Back to Journals » Drug Design, Development and Therapy » Volume 14
Laquinimod Protects Against TNF-α-Induced Attachment of Monocytes to Human Aortic Endothelial Cells (HAECs) by Increasing the Expression of KLF2
Authors Jiang T, Zhang W, Wang Z
Received 24 December 2019
Accepted for publication 6 February 2020
Published 30 April 2020 Volume 2020:14 Pages 1683—1691
DOI https://doi.org/10.2147/DDDT.S243666
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Tiechao Jiang, 1– 3 Wenhao Zhang, 1 Zhongyu Wang 1– 3
1Department of Cardiovascular Medicine, The Third Hospital of Jilin University, Changchun 130033, People’s Republic of China; 2Jilin Provincial Precision Medicine Key Laboratory for Cardiovascular Genetic Diagnosis, The Third Hospital of Jilin University, Changchun 130033, People’s Republic of China; 3Jilin Provincial Engineering Laboratory for Endothelial Function and Genetic Diagnosis of Cardiovascular Disease, The Third Hospital of Jilin University, Changchun 130033, People’s Republic of China
Correspondence: Zhongyu Wang
Department of Cardiovascular Medicine, The Third Hospital of Jilin University, No. 126, Xiantai Street, Changchun 130033, People’s Republic of China
Tel +86-431-84995258
Email [email protected]
Introduction: As a worldwide health issue, the treatment and prevention of atherosclerosis present an important goal. Increased levels of proinflammatory cytokines such as TNF-α-associated chronic inflammatory response cause endothelial cells to lose their ability to regulate vascular function. Lipid-laden immune cells are recruited to the endothelium where they adhere to the endothelial wall and invade the intimal space, thereby leading to the development of atherosclerotic lesions, fatty plaques, and thickening of the arterial wall. In the present study, for the first time, we investigated the effects of laquinimod, an immunomodulatory agent used for the treatment of multiple sclerosis, on human aortic endothelial in a TNF-α-induced atherosclerotic microenvironment. At present, the mechanism of action of laquinimod is not well defined.
Methods: The effects of laquinimod on the gene expression of IL-6, MCP-1, VCAM-1, E-selectin, and KLF2 were measured by real-time PCR. ELISA assay was used to determine protein secretion and expression. Phosphorylation of ERK5 and the protein level of KLF2 were measured by Western blot analysis. The attachment of monocytes to endothelial cells was assayed by calcein-AM staining and fluorescent microscopy.
Results: Our findings demonstrate that laquinimod reduced the expression of key inflammatory cytokines and chemokines, including IL-6, MCP-1, and HMGB1. We further demonstrate that laquinimod significantly reduced the attachment of monocytes to endothelial cells, which is mediated through reduced expression of the cellular adhesion molecules VCAM-1 and E-selectin. Here, we found that laquinimod could significantly increase the expression of KLF2 through activation of ERK5 signaling. The results of our KLF2 knockdown experiment confirm that the effects of laquinimod observed in vitro are dependent on KLF2 expression.
Conclusion: Together, these findings suggest a potential antiatherosclerotic capacity of laquinimod. Further research will elucidate the underlying mechanisms.
Keywords: atherosclerosis, laquinimod, endothelial cells, TNF-α, VCAM-1, E-selectin, KLF2
Introduction
Atherosclerosis is one of the most common cardiovascular diseases in the world, and its manifestations can be deadly. Characterized by vascular endothelial cell dysfunction and the formation of fatty plaques on the arterial endothelium, atherosclerosis is a complex disease involving diverse processes. In the early stages, atherosclerosis is nearly undetectable. However, if left untreated, advanced atherosclerosis can lead to plaque rupture or arterial occlusion, which results in heart attack or stroke.1–3 Inflammation is recognized as a key factor influencing stroke pathogenesis, and inhibition of the inflammatory response has been suggested as a treatment strategy.4 Tumor necrosis factor-α (TNF-α) is secreted by monocyte-macrophages and plays a crucial role in both the initiation and pathogenesis of atherosclerosis by triggering the activation of endothelial cells and endothelial dysfunction. Activated endothelial cells release proinflammatory cytokines, chemokines, and cellular adhesion molecules, which are key factors in atherogenesis.5 Patients with atherosclerosis have been found to have significantly increased serum levels of TNF-α.6 Inhibition of TNF-α has been suggested as a potential treatment approach, but recent research shows that inhibition of TNF-α alone may not be sufficient.7,8 Thus, other treatment options are actively being sought. The proinflammatory cytokine interleukin-6 (IL-6) is recognized as a central inflammatory mediator in atherosclerosis and a risk factor for cardiovascular events.9 IL-6 signaling initiates the acute phase inflammatory response, and inhibition of IL-6 has been shown to reduce the risk of atherothrombotic events.10 Monocyte chemoattractant protein-1 (MCP-1) is a CC chemokine that recruits lipid-laden monocytes and macrophages to the endothelial wall, where they infiltrate the intimal space and contribute to the development of atherosclerotic lesions. Studies involving MCP-1-deficient mice found reduced accumulation of immune cells in the aortic walls.11 High-mobility group box 1 (HMGB1) protein also plays a vital role in atherogenesis. Under normal conditions, HMGB1 resides in the nucleus where it regulates DNA transcription and protein assembly, among other things. However, in atherosclerosis, TNF-α causes HMGB1 to be actively secreted from cells, where it induces an inflammatory response and drives disease progression.12 Suppressing chronic inflammation is a vital part of treating and preventing atherosclerosis.
