")
Back to Journals » International Medical Case Reports Journal » Volume 15
Late-Onset Hereditary Transthyretin Amyloidosis Val30Met in an Elderly Person in a Non-Endemic Area
Authors Wang S , Sun J, Lu Q, Li H, Zhang Y
Received 26 January 2022
Accepted for publication 18 May 2022
Published 16 June 2022 Volume 2022:15 Pages 299—306
DOI https://doi.org/10.2147/IMCRJ.S357236
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Shun Wang,1,* Jingping Sun,2,* Qun Lu,1 Hao Li,3 Yun Zhang4
1Department of Cardiology, The First Affiliated Hospital of Xi’an Jiaotong University, Xian, Shaanxi, People’s Republic of China; 2Department of Cardiology, The Cleveland Clinic Foundation, Cleveland, OH, USA; 3ICU, The First Affiliated Hospital of Xi’an Jiaotong University, Xian, Shaanxi, People’s Republic of China; 4Department of Imaging Medicine, The First Affiliated Hospital of Xi’an Jiaotong University, Xian, Shaanxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shun Wang, Department of Cardiovascular Medicine, The First Affiliated Hospital of Xi’an Jiao Tong University, Xi’an, People’s Republic of China, Email [email protected] Jingping Sun, Cardiology Department, The Cleveland Clinic Foundation, Cleveland, OH, USA, Tel +1 346-719-7412, Email [email protected]
Introduction: Patients with late-onset transthyretin Val30Met-associated hereditary transthyretin amyloidosis (hATTR) in non-endemic areas still remain undiagnosed because of diverse clinical presentations and various non-specific symptoms.
Case Presentation: A 76-year-old male patient presented with progressive numbness, pain and weakness in his limbs, sweating, constipation and unexplained weight loss over the past seven years. He has shortness of breath, edema and hypotension for one month. The low QRS voltage on limb leads was not consistent with left ventricular hypertrophy, which is an important clue of cardiac amyloidosis (CA). The results of echocardiography speckle tracking imaging were consistent with CA. Serum immunofixation electrophoresis was negative, and serum-free light chain Fκ/Fλ ratio is normal or close to normal (0.26– 1.65) for the patient, so AL amyloidosis can be excluded. A missense mutation c. 148 G-A Val30Met (p.Val50Met) was detected in TTR gene sequencing. The genetic finding confirmed hATTR Val30Met, familial amyloid polyneuropathy (FAP) and CA for the patient. The treatment effect was poor, and he died of cardiac involvement.
Conclusion: It is challenge to make early diagnosis in patients with hATTR, due to the diversity of symptoms. Echocardiography is a vital tool in initial diagnosis. Genetic testing played vital roles in the definitive diagnosis of this disease. Raising awareness is critical for early diagnosis and provides opportunities for early treatment.
Keywords: hereditary transthyretin amyloidosis, familial amyloid polyneuropathy, cardiac amyloidosis, speckle tracking imaging
Case Presentation
A 76-year-old male complained with shortness of breath, systemic edema for one month. He had been diagnosed as “multiple peripheral neuropathy” in several hospitals due to progressive numbness, pain and weakness of limbs, sweating, constipation and unexplained weight loss for 7 years. He had a history of hypertension for 13 years, but his blood pressure is normal without therapy in the recent two years. The patient had no history of diabetes, smoking, alcohol over consumption in the past. His father had a history of limb numbness and systemic edema. The patient had no other family history of genetic diseases.
On physical examination, malnourished, anemic, lethargic, limb muscle weakness, decreased muscle tone, weakened tendon reflex, sacroiliac decubital necrosis and severe edema.
