Back to Journals » OncoTargets and Therapy » Volume 12
Knockdown of EIF3C promotes human U-2OS cells apoptosis through increased CAS P3/7 and Chk1/2 by upregulating SAPK/JNK
Authors Gao WL, Hu Y ![]() , Zhang ZQ, Du GW, Yin L, Yin ZS
, Zhang ZQ, Du GW, Yin L, Yin ZS
Received 11 September 2018
Accepted for publication 17 January 2019
Published 14 February 2019 Volume 2019:12 Pages 1225—1235
DOI https://doi.org/10.2147/OTT.S187209
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Takuya Aoki
Weilu Gao,1 Yong Hu,1 Zhengqin Zhang,2 Gongwen Du,1 Li Yin,1 Zongsheng Yin1
1Department of Orthopaedics, The First Affiliated Hospital of Anhui Medical University, Hefei 230022, China; 2Department of Anesthesiology, The First Affiliated Hospital of Anhui Medical University, Hefei 230022, China
Background: As a component of the EIF3 complex, EIF3C is essential for several steps in protein synthesis initiation. Recently, it has been addressed that EIF3C is overexpressed in several human cancers and plays a pivotal role in cell proliferation and tumorigenesis.
Materials and methods: Immunohistochemistry, quantitative real-time PCR (qPCR), and Western blotting assays were employed to determine the expression of EIF3C in osteosarcoma (OsC) tissues obtained from 60 patients. The levels of EIF3C mRNA and protein were assessed by qPCR and Western blotting, respectively. The effect of EIF3C knockdown on OsC cell proliferation was detected by MTT and colony formation assays, respectively. Cell apoptosis induced by EIF3C silencing was analyzed by flow cytometric analysis. PathScan stress and apoptosis signaling antibody array kit was used to analyze the potential effects of EIF3C knockdown on OsC cells.
Results: The levels of EIF3C were high in OsC tissues and cell lines. In addition, EIF3C knockdown by lentivirus-mediated shRNA targeting EIF3C significantly suppressed cell proliferation and colony formation and induced apoptosis in U-2OS cells. Moreover, EIF3C knockdown led to the upregulated expression of CASP3/7, Chk1/2, and SAPK/JNK, indicating that the downregulated expression of EIF3C might be associated with pro-apoptosis of U-2OS cells.
Conclusion: EIF3C may be a promising target for gene therapy of human OsC. However, the precise mechanisms behind the effect of EIF3C on OsC tumorigenesis require further analysis.
Keywords: apoptosis, caspase, checkpoint kinase, osteosarcoma, proliferation, SAPK/JNK, U-2OS
Introduction
Osteosarcoma (OsC), also known as “osteogenic sarcoma”, is the most frequent type of primary bone tumor. OsC is the second most leading cause of cancer-related deaths in adolescents and children and accounts for ~20% of all primary bone cancers.1–4 Treatment of OsC includes neoadjuvant and postoperative adjuvant chemotherapy, and although some improvements have been achieved in effectively curing the disease, OsC still remains a devastating disease with poor early diagnosis and multidrug resistance of OsC cells.5 For OsC patients, the 5-year survival rate is <40%.6,7 Therefore, it is of utmost importance to elucidate the molecular mechanisms underlying the development and progression of OsC, as well as to identify novel therapeutic targets and therapeutic approaches to treat this disease.
Translation is an essentially fundamental process that can be divided into three steps: initiation, elongation, and termination. During the initiation step, the EIF3 complex is responsible for stabilizing the 43S pre-initiation complex by interacting directly with eIF1, eIF2, eIF5, and the 40S ribosomal subunit.8,9 EIF3 is the largest mammalian scaffolding initiation factor and contains 13 subunits that are designated as EIF3A–3M.9 Among these subunits, EIF3C is an essential subunit that allows for the assembly of the EIF3 complex.10,11 Increasing evidence has revealed that alterations in the expression of EIF3C are associated with oncogenic properties;12 for instance, EIF3C was found to be overexpressed in seminomas13 and meningiomas.14 Additionally, it has been demonstrated that EIF3C is critical for proliferation of human colon cancer cells,15 glioma cells,16,17 and breast cancer cells.18 However, little is known about the role of EIF3C in human OsC.
In the present study, we first evaluated the expression of EIF3C in human OsC tissues and cell lines. Next, we used RNA interference technology in OsC U-2OS cells to determine the role of EIF3C in tumor proliferation, colony formation, and apoptosis. Finally, we used the PathScan stress and apoptosis signaling antibody array kit to determine the potential of EIF3C silencing to inhibit tumorigenesis in human OsC.
Materials and methods
Patients and samples
In the present study, 60 patients with OsC treated at the First Affiliated Hospital of Anhui Medical University between 2013 and 2016 were enrolled. The study was approved by the Medical Ethics Committee of the First Affiliated Hospital of Anhui Medical University. All the patients provided written informed consent, and the study was conducted in accordance with the Declaration of Helsinki. Tumor specimens and para-carcinoma tissues (referred to as normal tissues, at least 1.0 cm apart from the visible cancerous tissues) were used for detecting the levels of EIF3C expression by immunohistochemistry (IHC; paraffin-embedded tissues) and quantitative real-time PCR (qPCR; frozen tissues) assays as described below.
Immunohistochemistry
IHC was carried out on formalin-fixed, paraffin-embedded tissues. The slices (5 μm thick) were prepared for incubating sequentially with primary EIF3C antibody (1:500, catalog no PA5-62137; Thermo Fisher Scientific, Waltham, MA, USA). The experimental procedure and immunostaining scoring of EIF3C expression were conducted as previously described.19
Cell lines
Two human OsC cell lines, U-2OS and Soas-2, were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). Human lung fibroblasts (MRC-5 cell line) were provided by Prof Shengquan Zhang (Department of Biochemistry and Molecular Biology, Anhui Medical University) as a gift. Cells were maintained in Roswell Park Memorial Institute 1640 medium (Thermo Fisher Scientific) supplemented with 10% FBS, 2 mM L-glutamine, penicillin (100 U/mL), and streptomycin (100 μg/mL) (Sangon Biotech, Shanghai, China). Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Construction and transfection of shEIF3C lentivirus
For manipulating EIF3C expression, a lentiviral system containing shRNA was used. A candidate siRNA specifically targeting human EIF3C (GenBank Accession No NM_003752) was designed to target the sequence 5′-CCA TCC GTA ATG CCA TGA A-3′. In addition, a scramble sequence that served as the negative control shRNA was included in the study: 5′-TTC TCC GAA CGT GTC ACG T-3′. Stem-loop DNA oligonucleotides were synthesized and cloned into the lentiviral pGV115-GFP vector (GeneChem, Shanghai, China). An EIF3C shRNA-expressing lentivirus (shEIF3C) and scramble control siRNA-expressing lentivirus (shCtrl) were prepared using a Lentivector Expression System (GeneChem).
