the photoreceptor layer in certain areas.5,13,14,16,20 The deposit may show various patterns described as a spherical shape, multilayered, coalescent, or linearly packed within the BM-RPE complex. In some regions, the RPE may demonstrate depigmentation, thinning, or abrupt discontinuation, with melanin deposits from broken cells being found in other retinal sites, including the far retinal periphery.5,13,14,16,20
Biochemically, the sub-RPE deposits were demonstrated to measure around 50µm and carry a rich content of esterified and non-esterified cholesterol, protein, and calcium.14,16 The deposits also tend to present initially greater thickness in the mid-peripheral areas where photoreceptor loss and reduction in DA are most prominent.5,16 Progressive BM-RPE separation caused by the sub-RPE accumulation on histopathology correlates to the trilaminar pattern found on OCT images from L-ORD eyes, similarly observed in other autosomal dominant macular dystrophies, such as DD and SFD.4,10 However, measuring BM-RPE separation in L-ORD is difficult due to early atrophy.
The presence of CNV can be observed in areas of subretinal deposits associated with capillary formation from the choriocapillaris and a fibrovascular layer between the deposits and the elastin layer of BM.6,16 Characterization of areas of CNV has been further made recently by incorporation of multimodal imaging.6 Clinical evidence of atrophy formation without retinal deposits visible on fundoscopy in cases of Pro188Thr variant is a recent new finding contrasting to the conventional deposit-to-atrophy mechanism classically seen in Ser163Arg.2 Although sub-RPE deposits have been well characterized in L-ORD,5,14,16 the cause of formation is still not well established, with recent studies suggesting defective lipid metabolism mediated by AMPK as a cause.42
Clinical Findings
Laterality
L-ORD tends to affect both eyes with some level of symmetry which applies to the anatomic and functional changes, similarly to AMD.1,2,4–26 Previous investigations have shown a high level of agreement between eyes for different clinical markers of L-ORD,1 including morphology, extent, and pattern of LAZs, iris atrophy, cataract formation, sub-RPE deposits, retinal and choroidal structural changes (thickness and volume), vascular arcades thinning, and development of end-stage atrophic lesions.1–5,7–12,19,21,23,25,46,47
Functionally, L-ORD symmetry is noted on the degree of visual impairment in each eye tested by visual acuity, visual field, or microperimetry.1,2,8,9,17,19,24,46 In early or subclinical stages, L-ORD can affect eyes asymmetrically until the development of the full spectrum disease with characteristic fundus changes. Limitations from the current classification methods have been acknowledged in particular heterogeneous cases.1–3 To the best of our knowledge, no case of unilateral L-ORD has been identified.
Visual Acuity
The visual acuity in patients with L-ORD is mostly preserved at early ages with no macular atrophy. In the 30s and 40s, individuals may present only with nyctalopia while vision remains intact on evaluation with conventional reading charts.1–3,5,6,10–12,16,17,20,21,23 With disease progression and definitive atrophy involving the fovea, BCVA can rapidly decline to complete blindness.1–3,5,6,10–12,16,17,20,21,23 Different L-ORD variants may influence the pace of visual changes, although this characterization is still unclear. L-ORD patients generally manifest a reduction in visual performance over one to two decades, contrasting vision loss in non-neovascular AMD, which generally takes a steady and slower course starting from the 60s.1–7,9–12,14–17,19–21,24,28 The pace of visual reduction during the time of progressing disease is not fully understood in L-ORD individuals but is assumed to accelerate from clinical observations.1,2,12,20
Vision loss in L-ORD may also be dependent on the development of other ocular complications, including CNV2,3,6,11,22,25,26 and cataracts.3,8 CNV is treatable in L-ORD, and as a result, patients should be advised to return urgently in cases of sudden visual loss in a similar manner to non-neovascular AMD patients. Patients may also experience increasing photophobia from retinal degeneration and iris atrophy with associated pupillary dysfunction.2,20,21 At end-stage disease, variabilities from the vision standpoint can be seen amongst L-ORD populations, with cases retaining islands of peripheral vision. In contrast, others may progress to complete loss of central and peripheral vision.1,2,4,10–12,17,19,20
Pupillary Reaction Abnormalities
The pupillary reflex may become progressively impaired during the natural history of L-ORD, following the course of iris changes, including at the level of the pupillary sphincter muscle.2,11,21,23 However, some patients carrying the Pro188Thr variant may present with changes in pupillary reaction in the absence of any visible structural changes on the anterior segment.2 In these patients, end-stage disease with fixed semi-dilated pupils has been observed.2 Comparatively, AMD is not known to induce immediate changes in the pupillary reactions.
Anterior Segment Changes
Cornea
No corneal abnormalities or corneal-related refractive changes have been reported in L-ORD.
Angle
Although the presence of LAZ is widely recognized in L-ORD2–4,6,11,20,21,47,48 and has been previously associated with pigment dispersion in the angle in normal eyes,49 pigmentary angle abnormalities have not been consistently observed in L-ORD. Unpublished data by our group looking at anterior segment findings have identified increased pigment at the angle in a few cases of the Ser163Arg variant but no particular change in the angle structures, which is supported by other groups looking at Pro188Thr in which no angle changes were found.2,47 No angle abnormalities pointing to narrow-angle appear related to the cases of glaucoma which have been related to some L-ORD patients.3
Iris
A series of iris changes have been listed as part of the L-ORD phenotype, mainly consisting of rarefaction defects, iris stromal atrophy, and pupillary ruff atrophy.2,3,11,20,21,23,47 Although these changes may be insidious and variable, they may affect visual function by promoting glare and photophobia.2 In a series of 26 cases with Pro188Thr, mid-peripheral patchy iris atrophy occurred in 7 cases, a moth-eaten pattern in the pupillary margin in 9, and both defects in 6. This observation suggests a preponderance of iris abnormalities in this phenotype as opposed to LAZs, which occurred in only 2 cases, neither of which had iris changes.2 A mechanism for iris abnormalities in L-ORD has been hypothesized with the presence of LAZs and mechanic disruption of the iris pigment epithelium; nonetheless, pigment dispersion syndrome has not been well-characterized as part of L-ORD.3,47,49 Importantly, iris changes in L-ORD are most noticeable on the clinical examination under transillumination before dilation.