One of the most significant events in atherogenesis is the release of cellular adhesion molecules, such as vascular cellular adhesion molecule-1 (VCAM-1) and endothelial selectin (E-selectin). These molecules induce monocytes to roll along the endothelial wall and adhere to endothelial cells, thereby contributing to plaque development and stiffening of the arterial wall.13,14 TNF-α significantly increases the expression of both VCAM-1 and E-selectin.15 Kruppel-like factor 2 (KLF2) is a zinc-finger transcriptional factor that plays a protective role in a variety of cardiovascular diseases, including atherosclerosis. KLF2 is activated by extracellular signal-related protein kinase 5 (ERK5). ERK5/KLF2 signaling has been shown to slow the development of atherosclerosis by negatively regulating the expression of VCAM-1, intercellular adhesion molecule 1, E-selectin, and MCP-1,16 making it a valuable treatment target for atherosclerosis and other vascular diseases.
Laquinimod is a novel oral immunomodulator with high bioavailability currently being developed as a treatment for multiple sclerosis, Huntington’s disease, and Crohn’s disease, among others.17–19 Researchers have also begun to explore the potential of laquinimod as an anti-inflammatory agent and to treat glaucoma.20,21 The mechanism of action of laquinimod remains incompletely understood, and its effect on the cardiovascular system is unclear. In the present study, we explored the effect of laquinimod in human aortic endothelial cells (HAECs) stimulated with TNF-α to simulate an atherosclerotic microenvironment. We found that laquinimod can modulate ERK5/KLF2 signaling and reduce the attachment of monocytes to endothelial cells. Therefore, laquinimod may be a potential therapeutic agent against the development of atherosclerosis.
Materials and Methods
Cell Treatment and Culture
HAECs were purchased from Lonza, Switzerland and used in all experiments. The cells were grown in 2% serum endothelial growth media (EGM2) in low passage numbers (< 10). Human monocyte cell line U937 cells used in the cellular adhesion assay were purchased from the American Type Culture Collection (ATCC) and grown in 10% fetal bovine serum containing DMEM media. The cell cultures were maintained in a 5% (v/v) CO2/95% (v/v) nitrogen incubator at 37 °C prior to experimentation. For KLF2 knockdown, 90% confluent HAECs were transfected with KLF2 shRNA using Lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, USA). Laquinimod (TEVA Pharmaceuticals Industries, Ltd (Israel)) (purity≥98%) was dissolved in purified water. To establish TNF-α insult, the cells were stimulated with TNF-α (10 ng/mL) for 12, 24, and 48 hrs. To access the protective effects of laquinimod against TNF-α in HAECs, cells were stimulated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (2.5, 5 μM) for 24 h. To determine the involvement of ERK5, cells were treated with TNF-α (10 ng/mL), laquinimod (5 µM), and the ERK5 inhibitor XMD8-92 (10 nM) for 24 h.
Real-Time Polymerase Chain Reaction (PCR)
The total RNA was extracted from HAECs using an RNeasy Micro Kit from Qiagen (Hilden, Germany) in accordance with the manufacturer’s instructions. A Nanodrop spectrophotometer (Cole-Parmer, Chicago, IL) was used to quantify the RNA concentrations. Briefly, a total of 1 μg RNA was used cDNA synthesis using iScript™ Reverse Transcription Supermix for RT-qPCR (Invitrogen, Carlsbad, CA). SYBR-based real-time PCR experiments were performed to detect the total mRNA transcripts of IL-6, MCP-1, VCAM-1, E-selectin, ERK5, and KLF2 on an ABI 7500 real-time PCR platform.
