Back to Journals » Clinical Interventions in Aging » Volume 10
Klotho, stem cells, and aging
Authors Bian A, Neyra J, Zhan M, Hu MC
Received 19 March 2015
Accepted for publication 22 May 2015
Published 4 August 2015 Volume 2015:10 Pages 1233—1243
DOI https://doi.org/10.2147/CIA.S84978
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Ao Bian,1,2 Javier A Neyra,3 Ming Zhan,4 Ming Chang Hu1,3
1Charles and Jane Pak Center for Mineral Metabolism and Clinical Research, University of Texas Southwestern Medical Center, Dallas, TX, USA; 2Department of Nephrology, First Affiliated Hospital of Nanjing Medical University, Nanjing, People’s Republic of China; 3Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, 4Methodist Hospital Research Institute, Weill Cornell Medical College, Houston, TX, USA
Abstract: Aging is an inevitable and progressive biological process involving dysfunction and eventually destruction of every tissue and organ. This process is driven by a tightly regulated and complex interplay between genetic and acquired factors. Klotho is an antiaging gene encoding a single-pass transmembrane protein, klotho, which serves as an aging suppressor through a wide variety of mechanisms, such as antioxidation, antisenescence, antiautophagy, and modulation of many signaling pathways, including insulin-like growth factor and Wnt. Klotho deficiency activates Wnt expression and activity contributing to senescence and depletion of stem cells, which consequently triggers tissue atrophy and fibrosis. In contrast, the klotho protein was shown to suppress Wnt-signaling transduction, and inhibit cell senescence and preserve stem cells. A better understanding of the potential effects of klotho on stem cells could offer novel insights into the cellular and molecular mechanisms of klotho deficiency-related aging and disease. The klotho protein may be a promising therapeutic agent for aging and aging-related disorders.
Keywords: aging, cell senescence, klotho, stem cells, Wnt
Introduction
Klotho, the gene encoding the antiaging protein called klotho, was discovered in 1997 when mice developed multiple organ failure and shortened life span resembling human premature aging after this gene was serendipitously silenced.1 The klotho-deficient phenotype can be rescued by overexpressing klotho via genetic manipulation2 or viral delivery.3 Subsequently, klotho genes in humans and rats were cloned (Table 1). The chromosomal localization of mouse klotho is on 13q12, encompassing 50 kb and consisting of five exons (Figure 1).1,4 Two transcripts that arise from a single klotho gene through alternative RNA splicing were identified, and are predicted to encode a membrane and a secreted protein. The membrane protein consists of 1,014 amino acids, with an extracellular domain of twofold internal repeats (termed KL1 and KL2 domains), each about 450 amino acids long with 20%–40% homology to β-glucosidases. The secreted protein derived from the alternative transcript is only 550 amino acids long without the transmembrane domain1,4 (Figure 1).
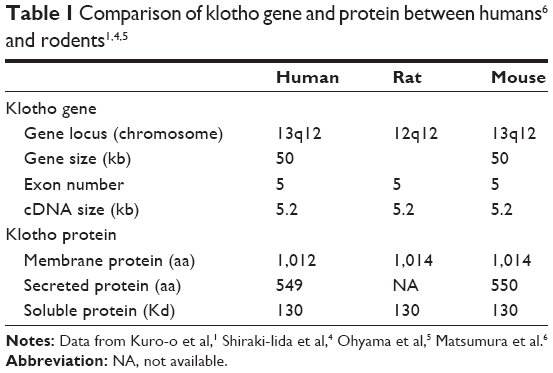 | Table 1 Comparison of klotho gene and protein between humans6 and rodents1,4,5 |
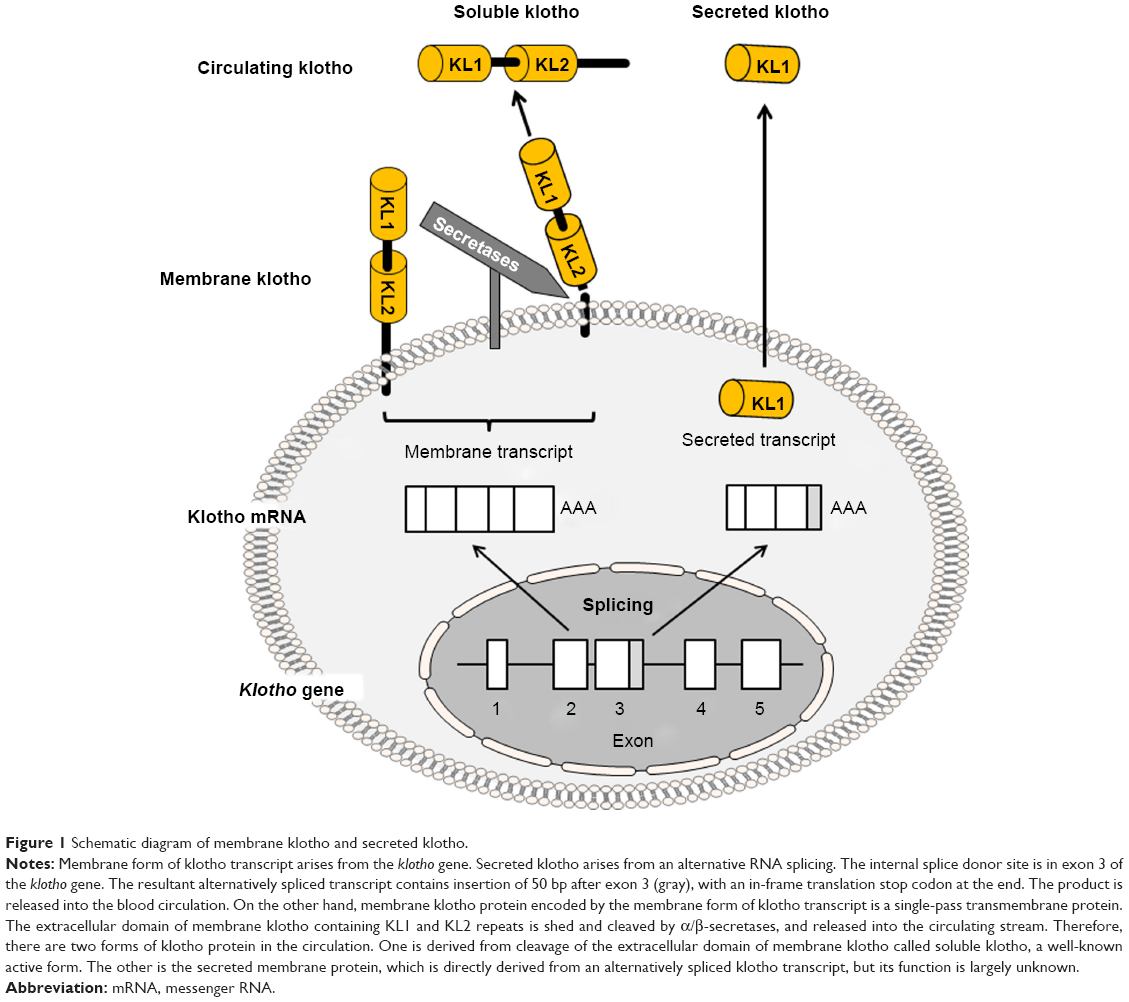 | Figure 1 Schematic diagram of membrane klotho and secreted klotho. |
The chromosomal localization of the rat klotho gene is on 12q12. The rat membrane klotho protein is also 1,014 amino acids long and is 94% and 85% homologous to those of mouse and human klotho proteins, respectively.5 The human klotho gene is localized on chromosome 13q12, encompassing 50 kb and consisting of five exons. Analogous to the mouse, two transcripts also arise from a single human klotho gene through alternative RNA splicing and encode a membrane (1,012 amino acids) or secreted (549 amino acids) protein.6
