Back to Journals » OncoTargets and Therapy » Volume 11
Knockdown of HOXA transcript at the distal tip suppresses the growth and invasion and induces apoptosis of oral tongue squamous carcinoma cells
Authors Mu M, Li Y, Zhan Y, Li X, Zhang B
Received 18 May 2018
Accepted for publication 16 August 2018
Published 13 November 2018 Volume 2018:11 Pages 8033—8044
DOI https://doi.org/10.2147/OTT.S174637
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Leo Jen-Liang Su
Mingkui Mu,1 Yue Li,1 Yuanbo Zhan,2 Xin Li,3 Bin Zhang2,4
1Department of Orthodontics, The Second Affiliated Hospital of Harbin Medical University, Harbin 150081, People’s Republic of China; 2Institute of Hard Tissue Development and Regeneration, The Second Affiliated Hospital of Harbin Medical University, Harbin 150081, People’s Republic of China; 3Department of Stomatology, The Fourth Affiliated Hospital of Harbin Medical University, Harbin 150001, People’s Republic of China; 4Heilongjiang Academy of Medical Sciences, Harbin 150001, People’s Republic of China
Background: Oral tongue squamous cell carcinoma (OTSCC) is an aggressive cancer which has high mortality rates. HOXA transcript at the distal tip (HOTTIP) is a lncRNA that can be used as a prognostic marker in multiple carcinomas. The expression of HOTTIP is found to be elevated in OTSCC tissues, and such elevation is correlated with poor prognosis. However, its functional role in regulating the growth and metastasis of OTSCC cells remains elusive and requires further investigation.
Methods: HOTTIP-silenced OTSCC cells were established by inhibiting HOTTIP expression via its exclusive shRNA. Whether HOTTIP knockdown affected the aggressive tumor behaviors of OTSCC cells was investigated in vitro and in vivo.
Results: We found that HOTTIP shRNA restrained the cell proliferation and arrested the cell cycle at G1 phase in TSCCA and TCA8113 cells. The expression levels of cyclins B, D1, and E were downregulated in HOTTIP-silenced cells. HOTTIP silencing suppressed the growth of xenograft tumors. Moreover, the silencing of HOTTIP triggered apoptosis in TSCCA and TCA8113 cells and altered the expression of a group of apoptosis-related molecules: downregulated Bcl-2, upregulated Bax, and enhanced the cleavage of caspase 3 and PARP. Knockdown of HOTTIP also suppressed the migration, invasion, and epithelial–mesenchymal transition (EMT) of both TSCCA and TCA8113 cell lines.
Conclusion: Our findings suggest that HOTTIP is required by the OTSCC cells to maintain their growth and metastasis in vitro. It may serve as a promising potential candidate for OTSCC therapy.
Keywords: HOTTIP, oral tongue squamous cell carcinoma, proliferation, metastasis, apoptosis, tumorigenicity
Introduction
Oral tongue squamous cell carcinoma (OTSCC) is one of the most common malignancies, which has an unfavorable prognosis.1 The incidence of OTSCC is high globally, especially in Asian countries.2 The morbidity and fatality rates of OTSCC are still high, although improvements have been made in the current treatments (surgery, chemotherapy, and radiotherapy).3 At the early stages of OTSCC, 20% of the patients die. The strongest pathological predictors for recurrence and mortality are the depth of invasion and the worst pattern of invasion.4 Finding novel molecules involved in the malignant behaviors of OTSCC cells will help improve the therapeutic strategy.
lncRNAs are, by definition, a new class of RNAs with the length of >200 nucleotides. They do not serve as templates for protein coding.5 Increasing evidence has suggested that the abnormal expression of lncRNAs is associated with the initiation of many pathological changes during carcinogenesis, such as the deregulated proliferation, apoptosis, migration, and invasion of cancer cells.6,7 Homo sapiens’ HOXA transcript at the distal tip (HOTTIP) located at the 5′ tip of the HOXA locus was initially identified in primary fibroblasts of vertebrates by Wang et al.8 It can promote the trimethylation of histone H3 lysine 4 and upregulate the expression of multiple 5′ HOXA genes in cis.9 The elevated expression of HOTTIP has been observed in a number of cancers, including gastric,10 papillary thyroid,11 and pancreatic cancers.12 HOTTIP overexpression can facilitate tumor growth and metastasis, while its silencing can inhibit tumor growth and metastasis and induce apoptosis.13,14 There was only one study by Zhang et al15 on OTSCC, which showed a high expression of HOTTIP in tumor tissues. The mechanisms underlying HOTTIP’s role in OTSCC development are unclear.
Abnormal cell proliferation is one of the main mechanisms in carcinogenesis.16 Previous studies have indicated that specific shRNA against HOTTIP inhibits cancer cell proliferation by hindering the cell cycle progression in multiple types of cancer cells, such as lung cancer cells17 and esophageal squamous carcinoma cells.18 Whether HOTTIP also affects the cell cycle progression of OTSCC cells is unknown and needs further investigation. Epithelial–mesenchymal transition (EMT) is required by tumor cells to invade and migrate to distal organs. Suppression of EMT may prevent the tumor cells from acquiring a migratory potential.19 Therefore, in this study, not only the role of HOTTIP in cell proliferation was explored but also its role in the migration, invasion, and EMT of OTSCC cells was investigated.
The TSCCA and TCA8113 cells with stable low expression of HOTTIP were established, and the proliferation, cell cycle progression, and mobility of these cells were assessed. We found that HOTTIP-silenced OTSCC cells acquired a less aggressive tumor phenotype.
Materials and methods
Cell culture
Human OTSCC cell lines CAL-27, TSCCA, and TCA8113 were obtained from Zhongqiaoxinzhou (Shanghai, People’s Republic of China) and were cultured in minimum essential medium eagle (Gibco; Carlsbad, CA, USA), eagle’s minimum essential medium (ScienCell; San Diego, CA, USA), and 1640 (Gibco) containing 10% FBS (Hyclone; Logan, UT, USA), respectively. The cells were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
Construction of HOTTIP-silenced OTSCC cells
HOTTIP-specific shRNA (target sequence, 5′- CGAAGCGCTCAGTAAGAAC 3′) and negative control (NC) shRNA were synthesized and inserted into a shRNA expression vector, pRNA-H1.1 (GenScript; Nanjing, People’s Republic of China). Transfection of shRNA plasmids was mediated by lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Stable transfectants were screened by selecting cells resistant to 500 μg/mL of G418 (Invitrogen). The knockdown efficiencies of HOTTIP shRNAs were determined by quantitative real-time PCR.
