Back to Journals » OncoTargets and Therapy » Volume 9
Knockdown of TMEM16A suppressed MAPK and inhibited cell proliferation and migration in hepatocellular carcinoma
Authors Deng L, Zhang J, Chen H, Ma B, Pan K, Su C, Xu F, Yang J
Received 8 September 2015
Accepted for publication 20 November 2015
Published 14 January 2016 Volume 2016:9 Pages 325—333
DOI https://doi.org/10.2147/OTT.S95985
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Liang Deng,1,* Jihong Yang,2,* Hongwu Chen,3 Bo Ma,4 Kangming Pan,1 Caikun Su,1 Fengfeng Xu,1 Jihong Zhang1
1Department of Hepatobiliary Surgery, The Eastern Hospital of the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, 2Department of General Surgery, The Affiliated Hospital of Hebei University, Baoding, 3Department of Emergency, 4Department of Gastroenterology, The Eastern Hospital of the First Affiliated Hospital of Sun Yat-sen University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: TMEM16A plays an important role in cell proliferation in various cancers. However, less was known about the expression and role of TMEM16A in hepatocellular carcinoma. We screened the expression of TMEM16A in patients’ hepatocellular carcinoma tissues, and also analyzed the biological function of hepatocellular carcinoma cells by knockdown of TMEM16A, as well as the expression of MAPK signaling proteins, including p38, p-p38, ERK1/2, p-ERK1/2, JNK, and p-JNK, and cell cycle regulatory protein cyclin D1 in TMEM16A siRNA-transfected SMMC-7721 cells by Western blot. Our results showed that TMEM16A was overexpressed in hepatocellular carcinoma tissues. Inhibition of TMEM16A suppressed the cell proliferation, migration, and invasion, and cell cycle progression but did not influence the cell apoptosis. TMEM16A siRNA-suppressed cancer cell proliferation and tumor growth were accompanied by a reduction of p38 and ERK1/2 activation and cyclin D1 induction, and were not influenced by other tested MAPK signaling proteins. In addition, inhibition of TMEM16A suppressed tumorigenicity in vivo. TMEM16A is overexpressed in hepatocellular carcinoma, and that inhibition of TMEM16A suppressed MAPK and growth of hepatocellular carcinoma. TMEM16A could be a potentially novel therapeutic target for human cancers, including hepatocellular carcinoma.
Keywords: TMEM16A, cell cycle, proliferation, apoptosis, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma is one of the most common malignancies. It is highly aggressive and has poor prognosis and results in high mortality throughout the world. It is estimated that 782,500 new liver cancer cases and 745,500 deaths occurred worldwide in 2012.1 People’s Republic of China alone accounted for ~50% of the total number of cases and deaths. Primary liver cancers are classified into three types based on the morphology and cytogenetic characteristics. They are hepatocellular carcinoma, intrahepatic cholangiocarcinoma, and combined hepatocellular and cholangiocarcinoma. Up to 75% of primary liver cancers are hepatocellular carcinoma.1 Although the use of antiviral treatments has reduced the risk of liver cancer, their use is still costly and unfeasible.2,3 Because of the rapid development, metastasis, and the lack of accurate diagnosis biomarkers at the initial stages of liver cancer, the prognosis and the survival rate for liver cancer patients remain poor.4 Furthermore, the effective adjuvant therapies for hepatocellular carcinoma are limited because of the poor understanding of the mechanism of hepatocellular carcinoma development.
TMEM16A (Anoctamin 1) is a member of the Ano family. Recently, it has been shown to be a calcium-activated chloride channel5 regulating cell proliferation.6 It is widely expressed in various tissues, such as smooth muscles, secretory epithelial cells, and sensory neurons.7 The upregulation of TMEM16A has also been shown in several cancers, such as breast and prostate cancer,8 head and neck squamous cell carcinoma (HNSCC),9 esophageal squamous cell cancer,10 and human colorectal cancer.11 It has been shown that TMEM16A activated the MAPK signaling proteins, and promoted the tumorigenesis and invasion of various cancers, such as HNSCC,6 breast cancer,12,13 and gastrointestinal stromal tumors.14 However, the expression and role of TMEM16A in hepatocellular carcinoma have not been evaluated to date.
To understand the effect of TMEM16A in the development of hepatocellular carcinoma and the underlying mechanism, we screened the expression of TMEM16A in patients’ tissues. We also analyzed the biological function of hepatocellular carcinoma cells by knockdown of TMEM16A, as well as the expression of MAPK signaling proteins, including p38, p-p38, ERK1/2, p-ERK1/2, JNK, and p-JNK, and cell cycle regulatory protein cyclin D1 in TMEM16A siRNA-transfected SMMC-7721 cells. Role of TMEM16A shRNA in tumorigenicity in vivo was also assessed.