Lens
C1QTNF5 is expressed in the lens, particularly in the lens capsule.8 Individuals with L-ORD have been noted to evolve with cataract formation as part of the phenotype.3,8 Phacoemulsification may be considered for relative visual recovery, which is dependable on the foveal status.8 Biometric specifics of the lens measurements in L-ORD versus the general population have not been established. LAZ has not been found to offer an increased rate of complications amongst L-ORD individuals undergoing cataract surgery, although previous results were non-comparative.3,8
Zonules
One of the anterior segment hallmarks of L-ORD is the presence of LAZs, usually noted on the ocular examination without dilation by retroillumination.1–11,15,17,20,21,23–25 LAZs are defined as zonule fibers extending closer to the central lens, usually beyond the 3-mm central area, causing reduction of the zonule-free zone, defined as the measured distance between the longest zonule to the lens center.34,49
LAZs may occur in the general population, being described in African-Americans in 2% in association with glaucoma, pigment dispersion syndrome, and hyperopia.34,50 None of these associated conditions have been clearly elucidated in L-ORD regarding LAZ. In a family with the Ser163Arg variant, LAZ occurred in 18 of 55 individuals, with 6 showing concomitant macular degeneration.20 While LAZs are primarily reported in older individuals, their presence below the 40s raises suspicion for L-ORD, with the youngest case reported as young as 24 years old.3,20 Data from our group, yet to be published, revealed that LAZs can occur in different patterns amongst L-ORD patients, including segmental, non-segmental, and mixed, similarly to other populations.47,49,51,52 Only 2 individuals of 26 in a cohort of Pro188Thr manifested LAZ despite presenting typical fundus changes for L-ORD.2
Posterior Segment Changes
Vitreous
No vitreous abnormalities have been associated with L-ORD.
Retina
C1QTNF5 is expressed by the RPE. The primary clinical features of L-ORD are found in the retina and macula. L-ORD is marked by extensive retinal atrophy involving all fundus topographies plus macula.1,2,4–12,15–26 The rate of progression for each of the retinal changes has been the scope of multiple prospective longitudinal investigations and correlational studies. Phenotypic variabilities in the retinal findings have challenged the establishment of disease categorization and uniform characterization of these changes.1–4,9,11 These challenges have been further reinforced by recent studies with new mutations revealing that genetic variabilities may account for discrepancies in clinical findings.1–3,29,30
The first retinal findings in L-ORD are the accumulation of yellowish subretinal deposits, which have been mainly found scattered in the mid-peripheral fundus areas but also involving the macula and extreme periphery, sometimes from the onset.1,2,4–26 These deposits were referred to as “drusenoid”, or “drusen-like”, or even simply “drusen” for their close resemblance on imaging to AMD-related drusen.1,2,4–26 These deposits are likely located in the subretinal space, showing a reticular or pseudodrusen-like pattern and being best localized by OCT, infrared and multicolor imaging.4,9,10,27,28 Fundoscopic distinction of each these deposits is challenging, as exemplified in Figure 2.1,10
 |
Figure 2 Multimodal imaging assessment of a 59-year-old man with early-stage disease (Ser163Arg) marked by subretinal deposits across the macula symmetrically in both eyes with relative foveal sparing (A and B). These pseudodrusen-like deposits are more easily visualized using 30-degree fundus autofluorescence (C and D) or infrared plus spectral-domain optical coherence tomography (E and F, arrowheads). |
Subretinal deposits in L-ORD tend to concentrate in the temporal and inferior areas close to the macula.1,2,4–26 A few cases with disease initiating in less favorable sites such as the nasal retina have been described, yet are much less representative.1,3,11 Patients are usually first diagnosed in the phase where deposits have already encroached the central macula, precluding precise observation of this process.1,2,4,7,9–13,16,19,20,23 De Zaeytijd et al described the fundoscopic changes in patients with the Pro188Thr variant within three phenotypic categories based on presentation: adjoining paving-stone-like atrophic patches, tiny yellow-white subretinal dots, or larger yellow, thick, round drusenoid deposits.2
With progression, the deposits may assume a coalescent pattern or remain discrete but promote disruption of outer retinal layers and neuroretina.2,4,14,16–18 In this stage, patients commonly reach the full spectrum of vision abnormalities, complaining of nyctalopia, difficulty in dark adaptation, and reduction in BCVA if the macular has been consistently affected.1,2,4–26 Findings may also be seen in color vision testing and dark-adapted full-field electroretinography (ffERG) tests.2,11,14–16,23,24 A case of L-ORD in the atrophic phase is shown in Figure 3.
 |
Figure 3 Multimodal imaging findings of the foveal-sparing, atrophic phase in a 67-year-old man with the Ser163Arg variant. Color fundus photos of both eyes (A and B) show small areas of atrophy, as indicated by the arrowheads on the color photographs (A and B) and fundus autofluorescence (C and D). Spectral-domain optical coherence tomography with scanning line cutting through the left fovea (E) reveals an impending area of atrophy characterized by complete loss of the retinal pigment epithelium and outer retinal atrophy (bracket), as well as diffuse choroidal thinning and reticular pseudodrusen-like changes (arrowheads) (F). |
The last phase of this process reaches its peak with partial and then complete disruption of the retinal structure with atrophy, often referred to as “geographic-like” or “geographic atrophy”.1,2,4,8,9,11,21,23 In this phase, CNV may be encountered in a number of cases, particularly in the periphery, where borderline zones between atrophy and normal retina are usually located.6,22,25,26 Keenan et al also identified large peripheral type 1 nonexudative CNV formations in all 4 L-ORD patients in their case series identified using wide-field fluorescein angiography.6
While most authors have used the term atrophy to describe end-stage L-ORD with scarring, the incorporation of OCT into clinical phenotyping has allowed a detailed interpretation of the L-ORD atrophic phase in light of recent AMD terminology.1,2,4,9,11,12 As such, terminologies such as “complete retinal pigment epithelium (RPE) and outer retinal atrophy” (cRORA) can be used to describe cases progression to atrophy which could be helpful in clinical trials.1,53,54 Borooah et al, in their prospectively collected series of 12 L-ORD patients, quantified the retinal changes over 2 years, confirming a high level of agreement between eyes (symmetry) and showing that patients may manifest a reduction in the central retinal thickness not reflected in the concomitant reduction of choroidal thickness.1 The authors also calculated the mean growth of atrophy to be 2.67 mm2/year (square rooted: 0.57 mm/year).1 Another group has also investigated the foveal-sparing atrophic phase confirming a monotonic growth of regions of atrophy within a range of approximately 0.53 to 1.97 mm/year (square-rooted values).9
Choroid
Choroidal changes in L-ORD have been mainly characterized by progressive thinning, affecting the inner layers and choriocapillaris, as well as CNV as a form of disease complication.1,2,6,15,23 Studies focused on the choroidal assessment in L-ORD have mainly relied on using OCT to detect these changes, focusing particularly on thickness metrics.1,4,23
Soumplis et al were one of the first to employ choroidal thickness measurements on the B-scan for L-ORD, proposing that reduction in thickness was probably involved in the disease mechanisms.23 Subsequently, comparative observations also indicated similar subfoveal choroidal thicknesses between L-ORD and AMD, suggesting a point of similarity between these two conditions, especially in cases with pseudodrusen.4
Choroidal volume and choroidal vascularity index have also been explored, though no changes were noted when compared to healthy, age-matched control eyes or prospectively over 2 years.1 A high level of symmetry between same-patient eyes for choroidal metrics was also noted.1 However, in a longitudinal study, no choroidal changes were noted in terms of thickness and volume over 2 years, despite reduced choroidal thickness at baseline compared to age-matched, normal control eyes.1 The authors acknowledged that uncontrolled factors inherently linked to modifying choroidal thickness might explain the lack of progression.1,55 Further clarification on the relationship between C1QTNF5 mutations and choroidal abnormalities is still required ideally with a prospective, longitudinal study controlling for factors affecting choroidal thickness.
