Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
Key Candidate Prognostic Biomarkers Correlated with Immune Infiltration in Hepatocellular Carcinoma
Authors Deng Z, Huang K ![]() , Liu D, Luo N, Liu T, Han L, Du D, Lian D, Zhong Z, Peng J
, Liu D, Luo N, Liu T, Han L, Du D, Lian D, Zhong Z, Peng J
Received 6 September 2021
Accepted for publication 18 November 2021
Published 18 December 2021 Volume 2021:8 Pages 1607—1622
DOI https://doi.org/10.2147/JHC.S337067
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Laura A. Dawson
Zenghua Deng,1,2,* Kanghua Huang,3,* Dongfang Liu,1 Nan Luo,1,2 Tingting Liu,1 Long Han,1 Dexiao Du,1 Dongbo Lian,1,2 Zhaohui Zhong,4 Jirun Peng1,2
1Department of Surgery, Beijing Shijitan Hospital, Capital Medical University, Beijing, 100038, People’s Republic of China; 2Ninth School of Clinical Medicine, Peking University, Beijing, 100038, People’s Republic of China; 3Department of Radiation Oncology, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, 310009, People’s Republic of China; 4Department of General Surgery, Peking University People’s Hospital, Beijing, 100044, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jirun Peng; Zhaohui Zhong Email [email protected]; [email protected]; [email protected]
Background: Hepatocellular carcinoma (HCC) is the most common subtype of primary liver cancer, which causes ∼ 800,000 deaths annually world-wide. Immune checkpoint inhibitor (ICI) has reformed cancer therapy and achieved unprecedented results in various malignancies, including HCC. However, the response rate of immunotherapy is very low in HCC. Considereing the complicated and unique immune status in liver, we hypothesize that critical molecules will affect prognosis and correlate with immune context in the tumor microenvironment of HCC.
Methods: Using Kaplan–Meier plotter, GEPIA2 and Integrative Molecular Database of Hepatocellular Carcinoma (HCCDB), survival genes and their prognostic value were estimated in HCC. Based on Tumor Immune Estimation Resource (TIMER), association between survival genes and immune infiltration was examined in HCC. FunRich and STRING were used to analyze gene ontology and protein–protein interaction (PPI) Network, qRT-PCR was used to measure mRNA level of candidates; and a Cell Counting Kit-8 was used to measure proliferation of HCC cell line.
Results: Using multiple databases, we identified 36 key prognostic genes highly expressed in HCC and associated with poor survival of patients. Meanwhile, the 36 gene signatures correlated with immune infiltration in HCC. Moreover, these genes were significantly associated with exhausted T cells and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) in HCC. Among the 36 key genes, SKA3, SGOL2, SPINDOC, TEDC2, TMCO3 and NUP205 were highly expressed in tumor samples compared with adjacent normal tissues in our HCC cohort (n=22). Additionally, proliferation of SMMC7721 cell line was inhibited when it interfered with SiRNA of each gene.
Conclusion: The 36 genes may serve as potential prognostic biomarkers and molecular targets to ameliorate tumor immune microenvironment (TIME) in HCC and therefore represent a novel avenue for individualized immunotherapy in HCC.
Keywords: HCC, prognosis, immune infiltrates, exhausted T cell, MDSC
Introduction
Hepatocellular carcinoma (HCC), which accounts for approximately 90% of primary liver cancers, is the sixth most common malignancy and the second leading cause of cancer-related mortality in the world.1,2 Although treatments with Sorafenib and Regorafenib show a modest survival benefit, their overall anti-tumor efficacies are still limited.3–6 Over recent decades, the landscape of cancer treatment has been revolutionized by immunotherapies.7 While such therapies as checkpoint blockade can lead to unprecedented clinical benefits for malignant melanoma, non-small cell lung cancer (NSCLC), bladder cancer and Hodgkin lymphoma, etc.,7–11 the response rate is low, and it is much lower in HCC,12 the underlying mechanisms remain to be elucidated. As a major immune organ, liver acts as a central immune-modulator to protect other organs and systems by maintaining immunologic tolerance.13 Sophisticated and unique immune sets in liver and HCC tumor microenvironment (TME) play important roles in virus infection, inflammatory reaction, immune evasion and response to immune therapy.13 Recently, various studies have identified that tumor-infiltrating lymphocytes in TME are associated with prognosis and immunotherapy response of HCC patients. For example, increased regulatory T cells (Treg) correlate with poor overall survival and resurgence-free time, while increased ratio values of B/T cells in TME correlate with longer survival in HCC.14,15 In addition, immune cell infiltration of the primary tumor affects individual response to checkpoint inhibitors in metastatic melanoma independent of PD-L1 status.16 Thus, there is an urgent need to uncover the molecular mechanisms of immune infiltrates in TME and to identify novel immune-related therapeutic targets in order to improve immunotherapy in HCC.
With rapid advances in next-generation sequencing (NGS) and bioinformatics, accumulated data have helped us to understand complicated biological characteristics of tumors in multiple dimensions. Simultaneously, more and more databases have been generated to analyze cancer pathogenesis comprehensively, such as Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and The Cancer Genome Atlas (TCGA, https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga),17 which have already molecularly characterized over 20,000 primary tumors and their matched normal samples among 33 types of cancer. According to the abundant data from TCGA and other databases, various related online visualized analyzing tools and software have also been created, such as GEPIA2 (http://gepia2.cancer-pku.cn/#index), which focuses on RNA sequencing data of 9736 tumors and 8587 normal samples from TCGA and GTEx projects. This can provide customizable functions, including differentially expressed survival-related genes in various cancers.18 Another tool, called Tumor Immune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/), is a comprehensive resource for systematic analysis of immune infiltrates across multiple cancer types.19,20 Such tools based on comprehensive databases extensively help us to uncover new potential prognostic markers in various cancer types as well as complicated associations between molecules and tumor immune microenvironment (TIME), so as to provide new potential targets for HCC immunotherapy, and to improve the precise treatment in HCC patients.
In our present study, we identified 36 key prognostic genes highly expressed in HCC and associated with poor survival of patients through multiple databases. Meanwhile, the 36 gene-signature correlated with immune infiltration in HCC. Moreover, these genes were significantly associated with exhausted T cells and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) in HCC. Among them, SKA3, SGOL2, SPINDOC, TEDC2, TMCO3 and NUP205 were highly expressed in tumor samples compared with adjacent normal tissues in our HCC cohort (n=22), Additionally, proliferation of SMMC7721 cell line was inhibited when it interfered with SiRNA of each gene.
Materials and Methods
Gene Analysis in GEPIA2
GEPIA2 is an updated version of GEPIA that analyzes RNA sequencing expression data of 9736 tumors and 8587 normal samples from TCGA and GTEx projects, using a standard processing pipeline. It provides customizable functions such as tumor/normal differential expression profiling, according to cancer types or pathological stages, patient survival analysis, similar gene detection, correlation analysis and dimensionality reduction analysis (http://gepia2.cancer-pku.cn/#index).18 In our study, we applied survival analysis to the most differential survival genes in hepatocellular carcinoma (HCC). We analyzed correlation between these genes and immune infiltrates in HCC through TIMER. We defined overall survival as prognosis and the median as group cutoff.
Correlation Analysis Through TIMER Database
Tumor Immune Estimation Resource (TIMER) is a comprehensive resource for systematic analysis of immune infiltrates across diverse cancer types (https://cistrome.shinyapps.io/timer/).19,20 Relative abundances of 6 immune infiltrates (B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages and dendritic cells) are estimated, which will be validated using pathological estimations.19 In our present study, we used TIMER to analyze association between the most differential survival genes and immune infiltrates in HCC. Moreover, associations between each gene and various markers of tumor-infiltrating immune cells were analyzed, including CD8+T cells, T cells (general), B cells, monocytes, TAMs, M1 macrophages, M2 macrophages, neutrophils, natural killer (NK) cells, dendritic cells (DCs), T-helper 1 (Th1) cells, T-helper 2 (Th2) cells, follicular helper T (Tfh) cells, T-helper 17 (Th17) cells, Treg, exhausted T cells, myeloid-derived suppressor cells (MDSC) and mucosal-associated invariant T (MAIT) cells, as referenced in prior studies.13,41,46,51–56 Then, Spearman correlation and estimated statistical significance of a pair of defined genes in HCC were generated through correlation modules. The targeted gene was applied to x-axis, and related markers were represented on y-axis. Relatively, gene expression level was displayed with log2 RSEM.
