Back to Journals » International Medical Case Reports Journal » Volume 11
Langerhans cell histiocytosis with hemorrhagic uveitis and exudative retinal detachment
Authors Kharel Sitaula R ![]() , Khatri A
, Khatri A ![]()
Received 9 January 2018
Accepted for publication 15 February 2018
Published 26 March 2018 Volume 2018:11 Pages 65—68
DOI https://doi.org/10.2147/IMCRJ.S162038
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Ranju Kharel Sitaula,1 Anadi Khatri2
1Uveitis Department, B.P. Koirala Lions Center for Ophthalmic Studies, Institute of Medicine, Tribhuvan University, Kathmandu, Nepal; 2Fellow of Vitreo-Retina Surgery, Lumbini Eye Institute, Siddharthanagar, Nepal
Abstract: Bilateral intraocular involvement in Langerhans cell histiocytosis (LCH) is uncommon. A 15-year-old boy presented with painless decreased vision in right and painful left red eye of 2 weeks duration. Visual acuity was 20/500 and 20/200, respectively. A fixed dilated pupil with exudative retinal detachment was present in the right eye and hemorrhagic iris nodules with hyphema and hypopyon were seen in the left eye. Intraocular pressure was 12 and 31 mmHg, respectively, in each eye. Ocular symptoms were preceded by fever with multiple skin rashes, subcutaneous nodules, and lymph node enlargement. The histopathological examination of skin and lymph node showed histiocytes positive for histiocytic S 100. He was treated with topical steroids and antiglaucoma eye drops along with intravenous vinblastine 6 mg and oral prednisolone (1 mg/kg). Hyphema and hypopyon were resolved, vision improved to 20/320 and 20/80, and intraocular pressure was under control. However, as multisystemic LCH has a poor prognosis, we were unable to save him. Hence, fatal conditions like LCH should also be considered in the differential diagnosis of any hemorrhagic uveitis.
Keywords: exudative retinal detachment, uveitis, langerhans cell histiocytosis, bilateral, IL-17
Introduction
Langerhans cell histiocytosis (LCH) is a multisystem disorder of unknown etiology characterized by the accumulation of histiocytes in various tissues.1 Langerhans cells (LCs) are dendritic cells and are normally only found in the skin and major airways. The LC is named after Paul Langerhans (1847–1888), a German pathologist.2 In LCH, the LCs are abnormal and cause damage and may be found in different parts of the body, including the eye.
The pathogenesis of LCH is still unclear. The benign morphology of its proliferating cells and its characteristic inflammatory infiltrates suggest that LCH may be an inflammatory disorder, and dysregulated expression of inflammatory cytokines, such as interleukin-17A, has been reported.3,4 However, the pathologic LCs in LCH are clonal.3 It has a variable clinical course, occasionally seen in adults, but predominantly occurs in children5 peaking during ages 1–4.6,7 LCH is more common in boys than girls.2 Unifocal and single-system disease dominates over multisystem disease in most studies,8–10 and multisystem LCH harbors a more unfavorable outcome.3,9,11,12
LCH was formerly known as “histiocytosis X,” and historically, it included three subgroups: eosinophilic granuloma, Hand–Schuller–Christian disease, and Letterer–Siwe disease. Ophthalmic manifestations in LCH are seen in 10% of cases,13 and most cases involved the orbit. Intraocular involvement is rare and is usually found in the subacute disseminated form.14 Bilateral intraocular involvement in LCH is exceptional. But, herein we report a case of acute multisystemic LCH with bilateral intraocular involvement. Written informed consent was taken from the patient’s father.