Optic nerve
Although not primarily affecting the optic nerve, L-ORD can still induce secondary neuropathy in the setting of glaucoma or end-stage retinal atrophy, causing an impact on the nerve fiber layer.1–4,9,11,12,15,20 The mechanism of glaucoma in L-ORD has not been completely elucidated, yet it is thought to involve a primary open-angle mechanism (POAG), as anterior chamber shallowing has not been identified in cases with increased intraocular pressure.20,47 One case showing pale and cupped optic nerve with normal ocular pressure has been described.19 Another case studied with histopathology and managed for increased pressure at the 60s revealed a pale optic nerve on specimen analysis.16 The effects of pigment in the trabecular meshwork found in some L-ORD individuals have not been confirmed to lead to raised intraocular pressure or secondary neuropathy.2,19,20 It is likely that POAG is unrelated to the C1QTNF5 as C1QTNF5 has not appeared in genome-wide association studies of glaucoma and that POAG is not fully penetrant, unlike most of the other features associated with L-ORD.
Vascular arcades
L-ORD does not particularly affect the retinal vascular arcades, although vessel attenuation may be seen in advanced disease following the diffuse compromise of the retina and choroid.1–3,15,17 Reduction in retinal vessel caliber has also been confirmed by histopathology.16 While vessel fading in advanced L-ORD is much more evident than in end-stage AMD, it is otherwise less prominent than in retinitis pigmentosa, which is classically marked by arteriolar attenuation and pigment clumping following the vascular bed.28,31,56 The mechanisms affecting retinal vasculature are possibly related to involutional changes in the retinal tissues, similar to what is observed at choroidal vessels assessed by the choroidal vascularity index.1,16
Systemic Associations
C1QTNF5 ribonucleic acid (RNA) expression is highest in the ciliary body and RPE within the eye.18,20 RNA-sequencing analysis has shown that C1QTNF5 RNA is expressed in many tissues, including adipose tissue, gastrointestinal tract, lung, and the brain.57 The C1QTNF5 protein has been shown to circulate in plasma. Animal studies have indicated that C1QTNF5 is a negative regulator of glucose metabolism and insulin sensitivity.40,42 However, currently, L-ORD has not been associated with any systemic condition.
Diagnostic Considerations
The diagnosis of L-ORD is based on a suggestive clinical and family history for IRD supported by ocular findings, which can be confirmed by genetic testing. The increasing use of free, at-point-of-service whole exome genetic testing in patients with a family history of early onset macular degeneration can help exclude L-ORD or other inherited macular diseases. In addition, imaging studies play a fundamental role in highlighting the ocular changes, guiding investigations and offering prognostic understanding into differentiating L-ORD from other similar IRDs. We describe here some of the most relevant complementary imaging and functional tests used to evaluate a patient with L-ORD.
Imaging
Fundus photography
Color fundus photography has been conventionally applied in studies to capture fundus changes in true color, especially at the macula, and to document findings for comparison.1–4,6–12,15,20,23,25 Similarly, wide-field techniques have been employed to assess for abnormalities in the retinal periphery, including the presence of subretinal deposits and CNV.6,10,26
Scanning laser ophthalmoscopy using multicolor imaging provides a relatively new strategy in L-ORD, allowing enhanced visualization of retinal pseudodrusen, deposits, and atrophy, especially in the infrared channel. It may better identify some of these features than color fundus photos.10,25
Fundus autofluorescence
Fundus autofluorescence (FAF) is considered an important technique for the assessment of IRDs such as L-ORD, revealing elements of the metabolic and structural status of the retina and RPE.1,2,4,9–12,23,31 FAF can provide information about the distribution and pattern of subretinal deposits, seen as hyperautofluorescent dots, and areas of RPE dysfunction usually accompanying those deposits as points of hypoautofluorescence.1,2,10,11,23 Moreover, FAF allows the visualization, delineation, and calculation of areas of atrophy or impending atrophy in retinal sites with or without deposits. In disease course, these areas are seen as small-to-large islands of hypoautofluorescence, which tend to enlarge as a consequence of partial or complete RPE and retinal disruption.1,2,4,9,11,12,23
Follow-up using FAF has been important for defining visual prognosis, phenotypic characterization, and potential endpoints for clinical trials. In multiple series investigating different L-ORD variants, patients were shown to evolve with different FAF features, some presenting with early central atrophy and others demonstrating initial islands of atrophy in the periphery, with progressive involvement of other retinal quadrants and macula.1,2,4,11,12,23 FAF can be used are a relative surrogate marker to study the rate of disease progression. By using FAF metrics, Borooah et al were able to calculate the rate of retinal atrophy expansion at approximately 2.7 mm2/year, which is comparable to AMD, although the study only covered foveal-sparing L-ORD over a short period of 2 years.1
Optical coherence tomography
OCT allows a cross-sectional view of the retinal and choroidal tissue (“light biopsy”) with significance for the study of disease biomarkers and prognostication.1,4,6,7,9,11,12,23 However, despite extensive dedicated research on these topics, there is a persistent gap in the consistent validation of OCT imaging markers in L-ORD.1–3,9
Along with other imaging modalities, OCT B-scans can help delineate the level, pattern, and extent of sub-RPE and subretinal deposits, revealing aspects of the outer retinal layer status which is detrimental to visual function.1,2,4,9–12,23 In one of the most extensive longitudinal studies employing OCT images, Borooah et al described a progressive reduction in retinal thickness over a 2-year period, which was not followed by a measurable reduction in choroidal thickness.1 The work by Cukras et al, which combined multimodal imaging changes in two L-ORD patients with preserved central vision, found disease progression in the form of confluent thickening of RPE and disorganization of EZ before neuroretinal disruption and atrophy.4 In another study involving 3 cases of L-ORD followed up for 4 years, progressive anatomic changes at the level of EZ by OCT directly correlated to loss of sensitivity in microperimetry.12 Cases with visual deterioration in the setting of preserved OCT findings, especially EZ, have also been recognized, suggesting that other factors may play a role in visual potential in these patients beyond findings seen on OCT.2,4,5,10,12
In addition, analysis by OCT can also outline areas of suspicion for secondary CNV, particularly in the transition zones between areas of atrophy and borderline retina.6,22,25,26 Lesions in the periphery consistent with type 1 CNV have been observed in four cases of L-ORD but were better evaluated with angiographic tests.6 In patients receiving anti-vascular endothelial growth factor (VEGF) drugs, OCT permits assessment of response to therapy.22,25
Angiography
