Back to Journals » Breast Cancer: Targets and Therapy » Volume 18
Keratin as a Potential Prognostic Biomarker for Breast Cancer Progression: An Evidence-Synthesizing Narrative Review
Authors Khairani AF ![]() , Alhaq KF, Herriyanto RI, Fauzi MB
, Alhaq KF, Herriyanto RI, Fauzi MB ![]() , Alfarafisa NM
, Alfarafisa NM ![]()
Received 4 November 2025
Accepted for publication 5 January 2026
Published 24 January 2026 Volume 2026:18 578887
DOI https://doi.org/10.2147/BCTT.S578887
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Astrid Feinisa Khairani,1 Karina Fitria Alhaq,2 Regina Indrasari Herriyanto,2 Mh Busra Fauzi,3 Nayla Majeda Alfarafisa1
1Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, Bandung, West Java, Indonesia; 2Faculty of Medicine, Universitas Padjadjaran, Bandung, West Java, Indonesia; 3Department of Tissue Engineering & Regenerative Medicine, Faculty of Medicine, Universiti Kebangsaan Malaysia, Kuala Lumpur, 56000, Malaysia
Correspondence: Astrid Feinisa Khairani, Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, Jalan Raya Bandung – Sumedang Km 21, Jatinangor, Sumedang, West Java, 45363, Indonesia, Tel +62-22-7795594, Email [email protected]
Purpose: Breast cancer (BC) remains a leading cause of mortality among females worldwide, making reliable biomarkers necessary for prognosis and personalized treatment. Keratins represent the largest and most diverse intermediate filament (IF) class utilized as marker proteins histopathologically because of their specific expression patterns related to epithelial differentiation. This narrative review explores the potential of keratin subtypes, which are extensively employed in the differentiation of specific BC subclasses, as prognostic markers.
Material and Methods: This narrative review used a structured literature search in PubMed and Scopus. Selected elements of the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) framework were adapted to improve transparency in the search and screening process. A total of 79 were included, covering eleven in vitro, six in vivo, and 62 human studies.
Results: The key findings highlight five significant keratin types: keratin 5/6 (K5/6), keratin 19 (K19), keratin 8/18 (K8/18), keratin 14 (K14), and keratin 17 (K17). These are expressed in specific BC subtypes and influence tumor progression through particular pathways. These keratin subtypes were selected for discussion as they are the most prevalent and clinically important keratins in BC and represent the strongest evidence base. In addition to their role in prognostic stratification, available evidence suggests that subtypes of keratin may serve to recognize metastatic potential early, influence adjuvant therapy decisions in high-risk subgroups, and monitor treatment responses dynamically. They serve as predictive biomarkers that help understand chemotherapy effectiveness, increase risk assessment in aggressive subsets of triple negative breast cancer (TNBC), and facilitate targeted therapeutic strategies, including K1-targeted peptide-doxorubicin conjugates demonstrating the potential for these factors to transition from merely predictive indicators to meaningful prognostic markers.
Conclusion: This review confirms that keratin actively contributes to tumor progression and emphasizes the importance of keratins in key oncogenic pathways, providing insights for the development of updated therapies.
Keywords: breast cancer, intermediate filaments, keratin, prognostic biomarkers, tumor markers
Introduction
BC manifests when a specific cell located in either ducts or lobules proliferates uncontrollably.1 This condition is of particular concern because female sex is considered one of the primary risk factors for developing BC, which has emerged as a leading cause of mortality worldwide.2 Recent global cancer statistics demonstrate that approximately 2,261,419 new cases of BC were diagnosed in 2020, with 685,000 deaths occurring worldwide in the same year.3
During tumor progression, malignant transformation induces a cascade of structural rearrangements, primarily driven by the EMT process. This process enables cells to acquire migratory and invasive properties that promote metastasis, a major cause of BC-related mortality. The EMT process involves cytoskeletal remodeling, enzyme secretion for extracellular matrix degradation, and altered microRNA expression, all of which facilitate cancer cell dissemination and therapy resistance.4
The prevailing diagnostic tools employed in the contemporary context to identify BC include imaging tests (eg, mammograms, ultrasounds, and magnetic resonance imaging) to examine breast tissue for any unusual findings. Markers such as hormone receptors, including estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), are found at the site where the tissue sample is extracted.5 However, the application of hormone markers has been demonstrated to be ineffective as hormone markers. First, they are not present in all BC subtypes, such as TNBC. TNBC is characterized by the absence of ER and PR expression, as well as HER2 amplification, resulting in a highly heterogeneous disease with limited well-defined molecular targets.6 Second, it is also possible that the expression of hormone receptors may change during disease progression. The study revealed a statistically significant change in 32.4% of patients in the ER group (P < 0.001), 40.7% of patients in the PR group (P < 0.001), and 14.5% of patients in the HER2 group (P = 0.44). Furthermore, females with ER+ primary tumors that changed to ER- tumors presented a substantial 48% elevated risk of mortality.7 Third, the presence of hormone receptors does not serve as an indicator of therapy resistance. Studies have shown that ER+ status is associated with a reduction in the efficacy of hormone therapy.8
Given the limitations of traditional hormonal biomarkers, recent attention has shifted toward cytoskeletal components specifically, intermediate filaments (IFs) such as vimentin, nestin, and keratin as alternative biomarkers. IFs maintain cell integrity and structure but are also dynamically remodeled during EMT, linking them directly to cancer aggressiveness, invasion, and therapeutic response. Alterations in the expression of these filaments have been correlated with BC progression, metastasis, and chemoresistance.4
Among cytoskeletal biomarkers, keratins are especially promising for clinical translation as they are lineage-specific, structurally stable, and routinely assessed in diagnostic pathology using immunohistochemistry (IHC). Different from hormone receptors, keratin expression depicts epithelial differentiation states and cytoskeletal remodeling that persist during disease progression, metastatic dissemination, and exposure to treatment. These features position keratins at the interface between tumor biology and clinical decision-making.
Here the role of keratin as a biomarker in BC was described in-depth, with significant interest in its prognostic and predictive effects in various molecular subtypes. The selected keratin subtypes addressed in this review (K5/6, K14, K17, K19, and K8/18) are considered the predominant basal and luminal keratin groups in BC and are the most clinically significant, with the strongest supporting evidence in the literature. The accumulating clinical and experimental data suggest that certain keratin subtypes are consistently associated with tumor aggressiveness, therapeutic response, and clinical outcomes that warrant their consideration beyond descriptive epithelial markers. Although the data for certain subtypes of keratin in predicting therapeutic response (predictive biomarkers) are limited, the growing evidence indicates that their consistent association with disease aggressiveness and clinical outcomes supports their broader role as prognostic markers. Current oncology frameworks emphasize that biomarker-guided therapy is best accomplished when a biomarker robustly connects tumor biology to actionable clinical decisions and not as a simple molecular feature only.
However, only a limited number of studies summarizing the use of potential keratin, both in the form of individuals and/or panels, as diagnostic and prognostic biomarkers for BC have been published. This review aims to address the need for clinical application to determine which keratin is the most relevant and accurate prognostic tool across various BC subtypes, contributing to a deeper understanding of the role of keratin as a biomarker for treatment approaches.
Materials and Methods
Eligibility Criteria
This review was conducted as a narrative synthesis and full PRISMA guidelines were not applied. Instead, selected elements of the PRISMA flow diagram were adapted to enhance transparency in the search, screening, and study selection processes. The literature search was performed in two screening phases. The first phase entailed abstract/keyword screening, whereas the second phase involved full-text screening. In the first phase, studies from PubMed were selected using medical subject heading (MeSH) terms and limited by date and text availability (Table 1). Additional studies were sourced from Scopus and filtered by year, subject, document type, language, and open access.