For lentiviral infection, U-2OS cells (1×105) were cultured in six-well plates and infected with shEIF3C or shCtrl lentivirus using a multiplicity of infection of 80. After 72 hours of infection, GFP expression was examined under a fluorescence microscope (Olympus XI-71; Tokyo, Japan). After 120 hours of infection, U-2OS cells were harvested and quantitative reverse transcription PCR (qRT-PCR) and Western blot analysis were performed to determine knockdown efficiency.
RNA extraction and qRT-PCR
Total RNA was extracted from human OsC tissues and cells using the TRIzol method (Thermo Fisher Scientific) under RNase-free conditions and according to the manufacturer’s guidelines. From each sample, 2 μg of total RNA was reverse-transcribed using M-MuLV reverse transcriptase and Oligo (dT) primers (Sangon Biotech) to generate single-stranded cDNA. To quantify EIF3C expression in U-2OS cells infected with shEIF3C lentivirus, qPCR was conducted using SYBR Green Premix Ex Taq (Takara, Dalian, China) at an ABI 7500 system (Thermo Fisher Scientific). The housekeeping gene GAPDH was used as an internal control. One microgram of cDNA was used as a template for PCR. The primers used for qPCR were as follows: GAPDH forward, 5′-TGA CTT CAA CAG CGA CAC CCA-3′ and reverse, 5′-CAC CCT GTT GCT GTA GCC AAA-3′ (product size: 121 bp); and EIF3C forward, 5′-AGA TGA GGA TGA GGA TGA GGA C-3′ and reverse, 5′-GGA ATC GGA AGA TGT GGA ACC-3′ (product size: 175 bp). The expression of EIF3C was normalized to GAPDH expression, and data analysis was conducted using the 2−ΔΔCT method.
Cell proliferation assay
Cell proliferation was measured using a multi-parametric high-content screening approach as described by Liu et al.20 After infection with either shEIF3C or shCtrl lentivirus, U-2OS cells in the logarithmic phase were reseeded at a density of 2×103 cells/well into 96-well plates. Cells were cultured at 37°C with 5% CO2 for 5 days. Subsequently, cells were counted on a daily basis using a Celigo® Cell Imaging Cytometer (Nexcelom Bioscience, Boston, MA, USA). At least 800 cells/well were analyzed for each experiment, and per-sample counting was performed in triplicate.
In addition, cell proliferation was evaluated by MTT assay as described previously.21 Briefly, at 1, 2, 3, 4, or 5 days postinfection, cells were incubated with 20 μL of MTT reagent (5 mg/mL; Gen-View Scientific Inc, Tallahassee, FL, USA) for 4 hours. Supernatants were removed by centrifugating at 1,800 rpm for 10 minutes at room temperature, and then the formazan product was dissolved using 150 μL/well of dimethyl sulfoxide (Sangon Biotech) for 10 minutes. The absorbance was read at 490 nm with ELx800 Absorbance Microplate Reader (BioTek Instruments, Winooski, VT, USA). All experiments were performed in triplicate.
Apoptosis assay
For the detection of apoptotic cells, an Annexin V-APC apoptosis detection kit (eBioscience, San Diego, CA, USA) was used. Briefly, U-2OS cells were infected with shEIF3C or shCtrl lentivirus, incubated for 96 hours, collected, and washed with PBS. The cells were adjusted to a final density of 1×106/mL in staining buffer, and 100 μL of the cell suspension was incubated with 5 μL APC-conjugated Annexin V for 15 minutes at room temperature in the dark. Cell apoptosis was analyzed by flow cytometry using an FACS Calibur (Becton-Dickinson, Franklin Lakes, NJ, USA). All experiments were performed in triplicate.
Colony formation assay
U-2OS cells transfected with shEIF3C or shCtrl lentivirus were cultured for 2 days until the logarithmic phase was reached. Subsequently, cells were reseeded at a density of 600 cells/well in six-well plates in triplicate and cultured for 2 weeks at 37°C with 5% CO2. Cells were fixed for 60 minutes in 4% paraformaldehyde at room temperature and stained with Giemsa stain (Dingguo Biotech, Shanghai, China) for 20 minutes. After rinsing with distilled H2O, images were taken using a fluorescence microscope (Olympus XI-71) and cell colonies were counted.
Western blotting analysis
To evaluate shEIF3C knockdown efficiency in U-2OS cells, Western blot analysis was employed to determine the levels of EIF3C expression. Briefly, U-2OS cells transfected with shEIF3C or shCtrl lentivirus were cultured for 96 hours, collected, and lysed in ice-cold lysis buffer (Sangon Biotech). Cell lysates were centrifuged at 12,000 rpm for 10 minutes at 4°C, and supernatants were harvested. Protein concentration was determined using the BCA protein assay kit (Sangon Biotech). For each sample, equal amounts of total protein were separated by SDS-PAGE using a 12.5% polyacrylamide gel and transferred to a polyvinylidene fluoride membrane (Sangon Biotech). Membranes were blocked with 5% skimmed milk in Tris-buffered saline containing Tween-20 for 1 hour and incubated overnight at 4°C with anti-EIF3C mouse antibody (catalog no E6283; Sigma-Aldrich Co., St Louis, MO, USA) or anti-GAPDH (catalog no sc-32233; Santa Cruz Biotechnology Inc., Dallas, TX, USA) at 1:2,000 dilution. Membranes were incubated using horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (catalog no sc-2005; Santa Cruz Biotechnology) and visualized using an enhanced chemiluminescence (ECL) reagent (catalog no 32106, Pierce™ ECL Western Blotting Substrate; Pierce Biotech, Inc., Rockford, IL, USA). Gray values of bands were measured using ImageJ software.