Laboratory tests (Table 1) revealed myocardial damage with persistent elevation of hs-TnI and NT-proBNP, neutrophilic leukocytosis, moderate anemia with decrease in erythrocytes and hematocrit, mild hypoxemia. Furthermore, other abnormal parameters are listed in Table 1. Chest X-ray showed bilateral pulmonary inflammation with pleural effusion (Figure 1). Electrocardiogram (ECG) indicated low QRS voltage of limb leads, abnormal Q wave of II, III and aVF, and poor increase of V1-V6 R wave (Figure 2). Echocardiography revealed cardiac hypertrophy, left ventricular diastolic dysfunction (grade II) and small amount of pericardial effusion (Figure 3). There was discordance between the low QRS voltage and left ventricular hypertrophy, which may be a vital clue to the diagnosis of cardiac amyloidosis (CA). The speckle tracking imaging showed reduced global longitudinal strain (GLS) with apical sparing and with an apical-to-basal strain ratio 2.7 and the ejection fraction (EF) to GLS ratio (EFSR) was 4.4, which were consistent with the ultrasonic characteristics of CA (Figure 3). A thorough diagnostic approach was followed, in order to detect the underlying pathological condition. Several computed tomography (CT) scans of the chest showed diffuse pulmonary interstitial changes, cystic changes and shadows of lung consolidation, gradually increased shadow of small calcification on the lung surface at the bottom of both lungs, which were typical manifestations of pulmonary amyloid deposition (Figure 4). Whether there is pulmonary inflammation and infection needs to be determined in combination with blood cell examination and C-reactive protein level. The CMR and nuclear medicine modalities (PYP) were not performed because of his critical condition.
