Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 19
Lactate Metabolism and Protein Lactylation in Diabetic Kidney Disease: A Narrative Review
Authors Sun L, Yang Y ![]() , Pan W
, Pan W ![]() , Zhang C
, Zhang C ![]() , Lu J, Guo S, Zhu Y, Zhu M
, Lu J, Guo S, Zhu Y, Zhu M ![]()
Received 7 February 2026
Accepted for publication 14 May 2026
Published 29 May 2026 Volume 2026:19 599060
DOI https://doi.org/10.2147/DMSO.S599060
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Lele Sun,1,2,* Yuanyuan Yang,1,2,* Wenyi Pan,1,2,* Chenghua Zhang,2 Jiawei Lu,2 Shaowen Guo,1,2 Ying Zhu,3 Meifeng Zhu2
1Department of Traditional Chinese Medicine, Nanjing University of Chinese Medicine, Nanjing, Jiangsu, People’s Republic of China; 2Department of Nephrology, Changzhou Affiliated Hospital of Nanjing University of Chinese Medicine, Changzhou, Jiangsu, People’s Republic of China; 3Department of Nephrology, Changshu Affiliated Hospital of Nanjing University of Chinese Medicine, Changshu, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Meifeng Zhu, Department of Nephrology, Changzhou Hospital Affiliated to Nanjing University of Chinese Medicine, No. 25 Heping North Road, Tianning District, Changzhou, Jiangsu, 213003, People’s Republic of China, Email [email protected] Ying Zhu, Department of Nephrology, Changshu Hospital Affiliated to Nanjing University of Chinese Medicine, No. 6 Huanghe Road, Suzhou, Jiangsu, People’s Republic of China, Email [email protected]
Abstract: Diabetic Kidney Disease (DKD) is one of the common and serious microvascular complications of diabetes, and the pathogenesis is complex and incompletely understood. Recent studies have shown that lactylation, as a novel form of protein post-translational modification, has been increasingly involved in the progression of DKD through the specific binding of lactate molecules to protein amino acid residues, particularly involving important pathological processes such as inflammatory regulation, cell death-related signaling pathways and fibrotic remodeling. This review summarizes current evidence on abnormal lactate metabolism in DKD and the mechanisms by which lactate-driven lactylation regulates signaling pathways associated with kidney injury. It further evaluates the potential of lactate as a clinical biomarker and discusses intervention strategies targeting lactate metabolism as well as enzymes involved in lactylation and delactylation. These findings provide new insights and future research directions for the prevention and treatment of DKD.
Plain Language Summary: Diabetic kidney disease is a major cause of chronic kidney failure in people with diabetes, yet current treatments do not fully prevent disease progression. Understanding the biological mechanisms that drive kidney damage remains essential for developing better therapies. This review examines how changes in cellular metabolism contribute to diabetic kidney disease, with a focus on lactate, a key product of glucose metabolism. Beyond its traditional role in energy production, lactate can modify proteins through a process known as lactylation, which influences gene activity and cellular behavior. Recent studies suggest that abnormal lactate metabolism and protein lactylation may promote inflammation, tissue scarring, and cell dysfunction in the diabetic kidney. We summarize current evidence from experimental and clinical studies linking lactate-related pathways to kidney injury and disease progression. We also discuss the limitations of existing research and highlight important gaps that require further investigation. By clarifying how metabolic signals influence kidney pathology, this review provides insight into potential therapeutic targets and supports future research aimed at improving outcomes for people with diabetic kidney disease.
Keywords: diabetic kidney disease, lactate, lactate metabolism, glycolysis, post-translational modification, protein lactylation, renal fibrosis
Introduction
Diabetic kidney disease is a serious complication of diabetes, which often develops into end-stage kidney disease, thus significantly reducing the life expectancy of patients.1 According to recent estimates from the International Diabetes Federation (IDF), the global prevalence of diabetes is approximately 583 million and is expected to rise to 853 million by 2050, with the majority of cases occurring in low- and middle-income countries. Among these individuals, approximately 40% may develop diabetic kidney disease, which is recognized as the leading cause of chronic kidney disease (CKD) worldwide.2,3 The pathogenesis of DKD is multifactorial, and it is generally believed that it involves hemodynamic changes, metabolic disturbances and signaling pathway disorders of the renin-angiotensin-aldosterone system.4 Although mineralocorticoid receptor antagonists (MRAs), RAAS inhibitors, and glucagon-like peptide-1 receptor (GLP-1R) agonists have been incorporated into standard clinical practice, their efficacy in delaying the progression of DKD remains limited, and they are unable to prevent progression to end-stage renal disease (ESRD).5 Therefore, there is an urgent need to explore novel therapeutic targets and intervention strategies. In 2019, Zhang et al identified a characteristic mass shift on lysine residues of proteolytic peptides using mass spectrometry, thereby revealing lysine lactylation as a novel protein post-translational modification (PTM), a finding that has expanded our understanding of epigenetic regulation.6 Subsequent studies have shown that lactate induces histone lactylation which accelerates epithelial-mesenchymal transition (EMT) in DKD, thus linking lactylation to DKD pathogenesis.7 Moreover, other epigenetic mechanisms, including DNA methylation, post-translational modifications (such as acetylation, methylation, ubiquitination, and phosphorylation), and non-coding RNAs, have also been implicated in DKD-related renal injury (Table 1).8–13 In addition, these epigenetic modifications exhibit dynamic crosstalk. For example, histone lactylation and acetylation competitively target lysine residues, with their relative levels determined by the intracellular balance of lactate and acetyl-CoA.14
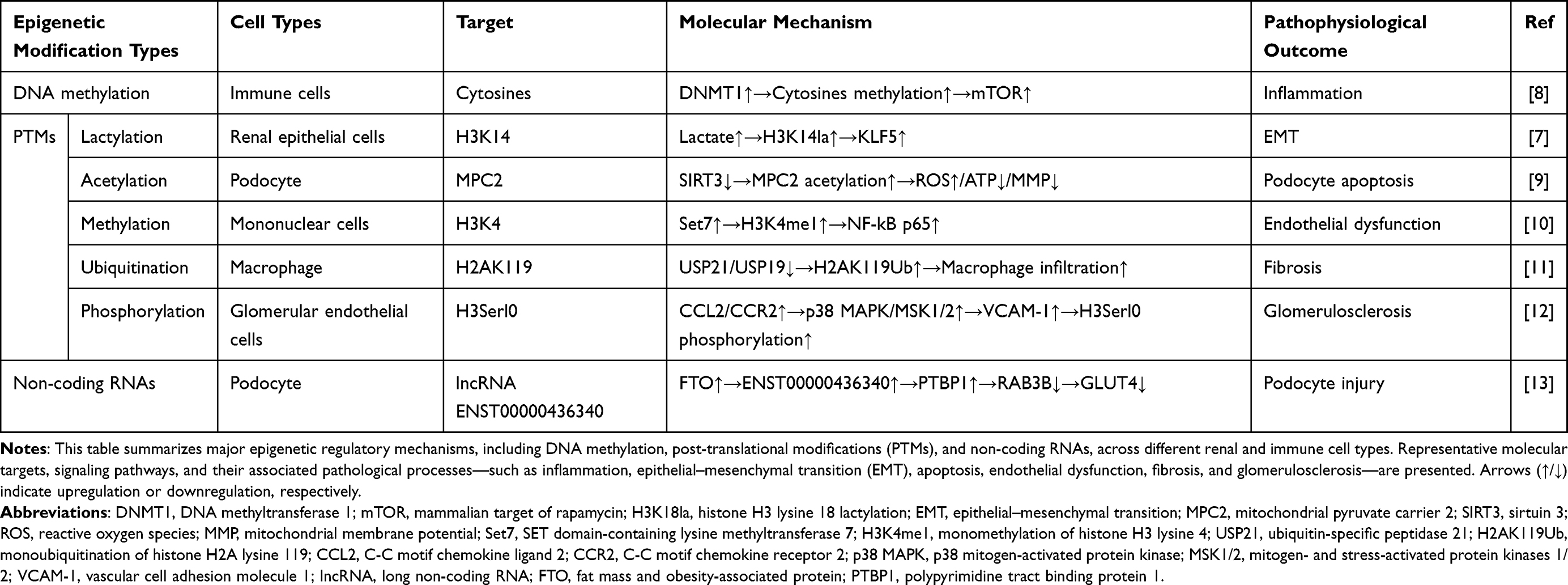 |
Table 1 Epigenetic Modifications Involved in the Pathogenesis of DKD |
As an emerging epigenetic modification, lactylation provides a mechanistic framework linking aerobic glycolysis-driven lactate accumulation to renal injury in DKD. Accumulating evidence has implicated lactylation not only in the development of diabetes but also in the progression of associated complications, including diabetic kidney disease and diabetic retinopathy.15 As a metabolite-derived epigenetic modification driven by intracellular lactate accumulation, lactylation links cellular metabolic states to gene regulation. Epigenetic modifications can be stably maintained as a form of cellular memory and exert long-term effects in DKD, thereby constituting a key pathological basis of the “metabolic memory” phenomenon.16 Metabolic memory refers to the persistence of renal injury in DKD despite subsequent normalization of glycemic levels.17 Epigenetic regulation contributes to this process by modulating chromatin accessibility and sustaining the activation of genes involved in inflammation, fibrosis, and apoptosis.18,19 In this context, lactylation may serve as a mechanistic link between transient metabolic disturbances and persistent transcriptional reprogramming. Moreover, a potential positive feedback loop between lactate accumulation and lactylation may further amplify lactylation-dependent transcriptional programs. Collectively, lactylation may function as a molecular bridge connecting metabolic reprogramming with epigenetic memory, thereby contributing to the sustained progression of renal injury. Emerging evidence suggests that modulation of lactate metabolism and lactylation-related pathways may represent a potential therapeutic strategy. Pharmacological interventions targeting key regulators of lactate production, transport, and lactylation may influence disease progression. However, its clinical translation remains limited due to the lack of standardized detection methods and targeted therapeutic strategies, with current evidence largely confined to preclinical studies. Addressing these gaps will be essential to establish clinical relevance and unlock the potential of lactylation for precision diagnostics and therapeutics in DKD. Accordingly, this review summarizes the current evidence implicating protein lactylation in the pathogenesis of DKD and highlights recent advances in lactylation-based therapeutic strategies and detection approaches.
Methodology
Literature for this review was identified through searches of major scientific databases, including PubMed, Web of Science, Scopus, and Google Scholar. The search strategy combined keywords related to lactylation, diabetic kidney disease, epigenetic regulation, metabolic reprogramming, and lactate using Boolean operators (AND/OR), as exemplified by the following query: (diabetic kidney disease OR DKD OR diabetic nephropathy OR DN) AND (lactylation OR histone lactylation OR protein lactylation) AND (epigenetic regulation OR epigenetics OR histone modification) AND (metabolic reprogramming OR glycolysis) AND (lactate OR lactate metabolism). The search was limited to articles published in English up to March 2026. Relevant studies were screened based on their relevance to the topic and their contribution to understanding the role of lactylation in DKD. Given the narrative nature of this review, study selection was not conducted according to a predefined systematic protocol, which may introduce potential selection bias.