PathScan analysis
PathScan analysis was performed using the PathScan® stress and apoptosis signaling antibody array kit (catalog no 12923; Cell Signaling Technology, Danvers, MA, USA) according to the manufacturer’s instructions. Briefly, cells transfected with shEIF3C or shCtrl lentivirus were lysed with 1X Cell Lysis Buffer (catalog no 7018; Cell Signaling Technology). After collection of whole-cell lysates, protein concentration was determined using a BCA protein assay kit (Sangon Biotech). Proteins, at equal concentration, were analyzed by a PathScan sandwich ELISA kit according to the manufacturer’s instructions. The array target map used can be found at the manufacturer’s homepage (http://www.cst-c.com.cn/products/12856.html). Slides were imaged using a ChemiScope5300 Pro Integrated chemiluminescence imaging system (CLiNX Science Instruments, Shanghai, China).
Statistical analyses
Data were analyzed using SPSS software version 16.0, and all experiments were performed in triplicate. Differences between groups were analyzed using Student’s t-test. Categorical data were evaluated by chi-squared test. P<0.05 was considered statistically significant.
Results
Relationship between EIF3C expression and clinicopathological characteristics of patients with OsC
High EIF3C mRNA expression was measured in OsC tissues (Figure 1A), and high EIF3C immunoreactivity was also observed in cancerous tissues with 70% of tumors expressing EIF3C (Figure 1B and Table 1). However, there was no significant relationship between EIF3C expression and clinicopathological features of patients enrolled in our study, including age, pathological grade, TNM stage, and lymph node metastasis (Table 1).
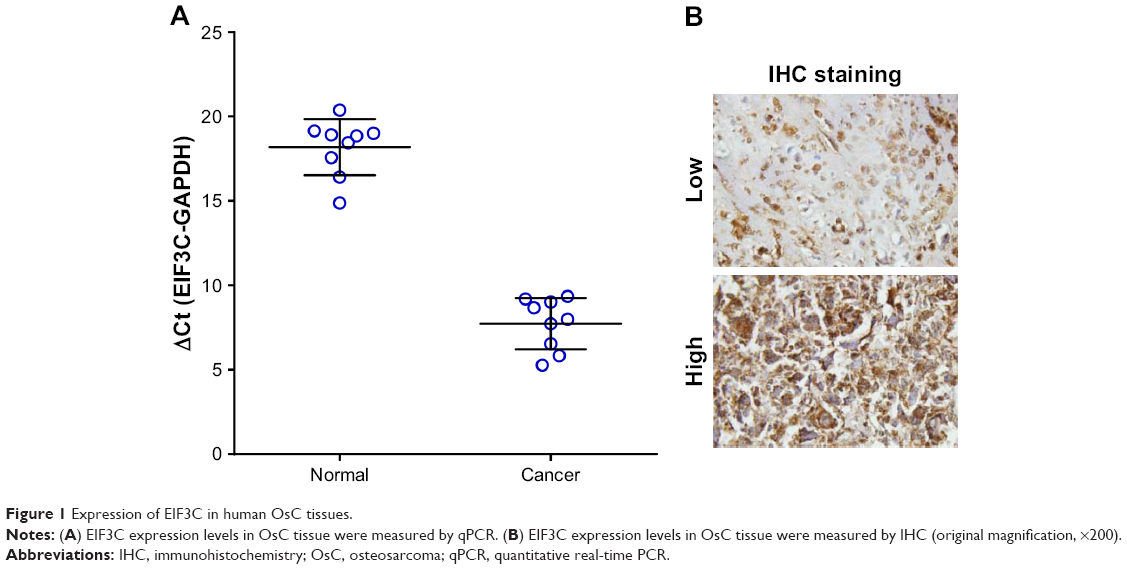 | Figure 1 Expression of EIF3C in human OsC tissues. |
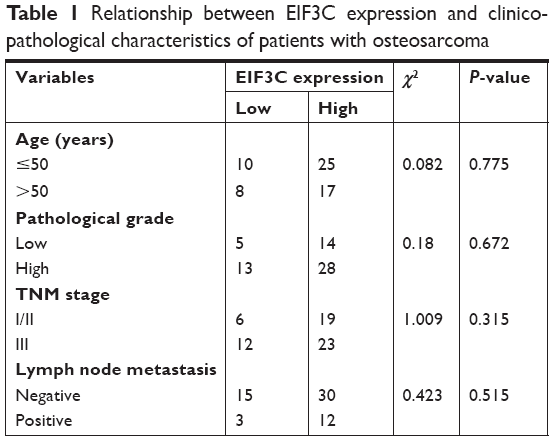 | Table 1 Relationship between EIF3C expression and clinicopathological characteristics of patients with osteosarcoma |
Lentivirus-mediated siRNA efficiently inhibits EIF3C expression in human OsC U-2OS cells
In malignant human OsC cell lines, U-2OS and Saos-2, high expression levels of EIF3C were detected by qPCR (Figure 2A) and verified by Western blotting (Figure 2B). To evaluate the effect of knocking down EIF3C expression using lentivirus-mediated siRNA, the OsC cell line U-2OS was transfected with shEIF3C or shCtrl lentivirus. Twenty-four hours after transfecting with shEIF3C or shCtrl lentivirus, cell proliferation was found to be unaffected (Figure 2D). EIF3C mRNA levels were determined using qPCR analysis, which demonstrated that U-2OS cells treated with shEIF3C lentivirus showed an ~52.7% reduction in EIF3C mRNA expression (Figure 2C). Western blot analysis was consistent with the mRNA data, and quantification of the protein bands also showed that shEIF3C lentivirus-treated cells had reduced EIF3C protein levels (Figure 2E and F). Together, these data demonstrated that EIF3C expression levels were successfully downregulated by shEIF3C lentivirus infection at both the mRNA and protein levels.