Cell viability assay
TSCCA or TCA8113 (3×103 cells/well) cells were seeded and cultured in a 96-well plate at 37°C for 12, 24, 48, or 72 hours. Cell viability was analyzed by using a Cell Counting Kit-8 (CCK-8; KeyGEN BioTECH; Nanjing, People’s Republic of China) following the manufacturer’s protocol.
Doubling time assay
TSCCA or TCA8113 cells were seeded in 24-well plates at a density of 5×104 in triplicate and allowed to grow for 24, 48, or 72 hours. Then, the number of cells was counted using an hemocytometer. The cell doubling rate at each time point was calculated via the following formula:20,21 (t−ti)×log2/(logNt−logNi), where Nt and Ni are the cell numbers at specific time points t and ti, respectively. The mean cell doubling time was averaged from the three time points.
2D clonogenic assay
Cells were digested and seeded in 35 mm dishes (500 cells/well) at 37°C. After 40 days, cells were fixed in 4% paraformaldehyde (Sinopharm; Shanghai, People’s Republic of China) for 20 minutes and stained with Wright–Giemsa staining solution (Nanjing Jiancheng Bioengineering Institute; Nanjing, People’s Republic of China) for 5 minutes. A colony image was acquired under an inverted phase-contrast microscope (Motic; Kowloon, Hong Kong). The colony formation rates were determined as formed colonies/500.
Flow cytometry
For cell cycle analysis, cells seeded in 6-well plates at a density of 1×105 cells/well were fixed in 70% cold ethanol for 2 hours at 4°C, washed with PBS, and stained with propidium iodide (PI) solution (Beyotime Institute of Biotechnology; Haimen, Jiangsu, People’s Republic of China). Apoptotic cells were assessed using a PI/Annexin V-FITC apoptosis detection kit (Beyotime Institute of Biotechnology) according to the manufacturer’s protocol. All cell samples were subjected to flow cytometry (BD Biosciences; Franklin Lakes, NJ, USA).
Hoechst and immunofluorescence staining
TSCCA or TCA8113 cells were grown on coverslips placed in 12-well plates at a density of 1×105 cells/well. For Hoechst staining, cells were incubated with Hoechst (Beyotime Institute of Biotechnology) for 5 minutes. For immunofluorescence staining, cells were fixed with 4% paraformaldehyde (Sinopharm) and permeabilized with 0.1% Triton-X 100 (Beyotime Institute of Biotechnology) at room temperature. Subsequently, cells were blocked with goat serum (Solarbio; Beijing, People’s Republic of China) and incubated with E-cadherin antibody (Cell Signaling Technology; Danvers, MA, USA) at 4°C overnight and then with Cy-3-conjugated goat anti-rabbit IgG secondary antibody (Beyotime Institute of Biotechnology) for 60 minutes at room temperature. DAPI was used to stain cell nuclei. Cell images were taken under a fluorescence microscope (Olympus; Tokyo, Japan).
Cell migration assay
Cancer cell migration ability was determined by scratch wound-healing assay. Cells were seeded in 6-well plates at a density of 1×105 cells/well, grown until reaching a confluence of 90%, and then were treated with mitomycin C (1 μg/mL; Sigma-Aldrich; St Louis, MO, USA) for 1 hour. For wound-healing assay, a linear wound was generated across the cell monolayer by a sterile 200 μL pipette tip. Thereafter, the cell monolayer was washed with serum-free medium to remove floating cells or debris, and then maintained in a humidified atmosphere of 5% CO2 at 37°C for additional 24 hours. Images were taken at 0 hour and 24 hours under an inverted phase-contrast microscope (Motic). The cell migration rate was calculated as the percentage of closure.
Cell invasion assay
Transwell inserts with 8 μm pores (Corning; Cambridge, MA, USA) were coated with 40 μL pre-diluted matrigel (BD Biosciences), and 200 μL of cell suspension (1×104 cells/well) was added to the upper chambers. The lower chamber was filled with medium containing 20% FBS. After 48 hours, cells that passed through the chamber membrane were fixed with 4% paraformaldehyde (Sinopharm) for 20 minutes and stained with 0.5% crystal violet (Amresco; Dallas, TX, USA) for 5 minutes. The number of invasive cells was counted under a microscope (Motic).
Qualitative real-time PCR
Total cell RNAs were obtained using TRIpure Reagent (BioTeke; Beijing, People’s Republic of China), and cDNAs using the RNAs as template were obtained via Super M-MLV reverse transcriptase (BioTeke). A qualitative real-time PCR was performed using SYBR Green Mastermix (Solarbio), and specific primer pairs on an Exicycler™ 96 real-time PCR machine (Bioneer; Daejeon, Korea). The primers were as follows: HOTTIP forward, 5′-AATGTAAGTGTCGCCCAATA-3′, HOTTIP reverse, 5′-AGTCAGGGAGAAGGTAAA-3′; E-cadherin forward, 5′-GAACGCATTGCCACATACAC-3′, E-cadherin reverse, 5′-TGGTGTAAGCGATGGCGGCA-3′; GAPDH forward 5′-GAAGGTCGGAGTCAACGGAT-3′, GAPDH reverse 5′-CCTGGAAGATGGTGATGGGAT-3′. The relative quantification values were calculated via 2−ΔΔCT and normalized to GAPDH.
Western blotting
Whole-cell lysates were obtained using RIPA buffer (Beyotime Institute of Biotechnology) for protein extraction. Protein samples (10–40 μg) were loaded onto the SDS-PAGE gel and then transferred onto polyvinylidene difluoride membranes (Millipore; Burlington, MA, USA) by electroblotting. For immunoblotting, the membranes were incubated with primary antibodies including Bcl-2 (BOSTER; Wuhan, People’s Republic of China), Bax (Proteintech; Wuhan, People’s Republic of China), cyclins B, D1, E, cleaved-caspase-3, cleaved-PARP, and E-cadherin (Cell Signaling Technology) at 4°C overnight and then with HRP-conjugated goat anti-mouse (Beyotime Institute of Biotechnology) or anti-rabbit IgG (Beyotime Institute of Biotechnology) at 37°C for 45 minutes. ECL reagent (Beyotime Institute of Biotechnology) was used for protein blot visualization, and the band intensities were quantified by Gel-Pro-Analyzer. GAPDH was used as the internal reference.