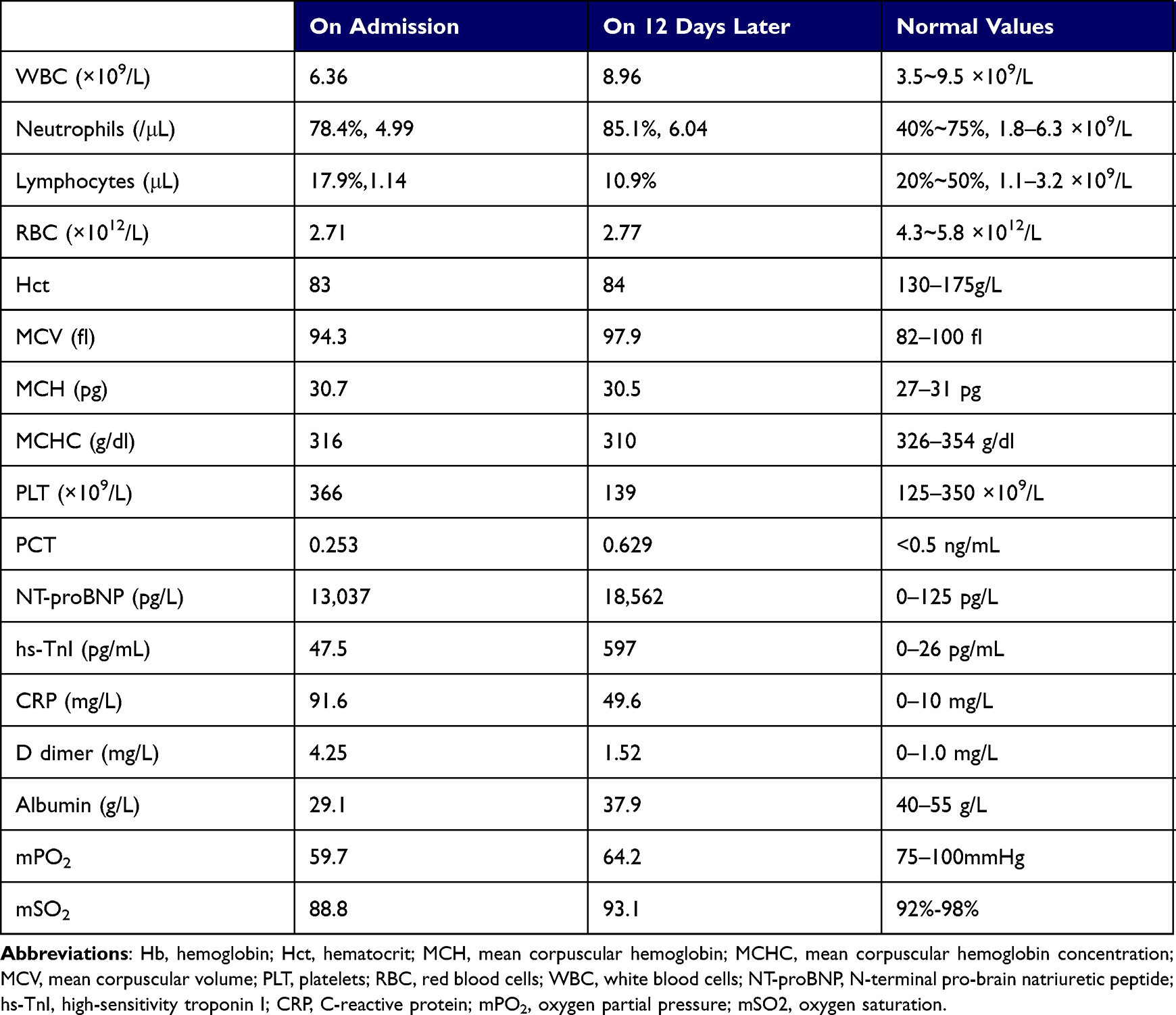 |
Table 1 Blood Laboratory Findings Revealed Myocardial Damage and Moderate Anemia |
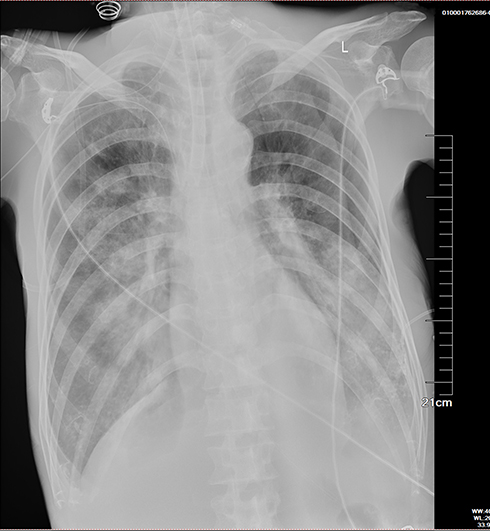 |
Figure 1 Chest X-ray indicated bilateral pulmonary inflammation and pleural effusion. |
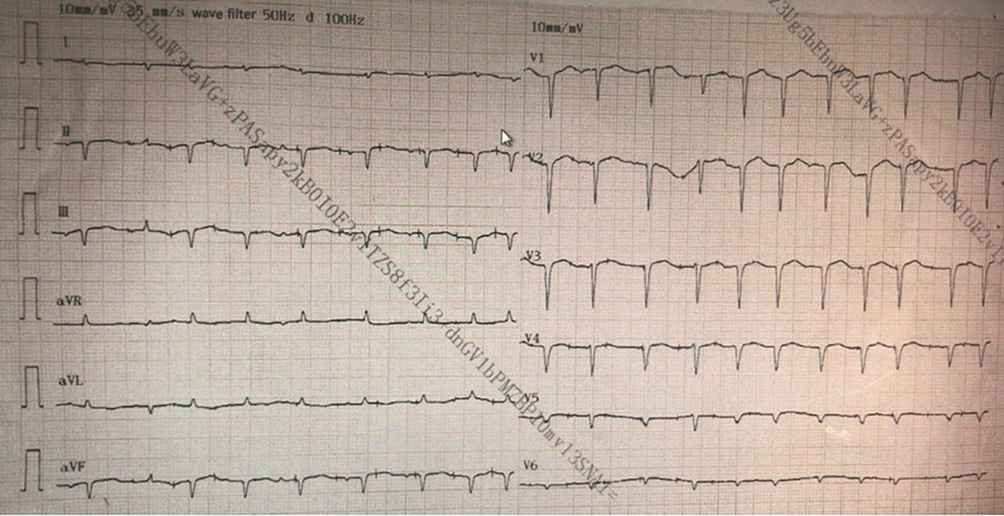 |
Figure 2 ECG showed sinus rhythm, left deviation of ECG axis, QRS low voltage on leads of limb, abnormal Q wave of II, III and AVF, and QRS presented as rS pattern in V1-V6 R. |
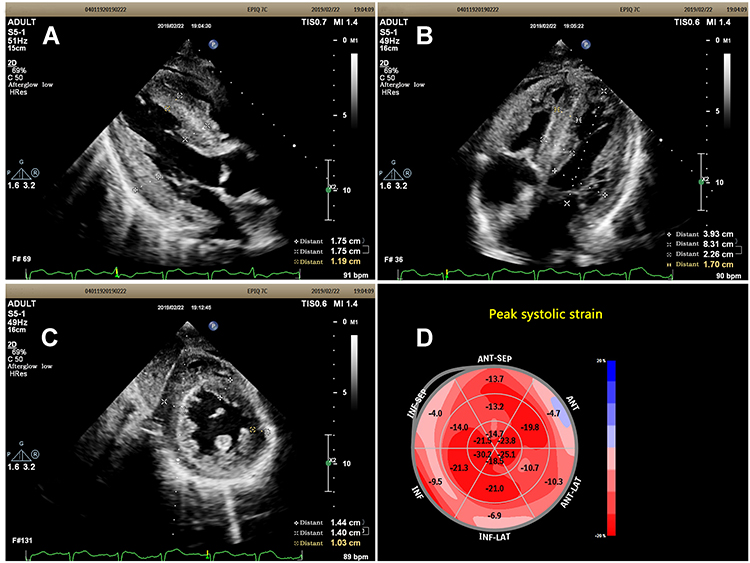 |
Figure 3 (A) Subcostal long-axis view showed the anterior ventricular septum and posterior wall were significantly thickened and a characteristic “granular sparkling” appearance of the thickened cardiac walls. (B) Subcostal 4-chamber view showed the ventricular septum and posterior lateral wall were significantly thickened and a characteristic “granular sparkling” appearance of the thickened cardiac walls. (C) Subcostal short axis view at the level of papillary muscle: all of the basal segments of ventricular wall were significantly thickened with a characteristic “granular sparkling” appearance. (D) The Bull’s eye diagram showed reduced global longitudinal strain (GLS) with apical sparing. |
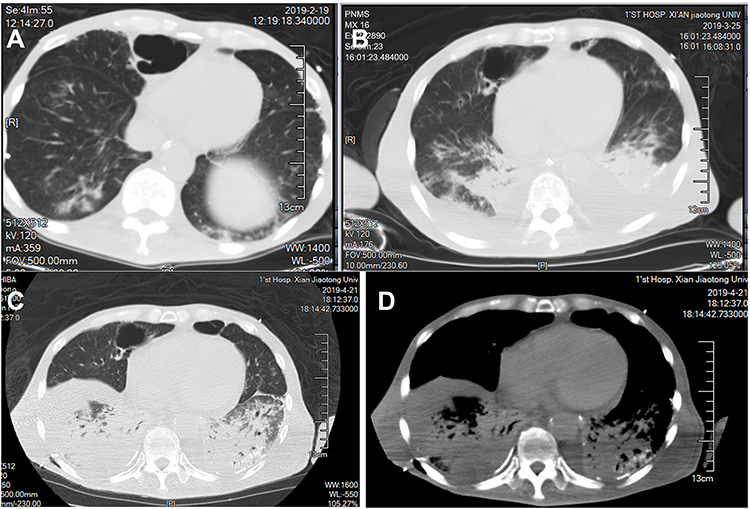 |
Figure 4 Display of chest CT images of the patient. Figure (A–C) were displayed on lung window at the top of the diaphragm at different times. The small calcification shadow on the lung surface at the base of both lungs gradually increased. In additional, they all showed diffuse pulmonary interstitial changes, cystic change and lung consolidation shadows. (D) Mediastinal window on the same plane as figure (A–C): fine sand granular calcification can be seen under the pleura in the lesion of the lower lobe of the left lung, which is clearer than that in the lung window. |