Gene Expression and Prognostic Analysis Through HCCDB Database
Integrative Molecular Database of Hepatocellular Carcinoma (HCCDB) was contributed by Tsinghua University and National Center for Liver Cancer & Shanghai Eastern Hepatobiliary Surgery Hospital. This database curated 15 public HCC expression datasets with up to ~4,000 clinical HCC samples and serves as a one-stop online resource for HCC gene expression and other comprehensive analyses, including prognostic and co-expression analysis (http://lifeome.net/database/hccdb/home.html).21 In our study, the most differential survival genes associated with immune infiltrates were analyzed by HCCDB, to compare the expression levels of these genes in HCC tumors with normal tissues, as well as their prognostic value in HCC.
Kaplan–Meier Plotter Database Analysis
The Kaplan–Meier plotter is capable of assessing potential effects of 54,000 genes on survival in 21 cancer types. Primary purpose is meta-analysis based discovery and validation of survival biomarkers (http://www.kmplot.com/analysis/index.php?p=background).57,58 In our study, we used this database to confirm prognostic value of selected key genes in HCC. The HR with 95% CI was marked.
Gene Ontology and Protein–Protein Interaction (PPI) Network Analysis
For better understanding of key prognostic genes associated with immune infiltrates in HCC, cellular component, molecular function and biological process were analyzed by Gene Ontology through FunRich software (http://www.funrich.org/). PPI network was analyzed by STRING online tool (https://string-db.org/cgi/input.pl?sessionId=VZH6o5N19gVM&input_page_show_search=on).
SPINDOC, TEDC2, NUP205, SGOL2, SKA3 and TMCO3 Expression Analysis
This study was approved by Ethical Committee of Beijing Shijitan Hospital and performed by complying with all relevant ethical regulations [No. sjtkyll-lx-2021(22)]. Written informed consent from each patient was obtained prior to study commencement. All processes were conducted in accordance with the Declaration of Helsinki.
About 10 mg HCC sample or adjacent normal tissue was disrupted and homogenized in 1 mL RNA-Solv Reagent (OMEGA R6934) containing 2% 2-mercaptoethanol. Total RNA was isolated using the E.Z.N.A Total RNA Kit II (OMEGA R6934-02) according to the tissue protocol. ReverTra Ace qPCR RT Kit (TOYOBO FSQ-101) was used to synthesize the first-strand cDNA. Real-time qPCR was done on Quant Studio 6 Real Time PCR System with SYBR (Thermo Fisher Scientific). 18S was used to normalize relative expression of target genes.
About 1×10^5 SMMC7721 cells were seeded into 12-well plates. Lipofectamine 3000 reagent was added to transfect SiRNA (final concentration=0.1MM) into cells at 70–80% confluence, according to standard protocol (Thermo Fisher Scientific). Cells were incubated in incubator with 5% CO2 at 37°C. Two days later, total RNA was collected using E.Z.N.A Total RNA Kit II (OMEGA R6934-02) according to cell protocol, followed by qPCR, as described previously.
Proliferation Assays
Two thousand SMMC7721 cells were seeded in 96-well plates. One day later, SiRNA (final concentration=0.1MM) was transfected into cells as described above. Cell Counting Kit-8 (YESEN 40203, Shanghai, China) was used to measure proliferation from the 1st to 5th day after interfered with SiRNA. Cell viability (%) = [OD450 (SiRNA)-OD450 (BLANK)]/[OD450 (SiRNA-NC)-OD450 (BLANK)].
Statistical Analysis
The survival curves were generated by GEPIA2, Kaplan–Meier plotter and HCCDB. Spearman correlation was used to evaluate association between gene expression and a specific parameter. The strength of correlation was defined by absolute value as follows: 0.00–0.19 “very weak”, 0.20–0.39 “weak”, 0.40–0.59 “moderate”, 0.60–0.79 “strong”, and 0.80–1.0 “very strong”. Statistical comparisons of mRNA levels between HCC tumors and adjacent normal tissues were conducted using paired t-tests or a one-way analysis of variance (ANOVA). Unpaired t-test was used to analyze viability of cells between NC and SiRNA group. A P-value of <0.05 was considered statistically significant.
Results
Identification of the Most Differential Survival-Related Genes and Their Association with Immune Infiltrates in HCC
Using TCGA database through GETIA2, the most differential survival-related genes were explored, which may serve as potential prognostic markers in HCC (Figure 1 and Supplementary Table 1). Five hundred genes were identified, including the top 10 genes HILPDA, CCDC58, B3GAT3, CLEC3B, CTB-147N14.6, SOCS2, LPCAT1, PES1, DNASE1L3 and AHSA1. Then, the association between each differential survival gene and immune infiltrates in HCC was analyzed through TIMER. We defined a relation between a gene and immune infiltration as existing when >4 types of immune cell (B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages and dendritic cells) were associated with a target gene. Consequently, 119 of 500 genes were identified as being associated with immune infiltrates in HCC (Figure 1 and Supplementary Table 2).
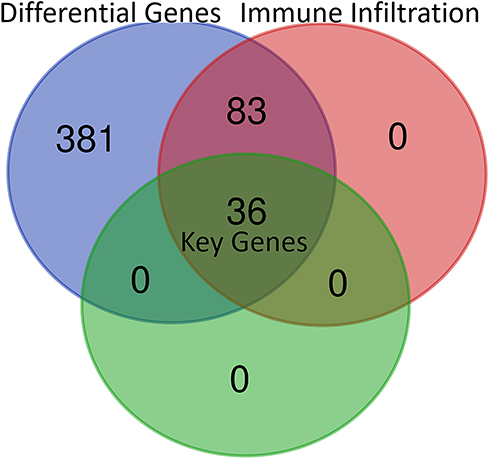 |
Figure 1 Thirty-six key genes were identified from multiple databases. Differential genes: The 500 most differential survival genes in HCC through TCGA database; Immune infiltration: the differential survival genes which are generally correlated with immune infiltration in HCC in TCGA through TIMER; Key genes: the differential expression genes with prognostic value which are correlated with immune infiltration in HCC from the 500 most differential survival genes. |
Identification of Key Candidate Prognostic Biomarkers Correlated with Immune Infiltrates in HCC
To identify new potential prognostic biomarkers in HCC, first we compared the expression levels of 119 genes (Supplementary Table 2) between HCC tumor and adjacent normal tissues, then we evaluated the prognostic values of these genes through the database Integrative Molecular Database of Hepatocellular Carcinoma (HCCDB), which has integrated 15 public datasets with up to 4000 clinical HCC samples and serves as a one-stop online resource for HCC gene expression and comprehensive analysis, including prognostic and co-expression analyses (http://lifeome.net/database/hccdb/home.html).21 From the above 119 genes correlated with immune infiltrates, 36 genes with significantly different expression between HCC and adjacent tissues were identified (Figure 2 and Supplementary Table 3). Further analysis indicated that the different expression levels of the 36 genes between HCC and adjacent tissues exhibited notable prognostic value in HCC; a higher level of gene expression was associated with much poorer survival (Table 1). In addition, we also confirmed each gene’s prognostic value in HCC through Kaplan–Meier plotter database analysis (http://www.kmplot.com/analysis/index.php?p=service&start=1) (Figure 3 and Supplementary Figure 1). The results suggested that the 36 genes may serve as new potential prognostic markers in HCC.