Materials and methods
Clinical sample collection
The study and all procedures performed involving human participants were approved by the ethics committee of The Eastern Hospital of the First Affiliated Hospital of Sun Yat-sen University. Written informed consents were obtained from the patients. Tissues were collected from 20 patients (male/female, 1/1) having hepatocellular carcinoma. Age of patients ranged from 30 to 50 years. In each patient, hepatocellular carcinoma and pericarcinous tissue were resected and collected during the operation. The tissue samples were directly placed into liquid nitrogen after surgical resection and were stored at −80°C until the measurement of the mRNA and protein expression of TMEM16A.
Cell culture
Human hepatoma cell line SMMC-7721 cells were purchased from Cell Bank of Shanghai Institute of Biochemistry & Cell Biology, Chinese Academy of Sciences. Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Invitrogen Inc., Carlsbad, CA, USA) and supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin at 37°C in 5% CO2.
Cell transfection
For knockdown of TMEM16A, the TMEM16A siRNA and negative siRNA (50 nM) were synthesized and purified by OriGene (OriGene, Rockville, MD, USA). SMMC-7721 cells were seeded in a six-well plate (2×105 per well) for 24 hours before transfection, and then were transfected with TMEM16A siRNA or negative siRNA using FuGENE® HD transfection reagent (Promega, Fitchburg, WI, USA) in accordance with the manufacturer’s instruction. For tumorigenicity assay, SMMC-7721 cells were stably transfected with TMEM16A shRNA (sc-76686-SH; Santa Cruz Biotechnology, Dallas, TX, USA). After transfection, the cells were grown in medium without antibiotics for 48 hours, and were used for further experiments.
Cell proliferation assay
SMMC-7721 cell survival was evaluated using MTT (3-(4,5-dimethyl-thiazol-2-y1)-2,5-diphenyl tetrazolium bromide; Sigma, St Louis, MO, USA) colorimetric assay. After transfection, cells were seeded in 96-well tissue culture plates at 2×104 cells per well for 0, 24, 48, 72, and 96 hours. Then, SMMC-7721 cells were washed with phosphate-buffered saline (PBS) and incubated in 100 μL of 5 mg/mL MTT solution (Invitrogen Inc.) for 3 hours. MTT is converted into purple-colored formazen in living cells which were then solubilized with dimethylsulfoxide (Invitrogen Inc.). Absorbance of this solution was measured at 450 nm using the microplate reader (Rayto Life and Analytical Science Co. Ltd, Shenzhen, People’s Republic of China).
Cell migration and invasion assay
SMMC-7721 cell migration and invasion were assayed using a transwell chamber (Millipore, Billerica, MA, USA). For the invasion assay, transwell chamber was coated with 30 μL of Matrigel and was placed into a 24-well plate and incubated for 30 minutes at 37°C. After 48 hours of transfection, SMMC-7721 cells were trypsinized and seeded in chambers at the density of 8×104 cells per well and cultured in RPMI-1640 medium with 2% serum, while 600 μL of 10% fetal bovine serum (FBS)–RPMI-1640 was added to the lower chamber. Twenty-four hours later, migrated cells were fixed with 100% methanol for 30 minutes and stained by crystal violet for 20 minutes. We used cotton swabs to remove non-migrated cells. Cell images were obtained under a phase-contrast microscope (Olympus, Tokyo, Japan).
Cell cycle assay
After transfection, cells were harvested following trypsinization and were rinsed three times with buffer solution at an adjusted concentration of 1×106 cells/mL and prepared using CycleTEST PLUS DNA Reagent Kit (Becton Dickinson, Franklin Drive, NJ, USA) according to the manufacturer’s instruction. Cell cycle status was analyzed by flow cytometer using propidium iodide (PI) as a specific fluorescent dye probe. The PI fluorescence intensity of 10,000 cells was measured for each sample using a Becton Dickinson FACSCalibur flow cytometer.
Cell apoptosis assay
Cell apoptosis was determined by Annexin V assay. After transfection for 48 hours, cells were harvested by trypsinization and washed with PBS, and suspended in Annexin V binding buffer. Fluorescein isothiocyanate-conjugated Annexin V and PI (KeyGen, Nanjing, People’s Republic of China) were added to the cells successively. After incubation, Annexin V binding buffer was added, and cells were analyzed by a FACScan (Becton Dickinson) flow cytometer. Annexin V(+)/PI(−) and Annexin V(+)/PI(+) represent the cells in early apoptosis and late apoptosis/necrosis, respectively.