Fluorescein and indocyanine angiography studies can further assist in revealing CNV and assessing the retinal and choroidal vascular integrity, including at the level of choriocapillaris.6,10,15,20 L-ORD may display vessel attenuation in mid-late disease, which may be highlighted by the fluorescein dye transit.2,3 Epithelial defects can be noted as points of early hyperfluorescence, while subretinal deposits tend to show progressive hyperfluorescence with staining.1,10,23 Indocyanine angiography helps in CNV assessment, especially in the periphery.6,10
Functional Tests
Color vision testing
Very few studies have reported color vision changes in patients with L-ORD, with mixed patterns of color deficiency.2,11,14,15 Abnormalities on the tritan spectrum using the Farnsworth Panel D-15 were reported in individuals carrying the Ser163Arg mutation. In contrast, changes in the red-green axis were predominant as part of Pro188Thr using Ishihara, Hardy-Rand-Rittler, and the D-15 scale.2,14,15 Deficits in red-green and blue-yellow axes were reported to co-occur in three cases of a different cohort carrying the Ser163Arg.11 Importantly, preserved color discrimination in the setting of retinal atrophy indicates that L-ORD does not affect cones selectively, especially in early disease.15 Using a similar analysis, the presence of changes in color perception in early disease, usually alongside macular changes, may favor the diagnosis of cone dystrophies as opposed to L-ORD. In a small subset of patients, color deficiencies may have resulted from advanced glaucomatous neuropathy.3,15
Visual fields
Visual field studies in L-ORD have been mostly based on the Goldman field testing in patients harboring the Ser163Arg variant. In these individuals, normal or near-to-normal perimetry was noted before fundus changes or until the fourth decade of life.2,4,10–12,15,16,20 Vincent et al described four patients in which variable baseline visual field results primarily indicated normality for stimulus targets to III4e to V4e, with most defects found in the nasal field; in the same cohort, other cases showing C-shaped defects, nasal constriction and scotomas to I4e target.11 Similar changes involving the nasal fields have been shown by other groups.10,12 In other cohorts, central scotomas involving the macula were seen from disease onset, with progression dependent on other factors, including the distribution of deposits and atrophy.12,16
In terms of longitudinal changes, Papastavrou et al reported on small changes in visual fields of two out of three L-ORD cases over 4 years, in mismatch to progressive structural changes seen on OCT or decreased retinal sensitivity by microperimetry; correspondence between perimetry and OCT changes was more clearly seen in one case with rapidly progressive disease.12 In most patients carrying the Pro188Thr variant, once retinal disease became visible around the third decade of life, the course of visual field defects showed initial loss of sensitivity in central and paracentral foveal areas with progression to coalescing scotomas in the mid-peripheral zones.2
While the pace of visual field changes seems to vary in early disease amongst different variants, areas of absolute central scotoma tend to form until complete loss of the entire visual field in all variants.1,2,12,17,20 Some remaining islands may persist until reaching an end-stage retinal compromise, although these may vary from case to case.2,11,17,20 Although data are limited on older patients beyond the 70s, extensive visual field loss is expected from the few published cases.2,16,17,19 In L-ORD cases with associated glaucoma, distinguishing visual field defects for this condition versus retina-related may be challenging.19 In certain instances, pupil dynamics secondary to iris atrophy or significant visual loss may hamper visual test completion or the reliability of results.2,20
Microperimetry
Microperimetry under mesopic light has been used in L-ORD as a sensitive and detailed method to monitor disease progression in addition to classic BCVA and Goldman kinetic perimetry.10,12 Three major studies focusing on microperimetry showed a reduced response in areas concentrating sub-RPE deposits and dense pseudodrusen and severe loss in sensitivity in areas with retinal atrophy.10,12,46 Microperimetry findings have been considered mostly symmetric and negatively correlated with age, confirming the loss of visual sensitivity over time.46 Papastavrou et al particularly demonstrated in three cases in which changes in mesopic microperimetry, in association with localized photoreceptor EZ loss in the para-macular area, may be a marker of disease progression, with similarities to the microperimetry pattern described in choroideremia.12 The authors were also able to calculate a mean loss of retinal sensitivity in L-ORD at 2.05 dB/year, which was comparatively higher than the rates observed in choroideremia.12 Alternatively, Alex et al calculated the yearly reduction in sensitivity at 0.66dB using 4-year interval data.46 Ideally, dark-adapted microperimetry may be preferred in L-ORD, although mesopic conditions have been mostly preferred thus far.10,12,46
Dark adaptometry
DA delay has been a well-recognized feature of L-ORD since the early clinical studies.3,5,14,16,17,20 Highest DA sensitivity has been achieved in L-ORD by a two-color chromatic method at a 30-degree angle of eccentricity.5,14 Dark adaptometry conducted by Kuntz et al while evaluating family members suggested that DA was delayed despite normal BCVA.14 Screening of L-ORD families in another series using DA for members at risk showed that DA was often present a decade before fundus features, with the youngest case being identified at 35 years old.16,17 Milam et al pointed to different degrees of rod dysfunction in L-ORD family members with unaffected ophthalmoscopy according to the area of retina sampled.16 In a report with anatomical-functional analysis, areas of subretinal deposits on OCT corresponded with a reduction in DA at the same fundus sites.5 DA was also employed as an endpoint in three studies trialing vitamin A, with variable DA results ranging from no improvement to partial improvement in DA kinetics.11,17,20 DA testing does not appear to have been carried out in any other variant thus far. Comparatively, AMD shows DA delay concentrated at the fovea, although this distinction may be challenging in some L-ORD cases.3
Full-field electroretinography
Full-field electroretinography (ffERG) can assess retinal function in L-ORD patients, revealing the degree of rod-specific dysfunction at early disease and late extension to cones during disease progression.2,11,14–16,23,24 Correlation between decreased ffERG response to fundus areas affected by drusenoid deposits and atrophy has been identified in L-ORD series.2,11,15,16,23,24 However, unaffected retinal areas have also displayed some degree of retinal dysfunction.2,24
Most ffERG protocols involved dark adaptation with prolonged dark adaptation normalizing ffERG findings.2,11,14–16,23,24 Photopic ffERG recordings are generally within normal limits in early disease, even in patients with fundus findings.2,11 FfERG findings have been mainly characterized as a reduction in the b- and a-waves, selectively or simultaneously, with predominant impact on the b-wave amplitude over the a-wave.11,23 The b/a ratio in L-ORD was calculated between 0.7 and 2.5 in a series of four affected individuals of the same family using serial ffERG evaluations (DA 0.01 and 2.29 stimulus), which pointed to a selective involvement of the b-wave amplitude in two cases suggestive of a greater involvement of bipolar cells over rod photoreceptors.11 In a different approach, Soumplis et al also performed pattern ERG in combination to ffERG on three cases manifesting foveal disease, in which severe macular dysfunction was confirmed on pattern ERG testing in addition to rod-cone dysfunction on ffERG.23