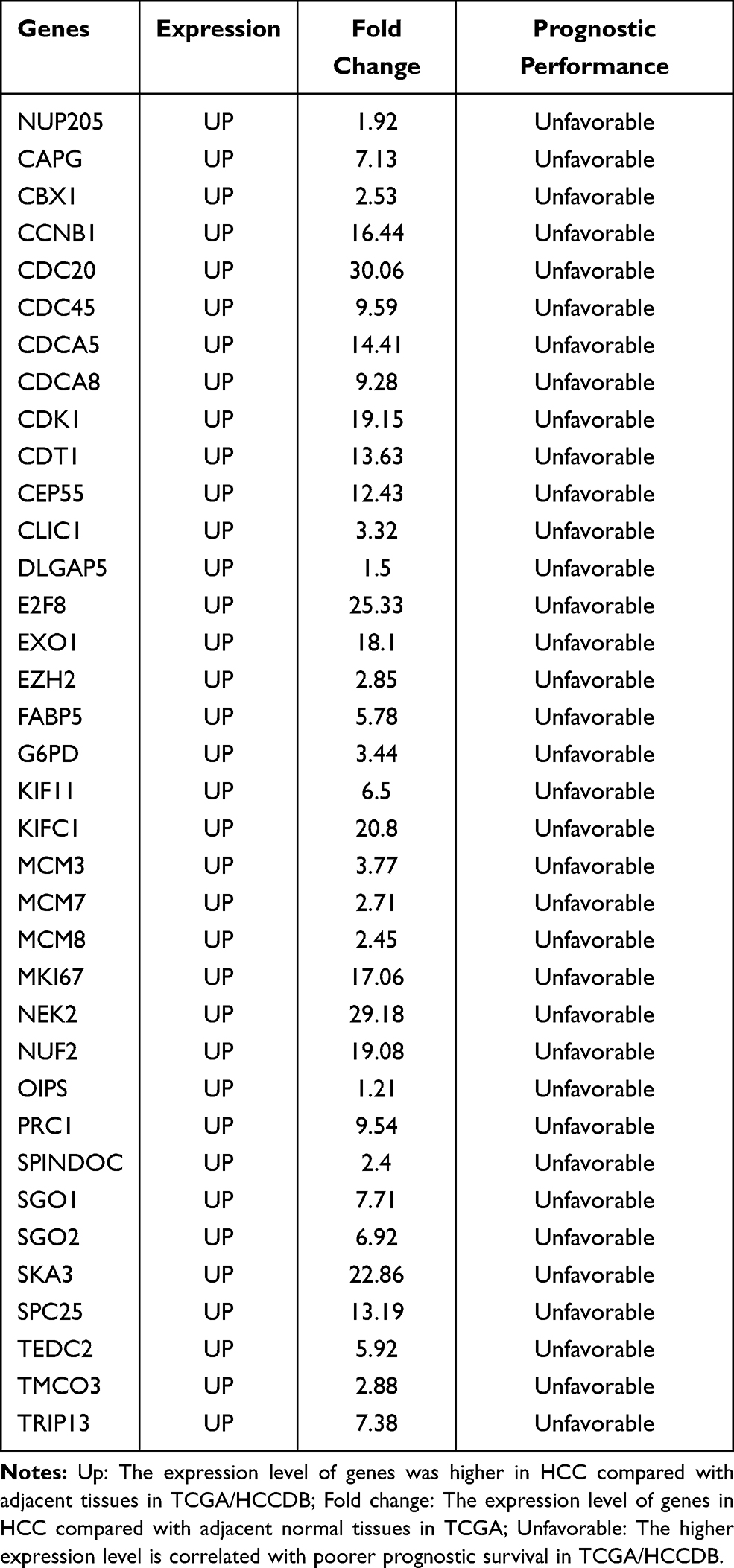 |
Table 1 Key Candidate Prognostic Biomarkers Correlated with Immune Infiltrates in HCC |
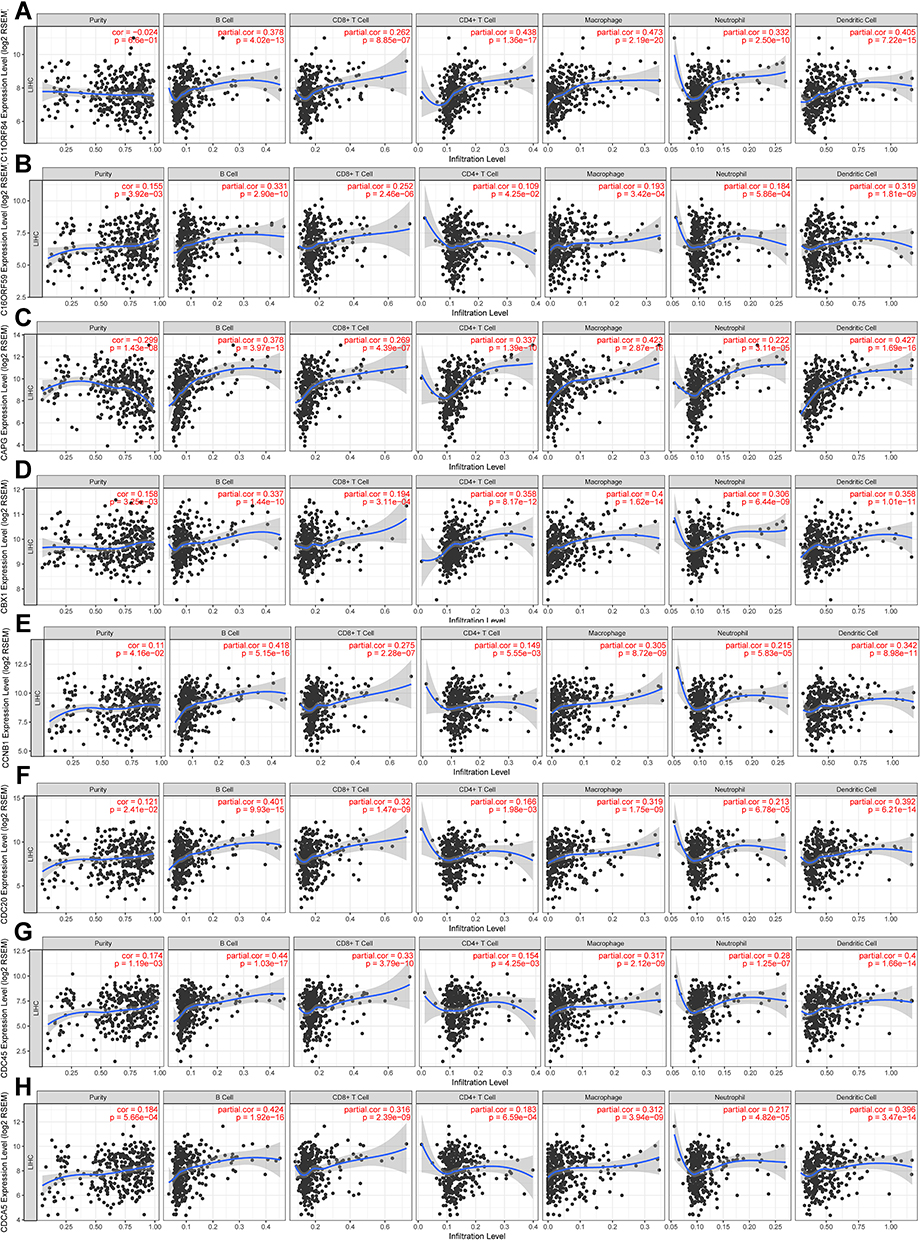 |
Figure 2 The key prognostic genes were correlated with immune infiltrates in HCC. “partial.cor” means tumor purity-corrected partial Spearman correlation. (A) C11ORF84 (SPINDOC), (B) C16ORF59 (TEDC2), (C) CAPG, (D) CBX1, (E) CCNB1, (F) CDC20, (G) CDC45, (H) CDCA5.The immune infiltrates correlation analysis results of the other 28 genes can be found in Supplementary Table 3 with a highly yellow marker. |
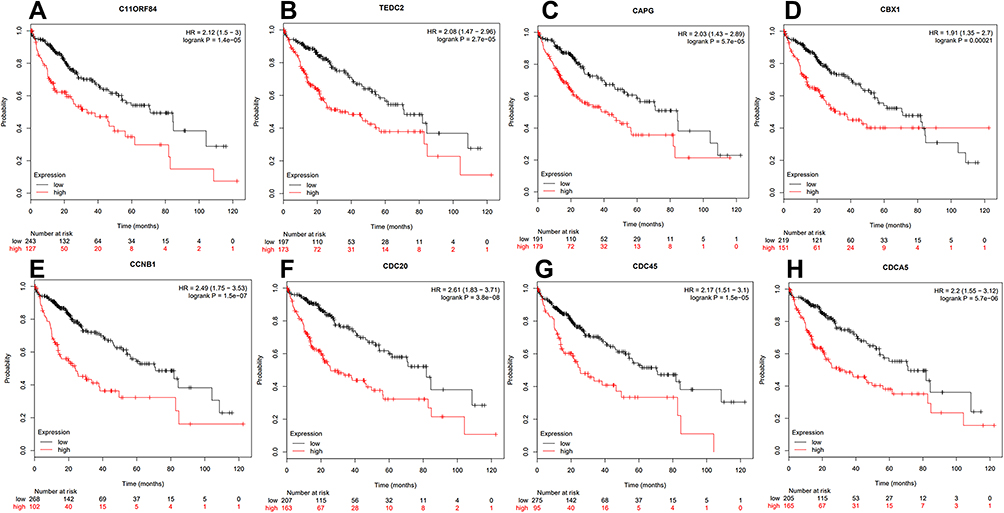 |
Figure 3 Kaplan–Meier survival curves were used to confirm the prognostic value of the key genes from Kaplan–Meier plotter database. Overall survival (OS) was used in all the genes to evaluate their prognostic value. (A) C11ORF84, (B) C16ORF59, (C) CAPG, (D) CBX1, (E) CCNB1, (F) CDC20, (G) CDC45, (H) CDCA5.The OS prognostic value analysis results of the other 28 genes can be found in Supplementary Figure 1. |
Correlation Between Key Biomarker Expressions and Immune Marker Sets in HCC
To our surprise, all 36 candidates were upregulated in HCC. We then applied TIMER to further explore molecular mechanisms underlying relationship between 36 key potential prognostic markers and TIME (Table 2, Supplementary Table 4). Interestingly, these 36 genes correlated with infiltration of CD8+T cells, general T cells, B cells, monocytes, tumor-associated macrophages (TAMs), and type 1 T helper cells (Th1). The strength of correlation varied from “very weak” to “moderate”. These genes showed no significant correlation with subtype of macrophages (M1/M2), natural killer cells, follicular helper T cells (Tfh), Th17 and mucosal-associated invariant T cells (MAIT). For neutrophils and dendritic cells (DCs), these genes showed significant correlation with ITGAM and ITGAX, respectively. For exhausted T cells, these genes were significantly positively correlated with PDCD1, CTLA4, HAVCR2 and TIGIT. These genes also showed general correlation with LAG3, BTLA, ENTPD1, CD27, TNFRSF9, WARS and CXCL13. Thus, these genes might be highly connected with T cell exhaustion in TME of HCC, which may therefore affect the survival of HCC patients.