Quantitative reverse transcription polymerase chain reaction
Total RNA was isolated using TRIZOL (Invitrogen Inc.). The RNA concentration was measured using GeneQuant II (Pharmacia, Uppsala, Sweden) at 260 nm. Reverse transcription reaction and cDNA synthesis was performed according to the manufacturer’s instructions (Invitrogen Inc.). Polymerase chain reaction analysis was performed on Applied Biosystems 7500 Sequence Detection system (ABI; Applied Biosystems, Foster City, CA, USA) using SYBR Premix Ex Taq GC kit (Takara, Japan). The primers were the following: for TMEM16A, 5′-ATTTCACCAATCTTGTCTCCATCA-3′ (forward) and 5′-TGATAACTCCAAGAACGATTGCA-3′ (reverse); for GAPDH, 5′-ACACCCACTCCTCCACCTTT-3′ (forward) and 5′-TTACTCCTTGGAGGCCATGT-3′ (reverse). Gene expression of TMEM16A was normalized to the level of GAPDH within each sample using the relative ΔΔCT method. Gene expression is shown as relative expression to control. The data shown are representative of three independent experiments.
Western blotting
Transfected cells were washed and then lysed on ice for 10 minutes in 50 mM Tris–HCl (pH 7.5), 10% glycerol, 2% sodium dodecyl sulfate, 0.1 M dithiothreitol, and 10 mM phenylmethylsulfonyl fluoride. Proteins were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and electroblotted onto a nitrocellulose membrane in 25 mM Tris base and 190 mM glycine at 50 V for 3 hours at 4°C. To detect the expression of MAPK signaling proteins (p38, p-p38, ERK1/2, p-ERK1/2, JNK, and p-JNK) and cell cycle regulatory protein cyclin D1 in TMEM16A siRNA-transfected SMMC-7721 cells, blots were incubated in 1:1,000 monoclonal antibodies against p38, p-p38, ERK1/2, p-ERK1/2, JNK, p-JNK, and cyclin D1, respectively. All antibodies were purchased from Santa Cruz Biotechnology. Proteins were detected by enhanced chemiluminescence as described by the manufacturer (Beyotime, Haimen, People’s Republic of China).
Tumorigenicity assay in nude mice
Six-week-old female BALB/c athymic nude mice (Vitalriver Laboratory Animals, Beijing, People’s Republic of China) were subcutaneously injected in the right flank with 2.0×106 cells in 0.1 mL of PBS. Once tumors were formed, tumor volume (V) was measured daily by caliper and calculated using the formula V = (L × W2)/2, where L was the length and W was the width of the tumor. The mice were randomly divided into two groups (n=6) for inoculation of TMEM16A shRNA-transfected SMMC-7721 cells and negative control (NC) shRNA-transfected SMMC-7721 cells for 42 days. Growth curves were plotted using average tumor volume within each experimental group every week. Six weeks later, the mice were euthanized, and the dissected tumors were collected and prepared for subsequent analyses. All animal experiments were approved by the animal center of the First Affiliated Hospital of Sun Yat-sen University.
Statistical analysis
For quantitative data, all results are expressed as the mean ± standard deviation. Statistical significance between groups was determined using one-way analysis of variance or an unpaired Student’s t-test using SPSS 18.0 (SPSS, Chicago, IL, USA). Each experiment was repeated at least three times. P<0.05 was considered statistically significant.
Results
Expression of TMEM16A is upregulated in hepatocellular carcinoma tissues
To investigate the role of TMEM16A in hepatocellular carcinoma, we compared the expression of TMEM16A between hepatocellular carcinoma and pericarcinous tissue (Figure 1). Both the mRNA (Figure 1A) and protein expressions (Figure 1B and C) of TMEM16A were upregulated by about threefold in hepatocellular carcinoma tissues, compared to pericarcinous tissue, suggesting an important role of TMEM16A in the development of human hepatocellular carcinoma. Then, a set of experiments was designed to detect the role of TMEM16A in the proliferation, cell cycle, and apoptosis in SMMC-7721 cells.
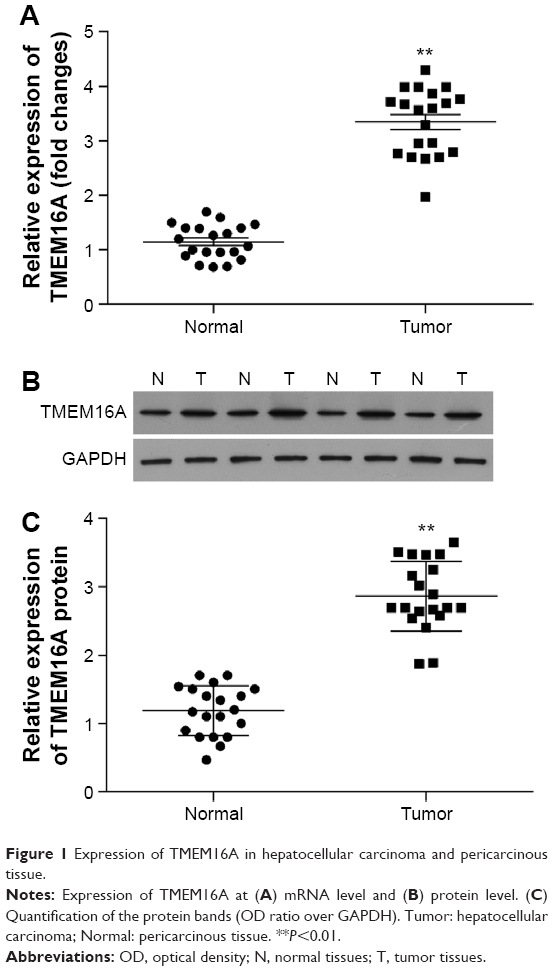 | Figure 1 Expression of TMEM16A in hepatocellular carcinoma and pericarcinous tissue. |
TMEM16A siRNA suppresses expression of TMEM16A
Transfection of TMEM16A siRNA almost abolished the mRNA expression of TMEM16A in SMMC-7721 cells (Figure 2A). At protein level (Figure 2B), the expression of TMEM16A in SMMC-7721 cells was significantly downregulated by TMEM16A siRNA in contrast to NC siRNA. Thus, the TMEM16A siRNA-transfected SMMC-7721 cells could be used to explore the role of TMEM16A in proliferation, migration, and invasion of hepatocellular carcinoma cells.