Xenografts
In vivo, TSCCA and TCA8113 cells transduced with HOTTIP shRNA or NC shRNA were subcutaneously injected into the right flank of BALB/c nude mice at 2×106 cells per mouse (n=6 in each group). Tumor size was measured every 3 days, and tumor volumes were calculated following V=a2×b×0.5, where a is the tumor width and b is the tumor length. Then mice were sacrificed, and the weight of each tumor was recorded. All animal experiments were approved by the Experimental Animal Ethics Committee of the Second Affiliated Hospital of Harbin Medical University, and all procedures conformed to the National Institutes of Health guidelines for the care and use of laboratory animals and laboratory procedures.
H&E staining
The tumors were harvested on day 21 after tumor cell injection, fixed in 4% paraformaldehyde (Sinopharm), and embedded in paraffin. The 5 μm-thick sections were stained with H&E according to the standard protocol.
Statistical analysis
Data were represented as mean ± SD and analyzed with GraphPad Prism 6. P-values were calculated using ANOVA for multiple comparisons. A P-value of <0.05 was considered to be statistically significant (presented in each figure legend as *P<0.05, **P<0.01).
Results
Establishment of HOTTIP-silenced OTSCC cells
HOTTIP expression levels in CAL-27, TSCCA, and TCA8113 were first determined with real-time PCR. The results showed that TSCCA and TCA8113 cells had higher expression of HOTTIP (Figure 1A). Therefore, we decided to use these two cell lines for the following loss-of-function assay of HOTTIP. TSCCA and TCA8113 cells transfected with HOTTIP shRNA or NC shRNA were selected with G418. Cells resistant to G418 were subjected to real-time PCR analysis. Our data showed that the HOTTIP-specific shRNA, but not the NC shRNA, significantly reduced HOTTIP expression in TSCCA and TCA8113 cells (Figure 1B and C).
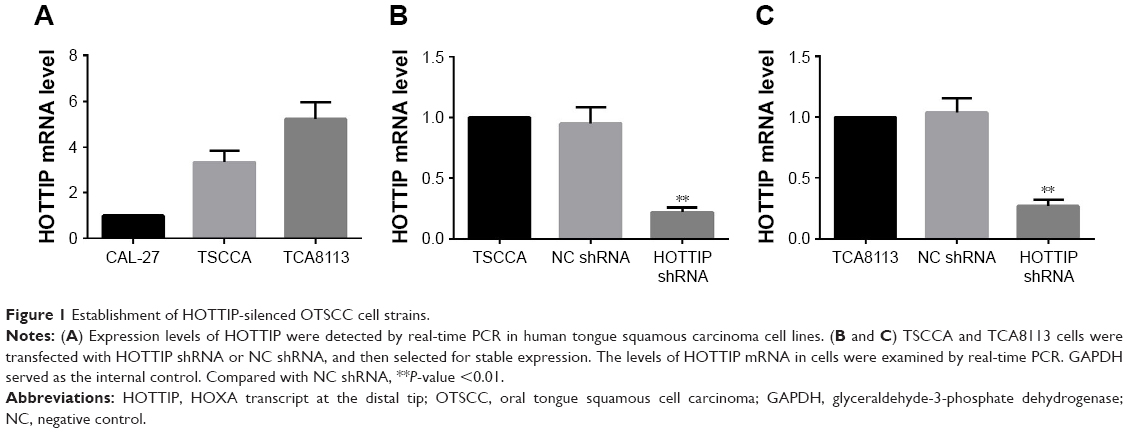 | Figure 1 Establishment of HOTTIP-silenced OTSCC cell strains. |
Suppression of HOTTIP inhibited OTSCC cell growth
The growth of TSCCA and TCA8113 cells transfected NC or HOTTIP shRNA was detected via CCK-8, cell counting, and colony formation assays. As shown in Figure 2A and B, knockdown of HOTTIP significantly delayed cell proliferation. HOTTIP silencing prolonged the cell doubling time from 27.13±3.02 hours to 43.45±2.83 hours in TSCCA cells and from 39.43±0.69 hours to 56.23±6.23 hours in TCA8113 cells (Figure 2C and D). Furthermore, inhibition of HOTTIP reduced the number of colonies by 70.20%±6.4% in TSCCA cells (Figure 2E) and by 74.93%±4.6% in TCA8113 cells (Figure 2F). In both cell lines, HOTTIP knockdown resulted in a delay of cell cycle progression. More cells were arrested in G1 phase when HOTTIP was knocked down (73.07%±2.27% vs 61.12%±4.34% = HOTTIP-silenced TSCCA cells vs NC cells, 69.11%±4.68% vs 57.72%±2.51% = HOTTIP-silenced TCA8113 cells vs NC cells; Figure 2G and H). In addition, Western blot analysis revealed that HOTTIP silencing reduced the protein expression of important cell cycle regulators, such as cyclin B (by 64.03%±3.90%), cyclin D1 (by 59.37%±9.55%), and cyclin E (by 78.98%±4.91%) in TSCCA cells (Figure 2I). Consistently, HOTTIP silencing downregulated the expression of cyclin B, cyclin D1, and cyclin E by 59.89%±4.78%, 63.75%±5.08%, and 70.33%±0.68% in TCA8113 cells (Figure 2J). These results demonstrated that HOTTIP silencing reduced the proliferation of OTSCC cells and arrested them at G1 phase.