Most ffERG studies involved cases of Ser163Arg,11,23,24 with one study characterizing families with Pro188Thr, in which similar ffERG findings were reported, with no variabilities amongst the three phenotypic subtypes for this variant.2 In this cohort, the median age in which patients reached extinguished ffERG responses over a 5-year-follow-up ranged between 60 and 68 years, with most results categorized within subnormal scotopic response with a normal photopic response.2
An important discovery in L-ORD relates to the possibility of partial reversion of rod dysfunction on ffERG using extended DA, even in patients at stages of the foveal-involving disease. In the original experiment, four of eight patients with delayed DA assessed by the standard protocol of 20 min manifested significant improvement in b-wave (DA 0.01) and a-wave (DA 3.0) after an extended DA of 16 hours. This observation confirmed a high level of rod dysfunction in L-ORD and suggested a window of opportunity for patient treatment even in moderate-to-advanced disease, in which some rod function still recordable despite anatomic damage.24
FfERG is not commonly used in diagnosing L-ORD as a normal ffERG should not exclude L-ORD, despite revealing abnormalities in early disease.3,15,23 ffERG findings have shown a high degree of symmetry between eyes.23 Considering the overlap of DA abnormalities and similar fundus changes amongst some IRDs, ffERG abnormalities should be appropriately judged in the context of other IRDs such as SFD and DD, which can similarly present with impaired rod thresholds.14
Multifocal electroretinography
Only one study explored multifocal electroretinography (mfERG) in L-ORD, aiming to measure cone response after light adaptation.10 Three patients showed reduced waveform amplitudes in areas of the presence of pseudodrusen, with correspondence to points of reduction in sensitivity mapped by microperimetry.10
Electrooculography
A single study has investigated L-ORD changes on electrooculography (EOG), which assesses the RPE-photoreceptor physiology.11 In this investigation, all three cases reported showed a normal light peak/dark trough (Arden) ratio, with one case further demonstrating abnormal dark trough amplitude in association with light peak delay and another revealing a delayed light peak.11
Differential Diagnosis
The diagnosis of L-ORD is permeated by the overlap with other chorioretinal conditions both in the spectrum of IRDs and degenerative conditions.28,32,56 A summary of the most relevant differential diagnoses of L-ORD with key points of distinction is outlined in Table 3.
 |
Table 3 Main Differential Diagnoses with Contrasting Elements from Late-Onset Retinal Degeneration |
As highlighted in the table, L-ORD can recapitulate AMD features, such as sub-RPE deposits, reticular pseudodrusen, CNV, and retinal atrophy. Notwithstanding, L-ORD can also resemble other rare autosomal dominant macular dystrophies which involve the macula, particularly SFD and DD, whose onset tends to be slightly earlier but similarly marked by nyctalopia and progression to diffuse deposits across the fundus.2,3,7,10 Distinction from these conditions may rely on correct identification of clinical signs, often requiring genetic testing for confirmation and family history.
In late-stage disease, with near complete retinal atrophy, L-ORD can be mistaken for end-stage RP and choroideremia, which will need to be differentiated by molecular testing. Importantly, in these conditions, a detailed clinical history disclosing the onset of visual symptoms in early childhood along with a slow pace of visual deterioration may be clues to guide the following clinical steps.56
Another important differential diagnosis consists of late-onset night blindness with peripheral flecks related to alterations in the retinoid cycle. Consequently, patients develop nyctalopia, mid-peripheral flecks, confluent late atrophic lesions, and extinguished ffERG.58 However, cases of this condition were not shown to present an autosomal dominant inheritance pattern or mutations in the C1QTNF5.58,59 One last diagnostic consideration in any case of L-ORD includes vitamin A deficiency, both of which may share a resemblance based on the rod dysfunction by ffERG, presence of nyctalopia, and relatively preserved neuroretina.5,11,20,60 Distinction is possible through a detailed clinical history revealing a familial pattern of visual loss and additionally testing for vitamin A levels.3,17,20,60
Management and Treatment
Despite advances in the overall understanding of IRDs and the mechanisms of L-ORD, no targeted therapy has proven effective in controlling the disease. Management options for L-ORD are primarily based on improving patient quality of life and controlling complications, such as glaucoma, cataract, and CNV.2,3,6,22,25 Visual recovery after these strategies remains highly dependent on the structural status of the fovea.1,3,4,9,12 After developing cataracts, patients may experience subjective benefits from phacoemulsification, which may enhance vision, especially at the islands of the preserved retina.3 In a study including 11 eyes of L-ORD cases undergoing cataract surgery, no complications were noticed after intraocular lens implantation at a variable follow-up time ranging from 6 to 84 months. This reinforces that LAZs could be redundant and not influence postoperative lens centration.8
Control of centrally-involved CNV by off-label intravitreal anti-VEGF therapy has been shown to prevent scar formation, yet results have all been single case-based.6,22,25,26 Spontaneous resolution of L-ORD-related CNV after closed observation has also been reported.11 Laser photocoagulation or photodynamic therapy was considered in the past for CNV, although the risk-benefit of these interventions is debatable.6,15
In the early 2000s, after reported functional similarities on ERG tests between L-ORD and vitamin A deficiency, supplementation with retinol/retinoic acid was attempted to restore visual response.5,17,20 In a 1-month trial of oral vitamin A 50,000 IU/day to 3 L-ORD individuals, variable improvement in the kinetics of DA was reached, with a reduction in the time delay but no normalization down to normal thresholds.17 Using an alternative regimen of vitamin A of 15,000 IU/day for 6 months, another group also confirmed the enhancement of the DA function, yet results were based during the time of administration and on a single treated case.20 Another treatment case with vitamin A received 15,000 IU/day with subjective improvement in color and contrast without improvement on the objective metrics.11
Although current recommendations for L-ORD treatment do not include metformin, recent studies on L-ORD RPE cells derived from human-induced pluripotent stem cells suggested that RPE dysfunction in L-ORD results from perturbation of AMPK signaling, one of the critical cellular metabolic regulators.42 Treatment of cells with AMPK modulator metformin re-sensitizes RPE cells in L-ORD to changes in ATP/AMP and hence allows control of cellular homeostasis and rescue in vitro disease-associated lesions, including sub-RPE deposits.42 However, caution must be taken when translating findings from in vitro studies directly to clinical practice until clinical trials test and confirm the efficacy of such strategies.