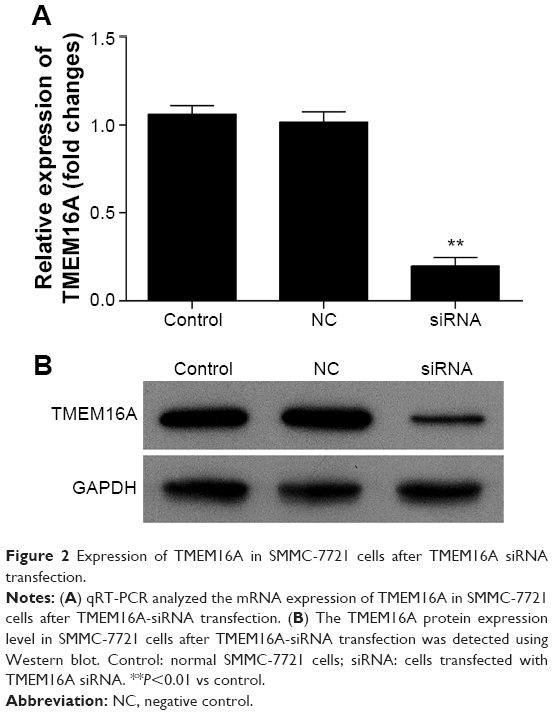 | Figure 2 Expression of TMEM16A in SMMC-7721 cells after TMEM16A siRNA transfection. |
TMEM16A siRNA suppresses the proliferation, migration, and invasion of SMMC-7721 cells
MTT and invasion assays were performed to investigate the biological function of TMEM16A in hepatocellular carcinoma cells (Figure 3). Results showed that transfection of NC siRNA did not influence the proliferation, migration, and invasion of SMMC-7721 cells. The knockdown of TMEM16A by its siRNA significantly attenuated the proliferation of SMMC-7721 cells after 48 hours (Figure 3A), and significantly inhibited the migration and invasion (Figure 3B and C) of SMMC-7721 cells.
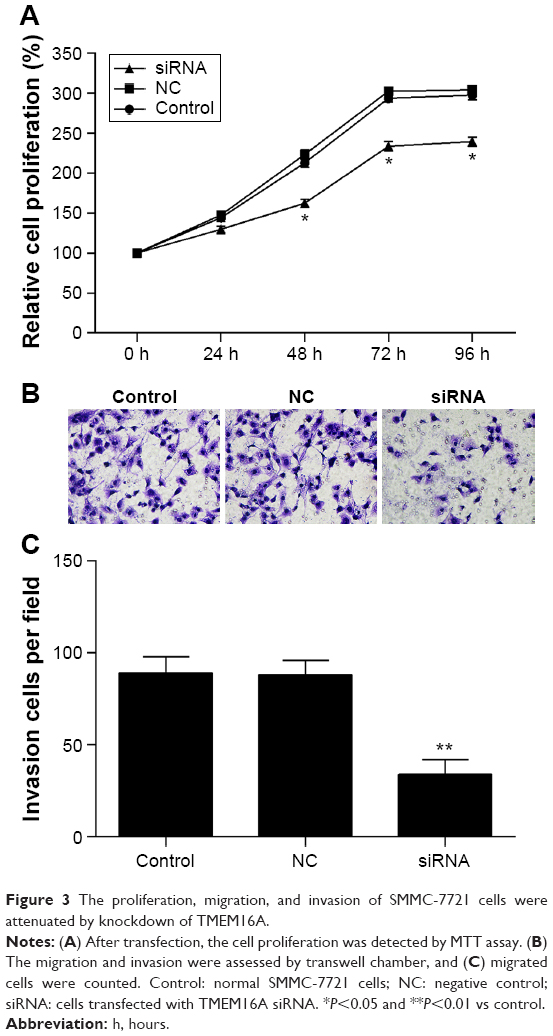 | Figure 3 The proliferation, migration, and invasion of SMMC-7721 cells were attenuated by knockdown of TMEM16A. |
Role of TMEM16A in cell cycle and apoptosis of SMMC-7721 cells
Cell cycle distribution was measured by flow cytometry with cell cycle staining kit (MultiSciences, Hangzhou, People’s Republic of China) (Figure 4A). The cell cycle phase is shown in a bar graph (Figure 4B) with the G0/G1, S, and G2/M phases. Results demonstrated that the G0/G1 phases in TMEM16A siRNA-transfected SMMC-7721 cells were significantly enhanced. In contrast, the S phase was significantly decreased, indicating the TMEM16A relation to the cell growth.