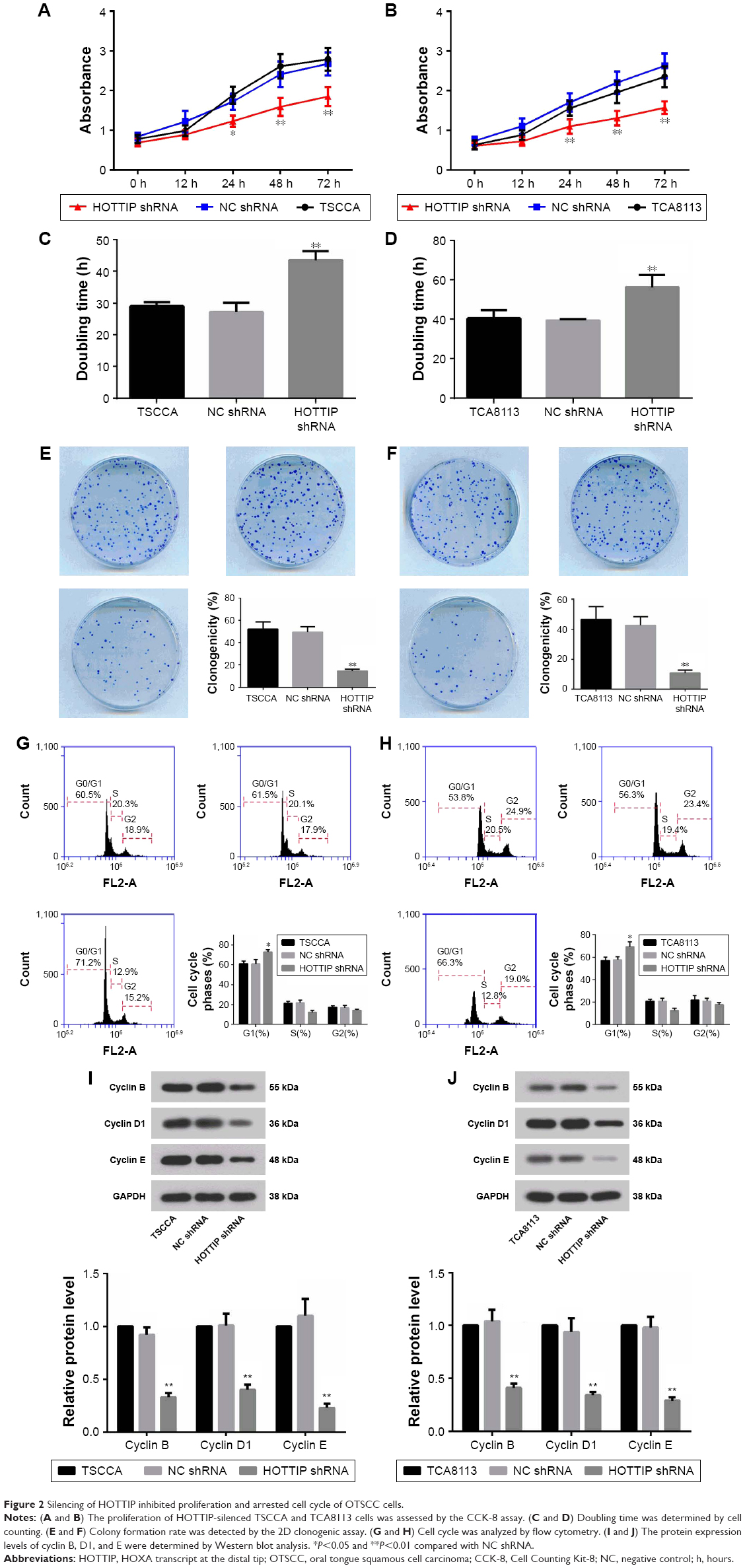 | Figure 2 Silencing of HOTTIP inhibited proliferation and arrested cell cycle of OTSCC cells. |
Silencing of HOTTIP induced cell apoptosis in OTSCC cells
Cell apoptosis was detected by flow cytometry using a PI/Annexin V-FITC apoptosis detection kit and by Hoechst staining. Figure 3A and B shows that knockdown of HOTTIP triggered apoptosis both in TSCCA and in TCA8113 cells. The percentage of apoptotic cells was increased from 3.12%±0.21% to 14.88%±2.03% in TSCCA cells and from 2.61%±0.27% to 21.22%±3.19% in TCA8113 cells when HOTTIP was silenced. Similarly, HOTTIP silencing robustly elevated the fraction of Hoechst-positive cells (Figure 3C and D). Furthermore, results from Western blot assay showed that silencing of HOTTIP increased the levels of pro-apoptotic proteins such as cleaved-caspase-3, Bax, and cleaved-PARP, whereas this downregulated anti-apoptotic Bcl-2 (Figure 3E and F).
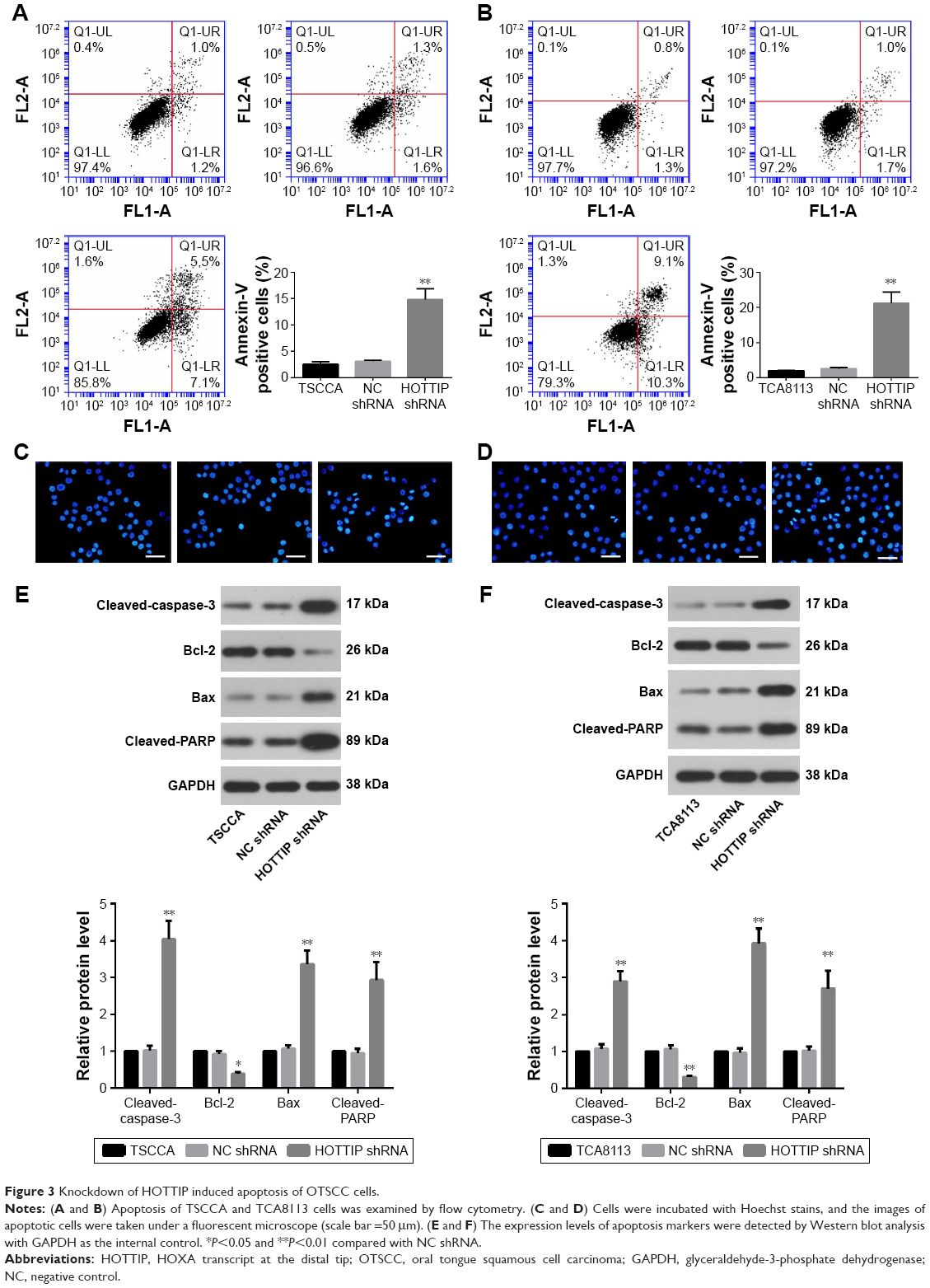 | Figure 3 Knockdown of HOTTIP induced apoptosis of OTSCC cells. |
Knockdown of HOTTIP inhibited cell migration and invasion in vitro
Scratch wound healing and matrigel-based transwell assays were used to analyze the motility and invasiveness of TSCCA and TCA8113 cells. As depicted in Figure 4A, HOTTIP shRNA significantly decreased the migratory and invasive ability of TSCCA cells by 55.83%±1.84% and 60.37%±7.80%, respectively. Similarly, knockdown of HOTTIP reduced motility by 30.93%±0.73% and invasiveness by 42.62%±8.92% in TCA8113 cells (Figure 4B). In addition, the expression of E-cadherin was measured by immunofluorescence assay and Western blotting. Compared with cells transfected with NC shRNA, E-cadherin expression was upregulated in the HOTTIP-silenced cells (Figure 4C–F).