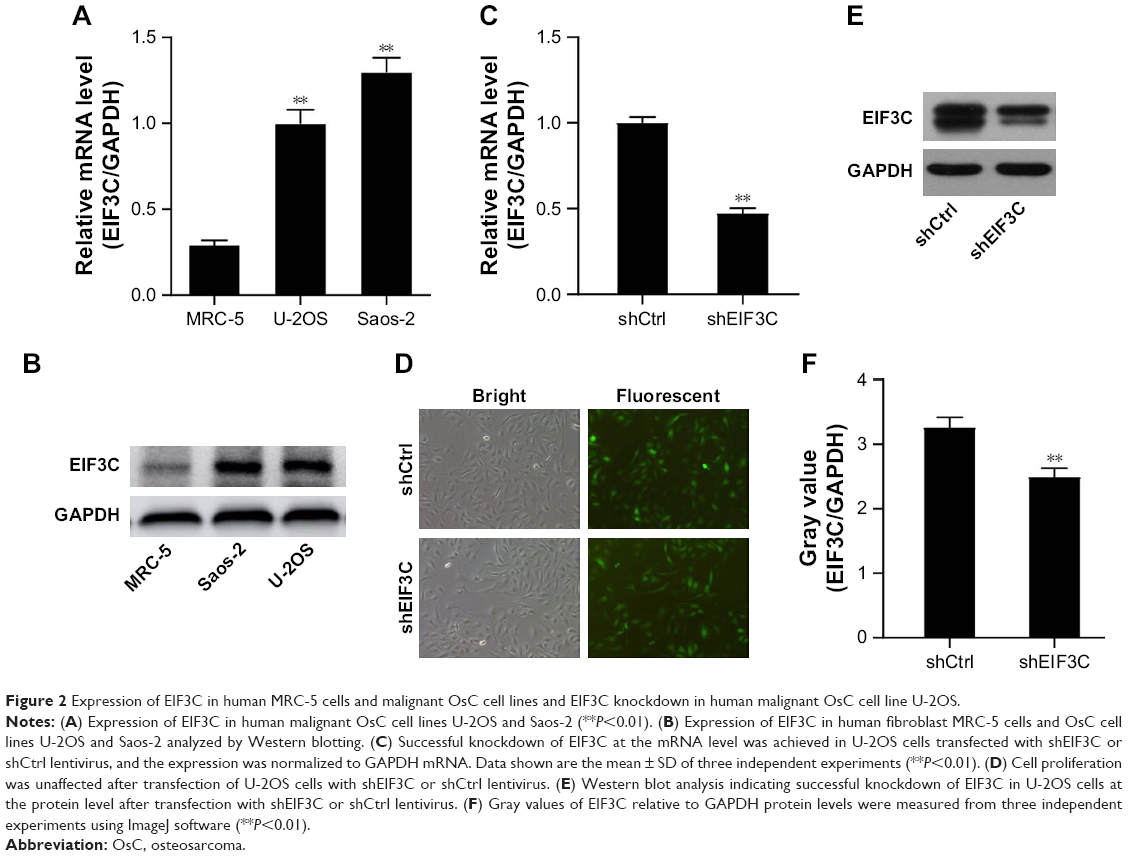 | Figure 2 Expression of EIF3C in human MRC-5 cells and malignant OsC cell lines and EIF3C knockdown in human malignant OsC cell line U-2OS. |
EIF3C knockdown suppresses U-2OS proliferation
To evaluate the role of EIF3C in the proliferation, U-2OS cells were transfected with shEIF3C or shCtrl lentivirus and cell count was continuously monitored over a 5-day period. In cells transfected with shEIF3C, statistically significant proliferation inhibition was observed after 96–120 hours when compared to cells transfected with shCtrl (P<0.01; Figure 3A and B). To verify the suppressive effect of shEIF3C knockdown on the proliferation of U-2OS cells, an MTT assay was performed and the OD490/fold ratio was determined 24 hours after plating of the cells. Consistent with the cell counting results, the OD490/fold ratio after 4 and 5 days of proliferation was significantly reduced in EIF3C-knocked down U-2OS cells when compared to controls (Figure 3C).
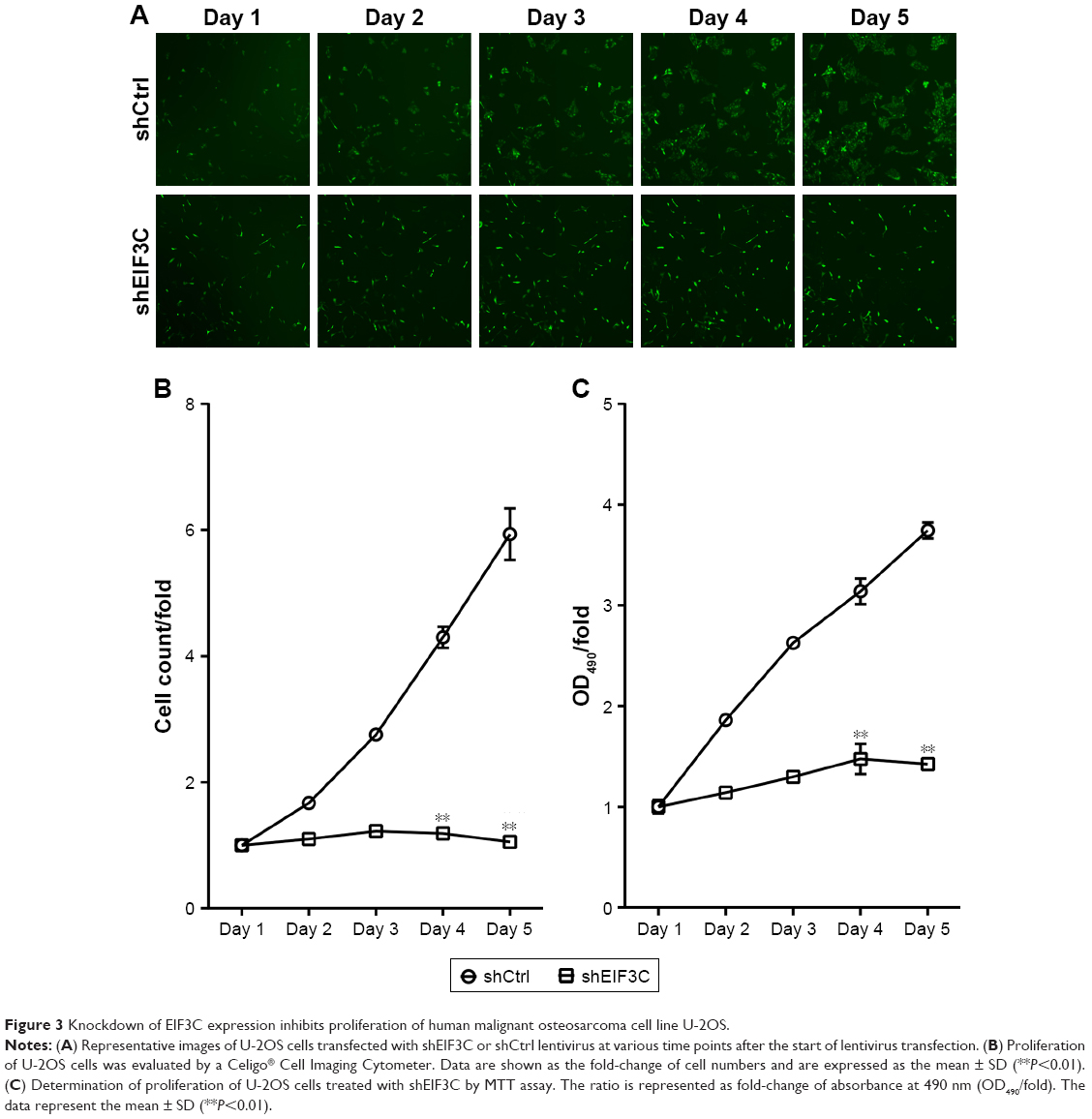 | Figure 3 Knockdown of EIF3C expression inhibits proliferation of human malignant osteosarcoma cell line U-2OS. |
EIF3C knockdown decreases the ability of U-2OS cells to form colonies