L-ORD is a potential candidate for gene therapy as rod photoreceptor response has been recovered upon extended DA, revealing some residual retinal function even in stages of advanced disease.24 To our knowledge, gene augmentation therapy studies have not been published even in the preclinical stage. Given the apparent dominant negative mechanism suggested by preclinical studies, it is unclear whether this strategy would be successful in L-ORD. A potential translational route could be to gene edit the mutant allele. Trials for clustered regularly interspaced short palindromic repeats (CRISPR) Cas9 gene editing have recently begun aimed at treating other IRD.32,61–66 The C1QTNF5 mutation in L-ORD is also potentially a target for treatment using CRISPR-Cas9 targeting the mutant alleles.67 Nonetheless, studies using this type of treatment have also not yet been published.
In the absence of alternatives to manage these patients, facilitating access to genetic testing is valuable in identifying families that can benefit from genetic counseling and complication surveillance.32,68,69 Genetic counseling will also be beneficial, particularly for those planning families, as in vitro fertilization and genetic testing pre-implantation of the blastocyst may prevent generational inheritance of L-ORD for patients affected by this condition.
Abbreviations
ADIPOR1, adiponectin receptor 1; AMD, age-related macular degeneration; AMPK, AMP-activated protein kinase; BCVA, best corrected visual acuity; BM, Bruch’s membrane; CNV, choroidal neovascularization; CRISPR, clustered regularly interspaced short palindromic repeats; DA, dark adaptation; EZ, ellipsoid zone; ffERG, full-field electroretinogram; HTRA1, HtrA serine peptidase 1; IRD, inherited retinal disease; LAZ, long anterior zonules; L-ORD, late-onset retinal degeneration; mfERG, multifocal electroretinogram; OCT, optical coherence tomography; RP, retinitis pigmentosa; RPE, retinal pigment epithelium; RNA, ribonucleic acid; SFD, Sorsby fundus dystrophy; SLO, scanning laser ophthalmoscopy; VEGF, vascular endothelial growth factor.
Acknowledgments
SB was supported by a Foundation Fighting Blindness Career Development Award and by the Nixon Visions Foundation
Funding
This work was supported in part by the University of California San Diego Vision Research Center Core Grant P30EY022589 and an unrestricted grant from Research to Prevent Blindness, NY. The funding organizations had no role in the design or conduct of this research.
Disclosure
The authors report no conflicts of interest in relation to this work.
References
1. Borooah S, Papastavrou VT, Lando L, et al. Characterizing the natural history of foveal-sparing atrophic late-onset retinal degeneration. Retina. 2021;41(6):1329–1337. doi:10.1097/IAE.0000000000003017
2. De Zaeytijd J, Coppieters F, De Bruyne M, et al. Longitudinal phenotypic study of late-onset retinal degeneration due to a founder variant c.562C>A p. (Pro188Thr) in the C1QTNF5 gene. Ophthalmic Genet. 2021;42(5):521–532. doi:10.1080/13816810.2021.1923041
3. Borooah S, Collins C, Wright A, Dhillon B. Late-onset retinal macular degeneration: clinical insights into an inherited retinal degeneration. Br J Ophthalmol. 2009;93(3):284–289. doi:10.1136/bjo.2008.150151
4. Cukras C, Flamendorf J, Wong WT, Ayyagari R, Cunningham D, Sieving PA. Longitudinal structural changes in late-onset retinal degeneration. Retina. 2016;36(12):2348–2356. doi:10.1097/IAE.0000000000001113
5. Jacobson SG, Cideciyan AV, Sumaroka A, Roman AJ, Wright AF. Late-onset retinal degeneration caused by C1QTNF5 mutation: sub-retinal pigment epithelium deposits and visual consequences. JAMA Ophthalmol. 2014;132(10):1252–1255. doi:10.1001/jamaophthalmol.2014.2059
6. Keenan TDL, Vanderford EK, de Silva T, Sieving PA, Cukras CA. Massive advancing nonexudative type 1 choroidal neovascularization in CTRP5 late-onset retinal degeneration: longitudinal findings on multimodal imaging and implications for age-related macular degeneration. Retina. 2021;41(11):2236–2245. doi:10.1097/IAE.0000000000003205
7. Khan KN, Borooah S, Lando L, et al. Quantifying the separation between the retinal pigment epithelium and Bruch’s membrane using optical coherence tomography in patients with inherited macular degeneration. Transl Vis Sci Technol. 2020;9(6):26. doi:10.1167/tvst.9.6.26
8. Papastavrou VT, Borooah S, O’Brien JM, et al. Cataract surgery in patients with late-onset retinal degeneration. J Cataract Refract Surg. 2017;43(8):1036–1043. doi:10.1016/j.jcrs.2017.05.041
9. Vanderford EK, De Silva T, Noriega D, Arango M, Cunningham D, Cukras CA. Quantitative analysis of longitudinal changes in multimodal imaging of late-onset retinal degeneration. Retina. 2021;41(8):1701–1708. doi:10.1097/IAE.0000000000003082
10. Borooah S, Papastavrou V, Lando L, et al. Reticular pseudodrusen in late-onset retinal degeneration. Ophthalmol Retina. 2021;5(10):1043–1051. doi:10.1016/j.oret.2020.12.012
11. Vincent A, Munier FL, Vandenhoven CC, Wright T, Westall CA, Héon E. The characterization of retinal phenotype in a family with C1QTNF5-related late-onset retinal degeneration. Retina. 2012;32(8):1643–1651. doi:10.1097/IAE.0b013e318240a574
12. Papastavrou VT, O’Brien JM, Regan AJ, Aftab AM, Browning AC. The progression of macular structural and functional changes in late-onset retinal degeneration. Retin Cases Brief Rep. 2020. doi:10.1097/ICB.0000000000001067
13. Duvall J, McKechnie NM, Lee WR, Rothery S, Marshall J. Extensive subretinal pigment epithelial deposit in two brothers suffering from dominant retinitis pigmentosa. A histopathological study. Graefes Arch Clin Exp Ophthalmol. 1986;224(3):299–309. doi:10.1007/BF02143075
14. Kuntz CA, Jacobson SG, Cideciyan AV, et al. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 1996;37(9):1772–1782.