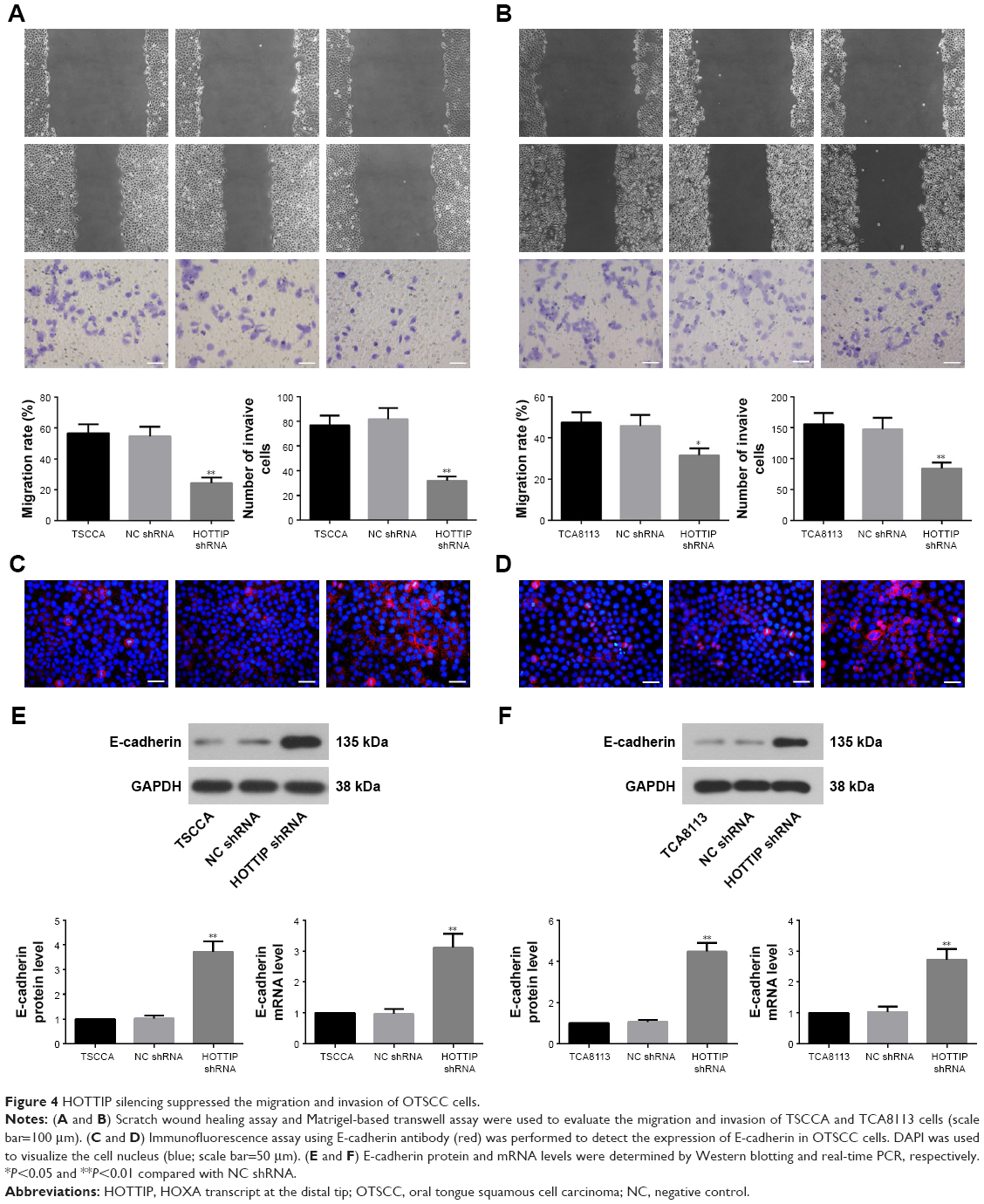 | Figure 4 HOTTIP silencing suppressed the migration and invasion of OTSCC cells. |
Silencing of HOTTIP suppressed xenograft growth in vivo
In order to validate the in vitro findings, the effect of HOTTIP downregulation on the growth of OTSCC cells was further analyzed in a xenograft tumor model. We found that HOTTIP silencing decreased the volumes of tumors formed by TSCCA cells by 81.73%±2.22% and those formed by TCA8113 cells by 69.02%±12.05% on day 21 post injection (Figure 5A and B). Meanwhile, tumor weight was also decreased in HOTTIP-silenced TSCCA and TCA8113 cells (Figure 5C and D). Furthermore, histological examination showed marked cytoplasm lysis and nuclear condensation in the tumor generated by HOTTIP-silenced cells (Figure 5E and F). Silencing of HOTTIP in the xenograft tumors was confirmed by qualitative real-time PCR (Figure 5G and H).