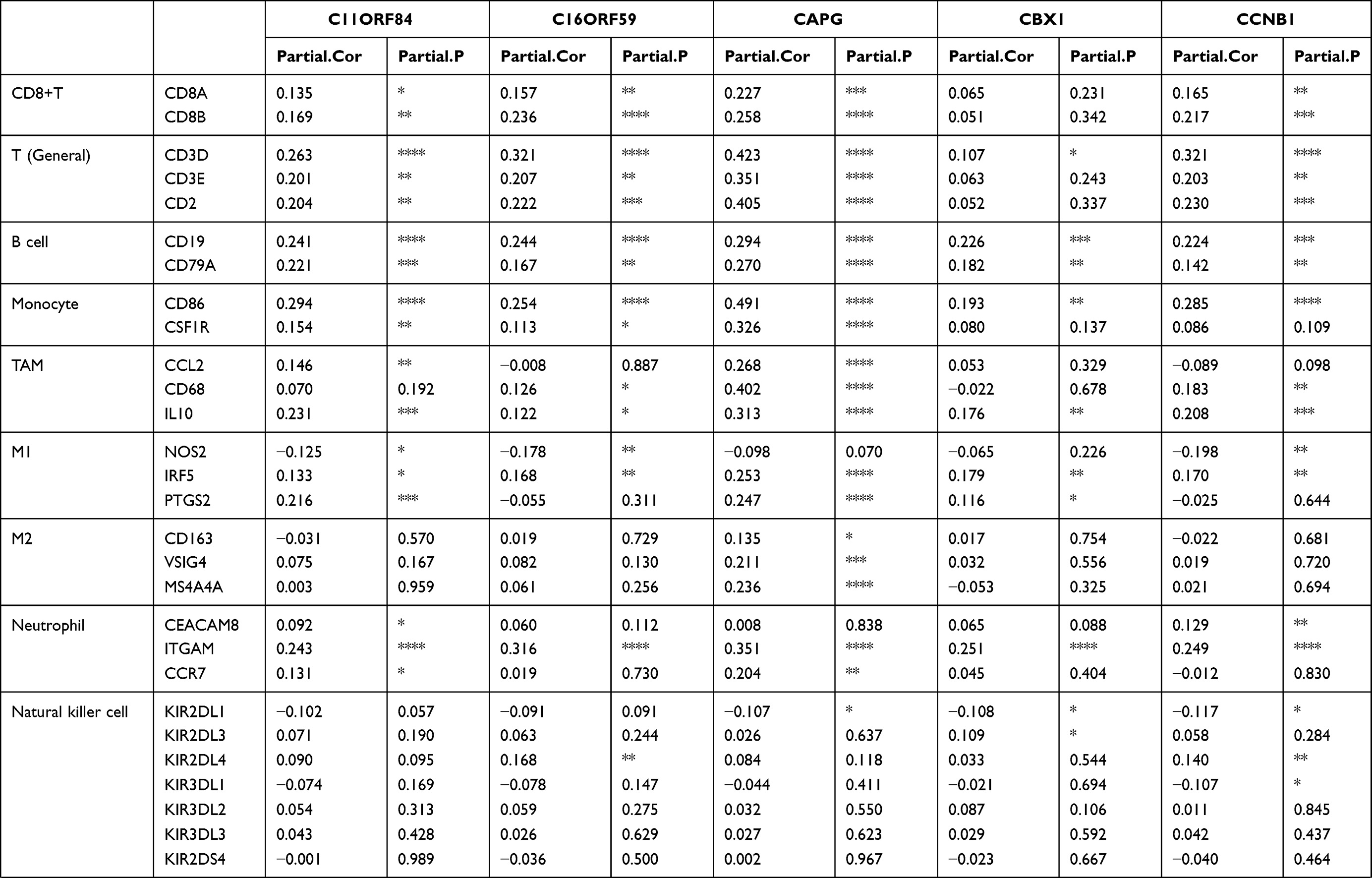 | 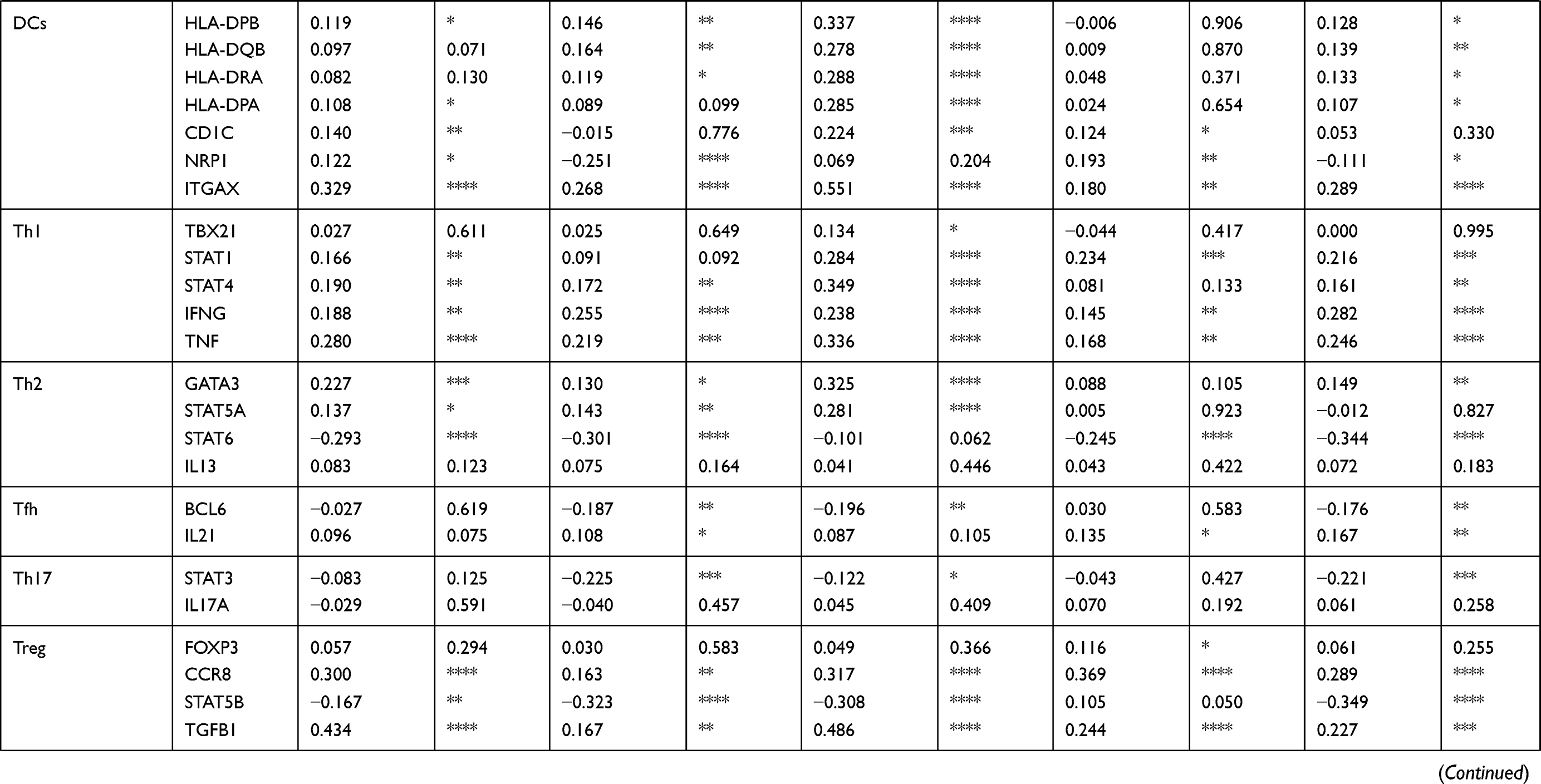 | 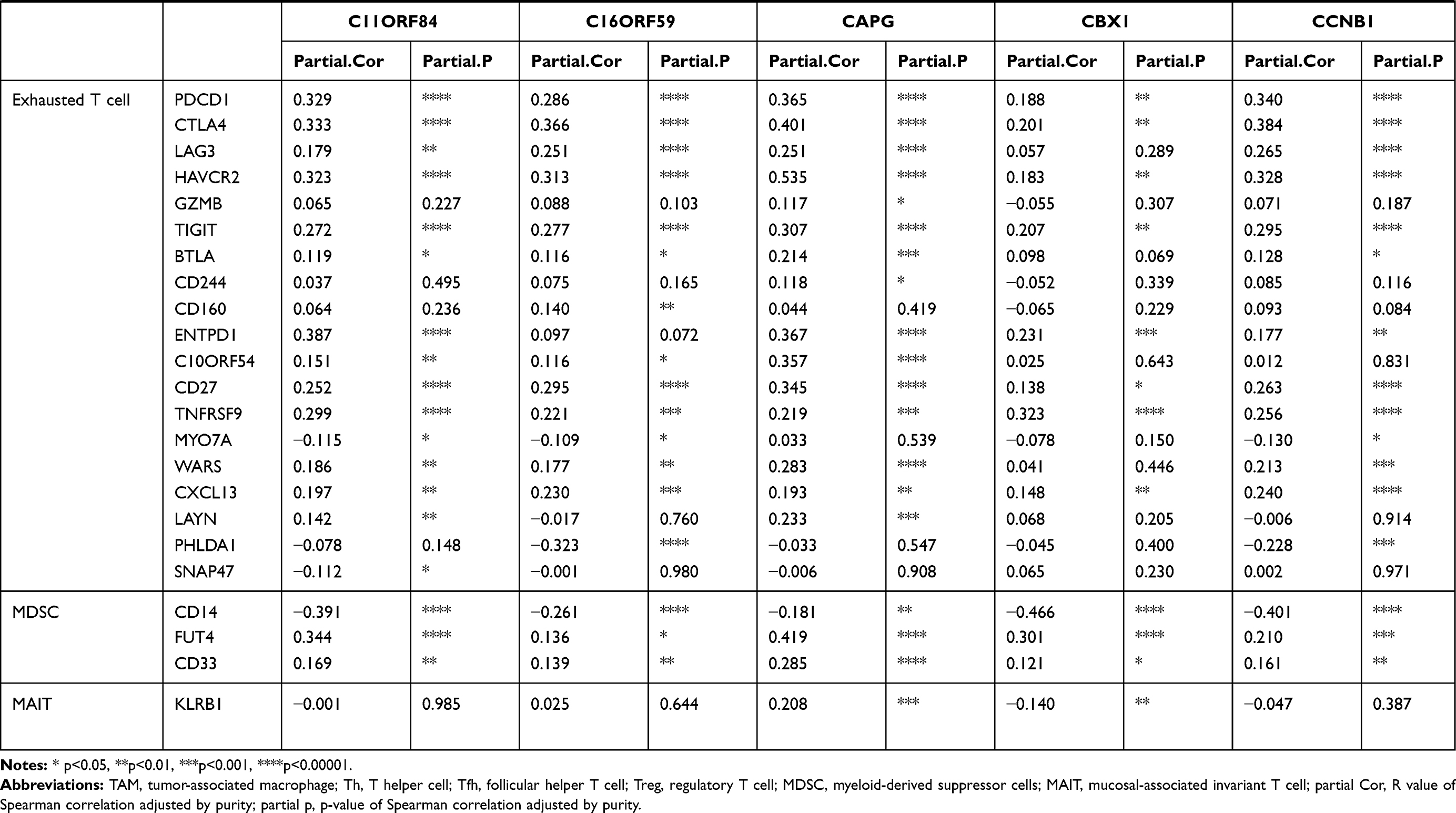 |
Table 2 Correlation Between Each Key Gene and Immune Cell Marker Sets in HCC |
In addition, for Treg cells, these genes showed very weak correlation with FOXP3, were negatively correlated with STAT5B and positively correlated with CCR8 and TGF-B1, suggesting these genes could probably influence Treg differentiation in HCC TME. Moreover, for MDSCs, these genes were significantly and positively associated with FUT4 (CD15, the molecular marker of PMN-MDSC) but negatively correlated with CD14 (the molecular marker of M-MDSC). Thus, considering their important role in the TIME, the other underlying mechanism behind the prognostic roles of these genes in HCC may be due to their positive correlations with Treg and MDSC.
Key Biomarkers' Gene Ontology and Protein–Protein Interaction (PPI) Network Analysis
To better understand potential roles of 36 key prognostic genes associated with immune infiltration, FunRich software was used to analyze cellular component, molecular function and biological process. Most of these genes can be localized in nucleus (82.4%) and nucleoplasm (58.8%) (Figure 4A). The most enriched biological processes were protein binding (91.7%), chromatin binding (22.2%) and DNA replication origin binding (11.1%) (Figure 4B). Moreover, these genes were associated with various functions, including cell division (45.7%), chromosome segregation (20%), and DNA replication (20%) (Figure 4C). In addition, based on PPI analysis, there were significant interactions among 36 proteins (avg. local clustering coefficient=0.774, PPI enrichment p-value <1.0e-16, Figure 5).
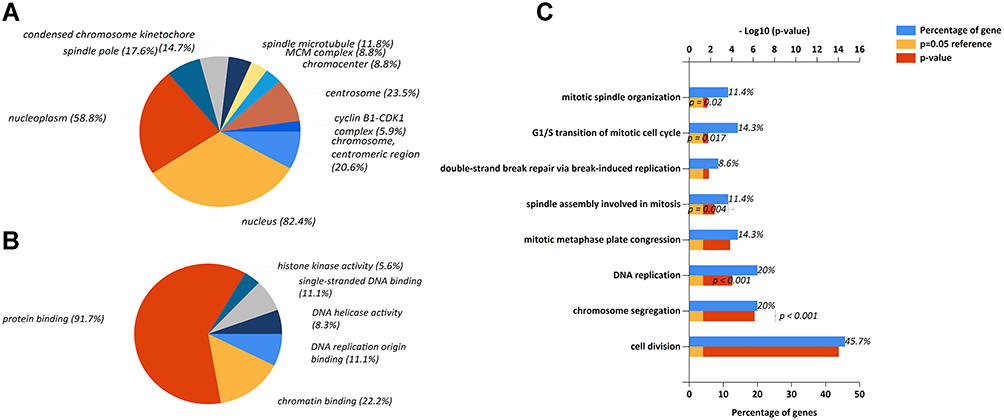 |
Figure 4 (A) Functional enrichment analysis of key prognostic markers. (A) Cellular components, (B) Biological processes, (C) Molecular functions in gene ontology analysis using FunRich software. p<0.05 was considered a statistically significant difference. |
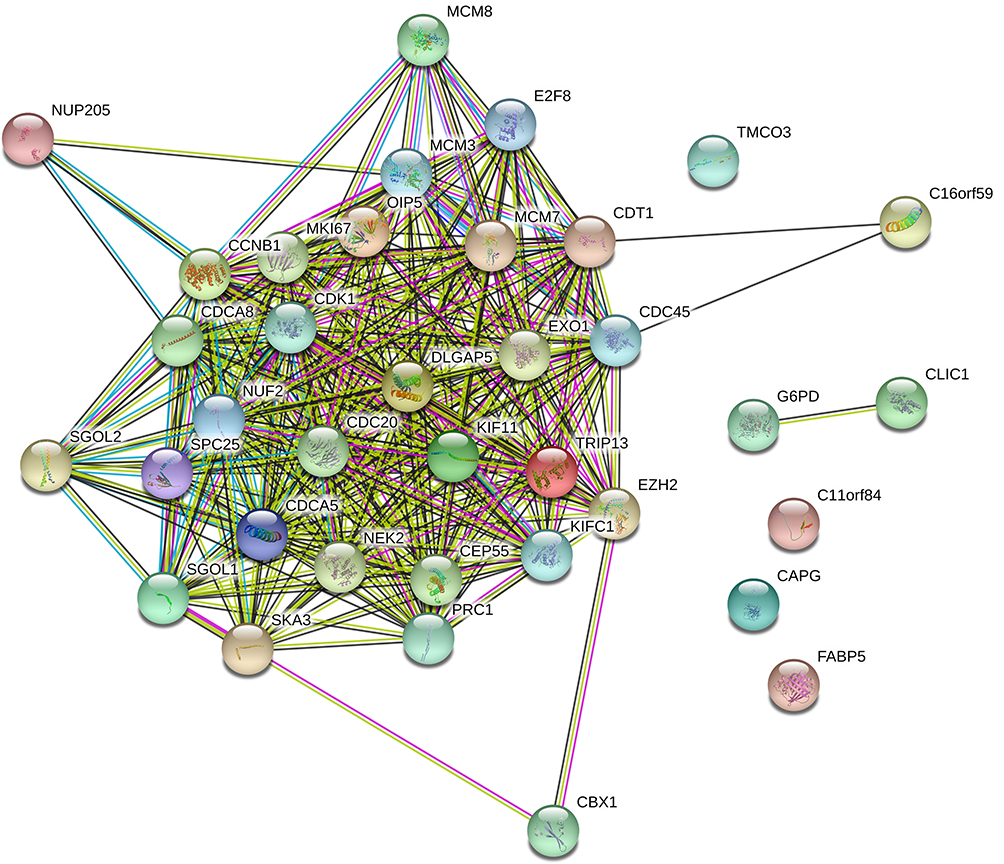 |
Figure 5 Protein–protein interaction (PPI) network for key potential prognostic genes in HCC. Those 36 key prognostic genes related to the PPI network are analyzed by STRING online tool (avg. local clustering coefficient=0.774, PPI enrichment p-value<1.0e-16). |
Expression of SPINDOC, TEDC2, NUP205, SGOL2, SKA3 and TMCO3 in HCC Samples
To confirm differential expression of key genes, we analyzed mRNA levels of SPINDOC, TEDC2, NUP205, SGOL2, SKA3 and TMCO3 in clinical samples. All these genes are highly expressed in HCC tumor tissues compared with adjacent normal tissues (Figure 6). The clinical information of the patients is summarized in Supplementary Table 7.