Lactate Metabolism and Protein Lactylation
Lactate metabolism represents a central metabolic process linking glucose utilization to cellular energy balance and signaling pathways. In the human body, lactate exists in the form of two stereoisomers, namely L-lactate and D-lactate, of which L-lactate is the main form of glycolysis.20 The traditional view is that lactate is only a metabolic waste of anaerobic glycolysis. However, with the introduction of the Warburg effect, the physiological function of lactate has been reconsidered. The Warburg effect was first found in tumor cells. Even under aerobic conditions, cells can produce lactate through glycolysis.21 Lactate is now increasingly considered to be involved in the pathological processes of various diseases, including inflammation, tumor progression, sepsis, and wound healing.22 In 2019, Zhang et al first reported the existence of lysine lactylation as a novel PTM of proteins.6 Lactylation is a new form of PTM derived from lactyl-CoA, which is characterized by the covalent connection of lactate and lysine residues in proteins.23 Although the Warburg effect does not directly regulate lactylation, the increase in glycolytic flux associated with this metabolic state will produce a large amount of lactate, and lactate can be used as a substrate for histone lactylation (Figure 1). Through this mechanism, lactate-associated lysine lactylation is believed to link metabolic reprogramming with epigenetic regulation, which represents an emerging focus of metabolic–epigenetic research.24 Studies have found that lysine lactylation is widely involved in the pathogenesis of a variety of kidney diseases, including renal cell carcinoma, acute kidney injury, diabetic kidney disease, and renal fibrosis.25
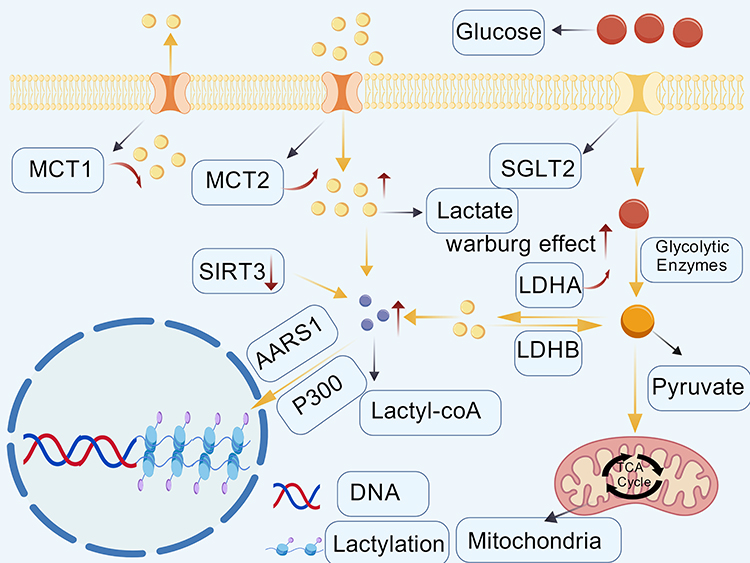 |
Figure 1 Schematic overview of lactate metabolism and protein lactylation. This schematic summarizes current understanding of the relationships among glucose metabolism, lactate production, transport, and protein lactylation. Enhanced glycolytic activity is shown to be associated with increased pyruvate-to-lactate conversion mediated by LDHA and LDHB, reflecting metabolic features commonly described as the Warburg effect. Lactate transport via MCT1 and MCT2, as well as its potential contribution to lactyl-CoA generation, are illustrated. Enzymes reported to participate in the regulation of protein lactylation, including the lactyltransferases p300 and AARS1, as well as the delactylase SIRT3, are indicated to highlight proposed regulatory nodes of this modification. Arrows represent proposed metabolic routes or reported associations rather than direct causal relationships.The figure was independently assembled by the authors using original graphical elements together with licensed BioGDP components. Created with BioGDP.com.26 |
Altered Lactate Metabolism in Diabetic Kidney Disease
The kidney plays an essential role in systemic lactate homeostasis, and alterations in renal lactate metabolism have been increasingly recognized in diabetic kidney disease. The kidneys are the second most important organ for lactate metabolism after the liver. The clearance of lactate by the kidneys depends on the functional division and coordination between the renal cortex and the renal medulla. The renal medulla mainly produces lactate through glycolysis, while the renal cortex efficiently absorbs and oxidizes lactate to meet energy requirements. Both are involved in regulating the glucose-lactate cycle.27 Under physiological conditions, lactate is freely filtered by the glomerulus and is almost completely reabsorbed in the proximal tubules, resulting in minimal urinary excretion and contributing to the maintenance of metabolic and acid–base homeostasis.28 In DKD, persistent hyperglycemia is accompanied by changes in cell metabolism, which is characterized by increased glycolysis dependence and reduced mitochondrial oxidative phosphorylation. These metabolic changes occur simultaneously with the increase in lactate levels in the renal tissue and are related to the progressive impairment of renal function.29 In the kidneys, the maintenance of lactate homeostasis depends on the synergy of lactate dehydrogenase (LDH), pyruvate dehydrogenase (PDH) and monocarboxylate transporters (MCTs) (Figure 2). The three together form a dynamic balance system, which regulates the concentration and distribution of lactate in the kidney.30 The production and utilization of lactate in renal tissue are mainly regulated by LDH and PDH. LDH is a core enzyme in glycolysis, which directly affects the production of lactate.31 Within the nephron, lactate dehydrogenase A (LDHA) is mainly located in proximal tubular cells, where it promotes the reduction of pyruvate to lactate. In contrast, lactate dehydrogenase B (LDHB) is higher in the distal segments of the nephron and participates in the reconversion of lactate into pyruvate. The two enzymes jointly maintain the metabolism of lactate.32 Evidence from kidney injury research shows that the concentration of lactate dehydrogenase in plasma and urine is increased. Therefore, lactate dehydrogenase can be used as a potential indicator to reflect the damage of specific nephron segments. In addition, persistent hyperglycemia can activate the polyol metabolic pathway, upregulate the expression of lactate dehydrogenase, and promote the accumulation of lactate in the renal tissue.33 PDH reduces lactate levels by facilitating the entry of pyruvate into mitochondrial oxidative metabolism, which is a key node for regulating cell energy metabolism and lactate homeostasis.34 Inhibiting PDH activity will enhance aerobic glycolysis, increase lactate generation, and may impair lactate clearance, thereby contributing to lactate accumulation in tissues.35,36 The transmembrane transport of lactate is another important mechanism to maintain its steady-state distribution, which is mainly mediated by the monocarboxylic acid transport protein family. In the kidney, MCT1 is mainly located in proximal tubular cells, while MCT2 is mainly expressed in the thick ascending branch and distal tubule, which participates in lactate transport together.37 It is reported that changes in MCT expression will affect glycolysis metabolism and cellular redox homeostasis.38 Under the condition of DKD, proximal renal tubular cells showed a decrease in MCT1 expression and an increase in MCT2 expression, which is associated with an increase in intracellular lactate levels.39 When lactate accumulation occurs in cells, the activity of MCT will be inhibited, thus hindering the transport of lactate from the inside of the cell to the outside of the cell, resulting in the further accumulation of lactate in the cell.40
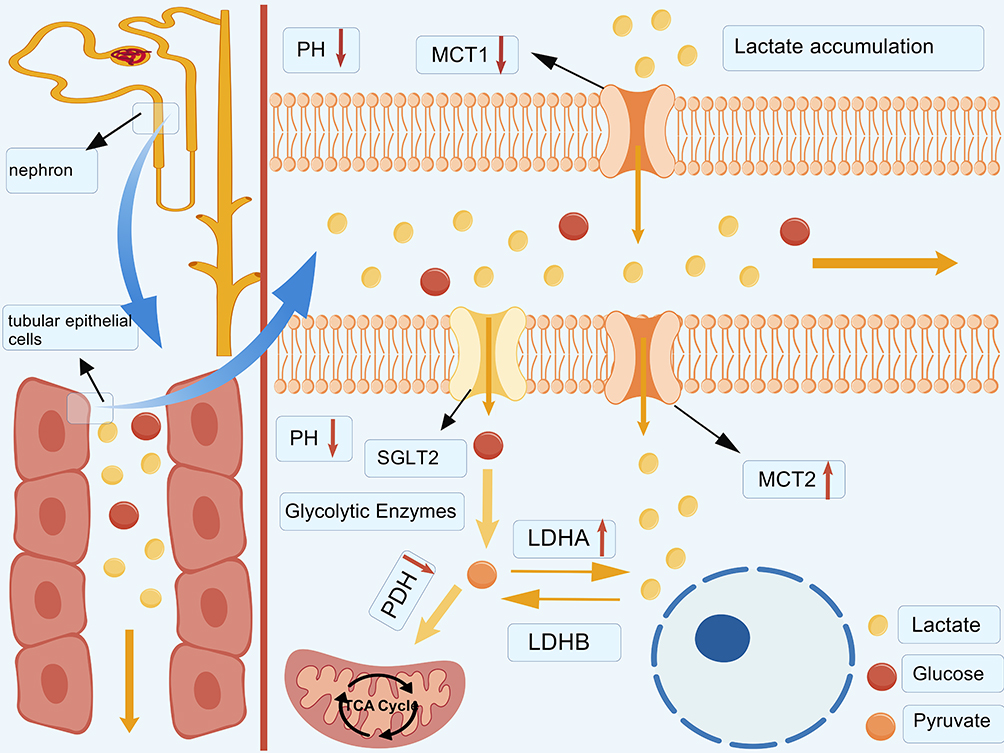 |
Figure 2 A schematic diagram of lactate metabolism in renal tubular epithelial cells under diabetic kidney disease. In diabetic kidney disease, glucose uptake by renal tubular epithelial cells via sodium–glucose cotransporter 2 (SGLT2) is associated with enhanced glycolytic activity and increased pyruvate generation. Pyruvate is preferentially converted to lactate by upregulated LDHA, while reduced activity of LDHB and PDH has been reported in association with further lactate accumulation. Intracellular lactate may be transported across the plasma membrane via monocarboxylic acid transporters, with altered MCT1- and MCT2-mediated fluxes described in diabetic kidney disease. Excessive lactate accumulation has been associated with changes in the local microenvironment, including altered pH, as well as with tubular epithelial cell injury and impaired renal function. Arrows indicate proposed metabolic routes or reported associations rather than direct causal relationships.This diagram was independently created by the authors using original graphical elements together with licensed BioGDP components. Created with BioGDP.com.26 |
Enzymatic Regulation of Protein Lactylation
The dynamic control of histone lactylation is achieved through the coordinated actions of three functional classes of enzymes, conventionally referred to as writers, erasers, and readers.41 Writers and erasers are responsible for catalyzing the installation and removal of lactyl groups on lysine residues of histones. Through these reversible modifications, they influence chromatin organization and subsequently regulate gene transcription. In contrast, reader proteins recognize lactylated lysine sites and facilitate downstream transcriptional responses. It is reported that acetyltransferase p300 can directly catalyze histone lactylation.6 Emerging evidence further suggests that these regulatory enzymes may exhibit cell-specific functions within the kidney, particularly in renal tubular epithelial cells, where metabolic reprogramming and lactate accumulation are prominent features of kidney injury. In sepsis-associated acute kidney injury, lactate induces H3K18la through the acetyltransferase p300. The elevated H3K18la subsequently promotes the transcription of sphingosine kinase 1 (SPHK1), which in turn mediates the phosphorylation of sirtuin 1 (SIRT1) and accelerates its degradation. The reduction of SIRT1 further enhances H3K18la accumulation, forming a positive feedback regulatory loop that ultimately drives tubular epithelial cell death. These findings reveal that p300 and SIRT1 cooperatively participate in tubular epithelial cell injury.42 In addition, alanyl-tRNA synthetase 1 (AARS1) has been identified as a lactyltransferase, which can directly catalyze histone lactylation, and its upregulation has been associated with aggravated renal dysfunction in DKD models.43 Recent studies further indicate that AARS1-mediated lactylation occurs predominantly in renal epithelial cells, where AARS1 mediates lactylation of STAT3 and p65, thereby regulating abnormal proliferation and fibrotic processes in renal tubular epithelial cells and ultimately promoting the progression of autosomal dominant polycystic kidney disease.44 Erasers, including histone deacetylases 1–3 (HDAC1–3) and sirtuins 1–3 (SIRT1–3), have been reported to remove lactyl modifications and dynamically regulate lactylation levels.45 Among these enzymes, the mitochondrial deacylase SIRT3 appears to play a particularly important role in proximal tubular epithelial cells, where mitochondrial metabolism is tightly coupled to lactate production and fibrotic signaling. In DKD, the impairment of SIRT3 function is associated with abnormal lactate accumulation and enhanced fibrotic reaction.46 In DKD, downregulation of SIRT3 in renal tubular cells promotes lactate accumulation, which subsequently enhances RUNX1 transcription through H4K12la, thereby inducing fibrotic responses in tubular epithelial cells.47 Furthermore, SIRT3 contributes to the maintenance of mitochondrial metabolic homeostasis in renal tubular epithelial cells by deacetylating Lys385 of pyruvate dehydrogenase E1α (PDHE1α), thereby preventing mitochondrial protein hyperacetylation, limiting metabolic reprogramming, and suppressing renal fibrosis.48 Collectively, these findings suggest that enzymes regulating protein lactylation exert cell-type–specific effects within the kidney and modulate renal cell injury by either delaying or promoting lactylation at specific target sites.