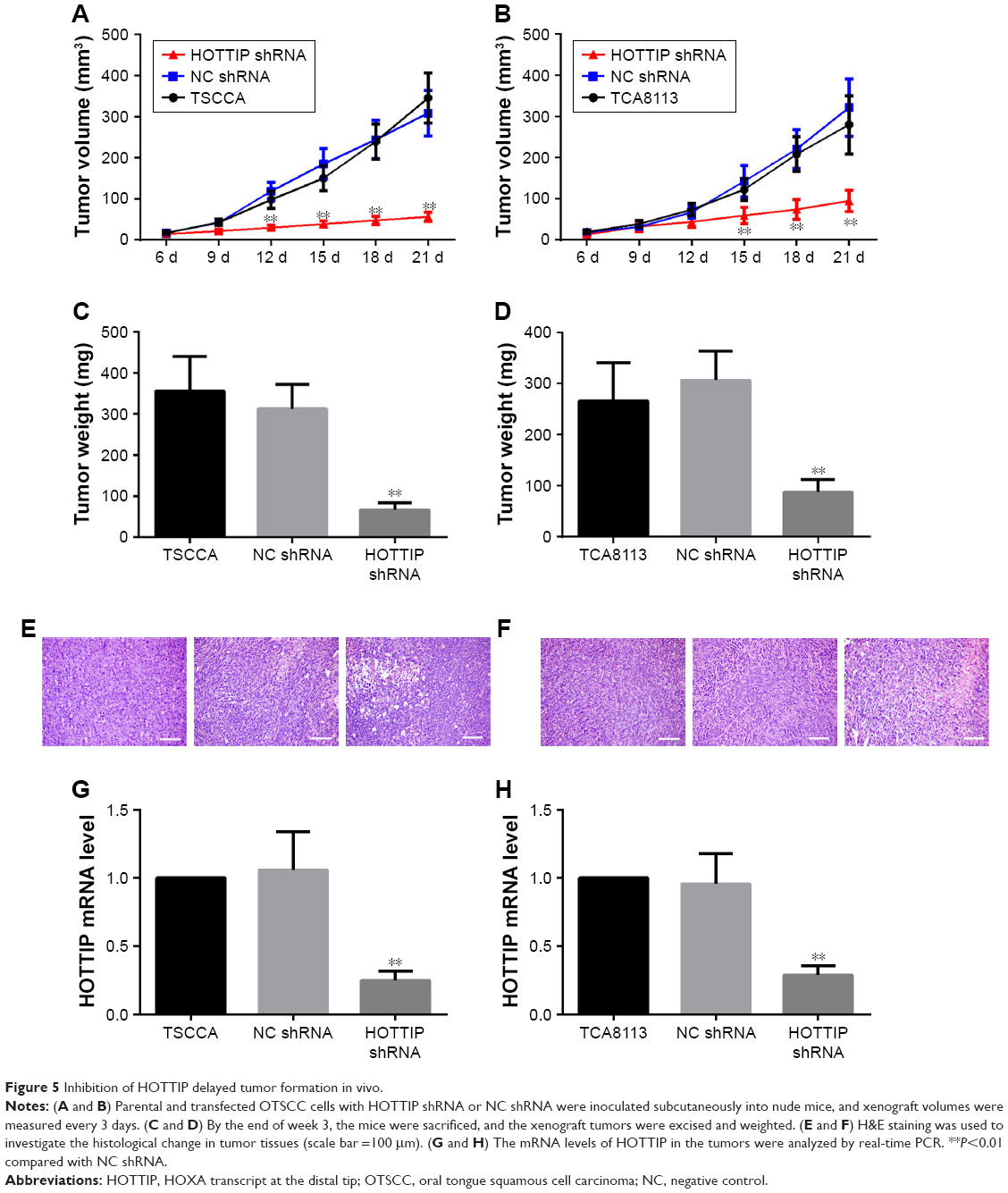 | Figure 5 Inhibition of HOTTIP delayed tumor formation in vivo. |
HOTTIP was a prognostic factor for patients with HNSC
OTSCC belongs to head and neck squamous carcinoma (HNSC). Gene expression profiling interactive analysis (GEPIA) was carried out to evaluate the potential of HOTTIP as a prognostic factor in HNSC. Gene expression profiling interactive analysis (GEPIA) was carried out, and the results were shown as boxplot and survival plot. As shown in Figure S1A, the expression level of HOTTIP was higher in tumor tissues than in nontumor tissues, although the data did not reach a significant level. Furthermore, as shown in Figure S1B, the overall survival rate of HNSC patients with high HOTTIP was significantly lower than those with low HOTTIP (P=0.016).
Discussion
Previous studies showed that HOTTIP is markedly increased in various cancer types, and such elevation is correlated with cancer progression. Liu et al13 reported that knockdown of HOTTIP inhibited colorectal cancer cell proliferation and migration by targeting SGK1. Lin et al14 identified HOTTIP as a potential diagnostic marker, and they demonstrated that HOTTIP modulated HOXA13 to facilitate the growth and metastasis of esophageal squamous carcinoma cells in vitro.HOTTIP has also been shown as a tumor cell growth-promoting molecule in lung adenocarcinoma22 and prostate cancer.23 The expression of HOTTIP was high in OTSCC tissues.15 Therefore, loss-of-function study was performed to investigate the role of HOTTIP in the development of OTSCC. We found that two OTSCC cell lines with stable low HOTTIP expression acquired a less malignant phenotype.
Excessive tumor cell proliferation due to abnormal cell cycle is the most important cause of mortality in cancer. The precise control of cell cycle – from gap phase 1 to DNA synthesis phase (G1/S) and from gap phase 2 to mitosis phase (G2/M) – ensures cells to proliferate and divide in a programmed manner. Cyclin family proteins, including cyclins B, D, and E, work in cooperation with cyclin-dependent kinases to orchestrate the cell cycle.24 HOTTIP silencing arrested osteosarcoma cells at G1 stage25 and decreased the expression of cyclins B1 and D1 in lung and prostate cancer cells.17,26 However, in glioma cells, the HOTTIP overexpression, but not its silencing, significantly decreased cyclin A expression and inhibited cell growth.27 These previous findings suggest that HOTTIP may have a discrepant regulatory role in various cancers. In OTSCC cells, our data were in accordance with the previous studies reporting a hindered cell cycle of HOTTIP-silenced cancer cells. To fully elucidate the role of HOTTIP in OTSCC progression, there is a need to upregulate its expression in OTSCC cells with low basal HOTTIP expression, which will be carried out in the near future by many groups. In mammary cancer cells28 and endometrial cancer,29 the forced expression of HOTTIP could accelerate cell cycle progression and enhance cell proliferation. As the silencing of HOTTIP had antiproliferative effects in OTSCC cells, it is possible that its overexpression may have pro-proliferative effects.