Western Blot Analysis
After the indicated treatment, HAECs were lysed using RIPA buffer supplemented with protease inhibitor cocktail. Then, 20 μg cell lysates were loaded onto 4–20% precasted PAGE gels to separate the proteins by size. The separated protein mixture was then transferred onto PVDF membranes and the corresponding protein levels were detected using their specific antibodies. Blots were quantified by densitometry analysis of three exposures using TotalLab TL100 image analysis software. The following antibodies were used in this study: p-ERK5 (1:500, #3371, Cell signaling technology, USA); ERK5 (1:2000, #ab196609, Abcam, USA); KLF2 (1:1000, #ab194486, Abcam, USA); tubulin (1:10,000, #ab210797, Abcam, USA); anti-rabbit IgG, HRP-linked Antibody (1:2000, #7074, Cell Signaling Technology, USA); anti-mouse IgG, HRP-linked Antibody (1:2000, #7076, Cell Signaling Technology, USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
After the indicated treatment, HAECs culture media was collected for analyses using ELISA kits (R&D Systems) in accordance with the manufacturer’s instructions. Briefly, 96-well plate reader spectrometry was used to collect the data. Absolute values were obtained using a standardized 4-PL curve.
shRNA Transfection
For KLF2 knockdown, a human KLF2 gene-specific silencing and control lentivirus vector short hairpin RNA (shRNA) was developed, and the corresponding lentivirus particles were produced. Then, the KLF2 and control knockdown lentiviruses were concentrated using a density gradient centrifuge and titrated to determine virus efficiency. The lentivirus was then transfected into 50% confluent HAECs for 48–72 h at 10 MOI. Knockdown efficiency was confirmed by immunoblotting with KLF2 antibody.
Monocyte–Endothelial Cell Adhesion Experiment
U937 monocytes were stained with 1 μM calcein-AM for 30 mins in the dark at 37 °C. After necessary treatment, 5 × 105 U937 monocytes were added to 1 × 105 confluent HAEC cells for 2 h. The unbounded cells were washed away. And the attached cells were visualized using a fluorescent microscope.
Statistical Analysis
All experiments were performed in triplicate. Experimental data are expressed as means ± standard error of measurement (SEM). Statistical analysis was performed using the SPSS statistical analysis software package. To determine statistical significance among different groups, one-way or two-way ANOVA was used, followed by Bonferroni’s test. A P-value of < 0.05 was considered statistically significant.
Results
Laquinimod Reduces TNF-α-Induced Inflammatory Response
First, we assessed the effect of laquinimod on TNF-α-induced increased expression of IL-6 and MCP-1, two key effectors of the inflammatory response in atherosclerosis. As shown in Figure 1A, TNF-α significantly increased the mRNA expression of IL-6 and MCP-1 6.7- and 4.3-fold, respectively. However, 2.5 and 5 µM laquinimod reduced these levels to only 2.7- and 1.7-fold. As shown in Figure 1B, the secretion of IL-6 and MCP-1 was increased more than 10-fold by TNF-α, while laquinimod mitigated this increase in a dose-dependent manner. Next, we measured the effects of laquinimod on the inflammatory mediator HMGB1. As shown in Figure 2, HMGB1 secretion was increased nearly 7-fold, while the two doses of laquinimod reduced this value in a dose-dependent manner.
 |
Figure 1 Laquinimod inhibited TNF-α-induced expression and secretion of pro-inflammatory cytokines in human aortic endothelial cells (HAECs). Cells were stimulated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (2.5, 5 μM) for 24 h. (A). mRNA of IL-6 and MCP-1 were measured by real-time PCR; (B). Secretion of IL-6 and MCP-1 was measured by ELISA (****P<0.0001 vs vehicle group; &P<0.01 vs TNF-α group; ##P<0.001 vs TNF-α group). |
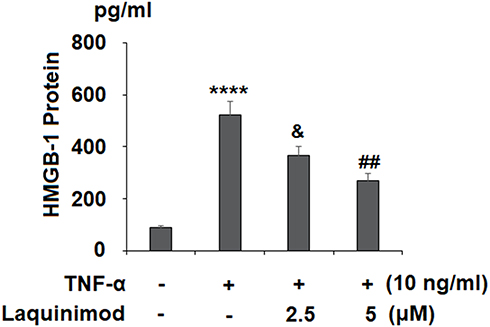 |
Figure 2 Laquinimod prevented TNF-α-induced secretion of HMGB1. Cells were stimulated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (2.5, 5 μM) for 24 h. Secretions of HMGB1 were measured by ELISA analysis (****P<0.0001 vs vehicle group; &P<0.01 vs TNF-α group; ##P<0.001 vs TNF-α group). |
Laquinimod Prevents the Attachment of Monocytes to Endothelial Cells
Next, we investigated the effects of laquinimod on the attachment of monocytes to endothelial cells. We first measured the expression of cellular adhesion molecules. As shown in Figure 3A and B, TNF-α significantly increased the expression of VCAM-1 and E-Selectin at both the mRNA and protein levels. However, treatment with laquinimod reduced them in a dose-dependent manner, suggesting an ability of laquinimod to prevent the attachment of immune cells to the arterial wall. To confirm this, we performed a cellular adhesion experiment using HAECs and U937 monocyte cell line. As shown in Figure 4, TNF-α induced a 3.6-fold increase in the number of attached monocytes, which was dose-dependently reduced to only 1.7-fold by laquinimod. Thus, laquinimod may significantly hinder the attachment of monocytes to endothelial cells.