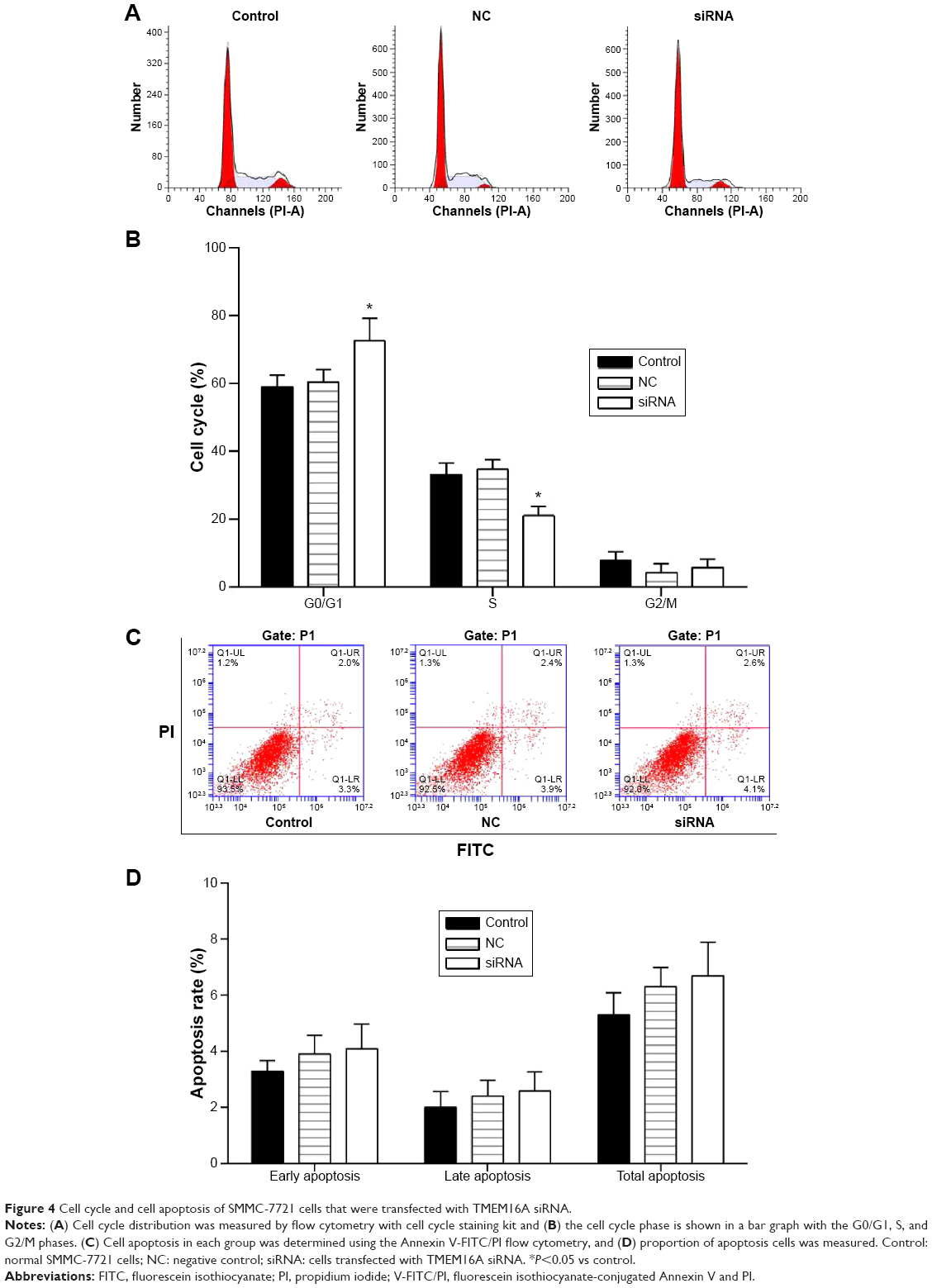 | Figure 4 Cell cycle and cell apoptosis of SMMC-7721 cells that were transfected with TMEM16A siRNA. |
The role of TMEM16A in the apoptosis of SMMC-7721 cells was investigated by flow cytometry (Figure 4C and D). There were no detectable changes in early, late, or total apoptosis by transfection of TMEM16A siRNA. Thus, TMEM16A siRNA arrested cell cycle progression at G0/G1 phase but did not influence the cell apoptosis.