Escape of apoptosis is regarded as a hallmark for therapy failure in cancer.30 Knockdown of HOTTIP has been shown to induce apoptosis by increasing Bax expression and decreasing Bcl-2 expression in prostate cancer cells.26 Sun et al31 revealed that HOTTIP acted as a competitive endogenous RNA for microRNA-216a, thus preventing Bcl-2 from binding microRNA-216a in lung cancer cells.Our study reveals that HOTTIP knockdown triggered cancer cell apoptosis and altered the expression of Bax and Bcl-2 which was in agreement with the earlier studies. The cleavage of caspase-3 and PARP indicates an activated apoptosis cascade.32,33 The elevation of cleaved-caspase-3 and cleaved-PARP in HOTTIP-silenced cells further confirmed the cell apoptosis results detected with PI/Annexin V-FITC and Hoechst assays. The underlying mechanisms require further investigation.
We next explored the migratory and invasive abilities of OTSCC cells in response to HOTTIP knockdown. Here, we found that silencing of HOTTIP inhibited the migration and invasion of TSCCA and TCA8113 cells. Similar results have also been reported in breast cancer cells34 and osteosarcoma cells,35 which support our current findings. The enhanced mobility of tumor cells is often associated with the accelerated EMT. EMT not only plays an important role in embryonic development but also drives the dissemination of cancer cells.36 Lin et al14 showed that HOTTIP promoted EMT by competitively binding to miR-30b and upregulated the expression of snail 1 in esophageal squamous carcinoma cells. Our study showed an E-cadherin upregulation in HOTTIP-silenced cells. This finding suggests that the inhibitory effects of HOTTIP shRNA on the migration and invasion of OTSCC cells at least attribute to EMT suppression.
As the knockdown of HOTTIP can impair the malignant behaviors of OTSCC cells, it is expected that the tumorigenicity of HOTTIP-silenced cells is decreased in vivo. As expected, we found that the growth of tumors in nude mice formed by the HOTTIP-silenced OTSCC cells was slower than that formed by the control cells. The anti-growth effects induced by HOTTIP silencing in vivo were also previously demonstrated in colorectal cancer cells.37 Therefore, HOTTIP may serve as a new therapeutic target in OTSCC.
The database of GEPIA integrates data from the Cancer Genome Atlas and Genotype-Tissue Expression. By analyzing the expression of HOTTIP with the clinic information of HNSC patients summarized in GEPIA, we found that the overall survival rate of HNSC patients with high HOTTIP was significantly lower than those with low HOTTIP. These data revealed HOTTIP as a prognostic indicator in HNSC.
Conclusion
HOTTIP plays a critical role in regulating cell proliferation, migration, invasion, apoptosis, and tumorigenesis of OTSCC cells. Our data define HOTTIP as a potential target for cancer therapy.
Acknowledgment
This study was supported by a grant from the Harbin Science and Technology Innovation Talent Project (grant no 2013RFQYJ084).
Disclosure
The authors report no conflicts of interest in this work.
References
Bello IO, Soini Y, Salo T. Prognostic evaluation of oral tongue cancer: means, markers and perspectives (I). Oral Oncol. 2010;46(9):630–635. | ||
Krishna Rao SV, Mejia G, Roberts-Thomson K, Logan R. Epidemiology of oral cancer in Asia in the past decade – an update (2000–2012). Asian Pac J Cancer Prev. 2013;14(10):5567–5577. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
Almangush A, Bello IO, Coletta RD, et al. For early-stage oral tongue cancer, depth of invasion and worst pattern of invasion are the strongest pathological predictors for locoregional recurrence and mortality. Virchows Arch. 2015;467(1):39–46. | ||
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–346. | ||
Chen X, Fan S, Song E. Noncoding RNAs: New Players in Cancers. Adv Exp Med Biol. 2016;927:1–47. | ||
Serghiou S, Kyriakopoulou A, Ioannidis JP. Long noncoding RNAs as novel predictors of survival in human cancer: a systematic review and meta-analysis. Mol Cancer. 2016;15(1):50. | ||
Wang KC, Yang YW, Liu B, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472(7341):120–124. | ||
Burgess DJ. Non-coding RNA: HOTTIP goes the distance. Nat Rev Genet. 2011;12(5):300. | ||
Zhao R, Zhang Y, Zhang X, et al. Exosomal long noncoding RNA HOTTIP as potential novel diagnostic and prognostic biomarker test for gastric cancer. Mol Cancer. 2018;17(1):68. | ||
Yuan Q, Liu Y, Fan Y, et al. LncRNA HOTTIP promotes papillary thyroid carcinoma cell proliferation, invasion and migration by regulating miR-637. Int J Biochem Cell Biol. 2018;98:1–9. | ||
Li Z, Zhao X, Zhou Y, et al. The long non-coding RNA HOTTIP promotes progression and gemcitabine resistance by regulating HOXA13 in pancreatic cancer. J Transl Med. 2015;13:84. | ||