The treatment for LCH is based on disease severity and a number of systems involved.1 Low to moderate doses of methotrexate, prednisone, and vinblastine are used as therapy. When intraocular LCH is part of widespread disease, treatment guidelines parallel those recommended for the multisystem disease.15 LCH with recurrent BRAF mutations respond well to an RAF pathway inhibitor.3 The prognosis and the ultimate outcome depend not only on the therapy but also upon several other factors, such as the age of presentation, multisystem involvement, and response to initial therapy. The mortality rate in children and patients with multisystem involvement is high because of failure of key organs such as bone marrow, lungs, and liver.1 The 5-year survival rate in children under 2 years, even with aggressive chemotherapy, is only 50%.16
Case report
A 15-year-old boy presented with profound painless progressive diminution of vision of right eye since 3 months. Suddenly, in the left eye, he developed a painful red eye during the last 15 days. On ocular examination, best-corrected visual acuity in the right eye was hand movement close to face with an inaccurate projection of rays and in the left eye was 6/60. The right eye pupil was 8 mm in size, dilated, and nonreacting (Figure 1A), and the retina had exudative retinal detachment around the disc with macular edema (Figure 1B). The left eye was congested and the cornea was edematous. The anterior chamber was filled with fibrinous exudates and hyphema (Figure 2). Hemorrhagic iris nodules were noted in the iris stroma without iris neovascularization. The pupil was 5 mm in size, irregular with multiple posterior synechiae, and sluggishly reacting and covered with fibrinous exudates. The lens appeared clear, but the details of vitreous and retina of the left eye could not be appreciated. An ultrasonography of right eye showed hyperechoic shadow in vitreous and areas of retinal detachment with subretinal fluid collection, but left eye ultrasonography was normal. Intraocular pressures in right and left eye were 12 and 31 mmHg.
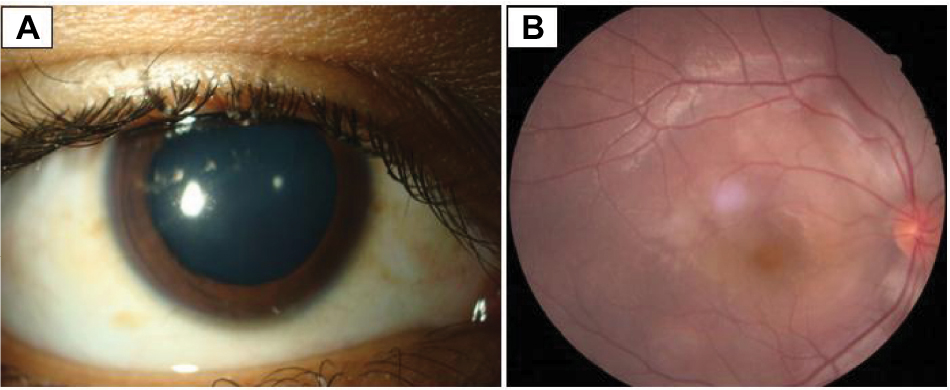 | Figure 1 Nonreacting, dilated pupil (A) and multifocal exudative retinal detachment with macular edema (B) in the right eye. |
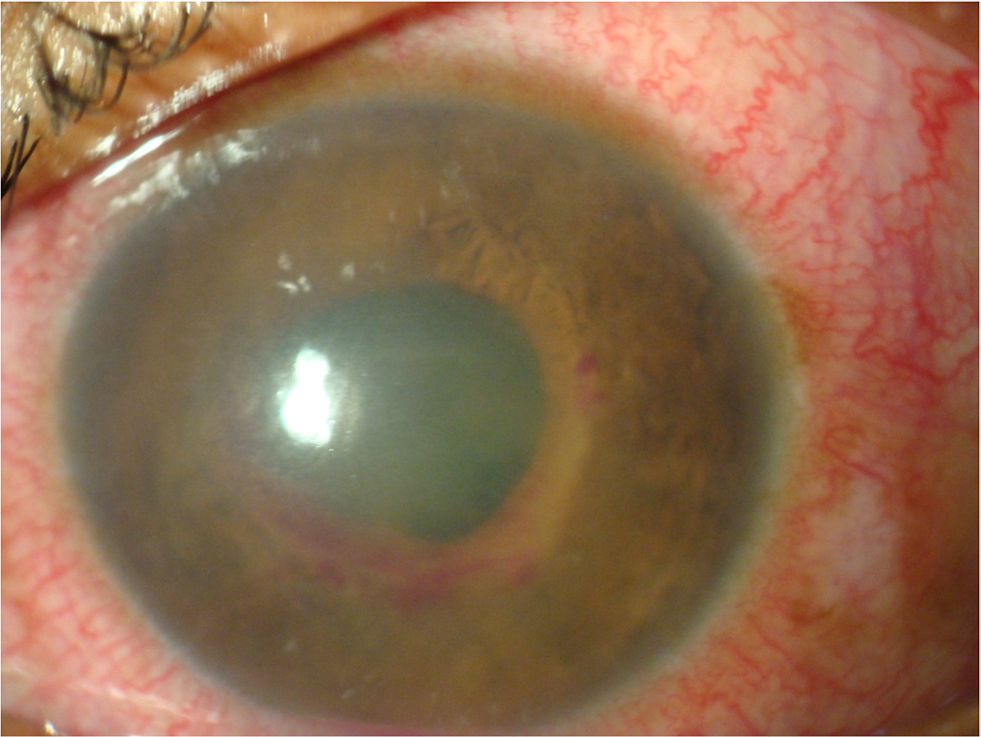 | Figure 2 Left eye with circumciliary congestion, edematous cornea, and fibrinous exudates with hyphema in anterior chamber. |
The episode was preceded by high-grade fever with chills/rigor and multiple skin rashes involving the face and trunks for the past 4 months. It was also associated with painful subcutaneous nodules over the scalp, trunk, and arms followed by painless enlargement of axillary and cervical lymph nodes.