Klotho is highly expressed in the kidney, brain, and to a lesser extent in other organs.1,7 The extracellular domain of membrane klotho can be cleaved and shed by secretases.8–10 This released extracellular domain is referred as “soluble klotho” in this manuscript to distinguish it from another short klotho protein containing only a KL1 domain called secreted klotho, which is directly encoded by secreted klotho transcript through alternative splicing (Figure 1). Soluble klotho is the main functional form present in the circulation.2,11–13 It is also present in the cerebrospinal fluid13,14 and urine of mammals.11,12,15,16 As a circulating substance, soluble klotho exerts biological actions on distant organs and multiple systems.17–20
Soluble klotho can function as a β-glucuronidase12,21–23 or sialidase24–26 to regulate sodium dependent phosphate cotransporters (NaPi), organic cation transporters, renal outer medullary K+ channel 1, and calcium-channel transient receptor potential vanilloid 5 in the kidney and maintain mineral homeostasis. Klotho deficiency may also decrease extracellular volume through downregulation of the Na-K-2Cl cotransporter in the loop of Henle, with consequent increases in antidiuretic hormone and aldosterone hormonal responses,27 both linked to the premature aging phenotype mediated by dehydration observed in klotho-deficient mice.28,29 Outside the kidney, klotho can also function as β-glucuronidase to enhance creatine transporter-protein activity and maintain neuronal function and survival,30 and to increase several K+ channel expression and activities: 1) KCNQ1/KCNE1, required for proper hearing and cardiac repolarization;31 2) voltage-gated K+ channel (Kv1.3),32 expressed in many tissues to regulate a wide variety of cellular functions, including excitability, cell proliferation, apoptosis, immune response, insulin sensitivity, and platelet function; 3) the cardiac K+ channel,33 a key channel for cardiac repolarization and deranged excitation following cardiac hypertrophy.34 Furthermore, soluble klotho can also modulate the IGF-12 and Wnt18,35 signaling pathways, playing a key role in antiaging, anti-tumor growth, and antifibrosis. Importantly, soluble klotho can suppress apoptosis and protect cells against a variety of insults, including hypoxia, hyperoxia, oxidative stress, and cytotoxic drugs.36,37
Aging is an inevitable and progressive biological process resulting in dysfunction and destruction of almost all tissues and organs. This is driven by a tightly regulated and complex interplay between genetic and acquired factors. Aging is typically characterized by an increase in senescence, a quantitative and qualitative decrease in stem cells, and abnormal structure at tissue levels. The final outcome of aging is death. In this context, klotho plays an important role in suppressing aging.
There are significant mineral-metabolism disturbances in klotho deficiency, and mineral imbalance per se can induce premature aging induced by klotho deficiency. Indeed, some of the premature aging features and early death seen in klotho-deficient mice can be rescued by reducing plasma phosphate via genetic deletion of NaPi2a in the kidney38 or a low-phosphate diet.39 Reduction of 1,25-(OH)2-vitamin D and its signaling pathway also rescued phenotypes in klotho-deficient mice.40,41 However, there may be additional mineral-independent effects of klotho that deserve some attention. There are sparse but definite data about the potential effects of the klotho protein on stem cells, a key player in aging and tissue-regeneration processes. This manuscript summarizes current data on how klotho deficiency induces stem cell senescence and depletion, and provides novel insights into the cellular and molecular mechanisms of how the klotho protein affects stem cells and aging. We propose the klotho protein as a potential candidate therapeutic agent to halt aging and aging-associated diseases.
Klotho deficiency induces premature aging
Klotho deficiency in hypomorphic klotho mice or silencing of the klotho gene led to similar phenotypes of premature aging and short life span.1 In addition, global or renal-specific conditional knockout of klotho also led to a similar phenotype.1,42 These experiments confirmed that destruction of the klotho gene or loss of klotho function leads to an accelerated aging and eventually results in death at 2–3 months, a survival of only a tenth of normal mouse life span, regardless of how klotho deficiency was induced.