To evaluate the colony formation ability of U-2OS cells after EIF3C knockdown, a colony formation assay was performed. The data showed that a reduction in the expression of EIF3C in U-2OS cells significantly impaired the colony formation ability, resulting in an ~90.5% reduction in colony formation. An average of 84 colonies was present on the plates of cells that were transfected with shCtrl lentivirus, whereas only eight clones were found on the plates of cells transfected with shEIF3C lentivirus (Figure 4A and B).
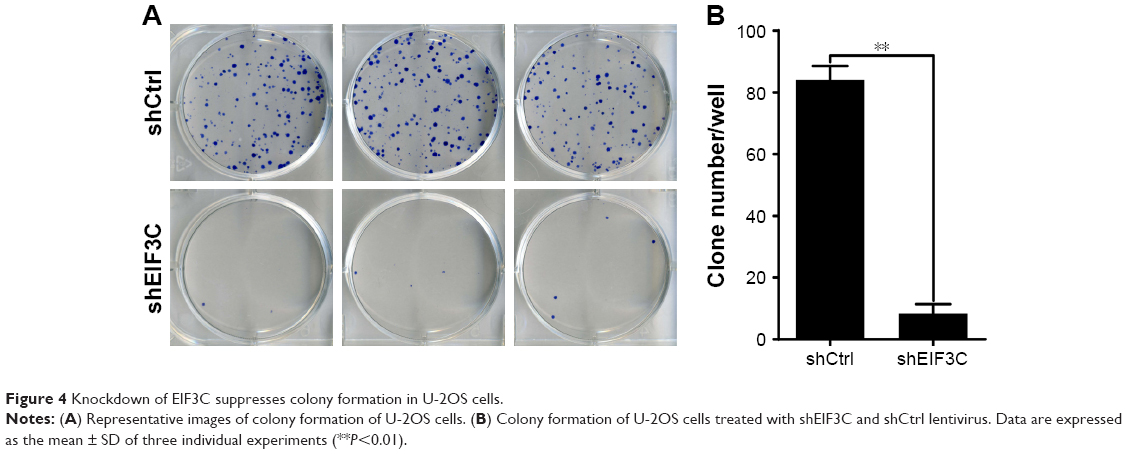 | Figure 4 Knockdown of EIF3C suppresses colony formation in U-2OS cells. |
Knockdown of EIF3C in U-2OS cells promotes apoptosis
To evaluate whether or not EIF3C expression affects apoptosis, we used the lentivirus approach and successfully knocked down EIF3C in U-2OS cells. Annexin V-APC staining was used to evaluate cell apoptosis. Apoptosis was observed in 16.39% of U-2OS cells treated with shEIF3C lentivirus; however, in U-2OS cells infected with shCtrl lentivirus, only 4.83% of the cells were apoptotic (Figure 5A and B). These results suggest that EIF3C knockdown induces apoptosis in U-2OS cells. Meanwhile, we found the EIF3C knockdown in human fibroblast MRC-5 cells also triggered enhanced apoptosis and decreased proliferation, while the proliferation inhibition and apoptosis rates were lower than that in cancerous cells after EIF3C knockdown (Figure S1).