On systemic examination, there were multiple papulonodular rashes over the face (Figure 3A) and trunk with a subcutaneous ulcerated nodule in the scalp, arm, and back of trunk (Figure 3B). The cervical and axillary lymph nodes were enlarged, firm, nontender, and not matted. Histopathological examination of skin and axillary lymph nodes revealed dense infiltrate of cells composed of histiocytes, lymphocytes, and occasional eosinophils. The histiocytes have scanty cytoplasm, a nucleus with coffee bean appearance and prominent nucleoli with mitosis (Figure 4) and were positive for histiocytic S 100, suggestive of LCH. MRI of the brain showed a well-defined enhancing lesion measuring 2×0.8 cm in the right scalp of superior temporal fossa which was isodense in T1 but hyperdense in T2 and fluid-attenuated inversion recovery with no obvious bony lesion. With the diagnosis of multifocal LCH, chemotherapy in form of injection of vinblastine 6 mg was started by an oncologist.
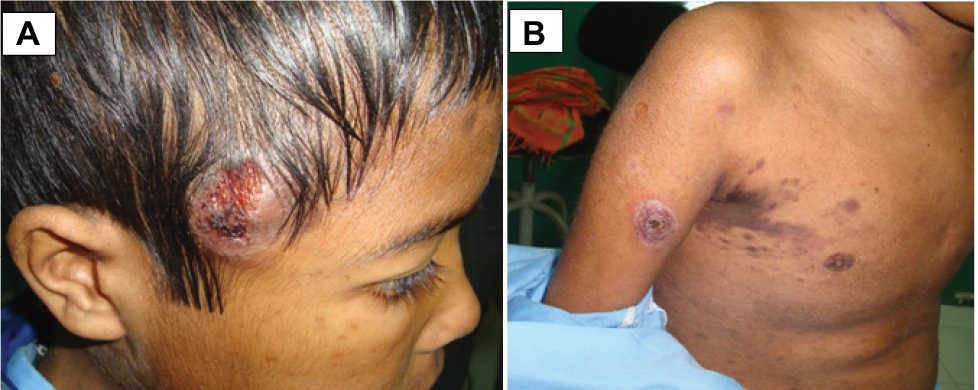 | Figure 3 Subcutaneous ulcerated nodule in the scalp (A) and papulomacular rashes and nodules on the arm and trunk (B). |
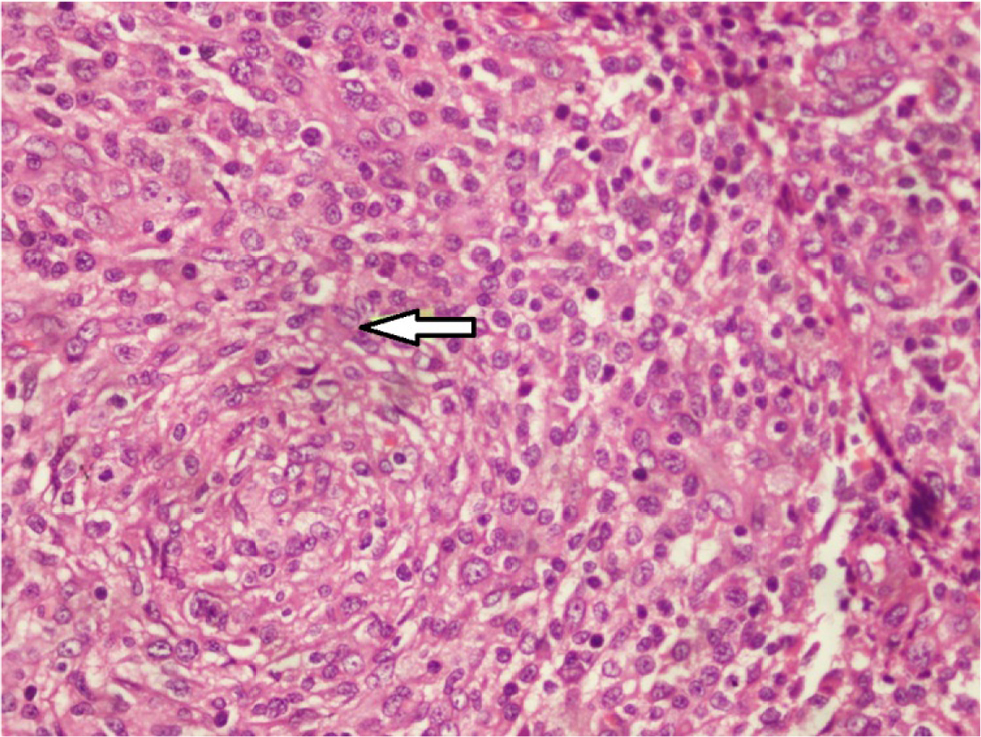 | Figure 4 Histopathological examination of skin showing dense cellular infiltrate composed of histiocytes, lymphocytes, and occasional eosinophils. Note: Arrow: the histiocytes have scanty cytoplasm, a nucleus with coffee bean appearance and prominent nucleoli with mitosis. |
For eyes, the patient was treated with prednisolone acetate 1% eye drop 6 hourly in the right eye and 1 hourly in left eye along with atropine 1% eye drop thrice and brimonidine tartrate 0.2% + timolol maleate 0.5% eye drop twice in the left eye. After consultation with an oncologist, a tapering dose of prednisolone 40 mg tablet once daily along with proton pump inhibitor therapy was started.
After 1 week, vision in right eye improved to counting finger at 1 foot with an inaccurate projection of rays and 6/24 in the left eye. No other significant changes were noticed in the right eye. In the left eye, the conjunctival congestion decreased, cornea became clear, and hypopyon and hyphaema were resolved and fundus glow became visible. The intraocular pressure was 16 and 13 mmHg in each eye. The dose of oral steroids was tapered, and the patient was kept under regular follow-up, but was lost to follow-up after 2 months. Later, the death of the patient due to multiple organ failure was confirmed by the patient’s guardian.