Expression of MAPK signaling proteins and cyclin D1 in TMEM16A siRNA-transfected SMMC-7721 cells
MAPK signaling proteins are important for the cell cycle. After knockdown of TMEM16A, p-p38 and p-ERK1/2 were significantly decreased, but p38, ERK1/2, p-JNK, and JNK were not changed (Figure 5). The cell cycle regulatory protein cyclin D1 was significantly decreased in TMEM16A siRNA-transfected SMMC-7721 cells. These results implicated that inhibition of p38 and ERK1/2 was associated with knockdown of TMEM16A and played an important role in TMEM16A-induced cell cycle arrest, whereas the JNK was not involved in TMEM16A-induced cell cycle arrest.
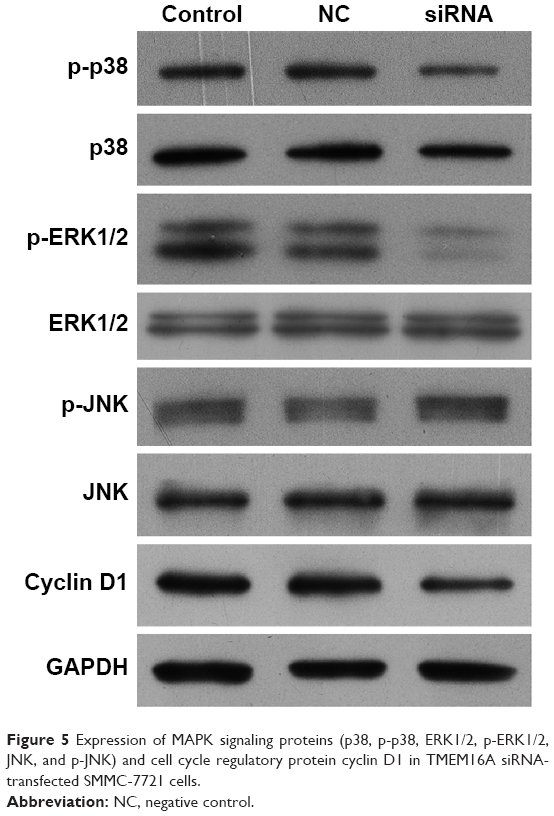 | Figure 5 Expression of MAPK signaling proteins (p38, p-p38, ERK1/2, p-ERK1/2, JNK, and p-JNK) and cell cycle regulatory protein cyclin D1 in TMEM16A siRNA-transfected SMMC-7721 cells. |
Cyclin D1 could be used to identify cancers that have an amplification of the q13 region of chromosome 11 (11q13). Cyclin D1 was significantly decreased with the knockdown of TMEM16A, indicating that the amplification of 11q13 is important for the TMEM16A overexpression.
TMEM16A siRNA suppresses tumorigenicity in vivo
We used an in vivo tumor model to confirm the above findings. We injected TMEM16A shRNA-transfected SMMC-7721 cells and NC shRNA-transfected SMMC-7721 cells into nude mice separately. Six weeks after injection, the group with TMEM16A shRNA formed substantially smaller tumors than the control group (Figure 6), suggesting that knockdown of TMEM16A could suppress the development of hepatocellular carcinoma in vivo.
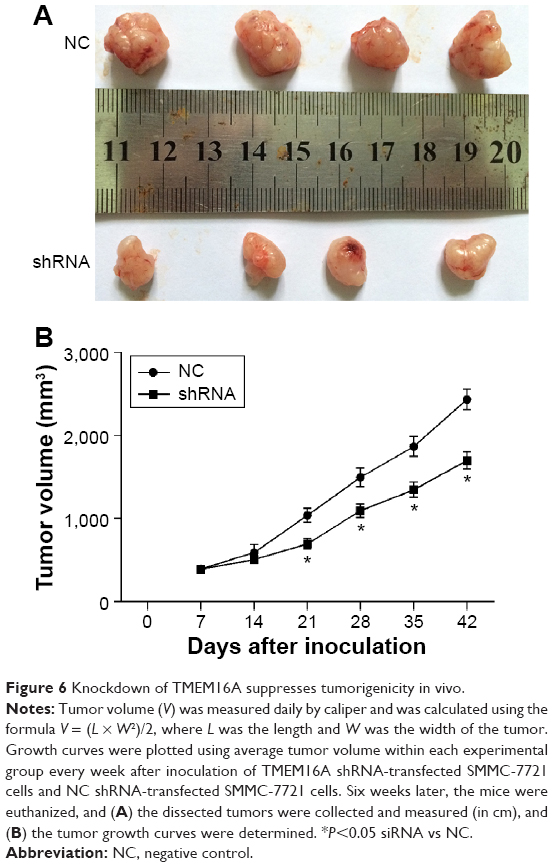 | Figure 6 Knockdown of TMEM16A suppresses tumorigenicity in vivo. |
Discussion
The poor understanding of hepatocellular carcinoma development mechanism has limited the effective adjuvant therapies for hepatocellular carcinoma.2,3 TMEM16A is a functional calcium-activated chloride channel5 that influences tumor growth and progression.6,15 It is widely overexpressed in various cancers, including breast and prostate cancer,8 HNSCC,9 esophageal squamous cell cancer,10 and human colorectal cancer.11 Consistent with studies mentioned above,2,3,5,6,8,10,11,15 we found that TMEM16A was overexpressed in hepatocellular carcinoma tissues compared with their pericarcinous tissue in all of 20 patients. However, the mechanism underlying the overexpression of TMEM16A remains unclear. The overexpression is possible due to the 11q13 amplification and signaling pathways or transcription factors during the tumorigenesis.16–18 Human cyclin D1 could be used to identify cancers that have an amplification of the 11q13.19 Our data show that the induction of TMEM16A is associated with the decrease of cyclin D1, indicating that the amplification of 11q13 is important for the TMEM16A overexpression.