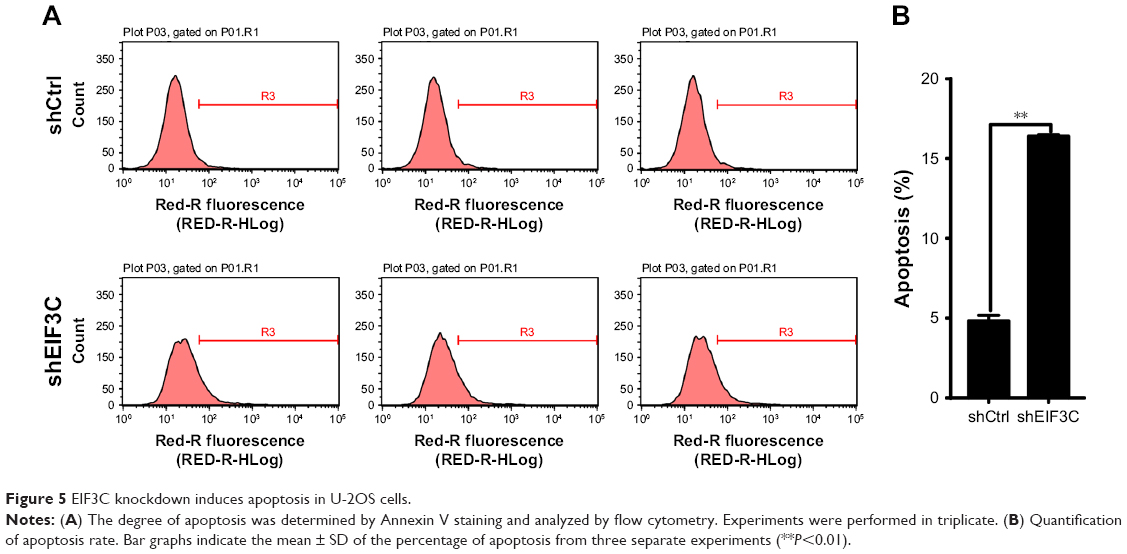 | Figure 5 EIF3C knockdown induces apoptosis in U-2OS cells. |
Silencing of EIF3C in U-2OS cells alters profiles of signaling molecules involved in stress and apoptosis
To investigate the signaling pathways in EIF3C-silenced U-2OS cells, we evaluated the profiles of signaling molecules involved in stress and apoptosis using a PathScan stress and apoptosis signaling antibody array kit. Using this kit, 19 signaling molecules involved in the regulation of stress response and apoptosis could be evaluated simultaneously (Figure 6A). The data showed that, compared to shCtrl-treated U-2OS cells, five signaling molecules were significantly enhanced in shEIF3C-treated U-2OS cells, including SAPK/JNK (Thr183/Tyr185), cleaved CASP3 (Asp175), cleaved CASP7 (Asp198), Chk1 (Ser345), and Chk1 (Thr68) (Figure 6B). All differentially expressed molecules were also verified by Western blotting (Figure 6C).
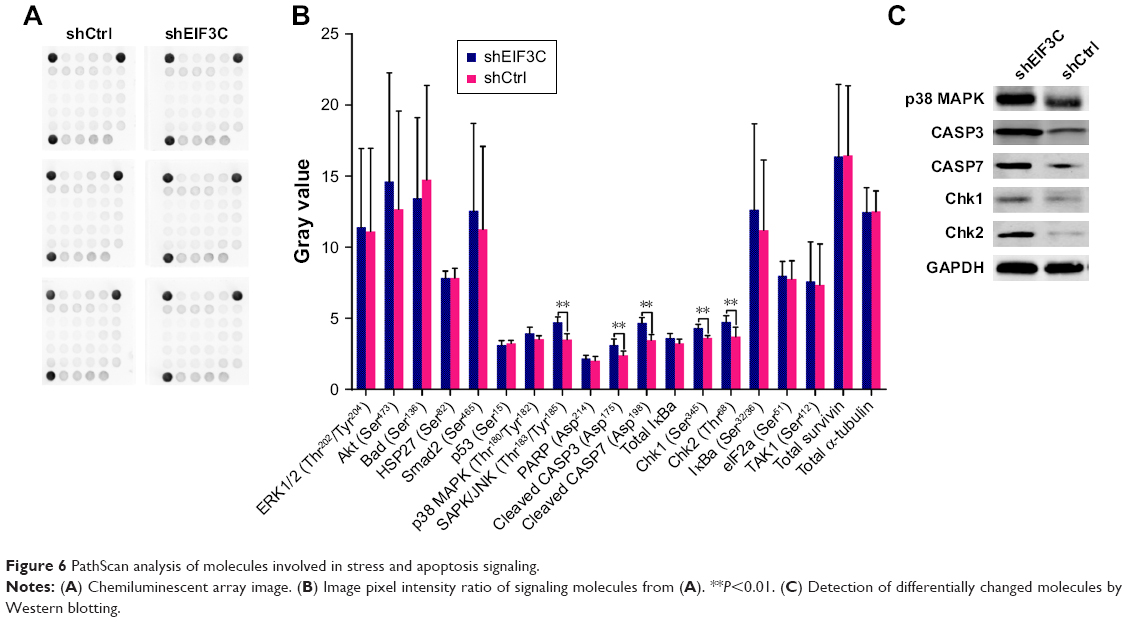 | Figure 6 PathScan analysis of molecules involved in stress and apoptosis signaling. |
Discussion
Previous studies have implicated a role of the EIF3 complex in functions other than translation and outside its general accepted role as a protein scaffold for the formation of initiation complexes.22 Aberrant EIF3 expression can result in translational dysregulation, which may lead to disease states, including tumorigenesis.23 It has been shown that mutated or inactivated EIF3 subunits are related to developmental defects.24,25 In addition, overexpression of EIF3 is associated with diverse types of cancer, including breast, prostate, and esophageal malignancies.12,23 Studies have demonstrated that in tumor types such as seminoma,13 meningioma,14 colon cancer,15 and glioma,16,17 expression of the EIF3C subunit of the EIF3 complex is upregulated. Recently, Lee et al demonstrated that overexpression of EIF3C augmented exosome secretion and promoted tumorigenesis of human hepatocellular carcinoma.18 These findings imply that EIF3C is closely related to human tumorigenesis;12,17 however, the role of EIF3C in human OsC has not yet been identified.
In the present study, we first detected the levels of EIF3C mRNA by qPCR and protein by IHC in human OsC tissues. Our data showed that the levels of EIF3C expression in cancer tissues were higher than those in normal tissues. Unfortunately, high EIF3C expression in cancer tissues was irrelevant to the clinicopathological characteristics of patients enrolled in our study, including age, pathological grade, TNM stage, and lymph node metastasis.