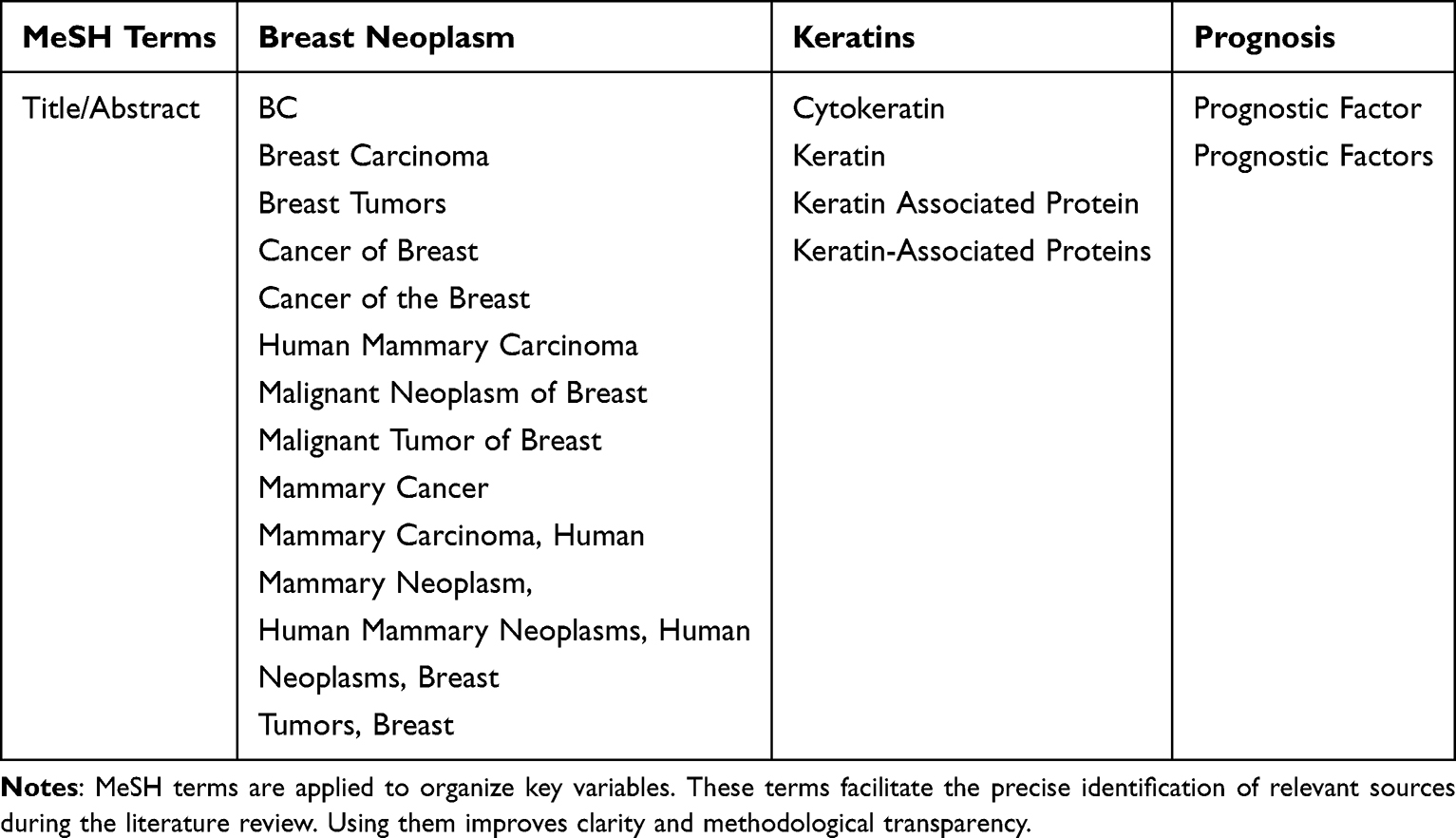 |
Table 1 Medical Subject Heading (MeSH) Terms |
Inclusion and Exclusion Criteria
The PICO framework for keyword selection in this study is structured as follows: population (P) focuses on the disease population and setting (DPS), with a specific focus on BC patients of all types within a global setting. The exposure (E) under consideration is keratin (or intermediate filaments). For comparison (C), the study evaluated various prognostic tools used for BC assessment. The outcome (O) aims to determine the expression of keratin and its prognostic value in BC patients, while the timeframe (T) for the study is set at 10 years, ensuring relevance to past trends and data. Finally, the design (D) of the research follows either a cross-sectional or human study approach, providing comprehensive insights into the relationship between keratin expression and BC prognosis.
Data Source and Search Strategy
In the review, Rayyan.ai was used to evaluate the eligibility of studies and to identify any potential duplications or irrelevances. Rayyan.ai categorizes articles as “include”, “exclude”, or “maybe” based on eligibility criteria. The inclusion criteria of this study are papers that are the results of original articles (human, in vivo, in vitro), titles, keywords and abstracts of sources that are consistent with the objectives of the study. In addition to the abstract, we included only journals that provided information about the sample analyzed, the investigated biomarkers, and the comparisons between groups shown in Figure 1 about identification of studies via databases and registers.
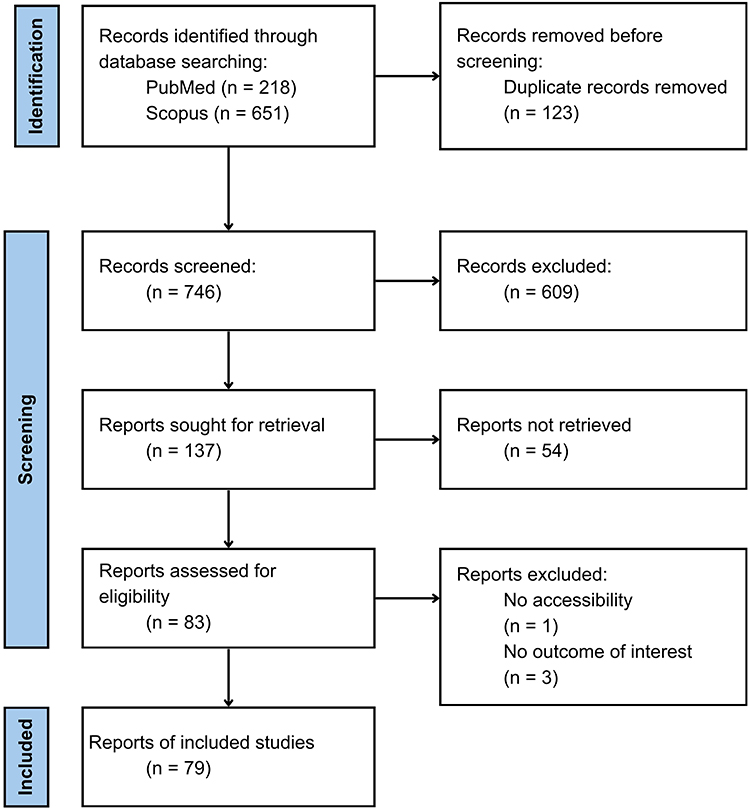 |
Figure 1 Literature Search Flowchart. |
Results
The studies (Tables 2–4) reviewed a total of 79 studies, which were categorized based on their design into three groups: in vitro, in vivo, and human studies. Each study addressed distinct scientific objectives. Among these studies, eleven in vitro investigations focused on elucidating keratin-associated signaling pathways, providing insights into how keratins drive cancer progression at the cellular level, and laying the foundation for clinical applications. Furthermore, six in vivo studies examined therapeutic interventions, utilizing preclinical models to test treatment strategies targeting keratin-expressing tumors. These studies included the evaluation of therapeutic agents delivered through keratin-targeting systems, which offers valuable guidance for future treatment development. In addition, 62 human studies investigated the prognostic significance of keratin subtypes in human BC. These studies analyzed keratin expression patterns to classify subtypes and predict clinical outcomes. Collectively, these studies contribute to a more comprehensive understanding of keratin function in cancer and its potential as a therapeutic and prognostic marker.