Enzymes involved in lactylation also play critical roles in other post-translational modifications, particularly acetylation, highlighting their dual regulatory functions.49 For example, p300 serves as a writer protein for lactylation and catalyzes lysine acetylation through its intrinsic HAT (histone acetyltransferase) domain, thereby modulating both modifications in response to the concentrations of various acyl-CoAs.50,51 HDAC1–3 and SIRT1–3 have been identified as dual-function deacylases that remove both lysine acetylation and lactylation.SIRT1–3 exhibit broad-spectrum deacylase activity, enabling the removal of multiple acyl modifications.52 It was found that SIRT1 is predominantly localized to the nucleus and cytoplasm, where it regulates both lactylation and acetylation, whereas SIRT3 is mainly localized to the mitochondria and modulates the same modifications on mitochondrial proteins.53 Similarly, HDAC1–3 function as both deacetylases and delactylases in mammals.54 Overall, these dual-function enzymes couple lactate metabolism to epigenetic regulation by utilizing shared catalytic domains and metabolite-dependent substrate selection, thereby coordinating acetylation and lactylation to modulate chromatin accessibility and transcription factor activity. Dynamic regulation of protein lactylation by these enzymes has been implicated in the modulation of protein function and transcriptional activity. However, the specific involvement of lactylation-related enzymes in the pathogenesis of DKD is not completely understood and requires further investigation.
Lactylation-Associated Pathological Mechanisms in Diabetic Kidney Disease
Inflammatory Activation
Inflammation is widely recognized as a central pathological feature of DKD.55 Lactate generated through glycolysis can modulate the release of immune cell activation, inflammatory mediators, and intracellular signaling pathways.56 Lactate metabolism has been shown to play a key role in driving macrophage polarization through mitochondrial pyruvate metabolism, a process central to the inflammatory response in DKD.57 In DKD, polarized macrophages accumulate in the glomerulus, amplifying renal inflammation and fibrosis through the release of pro-inflammatory cytokines.58,59 Lactate metabolism induces macrophage polarization by promoting H4K12 lactylation, further accelerating CKD progression.60 Meanwhile, macrophage polarization enhances glycolysis via the HIF-1α-HK2 signaling pathway, forming a positive feedback loop.61 These findings highlight a reciprocal interaction between lactate metabolism and macrophage polarization, linking metabolic reprogramming to inflammatory responses in DKD pathogenesis. Furthermore, epigenetic modifications such as histone acetylation and methylation regulate macrophage polarization and synergize with lactylation to drive macrophage-mediated inflammatory responses.62 Lactate produced by the metabolic conversion of pyruvate can activate the NLRP3 inflammasome through protein kinase R (PKR) phosphorylation. This activation pathway has been demonstrated to have pathological relevance and can significantly promote renal inflammatory responses and fibrosis.63,64 As research has progressed, the role of histone lactylation in the regulation of inflammatory responses has gradually been elucidated. In patients with infectious shock, high serum lactate levels and lactate-mediated histone H3 lysine 18 lactylation (H3K18la) can promote the release of procalcitonin (PCT) and a variety of inflammatory factors, suggesting that lactylation may be important in the regulation of systemic inflammation.65 The increase in H3K18la also facilitates binding to the promoters of NF-κB1 (p50) and RelA (p65), thus affecting NF-κB pathway activity and cytokine expression.66 Insulin-like growth factor binding protein 5 (IGFBP5) has been implicated in tissue inflammation by enhancing glycolysis and lactate production.67 Evaluation of renal tissues indicated that diabetic mice displayed substantially increased expression of endothelial–mesenchymal transition (EndoMT)–related markers along with IGFBP5 relative to control animals. In this context, loss of IGFBP5 was associated with a delayed course of renal injury progression. Further mechanistic studies found that IGFBP5 may promote lactate production by enhancing glycolytic activity, thereby inducing increased lactylation levels, among which H3K18la was the most prominent. Increased levels of H3K18la have been linked to enhanced activation of the NLRP3 inflammasome, accompanied by greater release of interleukin-1β, a process that appears to aggravate renal inflammation.68 Collectively, these observations suggest that histone lactylation is involved in DKD and may contribute to renal damage in association with heightened inflammatory responses. In addition, previous work has highlighted the glycolytic regulator PFKFB3 as a contributor to metabolic reprogramming in renal tubular cells. This shift is accompanied by increased lactate accumulation and elevated H4K12la, changes that coincide with transcriptional activation of NF-κB–related signaling and a heightened inflammatory state in the kidney.69 Lactate not only participates in the regulation of inflammation in DKD but also activates the NLRP3 inflammasome and the NF-κB pathway through histone lactylation, thereby promoting the release of inflammatory mediators and exacerbating inflammatory responses. This process links glycolytic metabolic reprogramming with renal inflammation and provides new insight into the interplay between metabolism and inflammation in DKD.
Cell Death
In the pathogenesis of DKD, programmed cell death of kidney cells induced by persistent hyperglycemic conditions is implicated in the occurrence and development of DKD.70 Under elevated glucose or pathological conditions, glucose metabolism in renal tubular cells is reprogrammed, tending towards the glycolysis pathway, leading to a large accumulation of lactate.71 This accumulated lactate can downregulate the expression of SIRT3 and AMP-activated protein kinase (AMPK) in renal tubular epithelial cells, thereby favoring the initiation of apoptotic processes.72 Lactate also exacerbates renal cell death by inducing protein lactylation. For example, lactate-associated histone lactylation is related to the activation of the RhoA/ROCK/NF-κB signaling pathway, which is associated with inflammatory responses and apoptosis.73 Moreover, lactate can promote abnormal mitochondrial fission by mediating lactylation of the mitochondrial fission-related protein Fis1, thereby inducing cell death and worsening renal dysfunction.74 ACSF2 is a ferroptosis-associated protein primarily localized in mitochondria in renal tubular cells. In DKD, enhanced glycolysis leads to lactate accumulation, which contributes to increased global lysine lactylation in renal tissues. Notably, lactylation of ACSF2 at lysine 182 (K182) has been shown to exacerbate mitochondrial injury. This modification promotes excessive accumulation of reactive oxygen species (ROS) and impairs mitochondrial function. The resulting oxidative stress may further drive ferroptosis, thereby contributing to tubular damage.75 In podocytes, lactate accumulation has been linked to the activation of mTORC1 signaling through lactylation of LARS1 at lysine 970, leading to impaired autophagy and increased apoptosis, thereby aggravating podocyte injury and proteinuria.76 Ferroptosis has also been implicated in DKD pathophysiology.77 Lactylation of proteins such as TRIM65 K206 has been reported to modulate the ubiquitination of iron response element-binding protein 2 (IREB2) and pyruvate dehydrogenase kinase 4 (PDK4), thereby facilitating ferroptotic responses and glycolytic dysfunction in renal tubular cells.78 In addition, AARS1 can induce H3K18la and STAT1, regulate the transcriptional expression of elongase-5 (ELOVL5), thus contributing to the process of ferroptosis and aggravating DKD-related renal dysfunction.43 Lactylation can also promote the expression of apoptosis-promoting genes by regulating gene transcription. Specifically, lactate from glycolysis can promote the lactylation of p53 and enhance its transcriptional activity, which further accelerates cell apoptosis.79 It is worth noting that the role of lactate-driven cell apoptosis is not limited to kidney disease, but has also been verified in other disease systems. For example, the increase in glycolysis reprogramming and the production of lactate in crystalline silicosis promotes the level of lactylation to drive the death of NLRP3-dependent cells, and inhibiting this reprogramming can significantly alleviate the occurrence of cell death.80 By modifying key proteins such as histones and mitochondrial factors, lactate has been implicated in the regulation of diverse programmed cell death processes, among them apoptosis, ferroptosis, and autophagy-related damage, changes that may contribute to ongoing renal parenchymal injury in DKD.