For the differential diagnosis between hereditary transthyretin cardiac amyloidosis and AL amyloidosis, this patient had three serum-free light chain assays and immunofixation electrophoresis. The first results were that Fκ and Fλ were 3.05 g/L (normal range: 3.3~19.4mg/L) and 1.37g/L (normal range: 5.7 ~ 26.3 mg/L), respectively. The second result was that elevated free light chain κ (90.9 mg/L, normal range: 3.3~19.4mg/L)) and free light chain λ (62.2 mg/L, normal range: 5.7 ~ 26.3 mg/L), and Fκ/Fλ ratio was 1.46 (normal range: 0.26~1.65). The third results were elevated free light chain κ (124.78 mg/L, normal range: 3.3~19.4mg/L) and free light chain λ (74.10mg/L, normal range: 5.7 ~ 26.3 mg/L), and Fκ/Fλ ratio was 1.68 (normal range: 0.26~1.65). Serum immunofixation electrophoresis was negative for the patient, and serum-free light chain Fκ/Fλ ratio is normal or close to normal (0.26–1.65), so AL amyloidosis can be excluded.1
Abdominal subcutaneous fat biopsy was negative, and no other tissue biopsy was done because of his critical condition. Although the patient is an elderly male, we did not consider the patient as senile wild-type amyloid transthyretin (wtATTR) amyloidosis and insisted on TTR genetic sequencing because the latter generally does not cause progressive polyneuropathy and autonomic dysfunction. A missense mutation c. 148 G-A (p. V50M, Val30Met) was detected (Figure 5, Table 2) in TTR genetic sequencing. It is the pathological mutation site of patients with hereditary amyloid transthyretin (hATTR) amyloidosis.