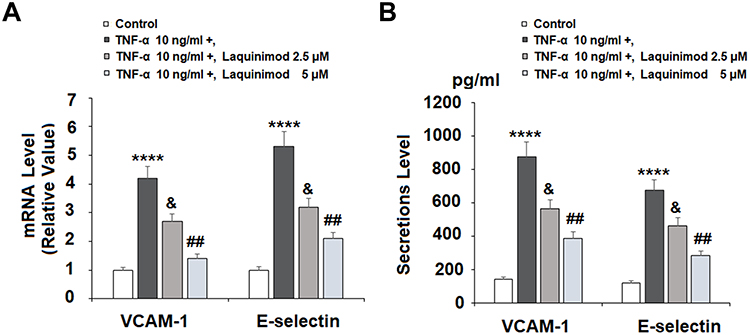 |
Figure 3 Laquinimod reduced TNF-α-induced expression of VCAM-1 and E-selectin. Cells were stimulated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (2.5, 5 μM) for 24 h. (A). mRNA of VCAM-1 and E-selectin as measured by real-time PCR; (B). Protein of VCAM-1 and E-selectin as measured by ELISA (****P<0.0001 vs vehicle group; &P<0.01 vs TNF-α group; ##P<0.001 vs TNF-α group). |
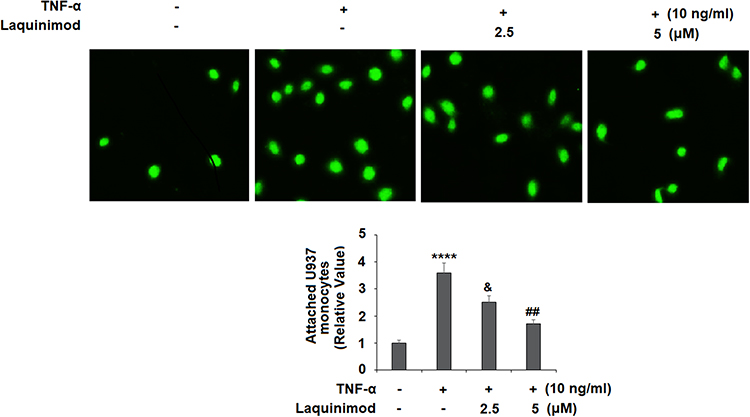 |
Figure 4 Laquinimod prevented TNF-α-induced attachment of U937 monocytes to human aortic endothelial cells (HAECs). Cells were stimulated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (2.5, 5 μM) for 24 h. Attached U937 monocytes were quantified (****P<0.0001 vs vehicle group; &P<0.01 vs TNF-α group; ##P<0.001 vs TNF-α group). |
Laquinimod Rescues KLF2 Expression Through ERK5
To determine the effects of laquinimod on the expression of the protective transcription factor KLF2, we first confirmed that TNF-α time-dependently reduces the expression of KLF2 in HAECs at both the mRNA and protein levels (Figure 5A and B). Next, as shown in Figure 6A and B, TNF-α reduced the expression of KLF2 by roughly half at both the mRNA and protein levels, while laquinimod remarkably rescued KLF2 expression to near baseline at the mRNA level and above baseline at the protein level. Thus, laquinimod has a potent capacity to rescue and increase KLF2 expression. Next, we investigated whether the ERK5 signaling pathway is involved in mediating the rescue of KLF2 by laquinimod. As shown in Figure 7A, TNF-α reduced the level of phosphorylated ERK5 by nearly 70%, which was rescued to only 11% below baseline in a dose-dependent manner. Additionally, we found that blockage of ERK5 with the specific ERK5 inhibitor XMD9-92 completely abolished the effects of laquinimod on KLF2 expression as we showed in Figure 7B and C.