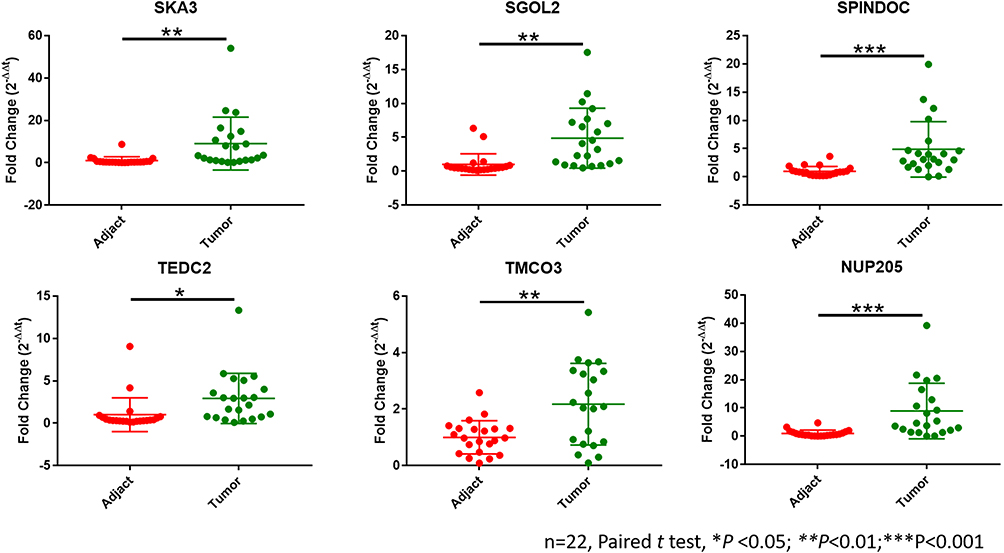 |
Figure 6 Twenty-two paired HCC tissues and adjacent normal tissues were collected from Aug. 2011 to Dec. 2019 in our hospital. The qPCR results showed that all these 6 genes were more highly expressed in HCC samples compared with adjacent normal tissues.*p<0.05, **p<0.01, ***p<0.001. |
Interfering mRNA Expression of SPINDOC, TEDC2, NUP205, SGOL2, SKA3 and TMCO3 Inhibit the Proliferation of the SMMC7721 HCC Cell Line
Considering that SPINDOC, TEDC2, NUP205, SGOL2, SKA3 and TMCO3 were highly expressed in HCC and higher expression is associated with poorer prognosis, we used specific SiRNA to knock down each gene respectively in human SMMC7721 HCC cell line. Interestingly, the proliferation of the SMMC7721 cell line was significantly inhibited when the expressions of SPINDOC, TEDC2, NUP205, SGOL2, SKA3 or TMCO3, respectively, were interfered with (Figure 7, Supplementary Figure 2 and Supplementary Table 6).
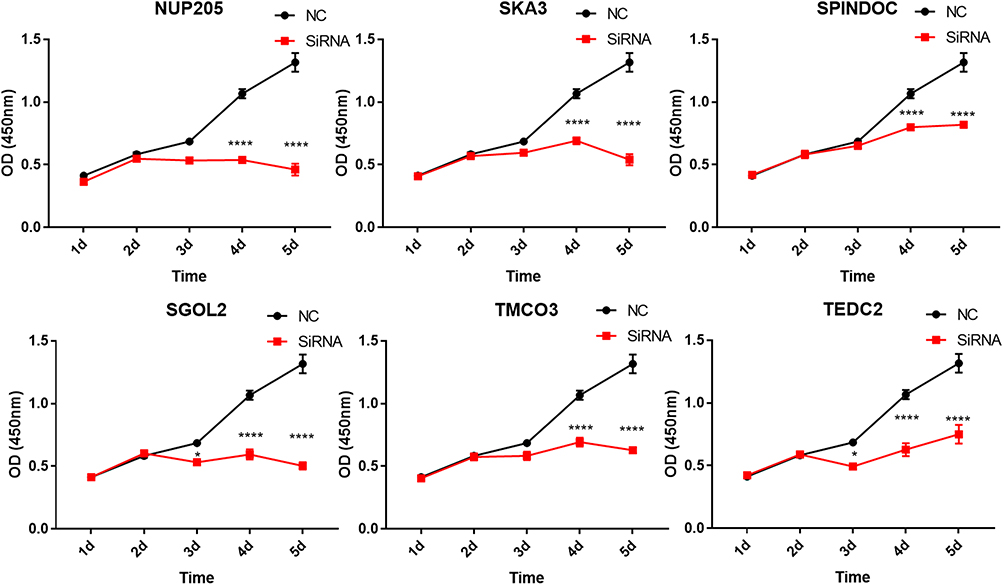 |
Figure 7 CCK8 reagents were used to assay the proliferation ability after SiRNA interference. The most effective SiRNA from Supplementary Figure 2 was used to interfere with the mRNA expression. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. |
Discussion
As the second leading cause of cancer-related death and the fifth most common cancer, primary liver cancer causes ~800,000 deaths each year, globally22. Generally, 80–90% of liver cancer cases are hepatocellular carcinoma (HCC).23 With advances in basic and clinical science, underlying mechanisms of HCC tumorigenesis and progression have been explored. The main risk factors for HCC are well known, including hepatitis B and C virus infection, alcohol consumption and aflatoxin.23 In addition, molecular pathogenesis of HCC was also revealed. Representative multiple targets drugs (Sorafenib and Regorafenib) have been widely applied in clinical practice and exhibit only a minor survival benefit, thus the prognosis of HCC remains very poor.3–6 Recently, immune checkpoint inhibitor (ICI) has reformed the paradigm of cancer therapy and achieved marvelous results in controlling several types of cancer. However, most cancer patients, especially those with HCC, show no response to ICI.12 Due to the complicated immune status in liver by itself, we hypothesize that some key molecules may affect its prognosis and correlate with the immune context in HCC.
In this study, we first analyzed the most differential survival-related genes in HCC using TCGA database and, through GEPIA2, 500 genes were identified (Supplementary Table 1). We then analyzed the relationships between these genes and immune infiltrates in HCC through TIMER, and 119 genes were found to correlate with the immune infiltrates in HCC (Figure 1 and Supplementary Table 2). To further estimate their prognostic values in HCC, we first compared the expression levels of the 119 genes between HCC tumor tissues and adjacent normal tissues, and then evaluated their prognostic values in HCC through another database, HCCDB, which specifically collected data from HCC-related studies and comprehensively analyzed the characteristics of HCC.21 Among the 119 genes, 36 key genes exhibited significantly differential expression in HCC tumors compared with adjacent tissues. Meanwhile, the 36 genes showed unfavorable prognostic value in HCC. A higher expression of 36 genes correlated with much poorer survival (Table 1). In addition, the gene ontology analysis revealed that these 36 molecules were mostly located in the important organs of the cell (nucleus and nucleoplasm), participating in significant biological processes (protein binding, chromatin binding and DNA replication origin binding), and were associated with various key functions (cell division, chromosome segregation, DNA replication) (Figure 4). Moreover, (PPI network analysis indicated that these 36 molecules had mutual correlations (Figure 5). As we all know, uncontrolled cell division is one of the most significant characteristics of cancer and the results of our gene ontology and PPI analysis may partly confirm the significant correlation between these 36 genes and the prognosis of HCC.