 |
Table 2 Gene Sequencing Revealed TTR Gene Missense Mutation |
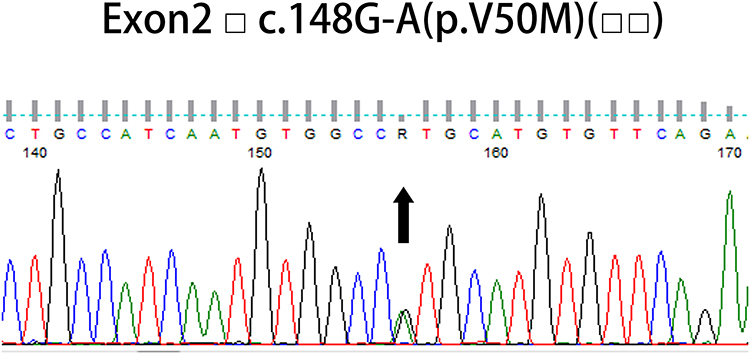 |
Figure 5 TTR gene sequencing: A missense mutation c. 148 G-A (p. V50M, Val30Met) was detected in TTR gene. |
The genetic finding confirmed late-onset transthyretin Val30Met-associated hereditary amyloid transthyretin (hATTR) amyloidosis, familial amyloid polyneuropathy (FAP) and cardiac amyloidosis (CA) for the patient. Recently, the patient’s first son developed limb numbness and found the same TTR gene abnormality.
The patient had received doxycycline + ursodeoxycholic acid, tafamidis (chlorobenzoic acid, a stable TTR tetramer). The patient had intractable intestinal obstruction on 7 days after treatment, and had to stop taking the medicine. After 80 days of treatment, the patient tragically died of cardiac involvement. We did not perform a biopsy of the heart and lung on autopsy to confirm ATTR amyloidosis deposits because the patient’s son refused.
Discussion
We present the case of an elderly male who was diagnosed as hATTR amyloidosis due to late onset Val30Met, FAP and CA. In this case, the diagnosis was established based on chronic progression of neuropathy without diabetes, limb numbness and pain, edema, hypotension, unexplained weight loss, persistent elevation of plasma hs-TnI and BNP, positive echo features of cardiac amyloidosis, pulmonary involvement, typical CT manifestations of pulmonary amyloid deposition, negative serum-free light chains and negative serum immunofixation, and presence of the unique TTR variant Val30Met.1 Although the results of tissue biopsy and PYP were not obtained, this did not affect the diagnosis of hATTR caused by late-onset val30met in this case.2 The patient took tafamidis orally. However, the treatment effect was poor because he was in an advanced stage. He died of cardiac involvement.
Hereditary ATTR (hATTR) is a rare autosomal dominant, systemic disorder characterized primarily by irreversible, progressive, and persistent peripheral nerve damage, with adult-onset onset,2,3 with cardiac and other organs involvement. Symptoms mostly appear before the age of 50 and are rare in the elderly. Mutations in the TTR gene (eg, replacement of valine with methionine at position 30 [Val30Met (p.Val50Met)]) cause TTR tetramers to destabilize and dissociate into variant TTR monomers that form amyloid fibrils, deposits in peripheral nerves and various organs, resulting in peripheral and autonomic neuropathy and several nonspecific symptoms.
Since hATTR is a progressive disease that can lead to largely irreversible tissue damage, prompt identification and diagnosis is critical for appropriate treatment and optimal outcome.4–6. Unfortunately, the diagnosis of hATTR is usually delayed by 3–4 years, which affects the function of patient’s vital organs and prognosis, cardiac involvement is often the cause of death7,8 Misdiagnosis frequently occurs due to the sporadic, late-onset, highly variable clinical presentation pattern of various TTR variants.9
There are only two main types of diagnostic tools in hATTR: TTR gene sequencing, which looks for amyloid gene variants in the TTR gene, and tools for detecting amyloid deposits, including classical biopsy and more recently, pyrophosphate bone scintigraphy (PYP). In addition, imaging examination is very important for the diagnosis of myocardial amyloidosis.