In addition to shortened life span, klotho-deficient mice demonstrated growth retardation; decreased physical activity; premature thymic involution; ectopic calcification; arteriosclerosis; pulmonary emphysema; osteoporosis; atrophy of skin, intestine, spleen, and gonads; and lipodystrophy.1 Early sudden death may result from cardiac arrhythmia or failure to respond properly to stress, possibly because of sinoatrial node dysfunction.43 Disturbed mineral metabolism is a prominent abnormal feature, including hypercalcemia, hyperphosphatemia, and hypervitaminosis D.1,12 The correction of phosphate and vitamin D levels rescues most of the premature aging phenotypes observed in klotho-deficient mice,40,41 indicating that klotho may suppress aging in mammals in large part through maintenance of mineral homeostasis or that mineral dysregulation (phosphate toxicity) may also play a causal role in the premature aging process (Figure 2). Interestingly, chronic kidney disease (CKD) in both human and animals has low levels of renal and circulating klotho and shares many clinical manifestations with klotho-deficient mice,44–46 suggesting that CKD may be an accelerated aging syndrome.47
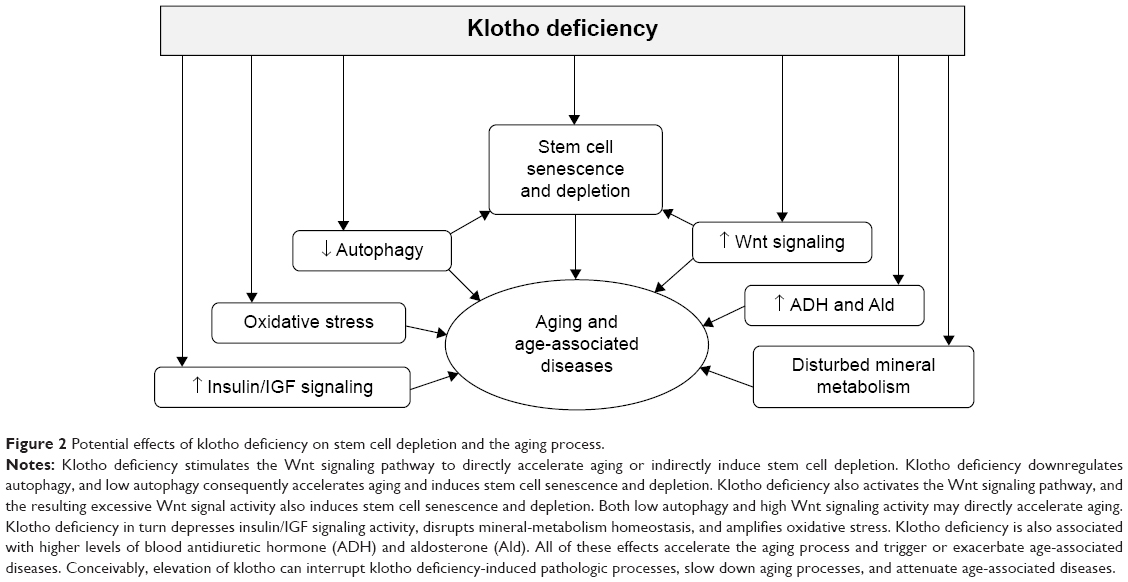 | Figure 2 Potential effects of klotho deficiency on stem cell depletion and the aging process. |
Klotho deficiency and cellular senescence
Cell senescence refers to physiological, structural, biochemical, and molecular changes that occur progressively during aging, culminating in the permanent cessation of cell division. Senescence is characterized by altered cellular morphology, increased activity of senescence-associated β-galactosidase, increased formation of senescence-associated heterochromatin foci and p16, permanent DNA damage, and chromosomal instability.48–52 As a state of permanent inhibition of cell proliferation, cellular senescence initiates and promotes chronic inflammation in multiple age-related chronic diseases, such as obesity, diabetes, atherosclerosis, Alzheimer’s disease, cancer, kidney disease, and degenerative disease.53–57
It is recognized that oxidative and genotoxic stress and mitochondrial dysfunction activate the senescence response by stimulating two pathways: p53/p21 and p16 pathways.51,52 The p53 protein is a tumor suppressor and can be activated by the ataxia telangiectasis-mutated kinase, and in turn activates p21 that effectively arrests cell proliferation and induces irreversible cell-cycle arrest in the G1- to S-phase transition. On the other hand, p16 is a tumor suppressor and another master regulator of cellular senescence58 that can be activated through p38 MAPK, enhancing senescence at later times.49,51,59,60 Klotho deficiency upregulates p53/p21 expression and increases the number of senescent cells,61 which can be attenuated by either knockdown of p53 or p21. Supplementation of klotho reduces H2O2-induced cell senescence and apoptosis through suppression of the p53/p21 signaling pathway.62 Klotho deficiency-induced cell senescence exacerbates endothelial damage and kidney-cell apoptosis triggered by oxidative stress.62,63
Klotho deficiency and autophagy
Autophagy is a “self-eating” process to maintain homeostasis. This process involves the sequestration of cytoplasmic components in double-membrane autophagosomes. Autophagy dysfunction is implicated in a variety of physiological and pathological processes, such as infection, cancer, metabolic and neurodegenerative disorders, and cardiovascular and pulmonary diseases, as well as physical exercise and aging.64–66 Whether klotho deficiency affects autophagy activity is a very important subject to be addressed.
In klotho-deficient mice, there is severe atrophy of skeletal muscle and activation of the autophagic-lysosomal pathway. Moreover, the signaling activity of mTOR, a suppressor of autophagy, is suppressed, presumably due to deficiency of essential amino acids in the klotho-deficient mice.67 Consistent with these findings in muscle, augmentation of autophagic markers, cleavage of light chain 3 (LC3), and autophagic ultrastructural alterations were found in the brain of klotho-deficient mice, similarly to those found in aged wild-type animals.68
In contrast, some experiments have shown stimulatory effects of klotho on autophagy in tumor cells.69,70 Tumor cells have low abundance of klotho expression and downregulation of autophagy. The restoration of klotho regulates IGF-1 receptor phosphorylation, activates downstream Akt-p70S6K and ERK signaling and autophagy, and subsequently suppresses tumor-cell proliferation and induces apoptosis.69 We recently found that transgenic mice overexpressing klotho had more cleaved LC3 and low levels of p62 in the kidney (unpublished data). Furthermore, an in vitro experiment showed that klotho induced puncta patterns of LC3 in cultured kidney cells, suggesting that klotho could activate autophagy flux in vivo and in vitro. It has been shown that defective autophagy predisposes the kidney to readily develop AKI after an ischemic insult and to more severe kidney impairment in senior versus general populations. Therefore, klotho-upregulated autophagy flux may be attributable to klotho-mediated renoprotection.
Aging of stem cells
Stem cells exist in most mammalian organs or tissues to maintain tissue homeostasis and participate in tissue repair or regeneration.71,72 Stem cells exhibit functional age-related changes, which manifest the declined homeostatic and regenerative activities of aging tissues.73–75 While some of the functional changes of stem cells arise intrinsically, others are imposed by age-related changes in the microenvironment or niche. The intrinsic changes have been observed at genomic, epigenomic, and proteomic levels. Some of the changes are potentially reversible, which may result in the rejuvenation of aged stem cells.