We then evaluated the expression levels of EIF3C mRNA in two different OsC cell lines and found that EIF3C was highly expressed in both cell lines. To assess the role of EIF3C in the OsC cell line U-2OS, we constructed the EIF3C-siRNA lentiviral vector to efficiently silence EIF3C in U-2OS cells. We found that EIF3C knockdown decreased both proliferation and the ability of U-2OS cells to form colonies. In addition, we verified that silencing EIF3C can induce apoptosis of U-2OS cells.
In eukaryotes, translational regulation is a fundamental process that is responsible for controlling cell development, homeostasis, and stress responses.26 Deregulation of translational control results in initiation and progression of cancer.27,28 A disrupted tumor suppressor mechanism causes apoptosis, which is a frequent event seen in cancer and contributes to tumorigenesis.29 Activation of key oncogenic pathways may alter the overall profiles of increased protein synthesis and specific regulatory networks for cellular transformation.28 As a major controller of translation initiation in protein synthesis, alteration of components in the EIF3C complex such as EIF3C leads to acceleration of protein synthesis involved in tumorigenesis and progression.30 Although EIF3C has been indicated as an important molecule, its mechanism of action in human OsC remains unclear.
To investigate the effect of EIF3C knockdown on shEIF3C-transfected U-2OS cells, we evaluated the stress and apoptosis profiles of various signaling molecules. The data showed that EIF3C knockdown significantly promoted the expression of SAPK/JNK (Thr183/Tyr185), cleaved CASP3 (Asp175), cleaved CASP7 (Asp198), Chk1 (Ser345), and Chk1 (Thr68). SAPKs, also known as JNKs, induce apoptosis by translocating to mitochondria with Bcl-xl interaction response to DNA damage.31 As effector caspases, both CASP3 and CASP7 cleave important cellular substrates to accomplish apoptosis.32 Both caspases are activated during Fas- and mitochondria-induced apoptosis.33,34 Downregulation of CASP3 or CASP7 is frequently seen in several forms of cancer.35–37 Inanami et al38 demonstrated that apoptosis is induced by CASP3 activation, which is required for de novo protein synthesis and operates through SAPK/JNK activation. DNA damage due to failure of repair results in cellular senescence or cell death.39 Phosphorylated Chk1/2 activates replication stress response, which is characterized by stalled replication forks40 and contributes to cancer cells senescence.41 The present study indicates that the increased apoptosis rate of U-2OS cells due to EIF3C knockdown may result in increased activation of SAPK/JNK, which is responsible for accelerating protein synthesis of CASP3/7 and Chk1/2. However, the detailed mechanism of action of pro-apoptosis due to EIF3C knockdown is currently unclear and requires further investigation.
Conclusion
Our study investigated the role of EIF3C in tumorigenesis in vitro. We found that EIF3C is involved in OsC proliferation and survival. Moreover, the inhibitory role of EIF3C knockdown in OsC proliferation was validated by a PathScan stress and apoptosis signaling antibody array kit, which suggested that the effect of EIF3C knockdown on tumor proliferation and apoptosis may involve the upregulation of SAPK/JNK, which promotes apoptosis by increasing the protein synthesis of CASP3/7 and Chk1/2.
Acknowledgment
This work was funded by Natural Science Foundation of Anhui Province (1608085QH204) and Youth Science Foundation Training Program of The First Affiliated Hospital of Anhui Medical University (2016KJ04).
Disclosure
The authors report no conflicts of interest in this work.
References
Ando K, Heymann M-F, Stresing V, Mori K, Rédini F, Heymann D. Current therapeutic strategies and novel approaches in osteosarcoma. Cancers (Basel). 2013;5(4):591–616. | ||
Clark JC, Dass CR, Choong PF. A review of clinical and molecular prognostic factors in osteosarcoma. J Cancer Res Clin Oncol. 2008;134(3):281–297. | ||
Sampo M, Koivikko M, Taskinen M, et al. Incidence, epidemiology and treatment results of osteosarcoma in Finland – a nationwide population-based study. Acta Oncol. 2011;50(8):1206–1214. | ||
Siclari VA, Qin L. Targeting the osteosarcoma cancer stem cell. J Orthop Surg Res. 2010;5(1):78. | ||
Mirabello L, Troisi RJ, Savage SA. Osteosarcoma incidence and survival rates from 1973 to 2004: data from the surveillance, epidemiology, and end results program. Cancer. 2009;115(7):1531–1543. | ||
Qureshi A, Ahmad Z, Azam M, Idrees R. Epidemiological data for common bone sarcomas. Asian Pac J Cancer Prev. 2010;11(2):393–395. | ||
Meyers PA, Schwartz CL, Krailo MD, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall Survival – A report from the children’s Oncology Group. JCO. 2008;26(4):633–638. | ||
Valásek L, Nielsen KH, Zhang F, Fekete CA, Hinnebusch AG. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol Cell Biol. 2004;24(21):9437–9455. | ||
LeFebvre AK, Korneeva NL, Trutschl M, et al. Translation initiation factor eIF4G-1 binds to eIF3 through the eIF3e subunit. J Biol Chem. 2006;281(32):22917–22932. | ||