Discussion
Bilateral intraocular involvement by LCH is rare and has been reported as both an isolated focus of LCH and as part of disseminated disease.2 Intraocular LCH can mimic iritis, iridocyclitis, or choroiditis.15 Secondary glaucoma can occur due to obstruction of aqueous outflow by LCs.15
LCH predominantly occurs in children and is occasionally seen in adults. Younger patients tend to have multifocal involvement17 and have a more aggressive clinical course compared with those with unifocal disease. Herein, we described an exceptional case of multifocal LCH with bilateral intraocular features sparing the orbit in an adult with unfavorable prognosis.
Letterer–Siwe variant of LCH typically has multifocal lesions with fever with a localized infection, otitis media, and maculopapular, eczematous, or purpuric skin lesions. There may be hepatosplenomegaly severe bone marrow involvement resulting in anemia, thrombocytopenia, and leukopenia and cystic lesions of the skull, pelvis, and long bones. The course is commonly fulminant and fatal, with few spontaneous remissions despite therapy.17 Our case was close to the Letterer–Siwe variant of LCH. Although LCs in LCH are clonal, their benign morphology and absence of reported recurrent genomic abnormalities have suggested that LCH may not be a neoplasm, but the oncogenic BRAF V600E mutation has been identified in few cases, which strongly suggests that LCH is a neoplasm.3 V600E BRAF mutations are more common in melanoma and respond well to therapeutic BRAF inhibitors like vemurafenib.18 Discovery of recurrent mutations affecting the MAPK and mTOR–AKT pathways in some of these histiocytoses has now led to the evolution of this condition from a primary inflammatory condition to that of a clonal neoplastic disease.19 However, we were not able to perform such cancer-associated mutation studies in our case due to unavailability of this facility.