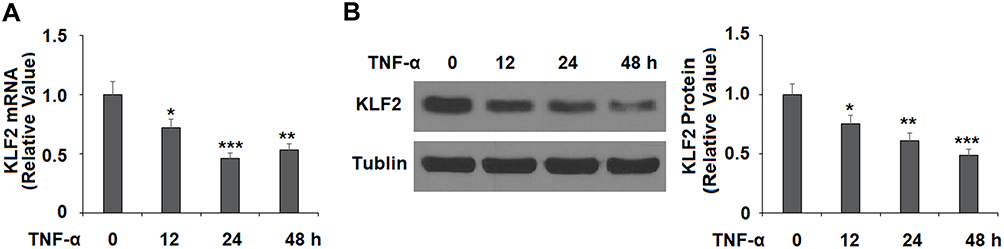 |
Figure 5 TNF-α reduced the expression of KLF2 in human aortic endothelial cells (HAECs). Cells were stimulated with TNF-α (10 ng/mL) for 12, 24, and 48 hrs. (A). mRNA of KLF2 was measured; (B). Protein expression of KLF2 was measured by Western blot analysis (*, **, ***P<0.05, 0.01, 0.005 vs vehicle group). |
 |
Figure 6 Laquinimod restored TNF-α-induced reduction of KLF2. Cells were stimulated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (2.5, 5 μM) for 24 h. (A). mRNA of KLF2 as measured by real-time PCR; (B). Protein of KLF2 as measured by Western blot analysis (****P<0.0001 vs vehicle group; &P<0.01 vs TNF-α group; ##P<0.001 vs TNF-α group). |
 |
Figure 7 The effects of laquinimod in promoting the expression of KLF2 are dependent on ERK5. (A). Laquinimod restored TNF-α-induced dephosphorylation of ERK5. Cells were stimulated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (2.5, 5 μM) for 24 h. Phosphorylation of ERK5 was measured Western blot analysis. (B,C). Blockage of ERK5 abolished the effects of laquinimod on KLF2 expression. Cells were treated with TNF-α (10 ng/mL) in the presence or absence of laquinimod (5 μM) or the ERK5 inhibitor XMD8-92 (10 nM) for 24 h. mRNA and protein levels of KLF2 were measured (*, #, $ P<0.01 vs previous column group). |
The Effects of Laquinimod are Mediated Through KLF2
Finally, we investigated whether the effects of laquinimod on cellular adhesion are mediated through KLF2. The KLF2 in HAECs was knockout by using lentiviral KLF2 shRNA as we showed in Figure 8A. As we demonstrated in Figure 8B, knockdown of KLF2 abolished the ability of laquinimod to reduce TNF-α-induced increased expression of VCAM-1 and E-selectin. Additionally, KLF2 knockdown abolished the ability of laquinimod to reduce the attachment of monocytes to endothelial cells, and in fact, further increased the number of attached U937 monocytes as we showed in Figure 8C. Therefore, laquinimod reduces the attachment of monocytes to endothelial through rescue of the ERK5/KLF2 signaling pathway.
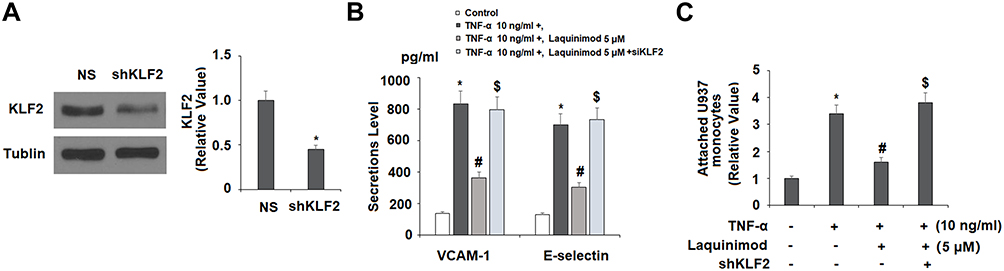 |
Figure 8 Silencing of KLF2 with KLF2 shRNA abolished the effects of laquinimod in inhibiting the expression of endothelial adhesion molecules and attachment of U937 monocytes to HAECs. Cells were transduced with lentiviral KLF2 shRNA or null control, followed by stimulation with TNF-α (10 ng/mL) in the presence or absence of laquinimod (5 μM) for 24 h. (A). Western blot results revealed the successful knockdown of KLF2; (B). Protein of VCAM-1 and E-selectin as measured by ELISA; (C). Attachment of U937 monocytes to human aortic endothelial cells (HAECs) (*, #, $P<0.01 vs previous column group). |
Discussion
Inhibiting the attachment of monocytes to endothelial cells presents a potential treatment strategy against atherosclerosis. In the present study, we employed HAECs and U937 monocyte cell line to investigate the effects of laquinimod, an immunomodulatory drug for the treatment of multiple sclerosis, in a TNF-α-induced atherosclerotic microenvironment. TNF-α insult is known to promote atherogenesis by upregulating the expression of adhesion molecules, such as VCAM-1, E-selectin, and inflammatory cytokines, including IL-6, MCP-1, and HMGB1.22–24 TNF-α and IL-6 are considered to be the main inflammatory cytokines involved in atherosclerosis. In pathological conditions, TNF-α and IL-6 are released from free cholesterol-laden macrophages, thereby promoting the development of vulnerable plaques and potentially life-threatening cardiovascular events.25 Previous in vivo research found that treatment with laquinimod downregulated the expression of TNF-α and IL-6 in splenocytes extracted from a chronic experimental autoimmune encephalomyelitis mouse model of MS.26 Here, we found that laquinimod could downregulate the expression of IL-6 induced by TNF-α in HAECs. MCP-1 contributes to inflammation and thrombosis by recruiting immune cells to sites of atherosclerotic lesions. A recent study found that MCP-1 levels could serve as an early biomarker for atherosclerosis.27 In a double-blind clinical trial, monocytes extracted from patients treated with laquinimod were found to excrete lower levels of MCP-1.28 Additionally, research suggests that laquinimod might reduce the expression of MCP-1 in the spinal cord.29 Our findings show that treatment with laquinimod dose-dependently reduced the expression of MCP-1 