15. Ayyagari R, Griesinger IB, Bingham E, Lark KK, Moroi SE, Sieving PA. Autosomal dominant hemorrhagic macular dystrophy not associated with the TIMP3 gene. Arch Ophthalmol. 2000;118(1):85–92. doi:10.1001/archopht.118.1.85
16. Milam AH, Curcio CA, Cideciyan AV, et al. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology. 2000;107(12):2256–2266. doi:10.1016/S0161-6420(00)00419-X
17. Jacobson SG, Cideciyan AV, Wright E, Wright AF. Phenotypic marker for early disease detection in dominant late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 2001;42(8):1882–1890.
18. Hayward C, Shu X, Cideciyan AV, et al. Mutation in a short-chain collagen gene, CTRP5, results in extracellular deposit formation in late-onset retinal degeneration: a genetic model for age-related macular degeneration. Hum Mol Genet. 2003;12(20):2657–2667. doi:10.1093/hmg/ddg289
19. Styles CJ, Dhillon B, Wright AF. The diagnosis of autosomal dominant late-onset retinal degeneration in two sisters. Eye. 2003;17(4):530–532. doi:10.1038/sj.eye.6700408
20. Ayyagari R, Mandal MNA, Karoukis AJ, et al. Late-onset macular degeneration and long anterior lens Zonules result from a CTRP5 gene mutation. Invest Ophthalmol Vis Sci. 2005;46(9):3363–3371. doi:10.1167/iovs.05-0159
21. Subrayan V, Morris B, Armbrecht AM, Wright AF, Dhillon B. Long anterior lens zonules in late-onset retinal degeneration (L-ORD). Am J Ophthalmol. 2005;140(6):1127–1129. doi:10.1016/j.ajo.2005.06.023
22. Aye KH, Gupta R, Talks SJ, Browning AC. Treatment of a choroidal neovascular membrane in a patient with late-onset retinal degeneration (L-ORD) with intravitreal ranibizumab. Eye. 2010;24(9):1528–1530. doi:10.1038/eye.2010.71
23. Soumplis V, Sergouniotis PI, Robson AG, et al. Phenotypic findings in C1QTNF5 retinopathy (late-onset retinal degeneration). Acta Ophthalmol. 2013;91(3):e191–195. doi:10.1111/aos.12010
24. Papastavrou VT, Bradshaw KR, Aye KH, Turney C, Browning AC. Improvement of retinal function in L-ORD after prolonged dark adaptation. Can J Ophthalmol. 2015;50(2):112–118. doi:10.1016/j.jcjo.2014.12.001
25. Mandal N, Lotery AJ. Multimodal imaging of late-onset retinal degeneration complicated by bilateral choroidal neovascularization. Eye. 2019;33(6):1020–1027. doi:10.1038/s41433-019-0348-8
26. Ramtohul P, Gascon P, Matonti F. Choroidal neovascularization in late-onset retinal macular degeneration. Ophthalmol Retina. 2019;3(2):153. doi:10.1016/j.oret.2018.11.010
27. Fleckenstein M, Grassmann F, Lindner M, et al. Distinct genetic risk profile of the rapidly progressing diffuse-trickling subtype of geographic atrophy in Age-Related Macular Degeneration (AMD). Invest Ophthalmol Vis Sci. 2016;57(6):2463–2471. doi:10.1167/iovs.15-18593
28. Fleckenstein M, Keenan TDL, Guymer RH, et al. Age-related macular degeneration. Nat Rev Dis Primers. 2021;7(1):31. doi:10.1038/s41572-021-00265-2
29. Stanton CM, Borooah S, Drake C, et al. Novel pathogenic mutations in C1QTNF5 support a dominant negative disease mechanism in late-onset retinal degeneration. Sci Rep. 2017;7(1):12147. doi:10.1038/s41598-017-11898-3
30. Borooah S, Stanton CM, Marsh J, et al. Whole genome sequencing reveals novel mutations causing autosomal dominant inherited macular degeneration. Ophthalmic Genet. 2018;39(6):763–770. doi:10.1080/13816810.2018.1546406
31. Broadgate S, Yu J, Downes SM, Halford S. Unravelling the genetics of inherited retinal dystrophies: past, present and future. Prog Retin Eye Res. 2017;59:53–96. doi:10.1016/j.preteyeres.2017.03.003
32. Georgiou M, Fujinami K, Michaelides M. Inherited retinal diseases: therapeutics, clinical trials and end points-A review. Clin Exp Ophthalmol. 2021;49(3):270–288. doi:10.1111/ceo.13917
33. Ayyagari R, Mandal MN, Karoukis AJ, et al. Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Invest Ophthalmol Vis Sci. 2005;46(9):3363–3371.
34. Roberts DK, Wilensky J. Long anterior lens zonules. Clin Experiment Ophthalmol. 2012;40(7):764–766. doi:10.1111/j.1442-9071.2012.02787.x
35. Bressler SB, Muñoz B, Solomon SD, West SK; Team tSEES. racial differences in the prevalence of age-related macular degeneration: the Salisbury Eye Evaluation (SEE) Project. Arch Ophthalmol. 2008;126(2):241–245. doi:10.1001/archophthalmol.2007.53
36. Chang MA, Bressler SB, Munoz B, West SK. Racial differences and other risk factors for incidence and progression of age-related macular degeneration: Salisbury Eye Evaluation (SEE) project. Invest Ophthalmol Vis Sci. 2008;49(6):2395–2402. doi:10.1167/iovs.07-1584
37. Shapiro L, Scherer PE. The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr Biol. 1998;8(6):335–338. doi:10.1016/S0960-9822(98)70133-2
38. Shu X, Tulloch B, Lennon A, et al. Disease mechanisms in late-onset retinal macular degeneration associated with mutation in C1QTNF5. Hum Mol Genet. 2006;15(10):1680–1689. doi:10.1093/hmg/ddl091
39. Tu X, Palczewski K. Crystal structure of the globular domain of C1QTNF5: implications for late-onset retinal macular degeneration. J Struct Biol. 2012;180(3):439–446. doi:10.1016/j.jsb.2012.07.011
40. Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J. 2008;416(2):161–177. doi:10.1042/BJ20081240
41. Tu X, Palczewski K. The macular degeneration-linked C1QTNF5 (S163) mutation causes higher-order structural rearrangements. J Struct Biol. 2014;186(1):86–94. doi:10.1016/j.jsb.2014.02.001
42. Miyagishima KJ, Sharma R, Nimmagadda M, et al. AMPK modulation ameliorates dominant disease phenotypes of CTRP5 variant in retinal degeneration. Commun Biol. 2021;4(1):1360. doi:10.1038/s42003-021-02872-x
43. Chekuri A, Zientara-Rytter K, Soto-Hermida A, et al. Late-onset retinal degeneration pathology due to mutations in CTRP5 is mediated through HTRA1. Aging Cell. 2019;18(6):e13011. doi:10.1111/acel.13011
44. Chavali VR, Khan NW, Cukras CA, Bartsch DU, Jablonski MM, Ayyagari R. A CTRP5 gene S163R mutation knock-in mouse model for late-onset retinal degeneration. Hum Mol Genet. 2011;20(10):2000–2014. doi:10.1093/hmg/ddr080
45. ClinVar VCV000813996.1. National Center for Biotechnology Information; 2018. Available from: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000813996.1.