With aging, stem cells exhibit a diminished capacity of self-renewal and proliferation, which results in increased apoptosis or senescence in the stem cell compartment and depletion of functional stem cells.76 Furthermore, stem cells show changes in lineage commitment. The cell fate is determined largely by the epigenome and mediated by various signaling pathways.77–83 Aging can alter the epigenome and pathways, which may lead to aberrant lineage specification of stem cell progeny, as demonstrated in such tissues as the skeletal muscle, tendon, and hematopoietic system.84–87 Accumulation of these abnormal progeny contributes to the gradual deterioration of tissue structure and function associated with aging. On the other hand, accumulation of DNA mutations is a common feature of stem cell aging. Elevated levels of DNA damage have been reported in aging epidermal stem cells and hematopoietic stem cells.88,89
With aging, the number of stem cells declines significantly in some cases.90,91 For example, depletion of melanocyte stem cells in the hair follicles and the appearance of mature pigmented melanocytes in the stem cell niche have been reported in both aged mice and humans,92,93 leading to one of the most visible phenotypic changes during aging – hair graying. Aging or genotoxic stress induces the accumulation of DNA damage in melanocyte stem cells that results in the loss of stem cell self-renewal.92 Depletion of neural stem cells, possibly also related to a specific loss of capacity for self-renewal, appears to be responsible for declining neurogenesis with age.94–96 However, in other cases, stem cells do not show a significant depletion with age.97,98
As a self-renewing cell population to assure proper function and normal tissue homeostasis across the life span, stem cells are more resistant to the same factors that lead to age-related changes in their replicative or postmitotic progeny.73,99,100 During DNA replication, aging-related mechanisms, such as telomere shortening, chromosome rearrangements, and single-base mutations,101 can occur and ultimately lead to cellular senescence.102 Stem cells possess defense and repair mechanisms that are relevant to both highly proliferative cells and to long-lived postmitotic cells.100 It has been observed that adult stem cells, particularly those in continuously renewing tissues, undergo many rounds of cell division to maintain normal tissue homeostasis.77 It has also been observed that telomeres in old stem cells are still longer than those in the other somatic cells in these tissues,103,104 as observed in the skin, small intestine, cornea, testis, and brain.104 These observations suggest that stem cells divide at a much slower rate than their proliferative progeny or that they have evolved mechanisms to protect against telomere shortening.
A distinction among intrinsic irreversible changes (eg, genomic mutations), intrinsic reversible changes (eg, epigenomic alterations), and extrinsic influences from the microenvironment or niche in stem cells is important in studying stem cell aging. A mechanistic understanding of stem cell aging will contribute greatly to stem cell therapeutics for diseases and disorders of aging.
Klotho deficiency induces stem cell senescence and depletion
As discussed earlier, adult tissue stem cells have the ability to adjust to environmental changes, affect the proliferation of neighboring cells, and consequently participate in tissue maintenance and regeneration. Aging impedes stem cell renewal and proliferation. Any factor that interferes with the capacity of stem cells to self-renew, proliferate, differentiate, and replace in adult tissues could accelerate aging (Figure 2).16,105–111 Therefore, dysfunction and depletion of stem cells and progenitor cells contribute to aging. Emerging data showed that some aging-related characteristics in klotho-deficient mice may result from senescence and/or depletion of stem cells or differentiation of stem cells to promote fibrosis.18,85
In the premature aging observed in klotho-deficient mice, skin atrophy is one of the gross phenotypes.1 The klotho-deficient mice had sparser hair than control mice. Histological examinations of the skin revealed a reduction in the number of hair follicles, and reduced dermal and epidermal thickness. The subcutaneous fat was barely detectable. Furthermore, there were a decreased number of stem cells, increased progenitor-cell senescence, and dramatic augmentation of Wnt protein and signaling activity in the skin,18 indicating that klotho is required for maintenance of both stem cell number and function. A coimmunoprecipitation study indicated that soluble klotho binds to various Wnt family members, including Wnt1, Wnt3, Wnt4, and Wnt5a, suppresses Wnt transcription, and inhibits Wnt biological activity in the skin. An overexpression of klotho effectively antagonizes the activity of endogenous and exogenous Wnt, which induces accelerated cell senescence both in vitro and in vivo.18 Therefore, overexpression of Wnt proteins may be one of the pathogenic factors to be implicated in aging, and klotho is a secreted Wnt antagonist. In the same context of Wnt effect on skin atrophy, enhanced Wnt activity is present in degenerative skeletal muscle of aged mice, which may have affected regeneration of skeletal muscle, impaired repair, and consequently increased tissue fibrosis. All of those alterations are associated with differentiation of muscular stem cells (satellite cells) from a myogenic to a fibrogenic lineage, whose conversion results from the activation of the canonical Wnt signaling pathway in aged myogenic progenitors and can be suppressed by Wnt inhibitors, including sFRP3 and DKK1. Therefore, the Wnt signaling pathway promotes muscular stem cell aging and increased tissue fibrosis.85 As a Wnt signaling antagonist, klotho conceivably rescues myogenic stem cells, improves muscle repair, and suppresses fibrosis. Therefore, klotho may be a promising therapeutic target for muscle regeneration and muscular dystrophies.
In contrast, a mouse model of X-linked hypophosphatemia with deficient Wnt coreceptor low-density lipoprotein receptor-related protein 6 and consequent reduced Wnt signaling did not alter FGF23-induced phosphaturia or reduced mineralization of the bone, suggesting a potential Wnt-independent pathway of phosphate homeostasis.112
Klotho replacement as a possible antiaging strategy
Given that changes of functionality and a decreased number of stem cells contribute to or accelerate aging, implantation of stem cells to replenish new functional stem cells would be one means to attenuate age-associated disease by rebuilding the tissue or organ. This has been shown to be effective in preclinical and clinical trials in some diseases, including multiple sclerosis,113 myocardial infarction,114 ischemic stroke,115 and cancer,116 and even for patients undergoing plastic surgery.108 However, long-term side effects of stem cell implantation are not fully recognized, and should be a concern in most cases in which stem cells are permanently injected into patients.108,115 For example, recipients of genetically altered bone marrow transplants developed leukemia years after their allegedly successful transplants had cured their severe combined immunodeficiency. Despite potential side effects, recent advances in stem cell research and technology have shown promise.
On the other hand, activation or stimulation of endogenous or resident stem cells is another strategy to abate aging and age-associated disease. Current data from animal and in vitro cell-culture studies clearly demonstrated that klotho deficiency is associated with stem cell senescence and depletion. Furthermore, klotho deficiency may not only be a trigger for aging but also a pathogenic intermediate for accelerated aging and development of age-associated diseases, including Alzheimer’s disease, hypertension, osteoporosis, cardiovascular disease, and CKD. Conceivably, any therapy that restores or stimulates endogenous klotho or administration of exogenous klotho might provide a novel treatment strategy for aging and age-associated diseases.