in HAECs stimulated with TNF-α. HMGB1 is another major proinflammatory cytokine that acts as a proinflammatory danger signal. Under normal circumstances, thrombomodulin blocks the expression of HMGB1, but in atherosclerosis, thrombomodulin is downregulated, thereby allowing unmitigated expression of HMGB1.27 Previous research has shown that TNF-α-mediated inflammatory response in HAECs involves HMGB1-mediated activation of Toll-like receptor 4.24 Additionally, HMGB1 triggers further upregulation of TNF-α, thereby initiating a sort of positive feedback loop.30 Thus, laquinimod may exert anti-inflammatory effects. We found that laquinimod could significantly reduce the expression of HMGB1. Together, our findings indicate that laquinimod may possess potent anti-inflammatory properties in HAECs.
The migration of immune cells to the endothelium and subsequent adhesion of monocytes to endothelial cells is a major driving force in atherogenesis. Presently, there is little research on the effects of laquinimod on cellular adhesion and the expression of related signaling molecules. An early study on laquinimod found that it could disrupt the ability of very late antigen-4 to bind with VCAM-1, thereby potentially mitigating the migratory capacity of T cells.31 A more recent study found that laquinimod could reduce the adhesiveness of the central nervous system endothelium through reduced expression of cellular adhesion molecules.32 Here, we found that laquinimod could indeed reduce the expression of VCAM-1 and E-selectin induced by TNF-α. We verified the functional outcome of reduced expression of these molecules by performing a cellular adhesion assay using U937 monocytes. Our findings indicate that laquinimod did indeed reduce the number of attached monocytes. Thus, laquinimod exhibits an anti-cellular adhesion capacity, which may of value in the treatment of atherosclerosis.
KLF2 is widely recognized as a key mechanosensitive atheroprotective factor. Reduced expression of KLF2 is associated with endothelial dysfunction and altered expression of genes that regulate vascular tone.33 Statins are a widely used class of vasoprotective agents that act, in part, through the ERK5 pathway to lower cholesterol, mediate angiogenesis, stabilize plaques, and suppress the immune response.34 Recent research has shown that rescue of KLF2 expression through ERK5 can prevent the attachment of monocytes to endothelial cells.35 While the role of KLF2 has been elucidated in numerous cell types, including myeloid cells, it is best recognized for its activity in endothelial cells.36 Presently, the effect of laquinimod on the ERK5/KLF2 pathway has not been elucidated. Here, we found that laquinimod could potently rescue TNF-α-induced reduced KLF2 expression through ERK5 signaling. Additionally, the results of our KLF2 shRNA transfection experiment demonstrate that the ability of laquinimod to prevent monocyte attachment in vitro is dependent on KLF2 signaling. Thus, laquinimod may exert atheroprotective effects by modulating the expression of the ERK5/KLF2 pathway, thereby suppressing inflammation and reducing the attachment of monocytes to endothelial cells. There are several limitations to our study. Firstly, our experiments lack a drug that can treat or actively improve atherosclerosis as a positive control. For example, statins, a family of 3-hydroxy-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors used for both primary and secondary prevention of coronary heart disease (CHD), have displayed their pharmacological functions in maintaining endothelial function by inhibiting the inflammatory response, preventing endothelial-leukocyte interactions, and increasing the expression of KLF2 [38, 39]. Therefore, statins could be used as a positive control in this study. Secondly, this preliminary research was only performed in vitro in an atherosclerotic microenvironment. It should be noticed that a diverse range of cell types, including endothelial cells, smooth muscle cells, fibroblasts, and monocytes, are involved in the pathogenesis of cardiovascular diseases. The effects of laquinimod on other organs and tissues have not been thoroughly studied. Therefore, subsequent in vivo experimentation is required to determine the safety and efficacy of laquinimod in the context of atherosclerosis. These findings help to shed light on the mechanism of action of laquinimod and set the foundation for further research.