46. Alex V, Papastavrou V, Browning AC, Dhillon B, Borooah S. Microperimetry in foveal sparing late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 2022;63(7):4532–F0319 .
47. Lando L, Nguyen AX-L, Li R, Megaw R, Dhillon B, Borooah S. Quantification of long anterior zonules in late-onset retinal degeneration. Invest Ophthalmol Vis Sci. 2022;63(7):4408–F0087 .
48. Mandal MN, Vasireddy V, Reddy GB, et al. CTRP5 is a membrane-associated and secretory protein in the RPE and ciliary body and the S163R mutation of CTRP5 impairs its secretion. Invest Ophthalmol Vis Sci. 2006;47(12):5505–5513. doi:10.1167/iovs.06-0312
49. Moroi SE, Lark KK, Sieving PA, et al. Long anterior zonules and pigment dispersion. Am J Ophthalmol. 2003;136(6):1176–1178. doi:10.1016/S0002-9394(03)00657-3
50. Roberts DK, Newman TL, Roberts MF, Teitelbaum BA, Winters JE. Long anterior lens zonules and intraocular pressure. Invest Ophthalmol Vis Sci. 2018;59(5):2015–2023. doi:10.1167/iovs.17-23705
51. Roberts DK, Yang Y, Morettin CE, Doan T, Newman TL, Wilensky JT. Quantification of long anterior lens zonules and their resulting zonule-free zone sizes. Clin Experiment Ophthalmol. 2015;43(8):773–775. doi:10.1111/ceo.12554
52. Roberts DK, Lo PS, Winters JE, Castells DD, Alexander CC, Teitelbaum BA. Prevalence of pigmented lens striae in a black population: a potential indicator of age-related pigment dispersal in the anterior segment. Optom Vis Sci. 2002;79(11):681–687. doi:10.1097/00006324-200211000-00005
53. Sadda SR, Guymer R, Holz FG, et al. Consensus definition for atrophy associated with age-related macular degeneration on OCT: classification of atrophy report 3. Ophthalmology. 2018;125(4):537–548. doi:10.1016/j.ophtha.2017.09.028
54. Shmueli O, Yehuda R, Szeskin A, Joskowicz L, Levy J. Progression of cRORA (Complete RPE and Outer Retinal Atrophy) in dry age-related macular degeneration measured using SD-OCT. Transl Vis Sci Technol. 2022;11(1):19. doi:10.1167/tvst.11.1.19
55. Caramoy A, Heindl LM. Variability of choroidal and retinal thicknesses in healthy eyes using swept-source optical coherence tomography - implications for designing clinical trials. Clin Ophthalmol. 2017;11:1835–1839. doi:10.2147/OPTH.S145932
56. Ben-Arie-Weintrob Y, Berson EL, Dryja TP. Histopathologic-genotypic correlations in retinitis pigmentosa and allied diseases. Ophthalmic Genet. 2005;26(2):91–100. doi:10.1080/13816810590968032
57. C1q and TNF related 5. National Center for Biotechnology Information; 2018. Available from: https://www.ncbi.nlm.nih.gov/gene/114902#gene-expression.
58. Tsunoda K, Fujinami K, Yoshitake K, Iwata T. Late-onset night blindness with peripheral flecks accompanied by progressive trickle-like macular degeneration. Doc Ophthalmol. 2019;139(3):171–184. doi:10.1007/s10633-019-09705-7
59. Yamamoto H, Simon A, Eriksson U, Harris E, Berson EL, Dryja TP. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet. 1999;22(2):188–191. doi:10.1038/9707
60. McBain VA, Egan CA, Pieris SJ, et al. Functional observations in vitamin A deficiency: diagnosis and time course of recovery. Eye. 2007;21(3):367–376. doi:10.1038/sj.eye.6702212
61. Ghazi NG, Abboud EB, Nowilaty SR, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a Phase I trial. Hum Genet. 2016;135(3):327–343. doi:10.1007/s00439-016-1637-y
62. Bainbridge JWB, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239. doi:10.1056/NEJMoa0802268
63. Cideciyan AV, Jacobson SG, Drack AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. 2019;25(2):225–228. doi:10.1038/s41591-018-0295-0
64. Russell SR, Drack AV, Cideciyan AV, et al. Intravitreal antisense oligonucleotide sepofarsen in Leber congenital amaurosis type 10: a phase 1b/2 trial. Nat Med. 2022;28(5):1014–1021. doi:10.1038/s41591-022-01755-w
65. Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010;18(3):643–650. doi:10.1038/mt.2009.277
66. Yu-Wai-Man P, Newman NJ, Carelli V, et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci Transl Med. 2020;12(573):573. doi:10.1126/scitranslmed.aaz7423
67. Gleditzsch D, Pausch P, Müller-Esparza H, et al. PAM identification by CRISPR-Cas effector complexes: diversified mechanisms and structures. RNA Biol. 2019;16(4):504–517. doi:10.1080/15476286.2018.1504546
68. Lam BL, Leroy BP, Black G, Ong T, Yoon D, Trzupek K. Genetic testing and diagnosis of inherited retinal diseases. Orphanet J Rare Dis. 2021;16(1):514. doi:10.1186/s13023-021-02145-0
69. Stone EM, Aldave AJ, Drack AV, et al. Recommendations for genetic testing of inherited eye diseases: report of the American Academy of Ophthalmology task force on genetic testing. Ophthalmology. 2012;119(11):2408–2410. doi:10.1016/j.ophtha.2012.05.047
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.