Fibrosis
Renal fibrosis is the core pathological feature of the progression of DKD. Its essence lies in the excessive deposition of the extracellular matrix, which eventually leads to renal failure.81 Aerobic glycolysis enhancement has been proven to promote collagen synthesis in fibroblasts and is closely linked to tissue fibrosis.82 As a prominent metabolite generated through glycolytic flux, lactate has been implicated in the activation of fibroblasts and the establishment of hypoxic, acidic microenvironments. Alongside these changes, lactate has been linked to enhanced transforming growth factor-β1 (TGF-β1) activity, a combination of effects that is thought to favor myofibroblast differentiation and drive fibrogenic responses.83–85 Lactate-induced protein lactylation has been implicated in EMT, a process associated with podocyte dysfunction and proteinuria in DKD.86 Increased lactate levels have been reported to correlate with the extent of podocyte EMT under hyperglycemic conditions.87 Experimental studies suggest that hyperglycemia-associated lactate accumulation promotes lactylation and EMT-related gene expression, which is associated with fibrotic remodeling and renal functional decline.88 Lactylation has also been reported to modulate transcription factors involved in EMT.89 In DKD, H3K14la has been linked to increased expression of the transcription factor KLF5 and enhanced EMT in renal epithelial cells, whereas inhibition of LDHA or KLF5 deficiency has been associated with reduced H3K14la levels and attenuation of fibrotic progression.7 In addition, H3K18la has been strongly associated with renal fibrosis in DKD. Downregulation of Glis1 weakens its interaction with the lactyl transferase KAT5, thus enhancing the level of H3K18la, promoting tubular epithelial cell senescence, and contributing to fibrosis.90 As a key glycolytic enzyme, the upregulation of pyruvate kinase M2 (PKM2) is linked to increased lactate production and H3K18la, and its expression level is correlated with the severity of fibrosis.91 During kidney stone development, increased galectin-3 (LGALS3) expression has been linked to PKM2 activation, accompanied by a shift toward enhanced glycolysis and greater lactate production. Accumulating lactate is associated with elevated H3K18la, which correlates with increased fibroblast growth factor receptor 4 (FGFR4) expression. Together, these changes may facilitate stone formation while also contributing to the progression of renal fibrosis.92 In chronic kidney disease, increased PKM2 activity is accompanied by lactate accumulation and higher levels of H3K18la, together with enhanced TGF-β1 transcription. In this setting, macrophages tend to adopt a profibrotic M2-like phenotype and participate in macrophage–fibroblast transition via Smad3 signaling, a pattern that parallels the development of renal fibrosis.93 Taken together, the available evidence supports an important involvement of H3K18la in processes underlying renal fibrotic remodeling. The development of renal fibrosis in DKD is closely related to disorders of lactate metabolism. Lactate-mediated protein lactylation regulates key pathways, including epithelial–mesenchymal transition, cell senescence, and fibrosis-related gene expression, thus activating fibrotic signaling pathways. Targeting lactate generation and its lactylation signaling pathways may provide new treatment strategies to slow down the progression of DKD fibrosis.
Lactate and Lactylation in DKD: Biomarkers and Therapeutic Implications
Lactate-Related Biomarkers
Lactate is an important metabolite, and its levels show significant changes in diabetic conditions.94 There is a clear positive correlation between the level of lactate and lactylation. Studies show that the increase in lactate levels in blood plasma, urine, and kidney tissue in diabetes is closely related to the decrease in glomerular filtration rate and proteinuria. Therefore, lactate can be used as a new biomarker to reflect the rate of decline in glomerular filtration and predict the risk of kidney disease.95,96 Lactate metabolism-related enzymes are also involved in disease progression and prognostic markers of DKD. In DKD patients, the expression of LDHA in the glomerulus and renal tubular epithelial cells is significantly increased, accompanied by a decrease in glomerular filtration rate (eGFR) and an increase in the expression of the fibrosis marker α-SMA.97 Elevated LDHA levels are not only an independent risk factor for the occurrence and development of DKD, but are also significantly associated with an increased risk of end-stage renal disease, increased cardiovascular mortality, and increased urinary albumin/creatinine ratio (ACR).98,99 It can be seen that the concentration of lactate and the expression of key metabolic enzymes are expected to be used as new biomarkers for the early diagnosis, disease assessment, progression prediction and monitoring of treatment response of DKD.
Detection and Clinical Potential of Protein Lactylation
Histone PTMs represent a central regulatory mechanism governing chromatin dynamics and gene transcription, and dysregulation of these modifications has been closely associated with the pathogenesis and progression of numerous human diseases. In 2019, the research group led by Yingming Zhao first identified lysine lactylation as a novel histone modification using a specific anti-H3K18la antibody combined with chromatin immunoprecipitation sequencing (ChIP-seq) and RNA sequencing analyses.6 Traditional methods for detecting histone PTMs, including Western blotting and ChIP-seq, rely largely on antibody-based recognition and are limited by relatively low throughput. Advances in high-resolution mass spectrometry have transformed the study of histone modifications, and mass spectrometry-based proteomics is now considered a powerful approach for identifying lactylation sites and performing global lactylome profiling.100
Although standardized clinical assays for lactylation detection remain lacking, several studies have begun to explore its detection in human tissues using antibody-based approaches. For example, immunohistochemical and immunoblot analyses using a monoclonal antibody against H4K5la revealed significantly elevated H4K5la levels in breast cancer tissues, with expression positively correlated with serum lactate and carcinoembryonic antigen levels.101 In parallel, label-free quantitative lactylome profiling strategies have been developed to systematically characterize lysine lactylation and investigate its metabolic regulation in diseases such as diabetes.102 Despite these advances, clinical detection of protein lactylation remains at an early stage. The development of high-sensitivity mass spectrometry platforms and highly specific anti-lactyllysine antibodies, together with optimized antibody-based detection protocols, will therefore be essential for establishing standardized assays and advancing lactylation as a clinically relevant biomarker.
Potential Therapeutic Strategies in DKD
With the deepening of the understanding of lactate metabolism and lactylation in DKD, the treatment strategies for these pathways have attracted increasing attention. Regulation of lactate production, related enzymes, and transport processes may provide potential opportunities for mechanism-based therapeutic intervention. Lactate production is closely associated with glomerular filtration, renal tubular sodium reabsorption, and urine flow. Loop diuretics can reduce lactate production by inhibiting the reabsorption of sodium in renal tubules.27 A clinical study involving 39 patients with DKD demonstrated that Sodium-glucose cotransporter 2 (SGLT2) inhibition reduced lactate production and restored oxidative phosphorylation in human proximal tubular cells.39 However, SGLT2 inhibitor use is associated with an increased risk of adverse events, particularly urinary tract infections, volume depletion, ketoacidosis, and fractures.103 LDHA is a key enzyme involved in lactate production. Modulating LDHA activity has been shown to limit lactate generation, thereby reducing the severity of renal injury. In the DKD mouse model, intervention with the LDHA inhibitor sodium oxalate (Oxa) can significantly reduce the serum lactate levels in mice and inhibit histone lactylation.88 Furthermore, research has also shown that angiotensin-receptor blocker (ARB) can reduce lactate accumulation by inhibiting the expression of LDHA in proximal renal tubules, thus exerting a protective role in the kidney.104 Nevertheless, inhibition of LDH triggers metabolic reprogramming that increases fatty acid catabolism. This metabolic shift is coupled with ROS accumulation and ATP depletion, leading to exacerbated DNA damage and impaired DNA repair. Collectively, these effects ultimately enhance cellular susceptibility to oxidative stress and promote cell death.105–107 Modulation of MCT activity has been shown to influence renal lactate handling. For example, the MCT inhibitor syrosingopine can block the transport of lactate, the end product of glycolysis, thereby attenuating tubular injury and fibrosis in unilateral ischemia–reperfusion injury (IRI) mice.108 Nevertheless, extensive inhibition of MCT expression may result in non-specific adverse effects. Clinical administration of the MCT1 inhibitor AZD3965 has been associated with adverse events, including retinopathy, cardiotoxicity, fatigue, gastrointestinal toxicities, and metabolic acidosis.109 Therefore, the development of highly selective MCT-targeted inhibitors is of great research significance. Lactate exerts diverse functions in metabolism. It acts as a critical glycolytic intermediate supplying the TCA cycle, regulates immune responses, and serves as a signaling molecule. It modulates cellular functions through lactate shuttling and receptor-mediated pathways. Accordingly, widespread inhibition of lactate generation or metabolism may disrupt these processes and lead to metabolic dysregulation.110–112
Current research indicates that therapeutic modulation of lactylation may be achieved through pharmacological intervention targeting lactylation “writers.” A-485 is a selective inhibitor of p300 that binds to its catalytic site and competes with acetyl-CoA, thereby suppressing histone modifications.113 Moreover, “erasers” have also been shown to regulate lactylation dynamics. Nicotinamide adenine dinucleotide (NAD⁺) regulates the deacylase activity of the sirtuin family.114 Honokiol (HKL) functions as a SIRT3 activator that markedly suppresses Cyclin E2 (CCNE2) lactylation through SIRT3 activation, ultimately driving hepatocellular carcinoma progression.115 Although no therapeutic strategy currently exists that specifically targets individual lysine lactylation sites in DKD, recent studies have demonstrated that genetic code expansion enables the site-specific incorporation of lysine lactylation, thereby laying the groundwork for the future development of targeted inhibitory strategies against this modification.116 Notably, there are currently no clinically available therapeutic strategies that directly and specifically target protein lactylation at defined lysine residues in DKD. Although pharmacological modulation of lactate metabolism may indirectly influence lactylation levels, such approaches lack specificity and may disrupt physiological metabolic homeostasis. Emerging evidence suggests that enzymes involved in lactylation dynamics, including lactylation “writers” and “erasers”, may represent potential targets for more selective intervention. However, the development of such strategies remains in its early stages, and their feasibility, specificity, and safety in the context of DKD require further investigation. In particular, whether targeting these enzymes can achieve site-specific modulation of lactylation without affecting other PTMs remains an important challenge.