Acknowledgment
This study is funded by the “National Natural Science Foundation of China (No.81570360)”.
Disclosure
There is no conflict of interest to disclose for all the authors in regard to this work.
References
1. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209(1):13–22. doi:10.1083/jcb.201412052
2. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. 2015;12(1):10. doi:10.1038/nrcardio.2014.173
3. Horie N, Tateishi Y, Morikawa M, et al. Acute stroke with major intracranial vessel occlusion: characteristics of cardioembolism and atherosclerosis-related in situ stenosis/occlusion. J Clin Neurosci. 2016;32:24–29. doi:10.1016/j.jocn.2015.12.043
4. Bäck M, Hansson GK. Anti-inflammatory therapies for atherosclerosis. Nat Rev Cardiol. 2015;12(4):199. doi:10.1038/nrcardio.2015.5
5. Qiu HN, Liu B, Liu W, Liu S. Interleukin-27 enhances TNF-α-mediated activation of human coronary artery endothelial cells. Mol Cell Biochem. 2016;411(1–2):1.
6. Xing J, Liu Y, Chen T. Correlations of chemokine CXCL16 and TNF-α with coronary atherosclerotic heart disease. Exp Ther Med. 2018;15(1):773–776. doi:10.3892/etm.2017.5450
7. Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-α reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24(11):2137–2142. doi:10.1161/01.ATV.0000143933.20616.1b
8. Oberoi R, Vlacil AK, Schuett J, et al. Anti-tumor necrosis factor-α therapy increases plaque burden in a mouse model of experimental atherosclerosis. Atherosclerosis. 2018;277:80–89. doi:10.1016/j.atherosclerosis.2018.08.030
9. Libby P, Rocha VZ. All roads lead to IL-6: a central hub of cardiometabolic signaling. Int J Cardiol. 2018;259:213–215. doi:10.1016/j.ijcard.2018.02.062
10. Ridker PM, Libby P, MacFadyen JG, et al. Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur Heart J. 2018;39(38):3499–3507. doi:10.1093/eurheartj/ehy310
11. Harrington JR. The role of MCP‐1 in atherosclerosis. Stem Cells. 2000;18(1):65–66. doi:10.1634/stemcells.18-1-65
12. De Souza AW, Westra J, Limburg PC, Bijl M, Kallenberg CG. HMGB1 in vascular diseases: its role in vascular inflammation and atherosclerosis. Autoimmun Rev. 2012;11(12):909–917. doi:10.1016/j.autrev.2012.03.007
13. Vogel ME, Idelman G, Konaniah ES, Zucker SD. Bilirubin prevents atherosclerotic lesion formation in low‐density lipoprotein receptor‐deficient mice by inhibiting endothelial VCAM‐1 and ICAM‐1 signaling. J Am Heart Assoc. 2017;6(4):e004820. doi:10.1161/JAHA.116.004820
14. Del Bo C, Marino M, Riso P, Møller P, Porrini M. Anthocyanins and metabolites resolve TNF-α-mediated production of E-selectin and adhesion of monocytes to endothelial cells. Chem Biol Interact. 2019;300:49–55. doi:10.1016/j.cbi.2019.01.002
15. Deng H, Song Z, Xu H, et al. MicroRNA-1185 promotes arterial stiffness though modulating VCAM-1 and E-selectin expression. Cell Physiol Biochem. 2017;41(6):2183–2193. doi:10.1159/000475576
16. Deng Y, Lei T, Li H, Mo X, Wang Z, Ou H. ERK5/KLF2 activation is involved in the reducing effects of puerarin on monocyte adhesion to endothelial cells and atherosclerotic lesion in apolipoprotein E-deficient mice. Biochim Biophys Acta. 2018;1864(8):2590–2599. doi:10.1016/j.bbadis.2018.04.021
17. Kaye J, Piryatinsky V, Birnberg T, et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc Natl Acad Sci. 2016;113(41):E6145–52. doi:10.1073/pnas.1607843113
18. Garcia-Miralles M, Yusof NA, Tan JY, et al. Laquinimod treatment improves myelination deficits at the transcriptional and ultrastructural levels in the YAC128 mouse model of Huntington disease. Mol Neurobiol. 2019;56(6):4464–4478. doi:10.1007/s12035-018-1393-1
19. Tarcic N, Haviv A, Blaugrund E, Kaye J, inventors; Teva Pharmaceutical Industries Ltd, assignee. Treatment of crohn’s disease with laquinimod. United States patent application US 15/703,496. 2018 Jan 4.