 |
Table 2 In vitro Studies |
 |
Table 3 In vivo Studies |
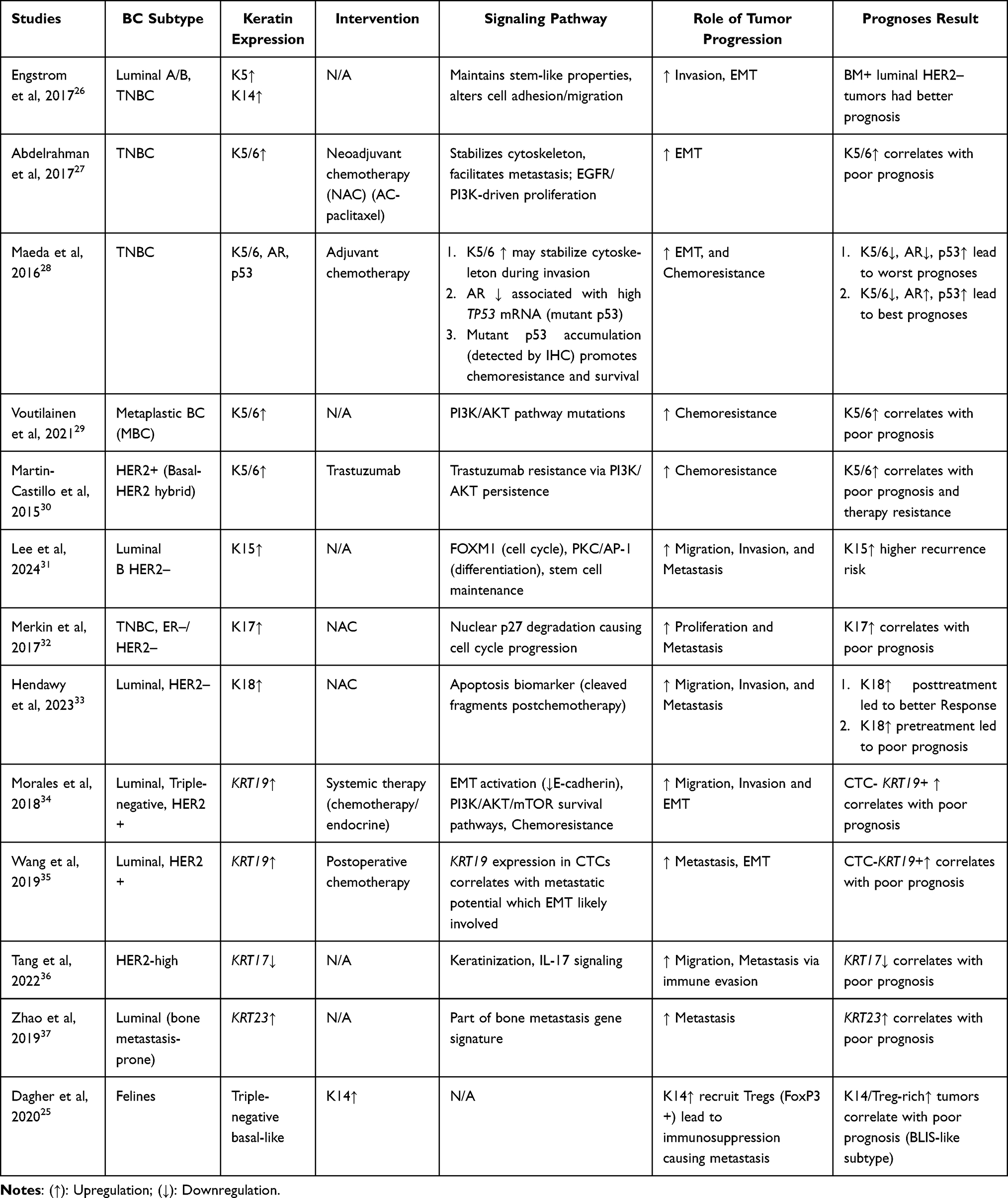 |
Table 4 Human Studies |
McGinn et al (2020) demonstrated that the upregulation of K5 activates the Wnt/β-catenin pathway, disrupts cell adhesion, and enhances endocytosis in the luminal (ER+), basal-like triple-negative and HER2+ BC subtypes. K5 stabilizes β-catenin through these pathways, enabling its nuclear translocation and TCF/LEF-mediated Wnt1 activation. This process sustains cancer stem cell (CSC) properties. K5/β-catenin interactions weaken E-cadherin, thereby reducing cell–cell adhesion and promoting invasion. Enhanced endocytosis and trafficking further alter signaling dynamics, thereby driving tumor progression. Taken together, these mechanisms increase invasion, CSC enrichment and EMT, and predicting a poor prognosis in ER+ and TNBC patients.9
Wang et al (2020) reported that, in TNBC and luminal basal-like subtypes, the simultaneous upregulation of K5, K6, K14, and K17 under anilin expression triggers pathways involving the regulation of nuclear actin, the induction of E-cadherin, and the maintenance of CSCs. Anillin modulates RNA polymerase II activity, affecting stemness genes, while its loss upregulates E-cadherin, reducing migration, and invasion. These pathways promote migration, invasion, metastasis, and cytoskeletal remodeling by sustaining CD44 (high)/CD24 (low) populations and mammosphere formation through TBX18 and SOX9 downregulation. High anillin levels are correlated with poor survival in TNBC patients.10
Verma et al (2022) and Padmanaban et al (2020) demonstrated that, in basal-like TNBC, high K14 expression activates H3K27me3-mediated transcriptional activation and Rho/ROCK/integrin signaling, thereby driving cytoskeletal reorganization and invasion. Specifically, H3K27me3 blocks the SPI repressor and allows RNA polymerase II recruitment, promoting KRT14 expression. This activation enhances migration, invasion, and metastasis, establishing K14 as a robust prognostic biomarker that predicts poor prognosis.11,12
Izdebska et al (2021) and Lin et al (2020) demonstrated that, in TNBC, luminal A (ER+) and luminal B (ER−/HER2+) cancers, the upregulation of K19 activates the HER2/ERK, Wnt/β-catenin, EMT, and ER stress signaling pathways. Matrix metalloproteinase (MMP)-9 degrades extracellular matrix (ECM) proteins, creating paths for metastasis, while the upregulation of vimentin and N-cadherin enhances EMT. K19 also stabilizes HER2/ERK and Wnt signaling, disrupts E-cadherin and modulates the unfolded protein response. Collectively, these processes promote migration, invasion, EMT, and cytoskeletal remodeling, all of which predict poor outcomes when MMP-9 levels are high.13,14
Zhao et al (2023) reported that in luminal TNBC, the downregulation of KRT7-AS and the subsequent activation of KRT7 stimulate the PTEN/PI3K/AKT pathway and degrade KRT7/FOXA1. Typically, KRT7-AS stabilizes PTEN, thereby suppressing AKT/NF-κB-driven proliferation and oncogenic FOXA1 expression. However, its loss enhances migration, invasion, metastasis, immune evasion, and chemoresistance, predicting a poor prognosis.15
Butler et al (2020) revealed that, in TNBC, KRT18 is silenced via DNMT3A-mediated promoter hypermethylation, which activates the Wnt/β-catenin and EMT pathways. Loss of KRT18 increases N-cadherin and reduces epithelial integrity, thereby increasing migration, invasion, and metastasis. Azacytidine counteracts this by inhibiting Wnt-3/4, GSK-3, and β-catenin, thereby reducing stemness and invasion. It also decreases VEGF secretion, which impairs tumor vascularization. Overall, DNMT3A-induced KRT18 silencing promotes EMT and an aggressive phenotype is associated with a poor prognosis.16
Saha et al (2018, 2019) demonstrated that KRT19 downregulation in TNBC activated the Src/GSK3β, Wnt/β-catenin/Notch, and HER2/ERK/SP1 pathways. The KRT19/β-catenin/RAC1 complex suppresses NUMB, thereby enabling Notch activation, enhancing stemness, and promoting invasion. KRT19 also stabilizes HER2 to promote proliferation and inhibits Src/GSK3β phosphorylation, thereby removing the brakes on stemness. These interactions increase migration, invasion, proliferation, stemness, EMT, and metastasis, predicting a worse prognosis.17,18
Yan et al (2024) demonstrated that, in TNBC, KRT81 upregulation activated focal adhesion signaling (FAK/Paxillin) and immune- and metabolic-related pathways, such as oxidative phosphorylation and glycolysis. These findings support migration, invasion, proliferation, and immune evasion, with elevated KRT81 predicting a poor prognosis.19
Key findings, including those of Saghaeidehkordi et al (2021), demonstrated that the upregulation of K1 enables a targeted drug delivery pathway in TNBC via NOD/SCID mice. A PDC binds to K1, which is expressed on TNBC cells and enters them via receptor-mediated endocytosis. This releases doxorubicin specifically in the acidic tumor environment. This pathway enhances tumor-specific drug delivery, reduces doxorubicin accumulation in healthy tissues, improves tumor suppression, and minimizes side effects. Consequently, high K1 expression in TNBC indicates a poor prognosis.However, the use of PDC improves therapeutic precision and could be adapted for other chemotherapies.20
Machado et al (2020) reported that in dogs with TNBL mammary tumors, high expression of K5/6 is associated with a signaling axis in which loss of ERβ promotes basal-like differentiation by driving EMT through loss of E-cadherin and upregulation of vimentin. K5/6 marks basal/myoepithelial cell layers and is associated with more aggressive tumor behavior, higher mitotic rates, and lymph node metastasis. Consequently, K5/6+ basal-like tumors predict a poor prognosis because of their rapid progression and metastasis. In contrast, triple-negative nonbasal-like tumors (K5/6-) are aggressive but less well characterized.21
Eivani et al (2016) reported on canine mammary carcinomas and examined the prognostic relevance of K5/6 and K7. While the basal marker K5/6 typically indicates aggressive, hormone receptor-negative tumors and poor outcomes in humans, this study revealed that neither K5/6 nor K7 had significant prognostic value in dogs. Furthermore, no clear link with EMT was observed, indicating species-specific differences in keratin-related tumor behavior.22