There is evidence that chloride channels regulate tumorigenesis and tumor progression.8,20–22 It has been reported that TMEM16A expression is linked with the growth of human HNSCC.6 TMEM16A also promoted the cell proliferation in a variety of cancer cell lines, such as breast cancer (MCF10A), pancreatic cancer (CFPAC-1), HNSCC (CAL33), and prostate cancer (PC-3).23 Similarly, our results have shown that the induction of TMEM16A directly impacted the cell proliferation and induced cell cycle arrest of SMMC-7721 cells. It is confirmed that TMEM16A functions as a proto-oncogene to drive the tumor growth via overexpression. However, it was demonstrated that there was no significant effect of TMEM16A knockdown on the proliferation of gastric cancer cells (AGS cells, BGC-823) and HNSCC cell line BHY.24 In vascular smooth muscle cells, knockdown of TMEM16A facilitated cell proliferation suggesting that TMEM16A served as a negative regulator, and overexpression of TMEM16A inhibited angiotensin-II-induced cell cycle transition and cell proliferation.25 Therefore, the expression of TMEM16A is tissue dependent.23,24,26
TMEM16A also plays an important role in cancer cell migration and invasion. It can promote gastric cancer cells migration and invasion by modulating TGF-β’s secretion.24 Inhibition of TMEM16A expression suppressed growth and invasion in human colorectal cancer cells11 and human prostate carcinoma.8,27 Our results also suggested the role of TMEM16A in metastasis of hepatocellular carcinoma. Moreover, recent studies revealed that TMEM16A contributed in tumorigenesis and cancer progression by serving as an important regulator of oncogenic signaling such as MAPK,6 CAMK,12 sonic hedgehog,28 and EGFR signaling pathway.12 Recently, it has been shown that the TMEM16A channel activity is associated with MAPK activation, such as ERK1/2 activation.6 The ERK inhibitor UO126 can inhibit the function of TMEM16A.29 Expression of cyclin D1 was positively associated with p-ERK1/2 and TMEM16A expression in human colorectal cancer cells.11 Here, we found similar changes in MAPK and cell cycle progression, cell proliferation, and invasion. Taken together, our results suggest that TMEM16A activates both ERK1/2 pathway and p38, thereby influencing cellular proliferation and metastasis. TMEM16A siRNA did not influence the JNK activation that also played an important role in development of hepatocellular carcinoma.30 The exact role of p38 and ERK1/2 in TMEM16A-induced cell cycle arrest should be further investigated in future.