Administration of exogenous klotho as a therapeutic agent
To date, klotho gene delivery is shown to effectively rescue many phenotypes observed in klotho-deficient mice, prolonging life span,117 attenuating the progression of hypertension and kidney damage in spontaneous hypertensive rats,118,119 ameliorating angiotensin II-induced kidney injury,120 improving endothelial function,121 and protecting from uremic cardiomyopathy.122 Although gene therapy is effective in animal studies, its safety is still questionable, and clinical application is not in proximity. There are few clinical trials testing gene therapy in specific diseases, usually genetic diseases, such as X-linked severe combined immunodeficiency,123 cancer treatment, or delivery of vaccines.124
Compared to viral delivery of the klotho gene in animals, administration of exogenous klotho protein is a safer, easier, and more direct modality to restore endocrine klotho deficiency. Similarly to the use of erythropoietin or erythropoiesis-stimulating agents to correct anemia in CKD patients and insulin to maintain normal glucose metabolism in type I diabetes, the administration of exogenous klotho protein may be a viable and effective option in the near future to dwindle aging. Klotho protein can potentially reverse or retard stem cell depletion and abate age-associated pathological processes.
To date, no studies of klotho protein administration in humans have been reported. In contrast, animal studies have already provided convincing and encouraging data to support the proof of concept that soluble klotho protein administration is safe and effective.11 We showed that soluble klotho protein attenuates kidney damage and preserves kidney function in an ischemia–reperfusion injury model causing acute kidney injury, which is a state of acute klotho deficiency.11 Furthermore, klotho protein inhibited renal fibrosis in a unilateral ureteral obstruction kidney-injury model, which is also a state of low klotho expression in the kidney.125 Interestingly, intraperitoneal injection every other day of soluble klotho protein effectively extended the life span of homozygous klotho-deficient mice, ameliorated premature aging-related phenotypes, such as growth retardation, premature thymus involution, and vascular calcification, and effectively reduced cellular senescence.126 Therefore, the preclinical data clearly support the therapeutic potential of soluble klotho protein for age-related disorders and klotho deficiency-associated diseases.
Activation of endogenous klotho expression
In the context of acute kidney injury, the renal tubules, where endogenous klotho is produced, are not fully destroyed, but are suppressed in their ability to produce klotho protein. In this context, strategies to increase endogenous production will be of therapeutic benefit. In particular, while klotho gene delivery is not yet implemented and klotho protein not yet available for clinical use, upregulation of endogenous klotho expression is of high clinical relevance.
To date, several categories of drugs in the market, including a PPARγ agonist,127–130 angiotensin II type I receptor antagonist (losartan),131–133 HMG-CoA reductase inhibitors (statin),134 and vitamin D active derivatives,135–138 have been shown to be effective in upregulating klotho expression in vivo and in vitro. The effect of upregulating klotho is definitely not associated with those drugs well-identified original pharmacologic targets.
Antioxidants and free radical scavengers may not directly play a role in the modulation of klotho expression. However, they can ameliorate oxidative stress, which is linked to aging and suppression of klotho expression in the kidney and cultured kidney cells.139,140 Klotho deficiency in turn increases oxidative stress and makes cells more susceptible to oxidative stress. Therefore, antioxidants are potentially useful in interrupting this spiral deterioration by upregulating klotho production and thus exerting antioxidant properties.141,142
Conclusion and future directions
Since klotho was serendipitously identified in 1997, our understanding of it as an aging suppressor has been continuously growing. Klotho protein has pleiotropic actions on many organs and tissues in mammals. However, very limited and premature data about klotho effects on stem cells are available. A better understanding of the effects of klotho on stem cells not only provides novel insights into the role of stem cells in antiaging processes but could also make a significant contribution to the advancement of regenerative medicine clinical practice.
Animal models have clearly demonstrated that klotho deficiency induces shortened life span and accelerated aging.1 Epidemiological data have also shown that soluble klotho is lower in elder than young adults, and that levels of soluble klotho are inversely correlated with age,143–145 indicating that aging is associated with klotho decline. Nonetheless, we do not know whether low soluble klotho is a prognostic biomarker for risk of earlier death in humans. Although we still do not fully understand why klotho is reduced with aging, maintenance of klotho levels by stimulation of endogenous klotho production or administration of exogenous klotho protein could be considered a potential therapeutic target to retard aging and attenuate age-associated diseases.
Thus far, animal experiments and in vitro cell-culture studies have shown the effects of soluble klotho protein on abating skin atrophy and skeletal muscle dystrophy during aging. It is anticipated that soluble klotho may play a pivotal role in regenerative medicine by preservation and activation of stem cells, particularly in heart tissue, where stem cells are very scarce or have low ability to replicate after injury. Therefore, if soluble klotho can activate stem cells or induce the replication of stem cells, klotho protein could be used as a promising therapeutic strategy for tissue repair and organ regeneration.
Acknowledgments
The authors acknowledge Drs Orson W Moe and Makoto Kuro-o for long-term collaborative work and support. MCH is in part supported by the NIH (R01-DK091392, R01-DK092461) and the Charles and Jane Pak Research Foundation, AB is in part supported by a Visiting Scholar Award from the National Natural Science Foundation of China (81170660H0509, 81270408H0220) and the Provincial Natural Science Foundation of Jiangsu, People’s Republic of China (BK2011849), and JAN is in part supported by the Ben J Lipps Research Fellowship Program of the American Society of Nephrology Foundation for Kidney Research and the Truelson Fellowship Fund at the Charles and Jane Pak Center of Mineral Metabolism and Clinical Research.
Disclosure
The authors report no conflicts of interest in this work.