Bado et al (2017) demonstrated that, in mice with K14CreERβp53 tumors (BLBC), high K14 expression emerges when ERβ is lost and p53 is knocked down. The loss of ERβ disrupts epithelial maintenance, shifting differentiation toward basal-like clones and promoting EMT, as evidenced by decreased E-cadherin expression and increased vimentin, N-cadherin, and α-SMA expression. This shift enhances invasion and metastatic potential, indicating a poor prognosis when ERβ levels are low and K14 levels are high. Conversely, maintaining ERβ alongside lower levels of K14 favors epithelial features and better outcomes.23
WalyEldeen et al (2024) reported that the interplay between K14, K18, and FBLN2 determines tumor behavior across claudin-low, luminal A, and basal/HER2+ BC subtypes in C57BL/6 mice and felines. FBLN2 interacts closely with KRT14 and integrins (ITGβ1/ITGα3) to maintain the basement membrane (BM) and suppress invasive potential by stabilizing the ECM. High FBLN2 expression upregulates KRT14 and downregulates KRT18, preserving a basal phenotype with a favorable prognosis in the early stages of the disease. In contrast, low FBLN2 expression shifts cells toward a luminal phenotype (KRT18+) with BM loss, promoting invasiveness and a poor prognosis in advanced stages.24
Dagher et al (2020) revealed that, in feline triple-negative basal-like carcinomas, high K14 expression is associated with increased infiltration of regulatory T cells (Tregs) marked by FoxP3, creating an immunosuppressed microenvironment. This suppresses antitumor immunity, facilitating tumor progression, escape, growth, lymph node metastasis, and advanced stages. Therefore, K14+ Treg-rich tumors, which are characteristic of the BLIS-like subtype, predict the worst outcomes, whereas luminal tumors with lower Treg infiltration are associated with longer survival.25
Engström et al (2017) demonstrated that, in luminal A/B and TNBC, the upregulation of K5 and K14 helps to maintain stem-like properties while also altering cell adhesion and migration. These pathways promote tumor invasion by driving cytoskeletal remodeling and EMT. The study also revealed that the status of the BM can be used to categorize luminal HER2- tumors, with BM+ luminal HER2- patients having a better prognosis than BM-patients do.26
Abdelrahman et al (2017) demonstrated that, in TNBC, high K5/6 expression stabilizes the cytoskeleton and facilitates metastasis through EGFR/PI3K-driven proliferation. K5/6 act as markers of high-risk TNBC subsets, promoting EMT via cytoskeletal remodeling and loss of epithelial markers, which correlates with aggressive clinical behavior. Thus, K5/6 upregulation predicts a poor prognosis.27
Maeda et al (2016) reported that, in TNBC, the coexpression of K5/6, AR, and p53 identifies high-risk patients. K5/6 upregulation may stabilize the cytoskeleton during invasion, whereas AR loss is associated with high mutant TP53 mRNA expression. Mutant p53 accumulation promotes chemoresistance and tumor survival. Together, these results indicate that tumors with low K5/6, low AR, and high p53 levels have the worst prognosis, whereas low K5/6, high AR, and high p53 levels predict better outcomes.28
Voutilainen et al (2021) reported that, in metaplastic BC (MpBC), high K5/6 expression is indicative of a basal-like phenotype associated with mutations in the PI3K/AKT pathway. This drives aggressive behavior and chemoresistance, resulting in a poor prognosis for basal-like MpBCs (K5/6/EGFR+). In contrast, nonbasal-like MpBCs (K5/6/EGFR–) did not relapse.29
Martin-Castillo et al (2015) demonstrated that high K5/6 expression contributes to trastuzumab resistance via persistent PI3K/AKT signaling in HER2+ basal-HER2 hybrid tumors. This pathway underpins poor prognosis and therapy resistance in K5/6+ HER2+ patients.30
Lee et al (2024) reported that increased K15 expression activates pathways involving FOXM1 (cell cycle), PKC/AP-1 (differentiation), and stem cell maintenance in Luminal B HER2-BC. K15 acts as a progenitor marker linked to self-renewal and basal-like signatures, enhancing migration, invasion, and metastasis. Consequently, K15 positivity is associated with shorter disease-free survival (DFS) and BC-specific survival (BCSS), indicating a poor prognosis.31
Merkin et al (2017) reported that, in TNBC and ER-/HER2– tumors, high K17 expression promotes nuclear p27 degradation, leading to cell cycle progression. By acting as a shuttle for p27, K17 promotes proliferation and coordinates with K16/K14 to disrupt the cytoskeleton synergistically, thereby enhancing invasion and metastasis. High K17 expression is associated with a poor prognosis and worse 5-year event-free survival (EFS) in advanced receptor-negative tumors.32
Hendawy et al (2023) reported that increased K18 expression after NAC reflects apoptosis in luminal HER2– BC, as cleaved K18 fragments circulate in the blood. While high K18 posttreatment indicates a better response to treatment, elevated K18 pretreatment is associated with a poorer prognosis owing to its influence on cell adhesion, motility, and aggressive tumor biology.33
Morales et al (2018) reported that, in luminal, TNBC and HER2+ BC, upregulation of KRT19 drives EMT by reducing E-cadherin and activating the PI3K/AKT/mTOR survival pathways. This promotes chemoresistance through upregulation of the stress response. Fragmented K19 disrupts the cytoskeleton, increasing invasion and metastasis. High CTC-KRT19+ levels are associated with shorter progression-free survival (PFS) and overall survival (OS), indicating a poor prognosis.34
Wang et al (2019) demonstrated that, in luminal and HER2+ cancers, the expression of KRT19 in CTCs is associated with metastatic potential and EMT activation. High CTC-KRT19 levels are associated with shorter distant metastasis-free survival (DMFS) and OS, whereas a reduction after treatment is associated with better PFS.35
Tang et al (2022) demonstrated that, in HER2-high BC, low KRT17 expression is associated with a poor prognosis. KRT17 plays a role in keratinization, cytokine interactions, and IL-17 signaling. Its loss promotes immune evasion, increasing migration, and metastasis.36
Zhao et al (2019) reported that, in luminal BC, which is prone to bone metastasis, high KRT23 expression is part of a bone metastasis gene signature. This indicates a greater risk of metastasis and worse outcomes.37
In Tables 2–4, bridging preclinical findings to clinical applications demands further research shown in Figures 2 and 3.