Discussion
As a narrative review, this study may be subject to potential selection bias, and future systematic analyses are warranted to provide a more comprehensive and unbiased synthesis of the available evidence. This review synthesizes current evidence on the critical involvement of lactate metabolism and lactylation in the pathogenesis of DKD, and explores their promising implications for clinical diagnosis and treatment. Traditionally, lactate has been regarded as a metabolic end product. However, the recognition of the Warburg effect has redefined lactate as an active metabolic regulator, thereby expanding its functional role in cellular processes. Metabolic reprogramming under hyperglycemic conditions promotes lactate accumulation, which acts as a key metabolic signal linking disordered energy metabolism to renal injury. Notably, lactate can serve as a substrate for histone lactylation, extending its role in epigenetic regulation. Emerging evidence further indicates that lactylation functions as a metabolite-driven signaling mechanism. It translates glycolysis-derived lactate accumulation into epigenetic regulation by inducing lysine lactylation on both histone and non-histone proteins. This modification regulates chromatin accessibility and facilitates the recruitment of transcriptional machinery, thereby modulating gene expression programs associated with disease progression. Histone lactylation is involved in multiple signaling pathways related to inflammation, cell death, and fibrosis, thereby contributing to the pathogenesis of DKD. In addition, lactylation of non-histone proteins further broadens the regulatory landscape of this modification in DKD. Collectively, these findings position lactylation as a central mediator that integrates glycolytic reprogramming with transcriptional control, ultimately contributing to DKD progression (Table 2 and Figure 3). Notably, while most studies consistently support a deleterious role of lactylation in DKD, the underlying mechanisms remain incompletely understood and may vary depending on cell type, disease stage, and experimental conditions. In addition to mechanistic insights, this review addresses the dysregulation of lactate metabolism in DKD, explores the potential of lactate as a biomarker and the clinical detection of lactylation, and summarizes experimental and clinical studies on targeted therapies for lactate metabolic disturbances, along with their associated adverse effects (Table 3). These observations further support the clinical relevance of lactate metabolism and lactylation in DKD.
 |
Figure 3 Overview of protein lactylation in diabetic kidney disease. This diagram illustrates reported pathways linking protein lactylation to pathological features of diabetic kidney disease, based on currently available evidence. Protein lactylation has been described in association with inflammatory activation, cell death, and renal fibrosis. Arrows represent reported associations rather than direct causal relationships, and upward arrows indicate relative increases under diabetic conditions. This diagram was independently created by the authors using original graphical elements together with licensed BioGDP components. Created with BioGDP.com.26 |
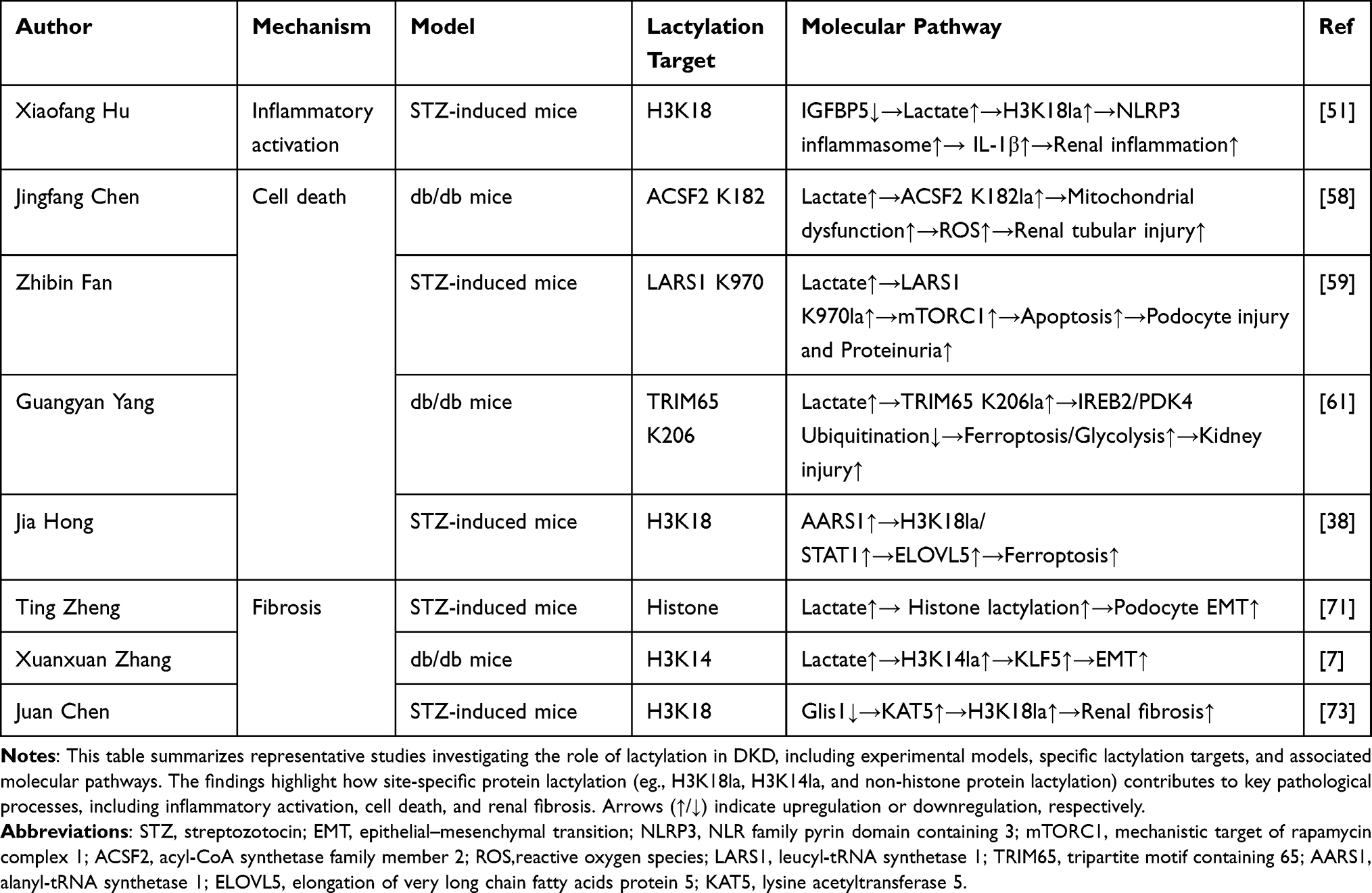 |
Table 2 Mechanistic Roles of Protein Lactylation in the Pathogenesis of DKD |
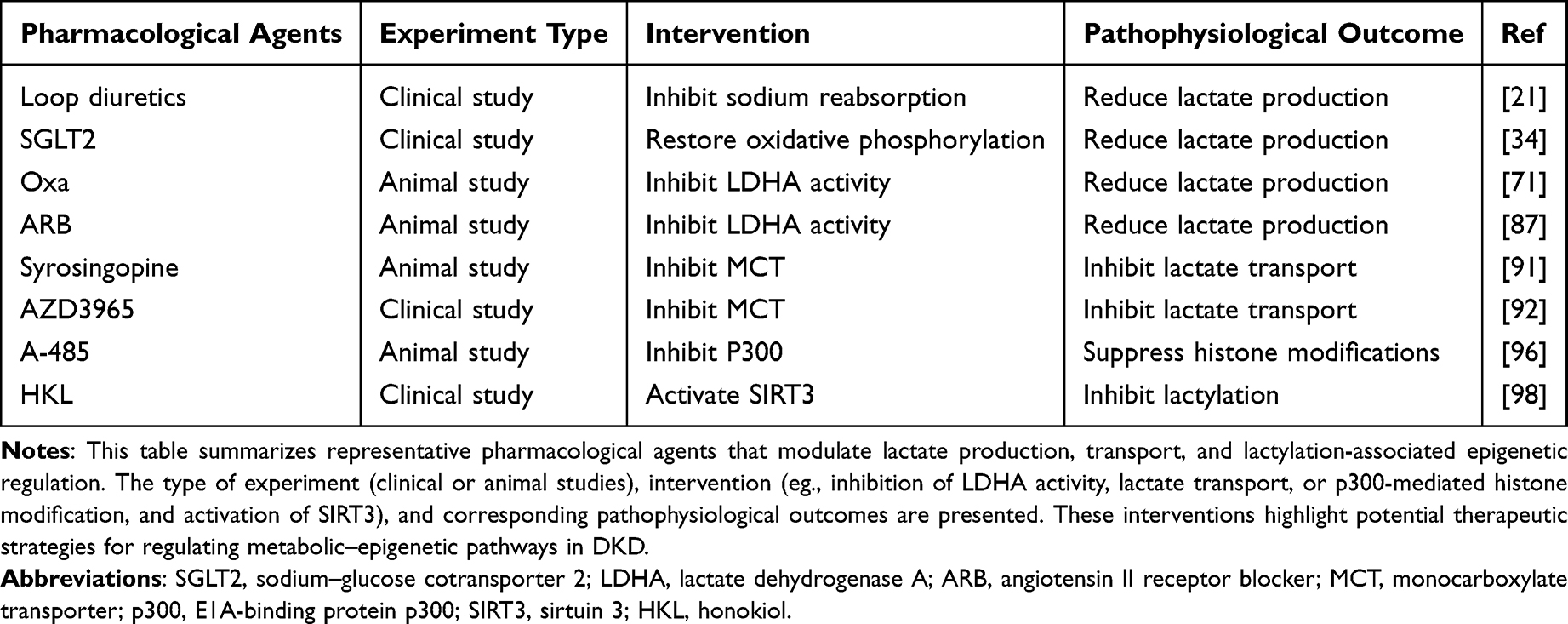 |
Table 3 Pharmacological Interventions Targeting Lactate Metabolism and Lactylation-Related Pathways |
Despite these advances, several limitations remain in the current research: First, most current evidence regarding protein lactylation in DKD is derived from experimental studies, including animal models and in vitro investigations. Whether the lactylation patterns identified in db/db mouse models accurately reflect those in human DKD remains uncertain, and direct clinical evidence from patients with DKD is still limited. Nevertheless, clinical studies in other diseases provide indirect support for the potential pathological relevance of lactylation. For instance, elevated levels of neutrophil H4K5la have been reported in patients with sepsis and have been proposed as a promising clinical biomarker.117 In ocular melanoma, histone lactylation has been shown to promote tumorigenesis by upregulating the expression of YTHDF2, which recognizes and degrades m6A-modified tumor-suppressor transcripts.118 Similarly, in hepatocellular carcinoma, H3K14la promotes chemoresistance by increasing NEDD4 expression, leading to PTEN ubiquitination and activation of the PI3K/Akt/mTOR signaling pathway.119 Furthermore, a clinical study titled “The mechanism of lactylation involved in the development of diabetes-related complications” (ChiCTR2500109301) is currently underway.120 Collectively, these findings highlight the potential mechanistic and clinical relevance of lactylation in human diseases and suggest that similar mechanisms may also exist in DKD. Therefore, large-scale studies involving human cohorts are urgently needed to determine whether the lactylation patterns observed in experimental models truly reflect the progression of DKD in patients. Secondly, current assessment of lactylation in clinical settings largely relies on lactate measurement, which serves only as an indirect and insufficient surrogate for actual lactylation levels. Although lactylation directed antibodies are available, they are primarily used in research settings and are limited by cost as well as suboptimal sensitivity and specificity. Furthermore, the limited availability of highly specific and well validated lactylation antibodies, together with the lack of high sensitivity mass spectrometry platforms, continues to hinder the clinical translation of lactylation as a reliable biomarker for diagnosis and prognosis. Finally, currently, no specific therapeutic agents directly targeting protein lactylation are available in clinical practice. Existing intervention strategies primarily focus on modulating lactate production and transport, as well as enzymes involved in lactate metabolism and lactylation. These include SGLT2 inhibitors, MCT inhibitors, LDHA inhibitors, and modulators of enzymes involved in lactylation and delactylation. Such approaches may indirectly influence lactylation levels by reducing intracellular lactate concentrations or by modulating enzymes associated with lactylation, thereby potentially attenuating the progression of kidney injury. However, these interventions may disrupt cellular energy homeostasis and lead to off-target metabolic effects, raising potential safety concerns.