Genetic Testing
Screening for TTR mutations by TTR gene sequencing is an important measure to identify sporadic cases with rapid idiopathic progressive axonal polyneuropathy of unknown origin or atypical CIDP.10 In patients with suspected ATTR-FAP, TTR genotyping should be performed to document specific pathogenicity TTR mutations; genotyping is the most reliable diagnostic method, and the absence of pathogenic variants precludes the diagnosis of ATTR-FAP.3 In all suspected cases, TTR gene positivity should be confirmed by DNA analysis.2,11
Histopathology
Tissue biopsy: Confirmation of amyloid deposition by tissue biopsy is critical to confirm the diagnosis of ATTR-FAP, especially in patients with no family history.2,12 Tissue biopsies stained with Congo red12 directly demonstrate amyloid deposits in involved tissues, including labial salivary glands and abdominal subcutaneous adipose tissue, gastrointestinal tract, neural tissue, and other organs with evidence of involvement. However, in the presence of typical signs and symptoms, negative biopsy results do not rule out ATTR-FAP.2
Imaging Examination
Echocardiography is an excellent first-line screening tool for cardiac amyloidosis (CA). The low QRS voltage is not consistent with the left ventricular hypertrophy, which may be a vital clue of cardiac amyloidosis (CA). An apical sparing pattern on global longitudinal strain (GLS) with an apical-to-basal strain ratio 2.7 (>2.1) and the ejection fraction (EF) to GLS ratio (EFSR) was 4.4 (>4.1) contribute to diagnosis CA. In the diagnosis of CA in patients with left ventricular hypertrophy, the cut-off value of EFSR index was 4.1 (95%, CI 3.6–4.1), the sensitivity was 89.7% (95%, CI 75.8–97.1), and the specificity was 91.7 (95%, CI 81.6–97.2), also among those with mild hypertrophy (12–16mm) and normal EF (>55%). The diagnostic value of EFSR index is not dependent on the underlying amyloidosis type (AL or ATTR).13
Cardiovascular magnetic resonance (CMR) and nuclear medicine modalities (including TcPYP and TcDPD as well as the more recent 11C Pittsburgh compound B PET/CT scans) can reveal the pathological substrate. PYP test can determine (if Grade 2–3) or exclude (if Grade 0) ATTR myocardial amyloidosis, and its clinical value was equivalent to endomyocardial biopsy. It should be noted that negative PYP test (Grade 0) cannot rule out AL myocardial amyloidosis for patients with suspected myocardial amyloidosis through clinical manifestations, echocardiography, CMR and ECG. Hematologic test positive and tissue biopsy positive can determine AL myocardial amyloidosis.14 However, PYP and CMR widespread use is still limited in grass-roots hospitals.
The pulmonary involvement part of the ATTR amyloidosis symptoms is in the case of this patient. Studies suggest that FAP with V30M mutation results in pulmonary involvement in elderly patients,15 and the extent of amyloid deposition in lungs increases as a function of age. We recommend screening for lung amyloid deposition in ATTR patients with the primary complaints of respiratory symptoms.
The differential diagnosis of hATTR included senile wtATTR amyloidosis, chronic acquired polyneuropathy, chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), idiopathic axonal polyneuropathy, multiple peripheral neuropathy, or hypertrophic cardiomyopathy. Senile wtATTR amyloidosis suggests that wild-type or normal transthyretin protein can form amyloid fibrils. Cardiac manifestations predominate in wtATTR. However, musculoskeletal involvement may also be observed that manifests clinically as carpal tunnel syndrome, spinal stenosis, and biceps tendon rupture, which were not found in our patient. In addition, wtATTR generally does not cause progressive polyneuropathy and autonomic dysfunction. Chronic acquired polyneuropathy is associated with diabetes, alcohol overconsumption, cytostatic drugs and cardiovascular disease, which were not found in our patient. Hypertrophic cardiomyopathy is characterized by asymmetric thickening, high dynamic contraction and high ECG voltage. Multiple peripheral neuropathy, CIDP, and idiopathic axonal polyneuropathy generally do not involve the heart.