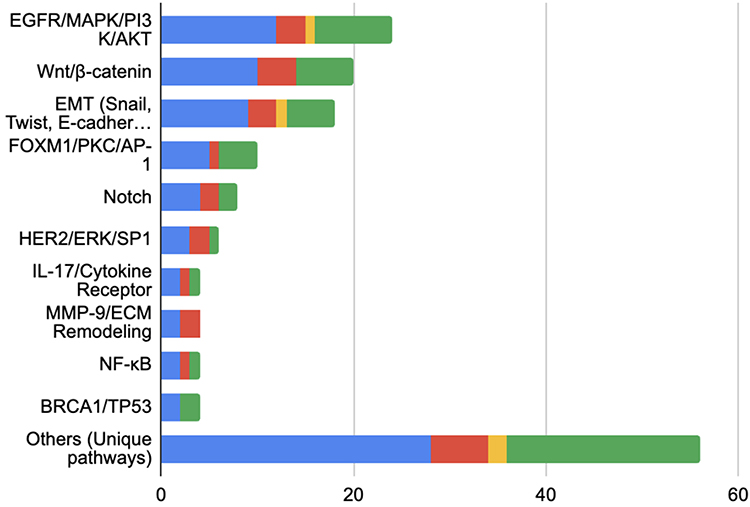 |
Figure 2 Major Keratin Signaling Pathway. Show the number of studies implicating each signaling pathway in BC, separated by in vitro (red), in vivo (yellow), dan human (green). The blue bar indicates the total number of studies for each pathway. |
 |
Figure 3 Dominant Keratin is Expressed in BC. Show the number of studies reporting specific keratin markers expressed in BC, separated by in vitro (red), in vivo (yellow), dan human (green). The blue bar indicates the total number of studies for each pathway. |
EGFR/MAPK/PI3K/AKT (12 studies) and Wnt/β-catenin (10 studies) are the most frequently investigated pathways, primarily in human and in vitro studies. Pathways such as EMT and FOXM1/PKC/AP-1 are also prominent but lack in vivo validation.
K5/6 dominated research (25 studies), especially in TNBC/basal-like subtypes, followed by K19 (12 studies), K14 (10 studies), K8/18 (8 studies), and K17 (6 studies). Most evidence comes from human studies, with 18, 9, 7, 5, and 4 studies identified for K5/6, K19, K14, K8/18, and K17, respectively.
Discussion
Keratin Signaling Pathways and Tumor Progression Mechanism
Keratin, the largest and most diverse class among IFs, serves as a critical marker for proteins in BC.38 Since most clinical evidence supporting keratin as a biomarker relies on IHC-based methods detection,27,28,30 keratin expression in BC has mainly been assessed using antibody-based assays as lineage-defining,5,26 prognostic, and therapy-stratifying markers.28 Across the included human studies, keratins such as K5/6, K14, K17, and K8/18 were mainly evaluated using semi-quantitative IHC scoring systems based on staining intensity and/or the proportion of positively stained tumor cells. These were often divided into high- and low-expression groups. However, differences in antibody clones, antigen retrieval methods, scoring thresholds, and inter-observer interpretation across studies may affect reproducibility and limit direct comparability, which is an important consideration for clinical application. In BC, keratin proteins play pivotal roles in tumor progression, with specific isoforms driving distinct signaling pathways that influence tumor behavior and therapeutic responses. For example, in K5/6+ tumors, the main pathway involved is the EGFR/MAPK/PI3K/AKT axis, especially in basal-like TNBC, where it promotes growth, survival, and metastasis.27 These cancers often have mutant p53, which makes them resistant to drugs.28,39 These findings emphasize the potential of keratin-specific pathway targeting for personalized therapies.
Keratin subtypes play pivotal roles in regulating critical signaling pathways that are involved in the progression of BC. These subtypes have been found to modulate tumor mechanisms, including metastasis, chemoresistance, immune evasion, and stemness, among others. This review systematically mapped the entire pathway into five major pathways across 79 studies shown in Figure 2. These pathways have been reported to be involved in keratin-associated signaling mechanisms in a variety of experimental studies, including in vitro, in vivo, and clinical studies in humans.
The consistent correlation between keratin expression and core oncogenic pathways suggests that individual keratin subtypes could serve as biomarkers associated with particular pathways,27,28,30 helping to identify tumors that depend on different signaling axes. This is consistent with translational techniques in which biomarkers can expedite drug development by classifying patients based on key pathway vulnerabilities rather than histology alone.40
Role of Keratin 5/6 as Prognostic Marker
K5/6 are basal keratins expressed in myoepithelial and progenitor cells and are associated with aggressive tumor phenotypes, including high-grade tumors, lymphovascular invasion, and chemoresistance,26,39 via several signaling pathways,26,27,39,41–43 as shown in Figure 4.
 |
Figure 4 Keratin 5/6 signaling pathway. The purple elements represent metastasis-related processes, yellow denotes therapy, pink indicates chemoresistance, and Orange corresponds to targeted therapy. |
K5/6 EGFR is a receptor tyrosine kinase that is activated by various ligands, including EGF or TGF-α. In patients with TNBC and BLBC, excess EGFR expression is frequently detected.44 Moreover, the MAPK pathway is characterized by a cascade of events that begin with the activation of EGFR, which is followed by the activation of RAS. Subsequently, RAF is activated, followed by the activation of MEK. The final activation is that of ERK, which translocates into the nucleus to activate the transcription of genes involved in proliferation and metastasis.45 Moreover, the PI3K/AKT pathway is also activated downstream of EGFR. Upon activation, EGFR stimulates PI3K to generate PIP3, which serves as a docking site for the activation of the AKT serine/threonine kinase.46 The activation of AKT has been demonstrated to promote cell survival by inhibiting apoptosis. This pathway plays a pivotal role in the progression of tumors, specifically in metastasis and chemoresistance. On the other hand, activation of the EGFR/MAPK/PI3K axis alters the dynamics of the cytoskeleton, thereby increasing cell motility. MAPK/ERK signaling activates pro-EMT transcription factors, including Twist, Snail, and Zeb1, which in turn lead to the suppression of E-cadherin and the upregulation of N-cadherin and vimentin. Concurrently, PI3K/AKT has been shown to increase the expression of MMP-2 and MMP-9, thereby promoting ECM degradation and facilitating invasion. Furthermore, AKT activation has been demonstrated to increase VEGF expression, thereby promoting angiogenesis and contributing to destabilization of the vascular endothelium.47 The mutant p53 protein has been shown to increase the activation of EGFR and the PI3K/AKT pathway, thereby increasing the resistance of cancer cells to chemotherapy agents such as anthracyclines or platinum-based drugs. Furthermore, mutant p53 disrupts HRR while activating alternative DNA repair pathways, thereby reducing tumor cell dependency on PARP and contributing to PARP inhibitor resistance.48
From a translational standpoint, the intense correlation of K5/6 expression with chronic EGFR/MAPK/PI3K/AKT activation in basal-like TNBC elevates K5/6 beyond that of a prognostic marker to that of a tumor subgroup that necessitates active signaling pathways reliance.27,28,30 Consequently, K5/6 expression may serve as an adjunct biomarker associated with tumors that are largely dependent on EGFR- or PI3K-driven survival signaling and therefore supportive of pathway-targeted or combination therapies.40
Role of Keratin 19 as Prognostic Marker
K19 in cancer progression, highlighting its dual function as an epithelial marker for detecting CTCs and micrometastases and its involvement in key oncogenic pathways, as shown in Figure 5.