With the continued advancement of research methodologies and deeper mechanistic investigations, studies on lactate-mediated protein lactylation are expected to further elucidate the pathogenesis of DKD and facilitate the development of novel therapeutic strategies.
Conclusion
In summary, lactate-mediated protein lactylation represents a novel link between metabolic reprogramming and epigenetic regulation in the pathogenesis of DKD. Although dysregulated lactate metabolism and enhanced lactylation have been associated with key pathological processes such as inflammation, cell death, and fibrosis, the exact mechanisms underlying these associations remain poorly understood and warrant further investigation. Future studies should focus on elucidating the interactions between lactylation and various pathogenic pathways in DKD, including inflammation, oxidative stress, metabolic dysregulation, and epigenetic modifications, to establish a more definitive link between lactylation and the progression of DKD. These insights highlight lactylation as a potential integrative mechanism linking metabolic and epigenetic dysregulation in DKD. Additionally, while clinical research on lactylation in diseases beyond diabetes and clinical trials exploring lactylation in diabetic complications have been initiated, most current evidence remains largely derived from preclinical studies, and the clinical application of lactylation as a biomarker in DKD is still limited due to challenges in detection methods. Therapeutic strategies targeting lactylation primarily focus on regulating lactate metabolism, transport, and enzymes involved in lactylation and delactylation to mitigate kidney damage caused by lactylation. However, these strategies may carry potential clinical side effects, and there is a lack of specific pharmacological agents targeting lactylation in DKD. Therefore, future efforts should prioritize clinical validation, the development of biomarkers, and the creation of specific drugs that target lactylation, which could ultimately translate these findings into novel diagnostic and therapeutic strategies for DKD.
Abbreviations
DKD, Diabetic kidney disease; CKD, chronic kidney disease; ESRD, end-stage renal disease; MRAs, mineralocorticoid receptor antagonists; GLP-1R, glucagon-like peptide-1 receptor; PTM, post-translational modification; EMT, epithelial-mesenchymal transition; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; MCTs, monocarboxylate transporters; LDHA, lactate dehydrogenase A; LDHB, lactate dehydrogenase B; SPHK1, sphingosine kinase 1; SIRT1, sirtuin 1; AARS1, alanyl-tRNA synthetase 1; HDAC1–3, deacetylases 1–3; SIRT1–3, sirtuins 1–3; PDHE1α, pyruvate dehydrogenase E1α; HAT, histone acetyltransferase; PKR, protein kinase R; H3K18la, H3 lysine 18 lactylation; PCT, procalcitonin; IGFBP5, insulin-like growth factor binding protein 5; EndoMT, endothelial–mesenchymal transition; AMPK, AMP-activated protein kinase; ROS, reactive oxygen species; IREB2, iron response element-binding protein 2; PDK4, pyruvate dehydrogenase kinase 4; ELOVL5, elongase-5; TGF-β1, transforming growth factor-β1; PKM2, pyruvate kinase M2; LGALS3, galectin-3; FGFR4, fibroblast growth factor receptor 4; eGFR, estimated glomerular filtration rate; ACR, albumin/creatinine ratio; ChIP-seq, chromatin immunoprecipitation sequencing; SGLT2, sodium-glucose cotransporter 2; Oxa, oxalate; ARB, angiotensin-receptor blocker; IRI, ischemia–reperfusion injury; NAD⁺, nicotinamide adenine dinucleotide; HKL, honokiol; CCNE2, cyclin E2.
Data Sharing Statement
No data was created or analysed in this study.
Author Contributions
Lele Sun: Conceptualization, Data curation, Writing – original draft, Visualization, Funding acquisition. Yuanyuan Yang: Conceptualization, Writing – original draft, Visualization, Writing – review and editing. Wenyi Pan: Data curation, Writing – original draft, Writing – review and editing. Chenghua Zhang: Supervision, Conceptualization, Writing – original draft, Funding acquisition, Writing – review and editing. Jiawei Lu: Data curation, Writing – review and editing, Supervision. Shaowen Guo: Supervision, Funding acquisition, Data curation, Writing – review and editing. Ying Zhu: Writing – original draft, Conceptualization, Writing – review and editing, Supervision. Meifeng Zhu: Conceptualization, Supervision, Funding acquisition, Writing – review and editing. Lele Sun, Yuanyuan Yang, and Wenyi Pan contributed equally to this work as co-first authors. All authors contributed to the article, gave final approval of the version to be published, agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Natural Science Foundation of Nanjing University of Chinese Medicine (Grant No. XZR2023018), the Jiangsu Province Graduate Research and Innovation Program (Grant Nos. SJCX25_0960 and SJCX25_0950), and the Science and Technology Project of the Changzhou Municipal Health Commission (Grant No. QN202325).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mogensen CE, Christensen CK, Vittinghus E. The Stages in Diabetic Renal Disease: with Emphasis on the Stage of Incipient Diabetic Nephropathy. Diabetes. 1983;32(Supplement_2):64–18. doi:10.2337/diab.32.2.S64
2. International Diabetes Federation. Brussels: international Diabetes Federation; Facts & figures. Available from: https://idf.org/about-diabetes/diabetes-facts-figures/.
3. Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: challenges, Progress, and Possibilities. Clin J Am Soc Nephrol. 2017;12(12):2032–2045. doi:10.2215/CJN.11491116
4. Lin YC, Chang YH, Yang SY, Wu KD, Chu TS. Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc. 2018;117(8):662–675. doi:10.1016/j.jfma.2018.02.007
5. Guo M, He F, Zhang C. Molecular Therapeutics for Diabetic Kidney Disease: an Update. Int J Mol Sci. 2024;25(18):10051. doi:10.3390/ijms251810051
6. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–580. doi:10.1038/s41586-019-1678-1
7. Zhang X, Chen J, Lin R, et al. Lactate drives epithelial-mesenchymal transition in diabetic kidney disease via the H3K14la/KLF5 pathway. Redox Biol. 2024;75:103246. doi:10.1016/j.redox.2024.103246
8. Chen G, Chen H, Ren S, et al. Aberrant DNA methylation of mTOR pathway genes promotes inflammatory activation of immune cells in diabetic kidney disease. Kidney Int. 2019;96(2):409–420. doi:10.1016/j.kint.2019.02.020
9. Feng J, Feng L, Yan Y, et al. SIRT3 deficiency aggravates mitochondrial metabolic disorder and podocyte injury in DKD via MPC2 acetylation. Cell Signal. 2025;135:112029. doi:10.1016/j.cellsig.2025.112029
10. Paneni F, Costantino S, Battista R, et al. Adverse Epigenetic Signatures by Histone Methyltransferase Set7 Contribute to Vascular Dysfunction in Patients With Type 2 Diabetes Mellitus. Circ Cardiovasc Genet. 2015;8(1):150–158. doi:10.1161/CIRCGENETICS.114.000671
11. Pandey A, Goru SK, Kadakol A, Malek V, Sharma N, Gaikwad AB. H2AK119 monoubiquitination regulates Angiotensin II receptor mediated macrophage infiltration and renal fibrosis in type 2 diabetic rats. Biochimie. 2016;131:68–76. doi:10.1016/j.biochi.2016.09.016
12. Alghamdi TA, Batchu SN, Hadden MJ, et al. Histone H3 Serine 10 Phosphorylation Facilitates Endothelial Activation in Diabetic Kidney Disease. Diabetes. 2018;67(12):2668–2681. doi:10.2337/db18-0124
13. Hu J, Wang Q, Fan X, et al. Long noncoding RNA ENST00000436340 promotes podocyte injury in diabetic kidney disease by facilitating the association of PTBP1 with RAB3B. Cell Death Dis. 2023;14(2):130. doi:10.1038/s41419-023-05658-7
14. Dai X, Lv X, Thompson EW, Ostrikov K. Histone lactylation: epigenetic mark of glycolytic switch. Trends Genet. 2022;38(2):124–127. doi:10.1016/j.tig.2021.09.009
15. Zhou M, Liu L, Sun Y, Wang X. Lactylation in diabetes mellitus and its complications: mechanisms of action and therapeutic potential - recent advances. Front Endocrinol. 2025;16:1710645. doi:10.3389/fendo.2025.1710645
16. Mimura I. Epigenetic memory in kidney diseases. Kidney Int. 2016;89(2):274–277. doi:10.1016/j.kint.2015.12.026
17. Ceriello A. Hypothesis: the “metabolic memory”, the new challenge of diabetes. Diabet Res Clin Pract. 2009;86:S2–S6. doi:10.1016/S0168-8227(09)70002-6
18. Zheng W, Guo J, Liu ZS. Effects of metabolic memory on inflammation and fibrosis associated with diabetic kidney disease: an epigenetic perspective. Clin Clin Epigenet. 2021;13(1):87. doi:10.1186/s13148-021-01079-5
19. Dong H, Sun Y, Nie L, et al. Metabolic memory: mechanisms and diseases. Signal Transduct Target Ther. 2024;9(1):38. doi:10.1038/s41392-024-01755-x
20. Connor H, Woods HF, Ledingham JGG. Comparison of the Kinetics and Utilisation of D(-)- and L(+)-Sodium Lactate in Normal Man. Ann Nutr Metab. 1983;27(6):481–487. doi:10.1159/000176723
21. Warburg O. The Metabolism of Carcinoma Cells. J Cancer Res. 1925;9(1):148–163. doi:10.1158/jcr.1925.148
22. Li X, Yang Y, Zhang B, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. 2022;7(1):305. doi:10.1038/s41392-022-01151-3
23. Izzo LT, Wellen KE. Histone lactylation links metabolism and gene regulation. Nature. 2019;574(7779):492–493. doi:10.1038/d41586-019-03122-1
24. Wan L, Zhang H, Liu J, et al. Lactylation and human disease. Expert Rev Mol Med. 2025;27:e10. doi:10.1017/erm.2025.3