References
Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. | ||
Kurosu H, Yamamoto M, Clark JD, et al. Suppression of aging in mice by the hormone klotho. Science. 2005;309(5742):1829–1833. | ||
Masuda H, Chikuda H, Suga T, Kawaguchi H, Kuro-o M. Regulation of multiple ageing-like phenotypes by inducible klotho gene expression in klotho mutant mice. Mech Ageing Dev. 2005;126(12):1274–1283. | ||
Shiraki-Iida T, Aizawa H, Matsumura Y, et al. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998;424(1–2):6–10. | ||
Ohyama Y, Kurabayashi M, Masuda H, et al. Molecular cloning of rat klotho cDNA: markedly decreased expression of klotho by acute inflammatory stress. Biochem Biophys Res Commun. 1998;251(3):920–925. | ||
Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;242(3):626–630. | ||
Kato Y, Arakawa E, Kinoshita S, et al. Establishment of the anti-klotho monoclonal antibodies and detection of klotho protein in kidneys. Biochem Biophys Res Commun. 2000;267(2):597–602. | ||
Chen CD, Tung TY, Liang J, et al. Identification of cleavage sites leading to the shed form of the anti-aging protein klotho. Biochemistry. 2014;53(34):5579–5587. | ||
Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104(50):19796–19801. | ||
Bloch L, Sineshchekova O, Reichenbach D, et al. Klotho is a substrate for α-, β- and γ-secretase. FEBS Lett. 2009;583(19):3221–3224. | ||
Hu MC, Shi M, Zhang J, Quinones H, Kuro-o M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010;78(12):1240–1251. | ||
Hu MC, Shi M, Zhang J, et al. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24(9):3438–3450. | ||
Imura A, Iwano A, Tohyama O, et al. Secreted klotho protein in sera and CSF: implication for post-translational cleavage in release of klotho protein from cell membrane. FEBS Lett. 2004;565(1–3):143–147. | ||
Semba RD, Moghekar AR, Hu J, et al. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci Lett. 2014;558:37–40. | ||
Hu MC, Shi M, Zhang J, et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22(1):124–136. | ||
Barker SL, Pastor J, Carranza D, et al. The demonstration of αklotho deficiency in human chronic kidney disease with a novel synthetic antibody. Nephrol Dial Transplant. 2015;30(2):223–233. | ||
Huang CL. Regulation of ion channels by secreted klotho: mechanisms and implications. Kidney Int. 2010;77(10):855–860. | ||
Liu H, Fergusson MM, Castilho RM, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317(5839):803–806. | ||
Goetz R, Nakada Y, Hu MC, et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-klotho complex formation. Proc Natl Acad Sci U S A. 2010;107(1):407–412. | ||
Carpenter TO, Insogna KL, Zhang JH, et al. Circulating levels of soluble klotho and FGF23 in X-linked hypophosphatemia: circadian variance, effects of treatment, and relationship to parathyroid status. J Clin Endocrinol Metab. 2010;95(11):E352–E357. | ||
Chang Q, Hoefs S, van der Kemp AW, Topala CN, Bindels RJ, Hoenderop JG. The β-glucuronidase klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310(5747):490–493. | ||
Tohyama O, Imura A, Iwano A, et al. Klotho is a novel β-glucuronidase capable of hydrolyzing steroid beta-glucuronides. J Biol Chem. 2004;279(11):9777–9784. | ||
Lu P, Boros S, Chang Q, Bindels RJ, Hoenderop JG. The β-glucuronidase klotho exclusively activates the epithelial Ca2+ channels TRPV5 and TRPV6. Nephrol Dial Transplant. 2008;23(11):3397–3402. | ||
Wolf MT, An SW, Nie M, Bal MS, Huang CL. Klotho up-regulates renal calcium channel transient receptor potential vanilloid 5 (TRPV5) by intra- and extracellular N-glycosylation-dependent mechanisms. J Biol Chem. 2014;289(52):35849–35857. | ||
Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro OM, Huang CL. Removal of sialic acid involving klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008;105(28):9805–9810. | ||
Cha SK, Hu MC, Kurosu H, Kuro-o M, Moe O, Huang CL. Regulation of renal outer medullary potassium channel and renal K(+) excretion by klotho. Mol Pharmacol. 2009;76(1):38–46. | ||
Fischer SS, Kempe DS, Leibrock CB, et al. Hyperaldosteronism in klotho-deficient mice. Am J Physiol Renal Physiol. 2010;299(5):F1171–F1177. | ||
Tamma G, Goswami N, Reichmuth J, De Santo NG, Valenti G. Aquaporins, vasopressin, and aging: current perspectives. Endocrinology. 2015;156(3):777–788. | ||
Tang C, Pathare G, Michael D, Fajol A, Eichenmüller M, Lang F. Downregulation of klotho expression by dehydration. Am J Physiol Renal Physiol. 2011;301(4):F745–F750. | ||
Almilaji A, Sopjani M, Elvira B, et al. Upregulation of the creatine transporter Slc6A8 by klotho. Kidney Blood Press Res. 2014;39(6):516–525. | ||
Almilaji A, Pakladok T, Muñoz C, Elvira B, Sopjani M, Lang F. Upregulation of KCNQ1/KCNE1 K+ channels by klotho. Channels (Austin). 2014;8(3):222–229. | ||
Almilaji A, Honisch S, Liu G, et al. Regulation of the voltage gated k channel kv1.3 by recombinant human klotho protein. Kidney Blood Press Res. 2014;39(6):609–622. | ||
Munoz C, Pakladok T, Almilaji A, et al. Klotho sensitivity of the hERG channel. FEBS Lett. 2013;587(11):1663–1668. | ||
Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K(+) channels: structure, function, and clinical significance. Physiol Rev. 2012;92(3):1393–1478. | ||
Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, Kashihara N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Renal Physiol. 2012;303(12):F1641–F1651. | ||
Ravikumar P, Ye J, Zhang J, et al. α-Klotho protects against oxidative damage in pulmonary epithelia. Am J Physiol Lung Cell Mol Physiol. 2014;307(7):L566–L575. | ||
Panesso MC, Shi M, Cho HJ, et al. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. 2014;85(4):855–870. | ||
Ohnishi M, Razzaque MS. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. FASEB J. 2010;24(9):3562–3571. | ||
Morishita K, Shirai A, Kubota M, et al. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J Nutr. 2001;131(12):3182–3188. | ||
Ohnishi M, Nakatani T, Lanske B, Razzaque MS. Reversal of mineral ion homeostasis and soft-tissue calcification of klotho knockout mice by deletion of vitamin D 1α-hydroxylase. Kidney Int. 2009;75(11):1166–1172. | ||
Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12(7):298–305. | ||
Lindberg K, Amin R, Moe OW, et al. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. 2014;25(10):2169–2175. | ||
Takeshita K, Fujimori T, Kurotaki Y, et al. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004;109(14):1776–1782. | ||
Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–533. | ||
Hu MC, Kuro-o M, Moe OW. Secreted klotho and chronic kidney disease. Adv Exp Med Biol. 2012;728:126–157. | ||
Hu MC, Kuro-o M, Moe OW. Klotho and chronic kidney disease. Contrib Nephrol. 2013;180:47–63. | ||