Emmanuel R, Weinstein S, Landesman-Milo D, Peer D. EIF3c: a potential therapeutic target for cancer. Cancer Lett. 2013;336(1):158–166. | ||
Obayashi E, Luna RE, Nagata T, et al. Molecular landscape of the ribosome pre-initiation complex during mRNA scanning: structural role for eIF3c and its control by eIF5. Cell Rep. 2017;18(11):2651–2663. | ||
Zhang L, Pan X, Hershey JW. Individual overexpression of five subunits of human translation initiation factor eIF3 promotes malignant transformation of immortal fibroblast cells. J Biol Chem. 2007;282(8):5790–5800. | ||
Rothe M, Ko Y, Albers P, Wernert N. Eukaryotic initiation factor 3 p110 mRNA is overexpressed in testicular seminomas. Am J Pathol. 2000;157(5):1597–1604. | ||
Scoles DR, Yong WH, Qin Y, Wawrowsky K, Pulst SM. Schwannomin inhibits tumorigenesis through direct interaction with the eukaryotic initiation factor subunit C (eIF3c). Hum Mol Genet. 2006;15(7):1059–1070. | ||
Song N, Wang Y, Gu XD, Chen ZY, Shi LB. Effect of siRNA-mediated knockdown of eIF3c gene on survival of colon cancer cells. J Zhejiang Univ Sci B. 2013;14(6):451–459. | ||
Hao J, Wang Z, Wang Y, et al. Eukaryotic initiation factor 3C silencing inhibits cell proliferation and promotes apoptosis in human glioma. Oncol Rep. 2015;33(6):2954–2962. | ||
Hao J, Liang C, Jiao B. Eukaryotic translation initiation factor 3, subunit C is overexpressed and promotes cell proliferation in human glioma U-87 MG cells. Oncol Lett. 2015;9(6):2525–2533. | ||
Lee HY, Chen CK, Ho CM, et al. EIF3C-enhanced exosome secretion promotes angiogenesis and tumorigenesis of human hepatocellular carcinoma. Oncotarget. 2018;9(17):13193–13205. | ||
Radhika K, Prayaga AK. Estrogen and progesterone hormone receptor status in breast carcinoma: comparison of immunocytochemistry and immunohistochemistry. Indian J Cancer. 2010;47(2):148–150. | ||
Liu H, Liang S, Yang X, et al. RNAi-mediated rpL34 knockdown suppresses the growth of human gastric cancer cells. Oncol Rep. 2015;34(5):2267–2272. | ||
Moodley S, Koorbanally NA, Moodley T, Ramjugernath D, Pillay M. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay is a rapid, cheap, screening test for the in vitro anti-tuberculous activity of chalcones. J Microbiol Methods. 2014;104:72–78. | ||
Lee AS, Kranzusch PJ, Cate JH. EIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature. 2015;522(7554):111–114. | ||
Hershey JW. Regulation of protein synthesis and the role of eIF3 in cancer. Braz J Med Biol Res. 2010;43(10):920–930. | ||
Choudhuri A, Maitra U, Evans T. Translation initiation factor eIF3h targets specific transcripts to polysomes during embryogenesis. Proc Natl Acad Sci U S A. 2013;110(24):9818–9823. | ||
Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3(4):e56. | ||
Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10(4):254–266. | ||
Stumpf CR, Ruggero D. The cancerous translation apparatus. Curr Opin Genet Dev. 2011;21(4):474–483. | ||
Mavrakis KJ, Wendel HG. Translational control and cancer therapy. Cell Cycle. 2008;7(18):2791–2794. | ||
Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432(7015):307–315. | ||
Silvera D, Arju R, Darvishian F, et al. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol. 2009;11(7):903–908. | ||
Kharbanda S, Saxena S, Yoshida K, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275(1):322–327. | ||
Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27(48):6194–6206. | ||
Hirata H, Takahashi A, Kobayashi S, et al. Caspases are activated in a branched protease cascade and control distinct downstream processes in Fas-induced apoptosis. J Exp Med. 1998;187(4):587–600. | ||
Slee EA, Harte MT, Kluck RM, et al. Ordering the cytochrome c–initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a Caspase-9–dependent manner. J Cell Biol. 1999;144(2):281–292. | ||
Volm M, Mattern J, Koomägi R. Inverse correlation between apoptotic (Fas ligand, caspase-3) and angiogenic factors (VEGF, microvessel density) in squamous cell lung carcinomas. Anticancer Res. 1999;19(3A):1669–1671. | ||
Palmerini F, Devilard E, Jarry A, Birg F, Xerri L. Caspase 7 downregulation as an immunohistochemical marker of colonic carcinoma. Hum Pathol. 2001;32(5):461–467. | ||
Soung YH, Lee JW, Kim HS, et al. Inactivating mutations of caspase-7 gene in human cancers. Oncogene. 2003;22(39):8048–8052. | ||
Inanami O, Takahashi K, Kuwabara M. Attenuation of caspase-3-dependent apoptosis by Trolox post-treatment of x-irradiated MOLT-4 cells. Int J Radiation Biol. 1999;75(2):155–163. | ||
Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408(6811):433–439. | ||
Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. | ||
Lee H, Kim Y, Jeong JH, Ryu JH, Kim WY. ATM/CHK/p53 pathway dependent chemopreventive and therapeutic activity on lung cancer by pterostilbene. PLoS One. 2016;11(9):e0162335. |
Supplementary material
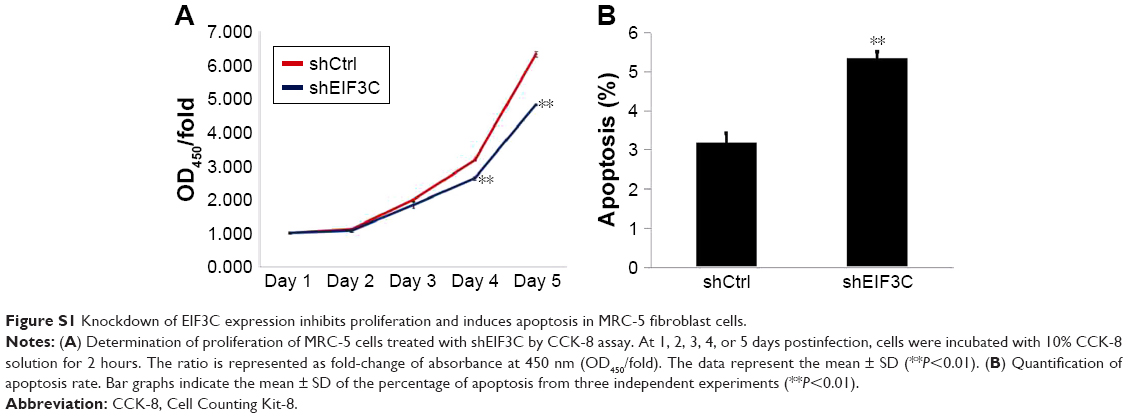 | Figure S1 Knockdown of EIF3C expression inhibits proliferation and induces apoptosis in MRC-5 fibroblast cells. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.