For hATTR patients, treatments in early-onset disease, stage I (patients can walk) and stage II (patients can walk with assistance) are available to help slow the progression of neuropathy and can improve the quality of life of patients.16,17 Outside Japan, the choice of drug therapy is first-line treatment, such as TTR stabilizing agent (tafamidis) and RNAi drug patisiran. However, liver transplantation remains a first-line treatment due to the positive results of liver transplantation in Japan (better life expectancy and higher survival rate).1
At present, the diagnostic rate of ATTR amyloidosis in China is still low. ATTR amyloidosis is often misdiagnosed or delayed due to factors such as low disease awareness and poor specificity of clinical symptoms. Because this disease is still in a process of being gradually recognized in China, and is limited to large hospitals, most grass-roots doctors do not know the disease, and its epidemiological status and incidence are not clear. In the 1990s, the Department of Neurology of Peking Union Medical College Hospital confirmed that the first ATTR-FAP family in China was Val30Met. However, from 2006 to 2019, only 20 probands were diagnosed with hATTR-FAP. Most of them were onset before the age of 50, and only 2 cases were late onset after the age of 50. It was found that val30met was not the dominant type of ATTR-FAP in China. Whether there is hot spot variation in Chinese patients is uncertain. Therefore, there is no data on the frequency of Val30Met mutation in China.
Conclusion
The prognosis of hATTR is poor, and diagnosis and treatment in early-onset disease may improve patient’s outcome, but it is often a challenge. Echocardiography is a noninvasive, reproducible method for assessing cardiac features and function in cardiac amyloidosis. Genetic testing played vital roles in the establishing diagnosis. Raising awareness is imperative as new treatments offer hope and the potential to change the trajectory of disease.
Ethics Approval and Informed Consent
The patient’s son provided informed consent. Ethical approval for this study was waived off by the Ethics Committee of the First Affiliated Hospital of Xi'an Jiaotong University as it is not considered a human medical research.
Consent for Publication
Written informed consent was obtained from the patient’s son for publication of this case report and any accompanying images.
Acknowledgments
We would like to thank RuiYong Yuan for his help in the diagnosis and treatment of the disease.
Disclosure
Shun Wang and Jing Ping Sun are co-first authors. The authors declare no conflicts of interest in this work.
References
1. Gertz MA. Immunoglobulin light chain amyloidosis: 2018 update on diagnosis, prognosis, and treatment. Am J Hematol. 2018;93(9):1169–1180. doi:10.1002/ajh.25149
2. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8(1):31. doi:10.1186/1750-1172-8-31
3. Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36(4):411–423. doi:10.1002/mus.20821
4. Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792. doi:10.1212/WNL.0b013e3182661eb1
5. Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–1854. doi:10.1097/TP.0000000000000574
6. Planté-Bordeneuve V. Update in the diagnosis and management of transthyretin familial amyloid polyneuropathy. J Neurol. 2014;261(6):1227–1233. doi:10.1007/s00415-014-7373-0
7. Coutinho P, Martins da Silva A, Lopas LJ. Forty years of experience with type 1 amyloid neuropathy. Review of 483 cases. In: amyloid and amyloidosis. Excerpta Med. 1980;1:88–98.
8. Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83(2):152–158. doi:10.1136/jnnp-2011-301299
9. Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109–2122. doi:10.1007/s00415-019-09688-0
10. Adams D, Lozeron P, Lacroix C. Amyloid neuropathies. Curr Opin Neurol. 2012;25(5):564–5723. doi:10.1097/WCO.0b013e328357bdf6
11. Quarta CC, Buxbaum JN, Shah AM, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(1):21–29. doi:10.1056/NEJMoa1404852
12. Arvanitis M, Koch CM, Chan GG, et al. Identification of transthyretin cardiac amyloidosis using serum retinol-binding protein 4 and a clinical prediction model. JAMA Cardiol. 2017;2(3):305–313. doi:10.1001/jamacardio.2016.5864
13. Pagourelias ED, Mirea O, Duchenne J, et al. Echo parameters for differential diagnosis in cardiac amyloidosis: a head-to-head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging. 2017;10(3):e005588. doi:10.1161/CIRCIMAGING.116.005588
14. Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on myocardial and pericardial diseases. Eur J Heart Fail. 2021;23(4):512–526. doi:10.1002/ejhf.2140
15. Koike H, Ando Y, Ueda M, et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287(1–2):178–184. doi:10.1016/j.jns.2009.07.028
16. Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi:10.1056/NEJMoa1716793
17. Benson MD, Dasgupta NR, Rao R. Diagnosis and screening of patients with hereditary Transthyretin Amyloidosis (hATTR): current strategies and guidelines. Ther Clin Risk Manag. 2020;14(16):749–758. doi:10.2147/TCRM.S185677
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.