Stenvinkel P, Larsson TE. Chronic kidney disease: a clinical model of premature aging. Am J Kidney Dis. 2013;62(2):339–351. | ||
Beltrami AP, Cesselli D, Beltrami CA. Stem cell senescence and regenerative paradigms. Clin Pharmacol Ther. 2012;91(1):21–29. | ||
Qian Y, Chen X. Senescence regulation by the p53 protein family. Methods Mol Biol. 2013;965:37–61. | ||
Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28(2):99–114. | ||
Bitto A, Crowe EP, Lerner C, Torres C, Sell C. The senescence arrest program and the cell cycle. Methods Mol Biol. 2014;1170:145–154. | ||
Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–496. | ||
Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21(6):354–359. | ||
Olivieri F, Recchioni R, Marcheselli F, et al. Cellular senescence in cardiovascular diseases: potential age-related mechanisms and implications for treatment. Curr Pharm Des. 2013;19(9):1710–1719. | ||
Campisi J, Robert L. Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol. 2014;39:45–61. | ||
Tian XL, Li Y. Endothelial cell senescence and age-related vascular diseases. J Genet Genomics. 2014;41(9):485–495. | ||
Zhu Y, Armstrong JL, Tchkonia T, Kirkland JL. Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr Opin Clin Nutr Metab Care. 2014;17(4):324–328. | ||
Li Q, Zhang Y, Fu J, et al. FOXA1 mediates p16(INK4a) activation during cellular senescence. EMBO J. 2013;32(6):858–873. | ||
Prieur A, Besnard E, Babled A, Lemaitre JM. p53 and p16(INK4A) independent induction of senescence by chromatin-dependent alteration of S-phase progression. Nat Commun. 2011;2:473. | ||
Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12(18):3063–3069. | ||
de Oliveira RM. Klotho RNAi induces premature senescence of human cells via a p53/p21 dependent pathway. FEBS Lett. 2006;580(24):5753–5758. | ||
Ikushima M, Rakugi H, Ishikawa K, et al. Anti-apoptotic and anti-senescence effects of klotho on vascular endothelial cells. Biochem Biophys Res Commun. 2006;339(3):827–832. | ||
Maekawa Y, Ohishi M, Ikushima M, et al. Klotho protein diminishes endothelial apoptosis and senescence via a mitogen-activated kinase pathway. Geriatr Gerontol Int. 2011;11(4):510–516. | ||
Schneider JL, Cuervo AM. Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol. 2014;11(3):187–200. | ||
Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24(1):92–104. | ||
Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368(7):651–662. | ||
Iida RH, Kanko S, Suga T, Morito M, Yamane A. Autophagic-lysosomal pathway functions in the masseter and tongue muscles in the klotho mouse, a mouse model for aging. Mol Cell Biochem. 2011;348(1–2):89–98. | ||
Shiozaki M, Yoshimura K, Shibata M, et al. Morphological and biochemical signs of age-related neurodegenerative changes in klotho mutant mice. Neuroscience. 2008;152(4):924–941. | ||
Shu G, Xie B, Ren F, et al. Restoration of klotho expression induces apoptosis and autophagy in hepatocellular carcinoma cells. Cell Oncol (Dordr). 2013;36(2):121–129. | ||
Xie B, Zhou J, Shu G, et al. Restoration of klotho gene expression induces apoptosis and autophagy in gastric cancer cells: tumor suppressive role of klotho in gastric cancer. Cancer Cell Int. 2013;13(1):18. | ||
Li L, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327(5965):542–545. | ||
Weissman IL. Stem cells: units of development, units of regeneration, and units in evolution. Cell. 2000;100(1):157–168. | ||
Jones DL, Rando TA. Emerging models and paradigms for stem cell ageing. Nat Cell Biol. 2011;13(5):506–512. | ||
Dorshkind K, Montecino-Rodriguez E, Signer RA. The ageing immune system: is it ever too old to become young again? Nat Rev Immunol. 2009;9(1):57–62. | ||
Bell DR, Van Zant G. Stem cells, aging, and cancer: inevitabilities and outcomes. Oncogene. 2004;23(43):7290–7296. | ||
Ruzankina Y, Pinzon-Guzman C, Asare A, et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1(1):113–126. | ||
van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. | ||
Rittié L, Stoll SW, Kang S, Voorhees JJ, Fisher GJ. Hedgehog signaling maintains hair follicle stem cell phenotype in young and aged human skin. Aging Cell. 2009;8(6):738–751. | ||
Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell. 2008;2(1):50–59. | ||
Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447(7143):407–412. | ||
Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nat Rev Mol Cell Biol. 2009;10(8):526–537. | ||
Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. | ||
Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125(2):315–326. | ||
Zhou Z, Akinbiyi T, Xu L, et al. Tendon-derived stem/progenitor cell aging: defective self-renewal and altered fate. Aging Cell. 2010;9(5):911–915. | ||
Brack AS, Conboy MJ, Roy S, et al. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science. 2007;317(5839):807–810. | ||
Taylor-Jones JM, McGehee RE, Rando TA, Lecka-Czernik B, Lipschitz DA, Peterson CA. Activation of an adipogenic program in adult myoblasts with age. Mech Ageing Dev. 2002;123(6):649–661. | ||
Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192(9):1273–1280. | ||
Sotiropoulou PA, Candi A, Mascré G, et al. Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol. 2010;12(6):572–582. | ||
Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature. 2007;447(7145):725–729. | ||
Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24(7):1726–1733. | ||
Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16(6):2027–2033. | ||
Inomata K, Aoto T, Binh NT, et al. Genotoxic stress abrogates renewal of melanocyte stem cells by triggering their differentiation. Cell. 2009;137(6):1088–1099. | ||
Nishimura EK, Granter SR, Fisher DE. Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science. 2005;307(5710):720–724. | ||
Renault VM, Rafalski VA, Morgan AA, et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5(5):527–539. | ||
Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf expression. Cell. 2008;135(2):227–239. | ||
Molofsky AV, Slutsky SG, Joseph NM, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443(7110):448–452. | ||
Giangreco A, Qin M, Pintar JE, Watt FM. Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell. 2008;7(2):250–259. | ||
Brack AS, Rando TA. Intrinsic changes and extrinsic influences of myogenic stem cell function during aging. Stem Cell Rev. 2007;3(3):226–237. | ||
Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8(9):703–713. | ||
Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441(7097):1080–1086. | ||
Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37(5):961–976. | ||
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. | ||