20. Katsumoto A, Miranda AS, Butovsky O, Teixeira AL, Ransohoff RM, Lamb BT. Laquinimod attenuates inflammation by modulating macrophage functions in traumatic brain injury mouse model. J Neuroinflammation. 2018;15(1):26. doi:10.1186/s12974-018-1075-y
21. Neumann R, Etzyoni R, inventors; Teva Pharmaceutical Industries Ltd, assignee. Treatment of glaucoma using laquinimod. United States patent application US 15/875,833. 2018 May 24.
22. Ohta H, Wada H, Niwa T, et al. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis. 2005;180(1):11–17. doi:10.1016/j.atherosclerosis.2004.11.016
23. Zhang F, Yu W, Hargrove JL, et al. Inhibition of TNF-α induced ICAM-1, VCAM-1 and E-selectin expression by selenium. Atherosclerosis. 2002;161(2):381–386. doi:10.1016/S0021-9150(01)00672-4
24. Yang WS, Han NJ, Kim JJ, Lee MJ, Park SK. TNF-α activates high-mobility group box 1-toll-like receptor 4 signaling pathway in human aortic endothelial cells. Cell Physiol Biochem. 2016;38(6):2139–2151. doi:10.1159/000445570
25. Li Y, Schwabe RF, DeVries-Seimon T, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6 model of nf-κb-and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280(23):21763–21772. doi:10.1074/jbc.M501759200
26. Moore S, Khalaj AJ, Yoon J, et al. Therapeutic laquinimod treatment decreases inflammation, initiates axon remyelination, and improves motor deficit in a mouse model of multiple sclerosis. Brain Behav. 2013;3(6):664–682. doi:10.1002/brb3.174
27. Basurto L, Gregory MA, Hernández SB, et al. Monocyte chemoattractant protein-1 (MCP-1) and fibroblast growth factor-21 (FGF-21) as biomarkers of subclinical atherosclerosis in women. Exp Gerontol. 2019;124:110624. doi:10.1016/j.exger.2019.05.013
28. Stasiolek M, Linker RA, Hayardeny L, Bar Ilan O, Gold R. Immune parameters of patients treated with laquinimod, a novel oral therapy for the treatment of multiple sclerosis: results from a double‐blind placebo‐controlled study. Immun Inflammation Dis. 2015;3(2):45–55. doi:10.1002/iid3.42
29. Mishra MK, Wang J, Silva C, Mack M, Yong VW. Kinetics of proinflammatory monocytes in a model of multiple sclerosis and its perturbation by laquinimod. Am J Pathol. 2012;181(2):642–651. doi:10.1016/j.ajpath.2012.05.011
30. Luan ZG, Zhang H, Yang PT, Ma XC, Zhang C, Guo RX. HMGB1 activates nuclear factor-κB signaling by RAGE and increases the production of TNF-α in human umbilical vein endothelial cells. Immunobiology. 2010;215(12):956–962. doi:10.1016/j.imbio.2009.11.001
31. West M, Miravalle A. Profile of oral laquinimod and its potential in the treatment of multiple sclerosis. Degener Neurol Neuromuscul Dis. 2011;1:25. doi:10.2147/DNND.S16374
32. Lühder F, Kebir H, Odoardi F, et al. Laquinimod enhances central nervous system barrier functions. Neurobiol Dis. 2017;102:60–69. doi:10.1016/j.nbd.2017.02.002
33. Lee JS, Yu Q, Shin JT, et al. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev Cell. 2006;11(6):845–857. doi:10.1016/j.devcel.2006.09.006
34. Komaravolu RK, Adam C, Moonen JR, Harmsen MC, Goebeler M, Schmidt M. Erk5 inhibits endothelial migration via KLF2-dependent down-regulation of PAK1. Cardiovasc Res. 2014;105(1):86–95. doi:10.1093/cvr/cvu236
35. Wang X, Wu Z, He Y, et al. Humanin prevents high glucose-induced monocyte adhesion to endothelial cells by targeting KLF2. Mol Immunol. 2018;101:245–250. doi:10.1016/j.molimm.2018.07.008
36. Shaked I, Ley K. Protective role for myeloid specific KLF2 in atherosclerosis. Circ Res. 2012;110(10):1266. doi:10.1161/CIRCRESAHA.112.270991
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.