 |
Figure 5 Keratin 19 signaling pathway. The red elements represent metastasis-related processes, green denotes migration, and pink indicates chemoresistance. |
This figure outlines two critical pathways.17,33,34,49–51 First, Wnt ligands (eg, Wnt3a) bind to the Frizzled receptor, thereby inhibiting the β-catenin destruction complex (which consists of GSK3β, APC, and Axin). Consequently, β-catenin accumulates in the cytoplasm and subsequently translocate into the nucleus, where it binds to TCF/LEF transcription factors to activate protumorigenic target genes, including c-Myc, Cyclin D1, and various MMPs.52 Second, the activation of the Notch receptor is initiated by the binding of ligands, such as Jagged or Delta-like proteins, resulting in the release of the NICD. The NICD translocates into the nucleus and activates genes, including Hes1 and Hey1, which play key roles in maintaining stemness and promoting EMT.53 During the metastatic process, a decrease in E-cadherin and an increase in vimentin serve as hallmarks of EMT. Nuclear β-catenin has been shown to induce the expression of transcription factors such as Snail, Twist, and Zeb1, which have been demonstrated to suppress CDH1 (the E-cadherin gene) and upregulate vimentin expression. Moreover, Notch signaling has been shown to increase the expression of MMP-2 and MMP-9, which in turn degrade the ECM and facilitate tumor cell invasion.54 K19+ cells that also exhibit active Wnt/Notch signaling have been shown to exhibit disrupted polarity and heightened motility, which renders them more susceptible to entering blood vessels or lymphatics.55 Moreover, the activation of the PI3K/AKT pathway by K19 fragments (eg, CYFRA 21–1) further supports chemoresistance by upregulating antiapoptotic proteins such as Bcl-2 and Survivin. Furthermore, nuclear β-catenin has been shown to increase the expression of ABC transporters, such as P-glycoprotein, which play a pivotal role in the active efflux of chemotherapeutic agents, thereby contributing to multidrug resistance. Furthermore, K19+ cells exhibiting active Notch signaling have been shown to acquire characteristics of CSCs, thereby conferring enhanced resistance to chemotherapy and radiation therapy.56
Likewise, K19-driven crosstalk across Wnt/β-catenin, Notch, and PI3K/AKT signaling determines a keratin-associated tumor state with defined features of stemness, epithelial–mesenchymal transition, and therapeutic resistance.17,18,34,35 These features are consistent with the biomarker–pathway axes commonly employed in companion diagnostic approaches, which suggest K19-positive tumors are a biologically defined subgroup that can be targeted towards pathway-based therapeutic modulation rather than uniform cytotoxic therapy.40
Role of Keratin 8/18 as Prognostic Marker
K8/18 is a luminal keratin marker with context-dependent functions in BC progression. In luminal BC, K8+ lymph node metastases correlate with better survival, whereas loss of K8/18 in primary lymph node tumors is associated with EMT and worse outcomes. In TNBC, cytoplasmic K8/18 promotes proliferation and poor prognosis by activating the PI3K/AKT/ERK pathways and its plasminogen receptor function enhances invasiveness, as shown in Figure 6.
 |
Figure 6 Keratin 8/18 signaling pathway. The purple elements represent metastasis-related processes and blue indicates invasion. |
K8/18 plays a multifaceted role in BC progression.16,24,49,57,58 Initially, PI3K is activated by receptor tyrosine kinases or the ER. Once activated, it converts PIP2 into PIP3, which in turn serves as a docking site for AKT recruitment and activation.59 Second, AKT is phosphorylated by PDK1 and mTORC2 following binding to PIP3. The substance plays a pivotal role in the suppression of apoptosis and the promotion of cell proliferation through several mechanisms, including the inhibition of proapoptotic proteins (eg, BAD and caspase-9) and the activation of transcription factors (eg, NF-κB).60 Third, ERK is part of the MAPK pathway, but in this context, it is activated through crosstalk with PI3K/AKT signaling. ERK has been demonstrated to regulate the expression of genes associated with cell cycle progression (eg, Cyclin D1) and cell motility.61 Via this pathway, the process of invasion is initiated via apoptosis inhibition and cell cycle progression. AKT has been shown to phosphorylate and inactivate proapoptotic proteins, such as BAD and FoxO, thus enabling cancer cells to survive despite DNA damage or microenvironmental stress. K8/18 has the capacity to reinforce this effect by stabilizing the PI3K/AKT complex. Furthermore, AKT activates mTOR, thereby enhancing protein synthesis and cell growth, whereas ERK increases Cyclin D1 expression, accelerating the G1-to-S phase transition. The result of the process under investigation is uncontrolled cell proliferation.62,63 Second, AKT and ERK have been demonstrated to increase the expression of MMPs, including MMP-9, which are involved in the degradation of the ECM, thereby facilitating tumor invasion.64,65 The PI3K/AKT pathway has also been demonstrated to activate NF-κB, thereby increasing the expression of adhesion molecules (eg, ICAM-1) that promote tumor cell interactions with the vascular endothelium.66,67 ERK further promotes partial EMT by downregulating E-cadherin, contributing to a more invasive phenotype.68,69
Role of Keratin 14 as Prognostic Marker
K14 contributes to BC progression, metastasis, and aggressive tumor behavior. K14 expression, driven by H3K27me3/SPI repression, promotes invasion through the Rho/ROCK/integrin pathway, as shown in Figure 7.
 |
Figure 7 Keratin 14 signaling pathway. The blue elements represent invasion processes and red indicates immune evasion. |
K14 significantly influences pivotal oncogenic pathways,9,11,12,43 including (a) Rho GTPases (eg, RhoA, Rac1, and Cdc42) are signaling proteins that play essential roles in the organization of the actin cytoskeleton and in cell motility. The activation of these processes is initiated by surface receptors, which include integrins and G protein–coupled receptors (GPCRs).70 (2) ROCK is a significant effector downstream of RhoA that phosphorylates substrates such as myosin II regulatory light chain (MLC), thereby promoting cytoskeletal contractility.71 (3) Integrins are cell adhesion receptors that link the ECM to the cytoskeleton.72 In this pathway, K14-driven tumor progression is initiated via invasion and immune evasion. Invasion via cytokeratin and cytoskeletal reorganization.73 The use of RhoA/ROCK signaling triggers MLC phosphorylation, thereby increasing actomyosin contractility and promoting the formation of stress fibers and invadopodia. These are cellular protrusions that degrade the ECM during cancer cell invasion.74,75 Integrins have been shown to activate FAK/Src pathways, which in turn promote invadopodia formation and the release of MMPs, thereby facilitating the breakdown of the ECM.76,77 Tumor immune evasion is generally associated with infiltration of Tregs and increased production of immunosuppressive cytokines (eg, TGF-β, IL-10).78 Furthermore, activation of Rho/ROCK signaling has been implicated in cytoskeletal remodeling and invasive behavior of tumor cells.71 Integrin αvβ3 signaling has been associated with formation of immunosuppressive tumor microenvironments,79 such as recruitment and functional polarization of regulatory immune cells.80 This phenomenon is facilitated by activated STAT3, which functions as an inducer of PD-L1 expression and integrin-mediated tumor cell adhesion to immune cells. The resulting effects include the inhibition of cytotoxic T-cell function and the promotion of immune evasion.81,82
In addition to its prognostic value, K14-mediated activation of Rho/ROCK and integrin signaling highlights a potential vulnerability of K14-high tumors to interventions targeting the cytoskeleton and mechanotransduction, particularly in invasive and immune-evasive disease states.11,12,25 This concept aligns with the increasing focus on reprogramming tumor ecosystems instead of causing direct cell death by targeting cytoskeletal dynamics, stromal interaction, and immune response modulation.83
Role of Keratin 17 as Prognostic Marker
K17 expression is correlated with poor prognosis in aggressive BC subtypes, particularly TNBC and basal-like tumors, as shown in Figure 8.