25. Xu Y, Li X, Mao Z, Xue C. Protein lactylation in kidney diseases. Front Cell Dev Biol. 2025;13:1533175. doi:10.3389/fcell.2025.1533175
26. Jiang S, Li H, Zhang L, et al. Generic Diagramming Platform (GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 2025;53(D1):D1670–D1676. doi:10.1093/nar/gkae973
27. Bartlett S, Espinal J, Janssens P, Ross BD. The influence of renal function on lactate and glucose metabolism. Biochem J. 1984;219(1):73–78. doi:10.1042/bj2190073
28. Höhmann B, Frohnert PP, Kinne R, Baumann K, Papavassiliou F, Wagner M. Proximal tubular lactate transport in rat kidney: a micropuncture study. Kidney Int. 1974;5(4):261–270. doi:10.1038/ki.1974.35
29. Cheng Y, Guo L. Lactate metabolism and lactylation in kidney diseases: insights into mechanisms and therapeutic opportunities. Ren Fail. 2025;47(1):2469746. doi:10.1080/0886022X.2025.2469746
30. Guan K, Zhang Y, Guo S, Ning X, Sun S. The new insights of lactate in various kidney diseases. Expert Rev Mol Med. 2025;27:e36. doi:10.1017/erm.2025.10023
31. Jacobsen NO. The histochemical localization of lactic dehydrogenase isoenzymes in the rat nephron by means of an improved polyvinyl alcohol method. Histochemie. 1969;20(3):250–265. doi:10.1007/BF00306013
32. Yanase H, Takebe K, Nio-Kobayashi J, Takahashi-Iwanaga H, Iwanaga T. Cellular expression of a sodium-dependent monocarboxylate transporter (Slc5a8) and the MCT family in the mouse kidney. Histochem Cell Biol. 2008;130(5):957–966. doi:10.1007/s00418-008-0490-z
33. Williamson JR, Chang K, Frangos M, et al. Hyperglycemic Pseudohypoxia and Diabetic Complications. Diabetes. 1993;42(6):801–813. doi:10.2337/diab.42.6.801
34. Jha MK, Lee IK, Suk K. Metabolic reprogramming by the pyruvate dehydrogenase kinase–lactic acid axis: linking metabolism and diverse neuropathophysiologies. Neurosci Biobehav Rev. 2016;68:1–19. doi:10.1016/j.neubiorev.2016.05.006
35. Luengo A, Li Z, Gui DY, et al. Increased demand for NAD+ relative to ATP drives aerobic glycolysis. Mol Cell. 2021;81(4):691–707.e6. doi:10.1016/j.molcel.2020.12.012
36. Kamel KS, Oh MS, Halperin ML. L-lactic acidosis: pathophysiology,classification, and causes;emphasis on biochemical and metabolic basis. Kidney Int. 2020;97(1):75–88. doi:10.1016/j.kint.2019.08.023
37. Becker HM, Mohebbi N, Perna A, Ganapathy V, Capasso G, Wagner CA. Localization of members of MCT monocarboxylate transporter family Slc16 in the kidney and regulation during metabolic acidosis. Am J Physiol-Ren Physiol. 2010;299(1):F141–F154. doi:10.1152/ajprenal.00488.2009
38. Halestrap AP, Meredith D. The SLC16 gene family—from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflüg Arch. 2004;447(5):619–628. doi:10.1007/s00424-003-1067-2
39. Darshi M, Kugathasan L, Maity S, et al. Glycolytic lactate in diabetic kidney disease. JCI Insight. 2024;9(11):e168825. doi:10.1172/jci.insight.168825
40. Uhernik AL, Tucker C, Smith JP. Control of MCT1 function in cerebrovascular endothelial cells by intracellular pH. Brain Res. 2011;1376:10–22. doi:10.1016/j.brainres.2010.12.060
41. Gao X, Pang C, Fan Z, Wang Y, Duan Y, Zhan H. Regulation of newly identified lysine lactylation in cancer. Cancer Lett. 2024;587:216680. doi:10.1016/j.canlet.2024.216680
42. Huang Y, Zhao E, Zhao G, et al. H3K18 lactylation-mediated SPHK1-SIRT1 feedback loop accelerates pyroptosis of tubular epithelial cells in sepsis-associated acute kidney injury. Theranostics. 2026;16(9):4768–4786. doi:10.7150/thno.122991
43. Hong J, Xu H, Yu L, et al. AARS1-mediated lactylation of H3K18 and STAT1 promotes ferroptosis in diabetic nephropathy. Cell Death Differ. 2025;33(3):589–604. doi:10.1038/s41418-025-01587-4
44. Tian L, Li X, Cheng S, Zhou X, Li X. AARS1 Mediates Lactylation of STAT3 and NF-kB Promotes Cyst Growth in ADPKD: FR-PO0662. J Am Soc Nephrol. 2025;36(10S). doi:10.1681/ASN.2025hwmfg6gd
45. Moreno-Yruela C, Zhang D, Wei W, et al. Class I histone deacetylases (HDAC1–3) are histone lysine delactylases. Sci Adv. 2022;8(3):eabi6696. doi:10.1126/sciadv.abi6696
46. Srivastava SP, Li J, Kitada M, et al. SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. 2018;9(10):997. doi:10.1038/s41419-018-1057-0
47. Shen S, Ying C, Fu X, et al. Delactylase effects of SIRT3 on a positive feedback loop involving the RUNX1-glycolysis-histone lactylation in diabetic kidney disease. Int J Biol Sci. 2026;22(4):1775–1792. doi:10.7150/ijbs.126011
48. Zhang Y, Wen P, Luo J, et al. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 2021;12(9):847. doi:10.1038/s41419-021-04134-4
49. Zhao S, Zhang X, Li H. Beyond histone acetylation—writing and erasing histone acylations. Curr Opin Struct Biol. 2018;53:169–177. doi:10.1016/j.sbi.2018.10.001
50. Liu X, Wang L, Zhao K, et al. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008;451(7180):846–850. doi:10.1038/nature06546
51. Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 2017;18(2):90–101. doi:10.1038/nrm.2016.140
52. Bheda P, Jing H, Wolberger C, Lin H. The Substrate Specificity of Sirtuins. Annu Rev Biochem. 2016;85(1):405–429. doi:10.1146/annurev-biochem-060815-014537
53. Du R, Gao Y, Yan C, et al. Sirtuin 1/sirtuin 3 are robust lysine delactylases and sirtuin 1-mediated delactylation regulates glycolysis. iScience. 2024;27(10):110911. doi:10.1016/j.isci.2024.110911
54. Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9(3):206–218. doi:10.1038/nrm2346
55. Barutta F, Bruno G, Grimaldi S, Gruden G. Inflammation in diabetic nephropathy: moving toward clinical biomarkers and targets for treatment. Endocrine. 2015;48(3):730–742. doi:10.1007/s12020-014-0437-1
56. Rabinowitz JD, Enerbäck S. Lactate: the ugly duckling of energy metabolism. Nat Metab. 2020;2(7):566–571. doi:10.1038/s42255-020-0243-4
57. Noe JT, Rendon BE, Geller AE, et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci Adv. 2021;7(46):eabi8602. doi:10.1126/sciadv.abi8602
58. Zhou W, Liu Y, Hu Q, Zhou J, Lin H. The landscape of immune cell infiltration in the glomerulus of diabetic nephropathy: evidence based on bioinformatics. BMC Nephrol. 2022;23(1):303. doi:10.1186/s12882-022-02906-4
59. Li HD, You YK, Shao BY, et al. Roles and crosstalks of macrophages in diabetic nephropathy. Front Immunol. 2022;13:1015142. doi:10.3389/fimmu.2022.1015142
60. Tang Y, Li Y, Yang X, et al. Intestinal metabolite TMAO promotes CKD progression by stimulating macrophage M2 polarization through histone H4 lysine 12 lactylation. Cell Death Differ. 2026;33(2):314–326. doi:10.1038/s41418-025-01554-z
61. Pei X, Lu L, Huang Z, et al. Activation of M1 macrophages promotes diabetic kidney disease by modulating glycolysis via HIF-1α-HK2 signaling pathway. Diabetol Metab Syndr. 2025;17(1):362. doi:10.1186/s13098-025-01894-3
62. Daskalaki MG, Tsatsanis C, Kampranis SC. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J Cell Physiol. 2018;233(9):6495–6507. doi:10.1002/jcp.26497
63. Lin HC, Chen YJ, Wei YH, et al. Lactic Acid Fermentation Is Required for NLRP3 Inflammasome Activation. Front Immunol. 2021;12:630380. doi:10.3389/fimmu.2021.630380
64. Yu F, Meng Y, Wang X, et al. Protein lactylation of citrate synthase promotes the AKI-CKD transition by activating the NLRP3 inflammasome. Cell Rep. 2025;44(8):116084. doi:10.1016/j.celrep.2025.116084
65. Chu X, Di C, Chang P, et al. Lactylated Histone H3K18 as a Potential Biomarker for the Diagnosis and Predicting the Severity of Septic Shock. Front Immunol. 2022;12:786666. doi:10.3389/fimmu.2021.786666
66. Wei L, Yang X, Wang J, et al. H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer’s disease through the NFκB signaling pathway. J Neuroinflammation. 2023;20(1):208. doi:10.1186/s12974-023-02879-7
67. Song C, Wang S, Fu Z, et al. IGFBP5 promotes diabetic kidney disease progression by enhancing PFKFB3-mediated endothelial glycolysis. Cell Death Dis. 2022;13(4):340. doi:10.1038/s41419-022-04803-y
68. Hu X, Chen W, Yang M, Li M, Li X, Ouyang S. IGFBP5 promotes EndoMT and renal fibrosis through H3K18 lactylation in diabetic nephropathy. Cell Mol Life Sci. 2025;82(1):215. doi:10.1007/s00018-025-05718-5
69. Wang Y, Li H, Jiang S, et al. The glycolytic enzyme PFKFB3 drives kidney fibrosis through promoting histone lactylation-mediated NF-κB family activation. Kidney Int. 2024;106(2):226–240. doi:10.1016/j.kint.2024.04.016
70. Zhou X, Xu C, Dong J, Liao L. Role of renal tubular programed cell death in diabetic kidney disease. Diabetes Metab Res Rev. 2023;39(2):e3596. doi:10.1002/dmrr.3596
71. Kanasaki K. The aberrant glycolysis in kidney proximal tubule: potential therapeutic target for DKD. Kidney Int. 2023;104(6):1056–1059. doi:10.1016/j.kint.2023.09.019
72. Tan C, Gu J, Li T, et al. Inhibition of aerobic glycolysis alleviates sepsis‑induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK‑regulated autophagy. Int J Mol Med. 2021;47(3):19. doi:10.3892/ijmm.2021.4852
73. Qiao J, Tan Y, Liu H, et al. Histone H3K18 and Ezrin Lactylation Promote Renal Dysfunction in Sepsis-Associated Acute Kidney Injury. Adv Sci. 2024;11(28):2307216. doi:10.1002/advs.202307216
74. An S, Yao Y, Hu H, et al. PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death Dis. 2023;14(7):457. doi:10.1038/s41419-023-05952-4