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8(3):311–323. | ||
Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22(5):654–667. | ||
Baker N, Boyette LB, Tuan RS. Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone. 2015;70:37–47. | ||
Peng Y, Xuan M, Leung VY, Cheng B. Stem cells and aberrant signaling of molecular systems in skin aging. Ageing Res Rev. 2015;19:8–21. | ||
Fry CS, Lee JD, Mula J, et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med. 2015;21(1):76–80. | ||
Salibian AA, Widgerow AD, Abrouk M, Evans GR. Stem cells in plastic surgery: a review of current clinical and translational applications. Arch Plast Surg. 2013;40(6):666–675. | ||
Florian C, Langmann T, Weber BH, Morsczeck C. Murine Müller cells are progenitor cells for neuronal cells and fibrous tissue cells. Biochem Biophys Res Commun. 2008;374(2):187–191. | ||
Gopinath SD, Rando TA. Stem cell review series: aging of the skeletal muscle stem cell niche. Aging Cell. 2008;7(4):590–598. | ||
Mazzoccoli G, Tevy MF, Borghesan M, Delle Vergini MR, Vinciguerra M. Caloric restriction and aging stem cells: the stick and the carrot? Exp Gerontol. 2014;50:137–148. | ||
Uchihashi K, Nakatani T, Goetz R, Mohammadi M, He X, Razzaque MS. FGF23-induced hypophosphatemia persists in Hyp mice deficient in the WNT coreceptor Lrp6. Contrib Nephrol. 2013;180:124–137. | ||
Derwenskus J, Lublin FD. Future treatment approaches to multiple sclerosis. Handb Clin Neurol. 2014;122:563–577. | ||
Lee S, Bick-Forrester J, Makkar RR, Forrester JS. Stem-cell repair of infarcted myocardium: ready for clinical application? Am Heart Hosp J. 2004;2(2):100–106. | ||
Qiao LY, Huang FJ, Zhao M, et al. A two-year follow-up study of cotransplantation with neural stem/progenitor cells and mesenchymal stromal cells in ischemic stroke patients. Cell Transplant. 2014;23 Suppl 1:S65–S72. | ||
Rahman M, Hoh B, Kohler N, Dunbar EM, Murad GJ. The future of glioma treatment: stem cells, nanotechnology and personalized medicine. Future Oncol. 2012;8(9):1149–1156. | ||
Shiraki-Iida T, Iida A, Nabeshima Y, et al. Improvement of multiple pathophysiological phenotypes of klotho (kl/kl) mice by adenovirus-mediated expression of the klotho gene. J Gene Med. 2000;2(4):233–242. | ||
Wang Y, Sun Z. Antiaging gene klotho regulates endothelin-1 levels and endothelin receptor subtype B expression in kidneys of spontaneously hypertensive rats. J Hypertens. 2014;32(8):1629–1636. | ||
Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54(4):810–817. | ||
Mitani H, Ishizaka N, Aizawa T, et al. In vivo klotho gene transfer ameliorates angiotensin II-induced renal damage. Hypertension. 2002;39(4):838–843. | ||
Saito Y, Nakamura T, Ohyama Y, et al. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem Biophys Res Commun. 2000;276(2):767–772. | ||
Xie J, Yoon J, An SW, Kuro-o OM, Huang CL. Soluble klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol. 2015;26(5):1150–1160. | ||
Williams DA. Curing genetic disease with gene therapy. Trans Am Clin Climatol Assoc. 2014;125:122–128. | ||
Heller R, Heller LC. Gene electrotransfer clinical trials. Adv Genet. 2015;89:235–262. | ||
Doi S, Zou Y, Togao O, et al. Klotho inhibits transforming growth factor-β1 (TGF-β1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem. 2011;286(10):8655–8665. | ||
Chen TH, Kuro OM, Chen CH, et al. The secreted klotho protein restores phosphate retention and suppresses accelerated aging in klotho mutant mice. Eur J Pharmacol. 2013;698(1–3):67–73. | ||
Yang HC, Deleuze S, Zuo Y, Potthoff SA, Ma LJ, Fogo AB. The PPARγ agonist pioglitazone ameliorates aging-related progressive renal injury. J Am Soc Nephrol. 2009;20(11):2380–2388. | ||
Zhang H, Li Y, Fan Y, et al. Klotho is a target gene of PPAR-γ. Kidney Int. 2008;74(6):732–739. | ||
Zhang R, Zheng F. PPAR-γ and aging: one link through klotho? Kidney Int. 2008;74(6):702–704. | ||
Chen LJ, Cheng MF, Ku PM, Lin JW. Rosiglitazone increases cerebral klotho expression to reverse baroreflex in type 1-like diabetic rats. Biomed Res Int. 2014;2014:309151. | ||
Zhou Q, Lin S, Tang R, Veeraragoo P, Peng W, Wu R. Role of fosinopril and valsartan on klotho gene expression induced by angiotensin II in rat renal tubular epithelial cells. Kidney Blood Press Res. 2010;33(3):186–192. | ||
Yoon HE, Ghee JY, Piao S, et al. Angiotensin II blockade upregulates the expression of klotho, the anti-ageing gene, in an experimental model of chronic cyclosporine nephropathy. Nephrol Dial Transplant. 2011;26(3):800–813. | ||
Karalliedde J, Maltese G, Hill B, Viberti G, Gnudi L. Effect of renin-angiotensin system blockade on soluble klotho in patients with type 2 diabetes, systolic hypertension, and albuminuria. Clin J Am Soc Nephrol. 2013;8(11):1899–1905. | ||
Narumiya H, Sasaki S, Kuwahara N, et al. HMG-CoA reductase inhibitors up-regulate anti-aging klotho mRNA via RhoA inactivation in IMCD3 cells. Cardiovasc Res. 2004;64(2):331–336. | ||
Lim K, Lu TS, Molostvov G, et al. Vascular klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012;125(18):2243–2255. | ||
Lau WL, Leaf EM, Hu MC, et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82(12):1261–1270. | ||
Forster RE, Jurutka PW, Hsieh JC, et al. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem Biophys Res Commun. 2011;414(3):557–562. | ||
de Borst MH, Vervloet MG, ter Wee PM, Navis G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J Am Soc Nephrol. 2011;22(9):1603–1609. | ||
Mitobe M, Yoshida T, Sugiura H, Shirota S, Tsuchiya K, Nihei H. Oxidative stress decreases klotho expression in a mouse kidney cell line. Nephron Exp Nephrol. 2005;101(2):e67–e74. | ||
Oh HJ, Nam BY, Lee MJ, et al. Decreased circulating klotho levels in patients undergoing dialysis and relationship to oxidative stress and inflammation. Perit Dial Int. 2015;35(1):43–51. | ||
Yamamoto M, Clark JD, Pastor JV, et al. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem. 2005;280(45):38029–38034. | ||
Kuro-o M. Klotho as a regulator of oxidative stress and senescence. Biol Chem. 389(3):233–241. | ||
Scholze A, Liu Y, Pedersen L, et al. Soluble α-klotho and its relation to kidney function and fibroblast growth factor-23. J Clin Endocrinol Metab. 2014;99(5):E855–E861. | ||
Pedersen L, Pedersen SM, Brasen CL, Rasmussen LM. Soluble serum klotho levels in healthy subjects. Comparison of two different immunoassays. Clin Biochem. 2013;46(12):1079–1083. | ||
Yamazaki Y, Imura A, Urakawa I, et al. Establishment of sandwich ELISA for soluble α-klotho measurement: age-dependent change of soluble α-klotho levels in healthy subjects. Biochem Biophys Res Commun. 2010;398(3):513–518. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.