The prognosis and the ultimate outcome depend not only on the therapy but also upon several other factors, such as the age of presentation, multisystem involvement, and response to initial therapy. The mortality rate in children and patients with multisystem involvement is high because of failure of key organs such as bone marrow, lungs, and liver. The 5-year survival rate for children under 2 years, even with aggressive chemotherapy, is only 50%.16
Conclusion
Intraocular LCH is rarely encountered in ophthalmic practice. Bilateral simultaneous intraocular manifestation is exceptional. Late onset of the disease with multifocal involvement is indicative of poor prognosis.
Consent
Written informed consent has been provided by the patient’s parent to have the case details and any accompanying images published.
Acknowledgment
We would like to thank Mr Suresh Raj Sharma, senior technical staff, for the acquisition of the clinical photographs and B-Scan.
Disclosure
The authors report no conflicts of interest in this work.
References
Das JK, Soibam R, Tiwary B, Magdalene D, Paul S, Bhuyan C. Orbital manifestations of Langerhans Cell Histiocytosis: a report of three cases. Oman J Ophthalmol. 2009;2(3):137–140. | ||
Herwig MC, Wojno T, Zhang Q, Grossniklaus HE. Langerhans cell histiocytosis of the orbit: five clinicopathologic cases and review of the literature. Survey Ophthalmol. 2013;58(4):330–340. | ||
Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–1923. | ||
McElligott J, McMichael A, Sangüeza OP, Anthony E, Rose D, McLean TW. Spontaneous regression of langerhans cell histiocytosis in a neonate with multiple bony lesions. J Pediatr Hematol Oncol. 2008;30(1):85–86. | ||
Moore A, Pritchard J, Taylor D. Histiocytosis X: an ophthalmological review. Br J Ophthalmol. 1985;69(1):7–14. | ||
Hamre M, Hedberg J, Buckley J, et al. Langerhans cell histiocytosis: an exploratory epidemiologic study of 177 cases. Med Pediatr Oncol. 1997;28(2):92–97. | ||
Laman JD, Leenen PJ, Annels NE, Hogendoorn PC, Egeler RM. Langerhans-cell histiocytosis ‘insight into DC biology.’ Trends Immunol. 2003;24(4):190–196. | ||
Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000–2004. Pediatr Blood Cancer. 2008;51(1):71–75. | ||
Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child. 2009;94(5):376–380. | ||
Stålemark H, Laurencikas E, Karis J, Gavhed D, Fadeel B, Henter JI. Incidence of Langerhans cell histiocytosis in children: a population‐based study. Pediatr Blood Cancer. 2008;51(1):76–81. | ||
Alston RD, Tatevossian R, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer. 2007;48(5):555–560. | ||
Jubran RF, Marachelian A, Dorey F, Malogolowkin M. Predictors of outcome in children with Langerhans cell histiocytosis. Pediatr Blood Cancer. 2005;45(1):37–42. | ||
Kim IT, Lee SM. Choroidal Langerhans’ cell histiocytosis. Acta Ophthalmol Scand. 2000;78(1):97–100. | ||
Tsai JH, Galaydh F, Ching SS. Anterior uveitis and iris nodules that are associated with Langerhans cell histiocytosis. Am J Ophthalmol. 2005;140(6):1143–1145. | ||
Margo CE, Goldman DR. Langerhans cell histiocytosis. Survey Ophthalmol. 2008;53(4):332–358. | ||
Stull M, Kransdorf M, Devaney K. Langerhans cell histiocytosis of bone. Radiographics. 1992;12(4):801–823. | ||
Konrad H, Clark BJ, Rose GE. Lymphocytic, plasmacytic, histiocytic, and hematopoietic tumors of the orbit. In: Tasman W, Jaeger EA, editors. Duane’s Clinical Ophthalmology. Philadelphia, PA: Lippincott Williams & Wilkins; 2000:30–32. | ||
Greaves WO, Verma S, Patel KP, et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagnostics. 2013;15(2):220–226. | ||
Haroche J, Cohen-Aubart F, Rollins BJ, et al. Histiocytoses: emerging neoplasia behind inflammation. Lancet Oncol. 2017;18(2):e113–e125. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.