75. Chen J, Feng Q, Qiao Y, et al. ACSF2 and lysine lactylation contribute to renal tubule injury in diabetes. Diabetologia. 2024;67(7):1429–1443. doi:10.1007/s00125-024-06156-x
76. Fan Z, Zhang Y, Yuan L, et al. LARS1 lactylation inhibits autophagy by activating mTORC1 to promote podocytes injury in diabetic kidney disease. Cell Signal. 2025;134:111955. doi:10.1016/j.cellsig.2025.111955
77. Li D, Jiang C, Mei G, et al. Quercetin Alleviates Ferroptosis of Pancreatic β Cells in Type 2 Diabetes. Nutrients. 2020;12(10):2954. doi:10.3390/nu12102954
78. Yang G, Liu X, Li Y, et al. TRIM65 as a key regulator of ferroptosis and glycolysis in lactate-driven renal tubular injury and diabetic kidney disease. Cell Rep. 2025;44(8):116091. doi:10.1016/j.celrep.2025.116091
79. Zhang L, Wang X, Zhou S, Feng Y. LDHA enhances brain injury and apoptosis after intracerebral hemorrhage by promoting P53 transcription through increasing P53 lactylation. Brain Res Bull. 2025;224:111292. doi:10.1016/j.brainresbull.2025.111292
80. You X, Xie Y, Tan Q, et al. Glycolytic reprogramming governs crystalline silica-induced pyroptosis and inflammation through promoting lactylation modification. Ecotoxicol Environ Saf. 2024;283:116952. doi:10.1016/j.ecoenv.2024.116952
81. Chen Y, Zou H, Lu H, Xiang H, Chen S. Research progress of endothelial-mesenchymal transition in diabetic kidney disease. J Cell Mol Med. 2022;26(12):3313–3322. doi:10.1111/jcmm.17356
82. Hu X, Xu Q, Wan H, et al. PI3K-Akt-mTOR/PFKFB3 pathway mediated lung fibroblast aerobic glycolysis and collagen synthesis in lipopolysaccharide-induced pulmonary fibrosis. Lab Invest. 2020;100(6):801–811. doi:10.1038/s41374-020-0404-9
83. Yin XN, Wang J, Cui LF, Fan WX. Enhanced glycolysis in the process of renal fibrosis aggravated the development of chronic kidney disease. Eur Rev Med Pharmacol Sci. 2018;22(13):4243–4251. doi:10.26355/eurrev_201807_15419
84. Li M, Jia F, Zhou H, Di J, Yang M. Elevated aerobic glycolysis in renal tubular epithelial cells influences the proliferation and differentiation of podocytes and promotes renal interstitial fibrosis. Eur Rev Med Pharmacol Sci. 2018;22(16):5082–5090. doi:10.26355/eurrev_201808_15701
85. Kottmann RM, Kulkarni AA, Smolnycki KA, et al. Lactic Acid Is Elevated in Idiopathic Pulmonary Fibrosis and Induces Myofibroblast Differentiation via pH-Dependent Activation of Transforming Growth Factor-β. Am J Respir Crit Care Med. 2012;186(8):740–751. doi:10.1164/rccm.201201-0084OC
86. Li Y, Kang YS, Dai C, Kiss LP, Wen X, Liu Y. Epithelial-to-Mesenchymal Transition Is a Potential Pathway Leading to Podocyte Dysfunction and Proteinuria. Am J Pathol. 2008;172(2):299–308. doi:10.2353/ajpath.2008.070057
87. Burns JE, Hurst CD, Knowles MA, Phillips RM, Allison SJ. The Warburg effect as a therapeutic target for bladder cancers and intratumoral heterogeneity in associated molecular targets. Cancer Sci. 2021;112(9):3822–3834. doi:10.1111/cas.15047
88. Zheng T, Gu YP, Wang JM, Huang TT, Gou LS, Liu YW. Lactate-triggered histone lactylation contributes to podocyte epithelial-mesenchymal transition in diabetic nephropathy in mice. Chem Biol Interact. 2025;408:111418. doi:10.1016/j.cbi.2025.111418
89. Huimin W, Xin W, Shan Y, et al. Lactate promotes the epithelial-mesenchymal transition of liver cancer cells via TWIST1 lactylation. Exp Cell Res. 2025;447(1):114474. doi:10.1016/j.yexcr.2025.114474
90. Chen J, He J, Wang X, et al. Glis1 inhibits RTEC cellular senescence and renal fibrosis by downregulating histone lactylation in DKD. Life Sci. 2025;361:123293. doi:10.1016/j.lfs.2024.123293
91. Ding H, Jiang L, Xu J, et al. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol-Ren Physiol. 2017;313(3):F561–F575. doi:10.1152/ajprenal.00036.2017
92. Ye Z, Sun Y, Yang S, et al. Lgals3 Promotes Calcium Oxalate Crystal Formation and Kidney Injury Through Histone Lactylation-Mediated FGFR4 Activation. Adv Sci. 2025;12(12):2413937. doi:10.1002/advs.202413937
93. Xiang T, Wang X, Huang S, et al. Inhibition of PKM2 by shikonin impedes TGF-β1 expression by repressing histone lactylation to alleviate renal fibrosis. Phytomedicine. 2025;136:156324. doi:10.1016/j.phymed.2024.156324
94. Talasniemi JP, Pennanen S, Savolainen H, Niskanen L, Liesivuori J. Analytical investigation: assay of d-lactate in diabetic plasma and urine. Clin Biochem. 2008;41(13):1099–1103. doi:10.1016/j.clinbiochem.2008.06.011
95. Broskey NT, Zou K, Dohm GL, Houmard JA. Plasma Lactate as a Marker for Metabolic Health. Exerc Sport Sci Rev. 2020;48(3):119. doi:10.1249/JES.0000000000000220
96. Zhao L, Gao H, Lian F, Liu X, Zhao Y, Lin D. 1 H-NMR-based metabonomic analysis of metabolic profiling in diabetic nephropathy rats induced by streptozotocin. Am J Physiol-Ren Physiol. 2011;300(4):F947–F956. doi:10.1152/ajprenal.00551.2010
97. Lee DY, Kim JY, Ahn E, et al. Associations between local acidosis induced by renal LDHA and renal fibrosis and mitochondrial abnormalities in patients with diabetic kidney disease. Transl Res. 2022;249:88–109. doi:10.1016/j.trsl.2022.06.015
98. Tang L, Yang Q, Ma R, et al. Association between lactate dehydrogenase and the risk of diabetic kidney disease in patients with type 2 diabetes. Front Endocrinol. 2024;15:1369968. doi:10.3389/fendo.2024.1369968
99. Xiao X, Zhang J, Lang Y, et al. Associations of lactate dehydrogenase with risk of renal outcomes and cardiovascular mortality in individuals with diabetic kidney disease. Diabet Res Clin Pract. 2023;203:110838. doi:10.1016/j.diabres.2023.110838
100. Zhao W, Zhang J, Chen K, Yuan J, Zhai L, Tan M. Mass spectrometry-based characterization of histone post-translational modification. Curr Opin Chem Biol. 2025;88:102622. doi:10.1016/j.cbpa.2025.102622
101. Zhu Y, Fu Y, Liu F, Yan S, Yu R. Appraising histone H4 lysine 5 lactylation as a novel biomarker in breast cancer. Sci Rep. 2025;15(1):8205. doi:10.1038/s41598-025-92666-6
102. Ran Y, Yang Q, Zhou L, Li H, Yang B, Zhang L. Protocol for label-free quantitative lysine lactylproteome profiling. STAR Protoc. 2025;6(2):103726. doi:10.1016/j.xpro.2025.103726
103. McGill JB, Subramanian S. Safety of Sodium-Glucose Co-Transporter 2 Inhibitors. Am J Cardiol. 2019;124:S45–S52. doi:10.1016/j.amjcard.2019.10.029
104. Azushima K, Kovalik JP, Yamaji T, et al. Abnormal lactate metabolism is linked to albuminuria and kidney injury in diabetic nephropathy. Kidney Int. 2023;104(6):1135–1149. doi:10.1016/j.kint.2023.08.006
105. Maeda M, Ko M, Mane MM, et al. Genetic and Drug Inhibition of LDH-A: effects on Murine Gliomas. Cancers. 2022;14(9):2306. doi:10.3390/cancers14092306
106. Yang Y, Chong Y, Chen M, et al. Targeting lactate dehydrogenase a improves radiotherapy efficacy in non-small cell lung cancer: from bedside to bench. J Transl Med. 2021;19(1):170. doi:10.1186/s12967-021-02825-2
107. Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci. 2010;107(5):2037–2042. doi:10.1073/pnas.0914433107
108. Tiwari R, Sharma R, Rajendran G, et al. Postischemic inactivation of HIF prolyl hydroxylases in endothelium promotes maladaptive kidney repair by inducing glycolysis. J Clin Invest. 2025;135(3):e176207. doi:10.1172/JCI176207
109. Halford S, Veal GJ, Wedge SR, et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin Cancer Res. 2023;29(8):1429–1439. doi:10.1158/1078-0432.CCR-22-2263
110. Hui S, Ghergurovich JM, Morscher RJ, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551(7678):115–118. doi:10.1038/nature24057
111. Choi EJ, Jang YY, Choi EJ, Oh CJ. The Role of Lactate in Immune Regulation: a Metabolic Rheostat via Transporters, Receptors, and Epigenetic Modifiers. Cells. 2025;14(14):1096. doi:10.3390/cells14141096
112. Brooks GA. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018;27(4):757–785. doi:10.1016/j.cmet.2018.03.008
113. Lasko LM, Jakob CG, Edalji RP, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550(7674):128–132. doi:10.1038/nature24028
114. Lu C, Thompson CB. Metabolic Regulation of Epigenetics. Cell Metab. 2012;16(1):9–17. doi:10.1016/j.cmet.2012.06.001
115. Jin J, Bai L, Wang D, et al.
116. Ren C, Wu Q, Xiao R, et al. Expanding the Scope of Genetically Encoded Lysine Post-Translational Modifications with Lactylation, β-Hydroxybutyrylation and Lipoylation. ChemBioChem. 2022;23(18):e202200302. doi:10.1002/cbic.202200302
117. Guo Y, Chu L, Shui W, et al. Histone lactylation in immune cells and its predictive role in sepsis progression: a prospective observational study. Shock. 2025. doi:10.1097/SHK.0000000000002659
118. Yu J, Chai P, Xie M, et al. Histone lactylation drives oncogenesis by facilitating m6A reader protein YTHDF2 expression in ocular melanoma. Genome Biol. 2021;22(1):85. doi:10.1186/s13059-021-02308-z
119. Zeng Y, Jiang H, Chen Z, et al. Histone lactylation promotes multidrug resistance in hepatocellular carcinoma by forming a positive feedback loop with PTEN. Cell Death Dis. 2025;16(1):59. doi:10.1038/s41419-025-07359-9
120. Chinese Clinical Trial Registry. The mechanism of lactylation modification involved in the development of diabetes-related complications is studied. 2025. Available from: https://www.chictr.org.cn/showproj.html?proj=235681.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.