The role of TMEM16A in the development of hepatocellular carcinoma was further evaluated by tumorigenicity assay in nude mice. Inhibition of TMEM16A significantly suppressed tumorigenicity in vivo. Thus, the development of small-molecule inhibitors against TMEM16A should be useful for treatment of human hepatocellular carcinoma. Our results have shown that TMEM16A was overexpressed in hepatocellular carcinoma, and that knockdown of TMEM16A suppressed MAPK and inhibited cell proliferation and migration in hepatocellular carcinoma. Although TMEM16A siRNA suppressed the cell proliferation, migration, and invasion and cell cycle progression, it did not influence the cell apoptosis. TMEM16A siRNA-suppressed cancer cell proliferation and tumor growth were accompanied by a reduction of p38 and ERK1/2 activation and cyclin D1 induction, and were not influenced by other tested MAPK signaling proteins. Inhibition of TMEM16A by its shRNA suppressed tumorigenicity in vivo. Thus, TMEM16A could be a potentially novel therapeutic target for human cancers, including hepatocellular carcinoma.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl:S2–S6. | ||
Lu T, Seto WK, Zhu RX, Lai CL, Yuen MF. Prevention of hepatocellular carcinoma in chronic viral hepatitis B and C infection. World J Gastroenterol. 2013;19(47):8887–8894. | ||
Zhu Z, Zhang X, Wang G, Zheng H. Role of microRNAs in hepatocellular carcinoma. Hepat Mon. 2014;14(8):e18672. | ||
Caputo A, Caci E, Ferrera L, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science (New York, N.Y.). 2008;322(5901):590–594. | ||
Duvvuri U, Shiwarski DJ, Xiao D, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72(13):3270–3281. | ||
Ferrera L, Caputo A, Galietta LJ. TMEM16A protein: a new identity for Ca(2+)-dependent Cl(−) channels. Physiology (Bethesda). 2010;25(6):357–363. | ||
Liu W, Lu M, Liu B, Huang Y, Wang K. Inhibition of Ca(2+)-activated Cl(−) channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 2012;326(1):41–51. | ||
Carles A, Millon R, Cromer A, et al. Head and neck squamous cell carcinoma transcriptome analysis by comprehensive validated differential display. Oncogene. 2006;25(12):1821–1831. | ||
Carneiro A, Isinger A, Karlsson A, et al. Prognostic impact of array-based genomic profiles in esophageal squamous cell cancer. BMC Cancer. 2008;8:98. | ||
Sui Y, Sun M, Wu F, et al. Inhibition of TMEM16A expression suppresses growth and invasion in human colorectal cancer cells. PLoS One. 2014;9(12):e115443. | ||
Britschgi A, Bill A, Brinkhaus H, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A. 2013;110(11):E1026–E1034. | ||
Choi EJ, Yun JA, Jabeen S, et al. Prognostic significance of TMEM16A, PPFIA1, and FADD expression in invasive ductal carcinoma of the breast. World J Surg Oncol. 2014;12:137. | ||
West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165(1):107–113. | ||
Habela CW, Ernest NJ, Swindall AF, Sontheimer H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J Neurophysiol. 2009;101(2):750–757. | ||
Wu H, Guan S, Sun M, et al. Ano1/TMEM16A overexpression is associated with good prognosis in PR-positive or HER2-negative breast cancer patients following tamoxifen treatment. PLoS One. 2015;10(5):e0126128. | ||
Ruiz C, Martins JR, Rudin F, et al. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS One. 2012;7(8):e43265. | ||
Dickson C, Fantl V, Gillett C, et al. Amplification of chromosome band 11q13 and a role for cyclin D1 in human breast cancer. Cancer Lett. 1995;90(1):43–50. | ||
Gillett C, Fantl V, Smith R, et al. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res. 1994;54(7):1812–1817. | ||
Li M, Wu DB, Wang J. Effects of volume-activated chloride channels on the invasion and migration of human endometrial cancer cells. Eur J Gynaecol Oncol. 2013;34(1):60–64. | ||
Chiang PC, Chou RH, Chien HF, Tsai T, Chen CT. Chloride intracellular channel 4 involves in the reduced invasiveness of cancer cells treated by photodynamic therapy. Lasers Surg Med. 2013;45(1):38–47. | ||
Leanza L, Biasutto L, Manago A, Gulbins E, Zoratti M, Szabo I. Intracellular ion channels and cancer. Front Physiol. 2013;4:227. | ||
Qu Z, Yao W, Yao R, Liu X, Yu K, Hartzell C. The Ca(2+)-activated Cl(−) channel, ANO1 (TMEM16A), is a double-edged sword in cell proliferation and tumorigenesis. Cancer Med. 2014;3(3):453–461. | ||
Lee FT, Mountain AJ, Kelly MP, et al. Enhanced efficacy of radioimmunotherapy with 90Y-CHX-A″-DTPA-hu3S193 by inhibition of epidermal growth factor receptor (EGFR) signaling with EGFR tyrosine kinase inhibitor AG1478. Clin Cancer Res. 2005;11(19 Pt 2):7080s–7086s. | ||
Wang M, Yang H, Zheng LY, et al. Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation. 2012;125(5):697–707. | ||
Pedemonte N, Galietta LJ. Structure and function of TMEM16 proteins (anoctamins). Physiol Rev. 2014;94(2):419–459. | ||
Atala A. Re: inhibition of Ca2+-activated Cl-channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. J Urol. 2013;189(6):2393. | ||
Reis-Filho JS, Savage K, Lambros MB, et al. Cyclin D1 protein overexpression and CCND1 amplification in breast carcinomas: an immunohistochemical and chromogenic in situ hybridisation analysis. Mod Pathol. 2006;19(7):999–1009. | ||
Almaca J, Tian Y, Aldehni F, et al. TMEM16 proteins produce volume-regulated chloride currents that are reduced in mice lacking TMEM16A. J Biol Chem. 2009;284(42):28571–28578. | ||
Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011;25(6):634–645. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.