Liu T, Yu T, Hu H, He K. Knockdown of the long non-coding RNA HOTTIP inhibits colorectal cancer cell proliferation and migration and induces apoptosis by targeting SGK1. Biomed Pharmacother. 2018;98:286–296. | ||
Lin C, Wang Y, Wang Y, et al. Transcriptional and posttranscriptional regulation of HOXA13 by lncRNA HOTTIP facilitates tumorigenesis and metastasis in esophageal squamous carcinoma cells. Oncogene. 2017;36(38):5392–5406. | ||
Zhang H, Zhao L, Wang YX, Xi M, Liu SL, Luo LL. Long non-coding RNA HOTTIP is correlated with progression and prognosis in tongue squamous cell carcinoma. Tumour Biol. 2015;36(11):8805–8809. | ||
Ait-Oudhia S, Mager DE. Array of translational systems pharmacodynamic models of anti-cancer drugs. J Pharmacokinet Pharmacodyn. 2016;43(6):549–565. | ||
Deng HP, Chen L, Fan T, Zhang B, Xu Y, Geng Q. Long non-coding RNA HOTTIP promotes tumor growth and inhibits cell apoptosis in lung cancer. Cell Mol Biol. 2015;61(4):34–40. | ||
Chen X, Han H, Li Y, Zhang Q, Mo K, Chen S. Upregulation of long noncoding RNA HOTTIP promotes metastasis of esophageal squamous cell carcinoma via induction of EMT. Oncotarget. 2016;7(51):84480–84485. | ||
Izdebska M, Zielińska W, Grzanka D, Gagat M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. Biomed Res Int. 2018;2018:4578373–13. | ||
Yang YK, Ogando CR, Wang See C, Chang TY, Barabino GA. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res Ther. 2018;9(1):131. | ||
Stepp MW, Doll MA, Carlisle SM, States JC, Hein DW. Genetic and small molecule inhibition of arylamine N-acetyltransferase 1 reduces anchorage-independent growth in human breast cancer cell line MDA-MB-231. Mol Carcinog. 2018;57(4):549–558. | ||
Zhang GJ, Song W, Song Y. Overexpression of HOTTIP promotes proliferation and drug resistance of lung adenocarcinoma by regulating AKT signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21(24):5683–5690. | ||
Yang B, Gao G, Wang Z, et al. Long non-coding RNA HOTTIP promotes prostate cancer cells proliferation and migration by sponging miR-216a-5p. Biosci Rep. 2018:BSR20180566. | ||
Sánchez I, Dynlacht BD. New insights into cyclins, CDKs, and cell cycle control. Semin Cell Dev Biol. 2005;16(3):311–321. | ||
Li Z, Zhao L, Wang Q. Overexpression of long non-coding RNA HOTTIP increases chemoresistance of osteosarcoma cell by activating the Wnt/β-catenin pathway. Am J Transl Res. 2016;8(5):2385–2393. | ||
Zhang SR, Yang JK, Xie JK, Zhao LC. Long noncoding RNA HOTTIP contributes to the progression of prostate cancer by regulating HOXA13. Cell Mol Biol (Noisy-le-grand). 2016;62(3):84–88. | ||
Xu LM, Chen L, Li F, et al. Over-expression of the long non-coding RNA HOTTIP inhibits glioma cell growth by BRE. J Exp Clin Cancer Res. 2016;35(1):162. | ||
Gao W, Wu XL, Li DZ, Liu HD. HOTTIP participates in mammary cancer by promoting cell proliferation via PI3K/AKT pathway. Eur Rev Med Pharmacol Sci. 2018;22(13):4181–4187. | ||
Guan Q, Zhang Q, Zhang C, Liu Q, Ren QL. HOTTIP regulates progression of endometrial cancer via activating PI3K/AKT pathway. Eur Rev Med Pharmacol Sci. 2018;22(12):3727–3733. | ||
Villanova L, Careccia S, De Maria R, Fiori ME. Micro-Economics of Apoptosis in Cancer: ncRNAs Modulation of BCL-2 Family Members. Int J Mol Sci. 2018;19(4):958. | ||
Sun Y, Hu B, Wang Q, et al. Long non-coding RNA HOTTIP promotes BCL-2 expression and induces chemoresistance in small cell lung cancer by sponging miR-216a. Cell Death Dis. 2018;9(2):85. | ||
Ola MS, Nawaz M, Ahsan H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol Cell Biochem. 2011;351(1–2):41–58. | ||
Soldani C, Scovassi AI. Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis. 2002;7(4):321–328. | ||
Sun Y, Zeng C, Gan S, et al. LncRNA HOTTIP-Mediated HOXA11 Expression Promotes Cell Growth, Migration and Inhibits Cell Apoptosis in Breast Cancer. Int J Mol Sci. 2018;19(2):472. | ||
Li F, Cao L, Hang D, Wang F, Wang Q. Long non-coding RNA HOTTIP is up-regulated and associated with poor prognosis in patients with osteosarcoma. Int J Clin Exp Pathol. 2015;8(9):11414–11420. | ||
Wei SC, Yang J. Forcing through Tumor Metastasis: The Interplay between Tissue Rigidity and Epithelial-Mesenchymal Transition. Trends Cell Biol. 2016;26(2):111–120. | ||
Lian Y, Ding J, Zhang Z, et al. The long noncoding RNA HOXA transcript at the distal tip promotes colorectal cancer growth partially via silencing of p21 expression. Tumour Biol. 2016;37(6):7431–7440. |
Supplementary material
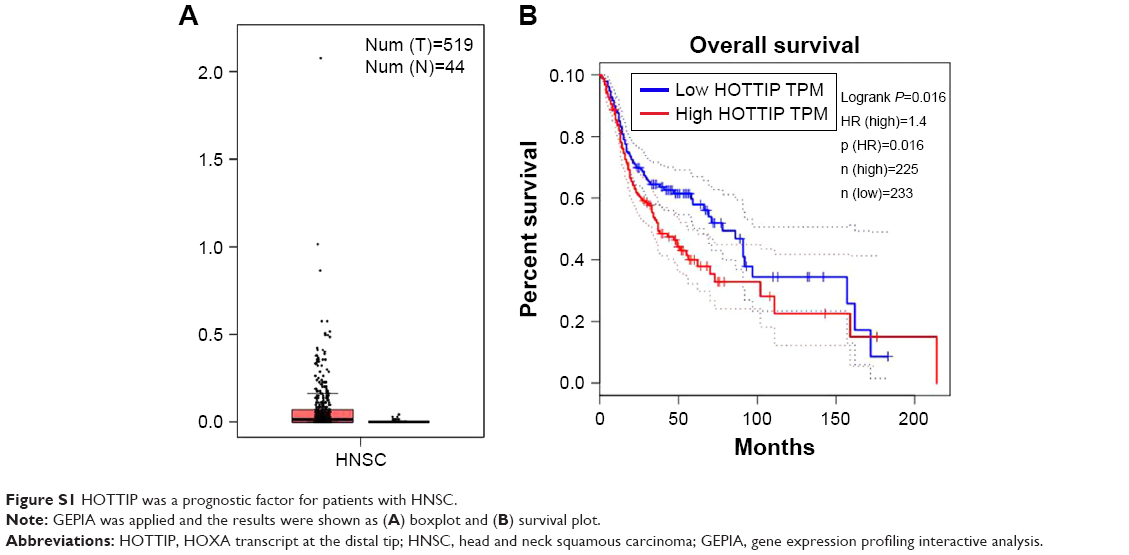 | Figure S1 HOTTIP was a prognostic factor for patients with HNSC. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.