 |
Figure 8 Keratin 17 signaling pathway. The pink elements represent chemoresistance. |
K17 drives tumor progression through multiple pathways.9,32,36 The IL-17/cytokine receptor pathway is a proinflammatory signaling pathway that involves the cytokine IL-17 and its receptor, IL-17R. IL-17 is produced by Th17 cells, mast cells, or even tumor cells themselves. The substance in question has been demonstrated to bind to IL-17R, which is expressed on the surface of both tumor cells and stromal cells.84,85 Upon binding, IL‑17R activates NF‑κB and MAPK, which in turn upregulates the expression of proinflammatory genes such as IL‑6 and MMP‑9.86 Furthermore, the activation of STAT3 results in increased tumor cell survival.87 IL-17 has been demonstrated to increase the production of MMP-9, which in turn degrades the ECM, thus facilitating tumor invasion. IL-17 has been shown to induce the release of TNF-α and IL-6, thereby creating a prometastatic microenvironment.88 IL-17 signaling induces the recruitment of myeloid-derived suppressor cells (MDSCs), which have been shown to suppress antitumor immunity and promote metastasis.89 Furthermore, the phenomenon of chemoresistance, which is induced by crosstalk with EGFR, can be attributed to the activation of antiapoptotic pathways via the same mechanism.90 In addition, IL-17 indirectly activates EGFR through the release of its ligands, including amphiregulin.91 EGFR activation has been demonstrated to stimulate the PI3K/AKT and MAPK pathways, thereby increasing cell survival.92 The resistance mechanism is initiated when STAT3, following activation by IL-17, results in the upregulation of the expression of Bcl-2 and multidrug resistance 1 (MDR1), thereby increasing the resistance of tumor cells to chemotherapy.93,94
On a more specific level, K17-driven activation of the IL-17-STAT3 inflammatory pathway mediates the association between keratin expression and cytokine-mediated survival, immune suppression, and chemoresistance in aggressive subtypes of BC.37,40 This context for inflammatory signaling is consistent with treatment paradigms driven by an ecosystem orientation that seek to break pro-tumor immune-cytokine loops.83
Significantly, many poor-prognosis keratin subtypes coalesce around inflammation-survival signaling networks primarily focused on NF-κB activation. NF-κB encapsulates epithelial-mesenchymal transition, immunological modulation, and apoptotic resistance through interactions with PI3K/AKT signaling, cytokine pathways, and cytoskeletal remodeling, enhancing aggressive tumor phenotypes.15,34 This is in line with data from other cancers that demonstrate that NF-κB-based programs favor invasion, survival, and therapy resistance, suggesting a valuable connection between keratin-driven phenotypes and stress-adaptive oncogenic modulation.95
While this review focuses primarily on individual keratin subtypes for convenience of interpretation, several studies from the dataset also indicate the biological role and clinical impact of multi-keratin expression. Examples encompass the combination of K5/6 with androgen receptor and p53 status in high risk TNBC,28 the combined expression profiling of K5, K6, K14, and K17,11 in stemness-associated tumor programs, as well as the K14-luminal (K18) balance regulated by ECM.23 These results suggest that function of keratin in BC is a result of the interaction of keratin networks and not a result of single markers by itself, thus are dynamic characteristics of tumor cell identity. While spatial transcriptomics and single-cell profiling were not used directly in the studies referenced, emerging data from these strategies are expected to optimize multi-keratin signatures via enhanced resolution of intratumoral heterogeneity, potentially gaining future utility for risk stratification and precision oncology.
Conclusion
This review confirms that keratin in BC not only acts as a diagnostic and prognostic biomarker but also plays an active biological role in tumor progression. Its natural functions include maintaining cytoskeletal stability and cell adhesion. However, specific subtypes, such as K5/6, K14, K17, K19, and K8/18, were selected because they are the most common and clinically significant in BC, have the most published evidence, and shown to promote oncogenic pathways. These include EGFR/MAPK/PI3K/AKT, Wnt/β-catenin, Rho/ROCK/integrin, and Notch. These pathways drive invasion, EMT, metastasis, chemoresistance, and immune evasion. Consequently, any therapeutic approach targeting keratin must meticulously consider the risk of disrupting its essential structural roles in normal tissues.
Indeed, the subtype-specific importance of keratin expression is especially pronounced in triple-negative and basal-like BC, with the most impactful prognostic value due to keratin classification. Basal keratins K5/6, K14, and K17 consistently identify TNBC and basal-like tumors and are associated with aggressive phenotypes characterized by EMT, invasion, metastasis, immune evasion, and chemotherapy resistance. On the other hand, K19 is less subtype specific and mainly serves as a marker of tumor dissemination, with circulating tumor cells in luminal, HER2-positive, and TNBC subtypes exhibiting metastatic tendency and poor clinical outcomes. Keratin 8/18 has a context-sensitive function as a marker of epithelial integrity and treatment response in luminal BC and promotes proliferation and aggressiveness through PI3K/AKT/ERK signaling when expressed aberrantly in TNBC. Overall, this approach suggests that keratin profiling is most useful when interpreted within specific BC subtypes, especially TNBC, rather than being applied uniformly across all molecular categories.
It is recommended that future research efforts concentrate on identifying keratin subtypes that exhibit specific dominant expression in tumor cells while displaying minimal or redundant functions in healthy epithelium. Examples of such keratins include K5/6 in basal-like TNBC and K14/K17 in tumors with proven invasive characteristics. These subtypes are more rational candidates for functional inhibition or pathway-specific knockdown. For subtypes whose natural functions are critical to tissue integrity, exploiting their overexpression as a targeting marker for precision drug delivery systems, such as PDC-K1-targeted therapy, may be safer. This dual strategy is designed to achieve a balance between the potential to suppress oncogenic signaling and the minimization of harm to normal tissue homeostasis.