Most of these genes (30/36) have been reported to correlate with the prognosis of HCC by other authors24 (Supplementary Table 5), and our results were very much in accordance with those of previous studies. In addition, our results indicated that all 36 genes had a significant correlation with the immune infiltration in HCC. This kind of relationship, to the best of our knowledge, hads never been reported before. Furthermore, for the 6 critical potential prognostic genes (NUP205, SKA3, SPINDOC, SGOL2, TMCO3 and TEDC2) no type of relationship with HCC, including with the prognosis of HCC, to our knowledge, has ever been reported before. Moreover, the 6 genes were confirmed to highly express in HCC tumors in our own cohort (n=22) (Figure 6). Notably, the proliferation of SMMC-7721 human hepatocellular carcinoma cell line was inhibited when the expressions of NUP205, SKA3, SPINDOC, SGOL2, TMCO3 and TEDC2, respectively, were interfered with (Figure 7). Their underlying molecular mechanism is worth further exploration to discover potential therapeutic targets for HCC.
In a previous study, SPINDOC was shown to directly bind Spindlin1 (SPIN1) and to strongly disrupt the histone methylation reading ability of SPIN1.25 SPIN1 is a transcriptional coactivator with critical functions in embryonic development and plays emerging roles in cancer. However, the interaction between SPINDOC with SPIN1 in HCC is worth deep exploration.
The functions of TEDC2 remain unexplored. Using a triple-level (gene-, transcript-, and exon-level) analysis in lung adenocarcinoma transcriptomes with 77 paired tumor–normal tissues, one previous study proposed TEDC2 as a differentially expressed gene.26 So far, the role of TEDC2 in HCC is unclear, and fundamental and clinical investigation is needed.
NUP205 (nucleoporin 205) encodes a nucleoporin, which can actively transport proteins, RNAs and ribonucleoprotein particles between nucleus and cytoplasm. One previous study identified NUP205 as a TMEM209-interacting protein. Mass spectrometric analysis indicated that, when stabilizing NUP205, c-Myc was upregulated in the nucleus. Overexpression of TMEM209 and TMEM209–NUP205 interaction could drive the proliferation of lung cancer.27 It is promising to explore its role and underlying mechanisms in HCC tumorigenesis.
SGO2 (shugoshin 2), also known as SGOL2 or TRIPIN, plays an important role in cell cycle progression during meiosis.28,29 SGO2 contains a MAD1/CDC20-like–MAD2 interaction motif and competes with MAD1 and CDC20 for binding to Mad2.29 CDC20, one key prognostic gene as identified in our present study, has also been reported previously.30–32 Moreover, it was recognized as a potential cancer therapeutic target by other researchers.33 Thus, further studies are required to elucidate the functions of SGO2/CDC20 in HCC.
SKA3 (spindle and kinetochore associated complex subunit 3), also called RAMA1 or C13orf3, participates in mitosis,34 and is considered one of the genes susceptible to prostate cancer metastasis.35 Overexpression of SKA3 contributes to the progression of colorectal adenoma to carcinoma progression.36 Thus, high expression of SKA3 associated with immune infiltration in HCC suggests it is a promising candidate for prognosis evaluation and therapy of HCC.
TMCO3 (transmembrane and coiled-coil domain 3), also known as C13orf11, was identified as one of the interaction partners of EGFR (epidermal growth factor receptor).37 Considering that the EGFR-related pathway plays an important role in HCC tumorigenesis,38–40 it is necessary to clarify whether TMCO3 affects the prognosis and immune status through the EGFR-related pathway.
In a previous study, we used single-cell sequencing to analyze the landscape of infiltrating T cells in liver cancer and revealed that subsets of exhausted T cells correlated with poorer prognosis in HCC patients.41 In present study, we analyzed the relationship between the 36 key potential prognostic genes and the immune sets in HCC with TIMER. The results showed that the 36 key potential prognostic genes were significantly associated with exhausted T cell markers, including PDCD1, CTLA4, HAVCR2 and TIGIT, suggesting these genes were closely related to T cell exhaustion in TME of HCC. Previous studies demonstrated that exhausted T cells in TME highly expressed inhibitory receptors, exhibited lower effector cytokine secretion and cytolytic activity and ultimately led to failure of tumor eradication. Reversing exhausted status of T cells represents a promising breakthrough in cancer immunotherapy.42,43 Thus, the 36 genes in this study may be used as potential targets to improve the prognosis of HCC, probably through reversing T cell exhaustion in TME.
Our results also showed that the expression of these 36 genes significantly and positively correlated with PMN-MDSC (CD14-CD11b+CD33+CD15+), whose phenotypes are similar to granulocytes, whereas it negatively correlated with M-MDSC, whose phenotypes are similar to monocytes.44 Previous studies indicated that subtypes of MDSC varied in different cancer types, depending on TME.45 A lower level of MDSC in cancer was related to favorable prognosis after immune checkpoint inhibitor therapy.44,46 In a spontaneous model of uveal melanoma, PMN-MDSC infiltrated in primary tumors could promote the dissemination of cancer cells through inducing epithelial-mesenchymal transition (EMT).47 Thus, the 36 key genes may regulate the differentiation of MDSCs in TME of HCC, and influence the prognosis through up-regulating PMN-MDSC. In addition, our TIMER analysis showed that the 36 genes positively correlated with CCR8 and TGF-B1, while they negatively correlated with STAT5B. CCR8, TGF-B1 and STAT5B are well-known Treg regulating molecules, therefore these genes may affect the prognosis of HCC by participating in the differentiation of Treg in TME of HCC.
Recently, immune infiltration in TME has been extensively explored. Tumor-infiltrating lymphocytes can affect the response to immunotherapy and prognosis.48,49 A recent study indicated that immune cell infiltration within primary tumors plays a more important role than PD-L1 status in response to checkpoint inhibition in metastatic melanoma.16 Additionally, pre-existing tumor-specific T cells may have limited reinvigoration capacity through checkpoint blockade therapy;50 however, the underlying mechanisms remains unknown. In our study, the 36 key genes related to the suppressive immune status in HCC may partly elucidate their connection with the poor survival of HCC patients. To our knowledge, although prognostic value was reported in most of these 36 key genes (Supplementary Table 5), their correlations with TIME in HCC were rarely explored. Better understanding of the correlation between these key prognostic genes and immune sets will open a promising avenue to ameliorate TIME and improve the immunotherapy in HCC.
Conclusions
In our study, 36 key potential prognostic genes are highly expressed in HCC and correlate with poor prognosis. These genes are significantly associated with suppressive immune status in HCC. These genes may serve as prognostic markers and potential targets to improve immunotherapy in HCC. Among them, SPINDOC, TEDC2, NUP205, SGOL2, SKA3 and TMCO3 are confirmed to be highly expressed in HCC tumors compared with adjacent normal tissues. These 6 genes may regulate proliferation of SMMC7721 (human HCC) cells. Collectively, these findings hugely enrich our knowledge of molecular characteristics and immune status in HCC.
Acknowledgments
We thank all the scientists for their efforts on the public databases which were used in our present study.
Funding
This study was supported by National Natural Science Foundation of China (No. 91742203, 82072601), Beijing Natural Science Foundation (No. 7182075) and Scientific Research Foundation for the Returned Overseas Chinese Scholars (No. 2110000021).
Disclosure
The authors declare no conflicts of interest.
References
1. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
2. Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33. doi:10.3322/caac.21654
3. Abou-Alfa GK, Schwartz L, Ricci S, et al. Phase II study of sorafenib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2006;24(26):4293–4300. doi:10.1200/JCO.2005.01.3441
4. Ben Mousa A. Sorafenib in the treatment of advanced hepatocellular carcinoma. Saudi J Gastroenterol. 2008;14(1):40–42. doi:10.4103/1319-3767.37808
5. Bruix J, Tak W-Y, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412–3419. doi:10.1016/j.ejca.2013.05.028
6. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet. 2017;389(10064):56–66. doi:10.1016/S0140-6736(16)32453-9
7. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi:10.1126/science.aaa8172
8. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. 2007;19(7):813–824. doi:10.1093/intimm/dxm057
9. Okazaki T, Chikuma S, Iwai Y, et al. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14(12):1212–1218. doi:10.1038/ni.2762
10. Berger KN, Pu JJ. PD-1 pathway and its clinical application: a 20year journey after discovery of the complete human PD-1 gene. Gene. 2018;638:20–25. doi:10.1016/j.gene.2017.09.050
11. Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi:10.1056/NEJMoa1200694
12. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, Phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi:10.1016/S0140-6736(17)31046-2
13. Ringelhan M, Pfister D, O’Connor T, et al. The immunology of hepatocellular carcinoma. Nat Immunol. 2018;19(3):222–232. doi:10.1038/s41590-018-0044-z
14. Garnelo M, Tan A, Her Z, et al. Interaction between tumour-infiltrating B cells and T cells controls the progression of hepatocellular carcinoma. Gut. 2017;66(2):342–351. doi:10.1136/gutjnl-2015-310814
15. Xue H, Lin F, Tan H, et al. Overrepresentation of IL-10-expressing B cells suppresses cytotoxic CD4+ T cell activity in HBV-induced hepatocellular carcinoma. PLoS One. 2016;11(5):e0154815. doi:10.1371/journal.pone.0154815
16. Kumpers C, Jokic M, Haase O, et al. Immune cell infiltration of the primary tumor, not PD-L1 status, is associated with improved response to checkpoint inhibition in metastatic melanoma. Front Med (Lausanne). 2019;6:27.
17. Teufel A. Bioinformatics and database resources in hepatology. J Hepatol. 2015;62(3):712–719. doi:10.1016/j.jhep.2014.10.036
18. Tang Z, Kang B, Li C, et al. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556–W560. doi:10.1093/nar/gkz430
19. Li B, Severson E, Pignon J-C, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17(1):174. doi:10.1186/s13059-016-1028-7
20. Li T, Fan J, Wang B, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77(21):e108–e110. doi:10.1158/0008-5472.CAN-17-0307
21. Lian Q, Wang S, Zhang G, et al. HCCDB: a database of hepatocellular carcinoma expression atlas. Genomics Proteomics Bioinformatics. 2018;16(4):269–275. doi:10.1016/j.gpb.2018.07.003
22. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86. doi:10.1002/ijc.29210
23. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi:10.1038/nrdp.2016.18
24. Xiang X-H, Yang L, Zhang X, et al. Seven-senescence-associated gene signature predicts overall survival for Asian patients with hepatocellular carcinoma. World J Gastroenterol. 2019;25(14):1715–1728. doi:10.3748/wjg.v25.i14.1715
25. Bae N, Gao M, Li X, et al. A transcriptional coregulator, SPIN.DOC, attenuates the coactivator activity of Spindlin1. J Biol Chem. 2017;292(51):20808–20817. doi:10.1074/jbc.M117.814913
26. Hsu M-K, Wu I-C, Cheng C-C, et al. Triple-layer dissection of the lung adenocarcinoma transcriptome - regulation at the gene, transcript, and exon levels. Oncotarget. 2015;6(30):28755–28773. doi:10.18632/oncotarget.4810
27. Fujitomo T, Daigo Y, Matsuda K, et al. Critical function for nuclear envelope protein TMEM209 in human pulmonary carcinogenesis. Cancer Res. 2012;72(16):4110–4118. doi:10.1158/0008-5472.CAN-12-0159
28. Huang H, Feng J, Famulski J, et al. Tripin/hSgo2 recruits MCAK to the inner centromere to correct defective kinetochore attachments. J Cell Biol. 2007;177(3):413–424. doi:10.1083/jcb.200701122
29. Rattani A, Wolna M, Ploquin M, et al. Sgol2 provides a regulatory platform that coordinates essential cell cycle processes during meiosis I in oocytes. Elife. 2013;2:e01133. doi:10.7554/eLife.01133
30. Long C, Chen J, Zhou H, et al. Diosgenin exerts its tumor suppressive function via inhibition of Cdc20 in osteosarcoma cells. Cell Cycle. 2019;18(3):346–358. doi:10.1080/15384101.2019.1568748
31. Kim Y, Choi J-W, Lee J-H, et al. Spindle assembly checkpoint MAD2 and CDC20 overexpressions and cell-in-cell formation in gastric cancer and its precursor lesions. Hum Pathol. 2019;85:174–183. doi:10.1016/j.humpath.2018.10.029
32. Cheng S, Castillo V, Sliva D. CDC20 associated with cancer metastasis and novel mushroomderived CDC20 inhibitors with antimetastatic activity. Int J Oncol. 2019;54(6):2250–2256. doi:10.3892/ijo.2019.4791
33. Wang L, Zhang J, Wan L, et al. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol Ther. 2015;151:141–151. doi:10.1016/j.pharmthera.2015.04.002
34. Daum JR, Wren JD, Daniel JJ, et al. Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Curr Biol. 2009;19(17):1467–1472. doi:10.1016/j.cub.2009.07.017
35. Lee M, Williams KA, Hu Y, et al. GNL3 and SKA3 are novel prostate cancer metastasis susceptibility genes. Clin Exp Metastasis. 2015;32(8):769–782. doi:10.1007/s10585-015-9745-y
36. Chuang T-P, Wang J-Y, Jao S-W, et al. Over-expression of AURKA, SKA3 and DSN1 contributes to colorectal adenoma to carcinoma progression. Oncotarget. 2016;7(29):45803–45818. doi:10.18632/oncotarget.9960
37. Deribe YL, Wild P, Chandrashaker A, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal. 2009;2(102):ra84. doi:10.1126/scisignal.2000576
38. Xu Y, Xu H, Li M, et al. KIAA1199 promotes sorafenib tolerance and the metastasis of hepatocellular carcinoma by activating the EGF/EGFR-dependent epithelial-mesenchymal transition program. Cancer Lett. 2019;454:78–89. doi:10.1016/j.canlet.2019.03.049
39. Komposch K, Sibilia M. EGFR signaling in liver diseases. Int J Mol Sci. 2015;17:1.
40. Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol. 2014;49(1):9–23. doi:10.1007/s00535-013-0907-x
41. Zheng C, Zheng L, Yoo J-K, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342–1356e16. doi:10.1016/j.cell.2017.05.035
42. Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6(6):e1792. doi:10.1038/cddis.2015.162
43. Philip M, Schietinger A. Heterogeneity and fate choice: t cell exhaustion in cancer and chronic infections. Curr Opin Immunol. 2019;58:98–103. doi:10.1016/j.coi.2019.04.014
44. Youn J-I, Nagaraj S, Collazo M, et al. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–5802. doi:10.4049/jimmunol.181.8.5791
45. Solito S, Marigo I, Pinton L, et al. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci. 2014;1319(1):47–65. doi:10.1111/nyas.12469
46. Park SM, Youn JI. Role of myeloid-derived suppressor cells in immune checkpoint inhibitor therapy in cancer. Arch Pharm Res. 2019;42(7):560–566. doi:10.1007/s12272-019-01165-6
47. Toh B, Wang X, Keeble J, et al. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011;9(9):e1001162. doi:10.1371/journal.pbio.1001162
48. Zhang H, Liu H, Shen Z, et al. Tumor-infiltrating neutrophils is prognostic and predictive for postoperative adjuvant chemotherapy benefit in patients with gastric cancer. Ann Surg. 2018;267(2):311–318. doi:10.1097/SLA.0000000000002058
49. Waniczek D, Lorenc Z, Śnietura M, et al. Tumor-associated macrophages and regulatory T cells infiltration and the clinical outcome in colorectal cancer. Arch Immunol Ther Exp (Warsz). 2017;65(5):445–454. doi:10.1007/s00005-017-0463-9
50. Yost KE, Satpathy AT, Wells DK, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25(8):1251–1259. doi:10.1038/s41591-019-0522-3
51. Pan JH, Zhou H, Cooper L, et al. LAYN is a prognostic biomarker and correlated with immune infiltrates in gastric and colon cancers. Front Immunol. 2019;10:6. doi:10.3389/fimmu.2019.00006
52. Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40(11):2969–2975. doi:10.1002/eji.201040895
53. Siemers NO, Holloway JL, Chang H, et al. Genome-wide association analysis identifies genetic correlates of immune infiltrates in solid tumors. PLoS One. 2017;12(7):e0179726. doi:10.1371/journal.pone.0179726
54. Danaher P, Warren S, Dennis L, et al. Gene expression markers of Tumor Infiltrating Leukocytes. J Immunother Cancer. 2017;5(1):18. doi:10.1186/s40425-017-0215-8
55. Sousa S, Maatta J. The role of tumour-associated macrophages in bone metastasis. J Bone Oncol. 2016;5(3):135–138. doi:10.1016/j.jbo.2016.03.004
56. Ling L, Lin Y, Zheng W, et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci Rep. 2016;6:20358. doi:10.1038/srep20358
57. Lanczky A, Nagy Á, Bottai G, et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat. 2016;160(3):439–446. doi:10.1007/s10549-016-4013-7
58. Nagy A, Lánczky A, Menyhárt O, et al. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8(1):9227. doi:10.1038/s41598-018-27521-y
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.