Abbreviations
BC, breast cancer; IF, intermediate filament; PRISMA, referred Reporting Items for Systematic Reviews and Meta-Analyses; K-number, keratin protein-number; KRT-number, keratin gene-number; ER, estrogen receptor; PR, progesterone receptor; HER2, human epidermal growth factor receptor 2; TNBC, triple negative breast cancer; BLBC, basal-like breast carcinoma; NAC, neoadjuvant chemotherapy; CSC, cancer stem cell; EMT, epithelial-mesenchymal transition; ECM, extracellular matrix; MMP, Matrix metalloproteinase; KRT7-AS, Keratin-7 antisense; PTEN, Phosphatase and tensin homologs; PI3K, Phosphoinositide 3-kinase; AKT, Ak strain/also known as protein kinase B; FOXA1, Fork head Box A1; NF-κB, Nuclear Factor kappa B; DNMT3A, DNA (cytosine-5)-methyltransferase 3 alpha; GSK-3, Glycogen synthase kinase 3; ERK, Extracellular signal-regulated kinases; SP1, Specificity protein 1; RAC1, Ras-related C3 botulinum toxin substrate 1; GSK-3β, Glycogen synthase kinase 3 beta; FAK, focal adhesion signaling; NOD, Non-Obese Diabetic background; SCID, Severe Combined Immunodeficiency mutation; PDC, Peptide-Doxorubicin Conjugate; TNBL, Triple-negative basal-like; α-SMA, alpha–smooth muscle actin; ERβ, Estrogen receptor beta; FBLN2, Fibulin 2; ITGβ1, Integrin beta 1; ITGα3, Integrin alpha 3; Tregs, regulatory T cells; FoxP3, Forkhead box P3; K14CreERβp53, K14-CreER; ERβ^F/F; p53^F/F transgenic mice; BM, Basement membrane; AR, Androgen receptor; p53, Tumor suppressor protein; MpBC, metaplastic BC; FOXM1, Forkhead Box M1; PKC, Protein Kinase C; AP-1, Activator Protein-1; mTOR, Mammalian target of rapamycin; EGFR, Epidermal growth factor receptor; MAPK, Mitogen-Activated Protein Kinase; AC, Adriamycin/doxorubicin + cyclophosphamide; TP53, Gene that encodes the p53 protein; Nuclear p27, Cyclin-Dependent Kinase Inhibitor p27Kip1; PFS, progression-free survival; OS, overall survival; EFS, event-free survival; DMFS, distant metastasis-free survival; IHC, Immunohistochemistry; TGF, Transforming Growth Factor; RAS, Rat sarcoma virus oncogene; RAF, Rapidly Accelerated Fibrosarcoma kinase; MEK, MAPK/ERK kinase (also called MAP2K); Twist/Snail/Zeb1, Transcription factors; VEGF, Vascular Endothelial Growth Factor; HRR, Homologous Recombination Repair; PARP, Poly(ADP-ribose) Polymerase; Wnt, Wingless/integrated; APC, Adenomatous Polyposis Coli; c-Myc, Cellular Myelocytomatosis oncogene; NICD, Notch Intracellular Domain; Hes1, Hairy and Enhancer of Split 1; Hey1, Hairy/Enhancer-of-split related with YRPW motif 1; CDH1, Cadherin 1; CYFRA, Cytokeratin Fragment; PIP3, Phosphatidylinositol (3,4,5)-trisphosphate; PDK1, 3-Phosphoinositide-Dependent Protein Kinase-1; BAD, Bcl-2–associated agonist of cell death; FoxO, Forkhead Box O; H3K27me3, Trimethylation of lysine 27 on histone H3; SPI, Family of transcription factors; Rho/ROCK, Ras homolog/Rho-associated, coiled-coil containing protein kinase; Cdc42, Cell division control protein 42 homolog; GTPases, Guanosine Triphosphatases; GPCRs, G protein–coupled receptors; MLC, Myosin II Regulatory Light Chain; STAT3, Signal Transducer and Activator of Transcription 3; PD-L1, Programmed Death-Ligand 1; Th17, T helper 17 cells; MDSCs, myeloid-derived suppressor cells; MDR1, multidrug resistance 1; Bcl-2, B-cell lymphoma 2.
Acknowledgments
We want to thank the Faculty of Medicine at Universitas Padjadjaran for their significant support in completing this study.
Author Contributions
All authors have made significant contributions to the work reported, whether through conception, study design, execution, data collection, analysis, or interpretation. They participated in drafting, revising, or critically reviewing the article, gave final approval of the version to be published, agreed on the journal to which the article has been submitted, and are committed to being accountable for all aspects of the work.
Funding
This work was supported by an internal grant (No. 5317/UN6.C/PT.02/2025) from Universitas Padjadjaran, West Java, Indonesia.
Disclosure
The authors report no conflicts of interest in this work.
References
1. CDC. Breast cancer basics. 2024. Available from: https://www.cdc.gov/breast-cancer/about/?CDC_AAref_Val=https://www.cdc.gov/cancer/breast/basic_info/what-is-breast-cancer.htm#print.
2. Kumar V, Abbas AK, Aster JC. Robbins Basic Pathology.
3. American Cancer Society. Key statistics for breast cancer. 2025. Available from: https://www.cancer.org/cancer/types/breast-cancer/about/how-common-is-breast-cancer.html#:~:text=Breast%20cancer%20is%20the%20second,in%2039%20about%202.5%25.
4. Shalannandia WA, Chou Y, Bashari MH, Khairani AF. Intermediate filaments in breast cancer progression, and potential biomarker for cancer therapy: a narrative review. Breast Cancer. 2024;16:689–20. doi:10.2147/BCTT.S489953
5. Leidy J, Khan A, Kandil D. Basal-like breast cancer: update on clinicopathologic, immunohistochemical, and molecular features. Arch Pathol Lab Med. 2014;138(1):37–43. doi:10.5858/arpa.2012-0439-RA
6. Kim MJ, Ro JY, Ahn SH, Kim HH, Kim SB, Gong G. Clinicopathologic significance of the basal-like subtype of breast cancer: a comparison with hormone receptor and Her2/neu-overexpressing phenotypes. Hum Pathol. 2006;37(9):1217–1226. doi:10.1016/j.humpath.2006.04.015
7. Lindström LS, Karlsson E, Wilking UM, et al. Clinically used breast cancer markers such as estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 are unstable throughout tumor progression. Clin Oncol. 2012;30(21):2601–2608.
8. Scientists discover how some advanced breast cancers become resistant to hormone therapy. The Institute Of Cancer Research.
9. McGinn O, Ward AV, Fettig LM, et al. Cytokeratin 5 alters β-catenin dynamics in breast cancer cells. Oncogene. 2020;39(12):2478–2492. doi:10.1038/s41388-020-1164-0
10. Wang D, Naydenov NG, Dozmorov MG, Koblinski JE, Ivanov AI. Anillin regulates breast cancer cell migration, growth, and metastasis by non-canonical mechanisms involving control of cell stemness and differentiation. Breast Cancer Res. 2020;22(1). doi:10.1186/s13058-019-1241-x
11. Verma A, Singh A, Singh MP, et al. EZH2-H3K27me3 mediated KRT14 upregulation promotes TNBC peritoneal metastasis. Nat Commun. 2022;13(1). doi:10.1038/s41467-022-35059-x
12. Padmanaban V, Tsehay Y, Cheung KJ, Ewald AJ, Bader JS. Between-tumor and within-tumor heterogeneity in invasive potential. PLoS Comput Biol. 2020;16(1). doi:10.1371/journal.pcbi.1007464
13. Izdebska M, Zielińska W, Krajewski A, et al. Downregulation of mmp-9 enhances the anti-migratory effect of cyclophosphamide in mda-mb-231 and mcf-7 breast cancer cell lines. Int J Mol Sci. 2021;22(23):12783. doi:10.3390/ijms222312783
14. Lin TY, Wang PW, Huang CH, Yang PM, Pan TL Characterizing the relapse potential in different luminal subtypes of breast cancers with functional proteomics. Int J Mol Sci. 2020;21(17):1–16. doi:10.3390/ijms21176077
15. Zhao Z, Meng M, Yao J, et al. The long non-coding RNA keratin-7 antisense acts as a new tumor suppressor to inhibit tumorigenesis and enhance apoptosis in lung and breast cancers. Cell Death Dis. 2023;14(4). doi:10.1038/s41419-023-05802-3
16. Butler C, Sprowls S, Szalai G, et al. Hypomethylating agent azacitidine is effective in treating brain metastasis triple-negative breast cancer through regulation of DNA methylation of keratin 18 gene. Transl Oncol. 2020;13(6):100775. doi:10.1016/j.tranon.2020.100775
17. Saha SK, Kim K, Yang GM, Choi HY, Cho SG. Cytokeratin 19 (KRT19) has a role in the reprogramming of cancer stem cell-like cells to less aggressive and more drug-sensitive cells. Int J Mol Sci. 2018;19(5):1423. doi:10.3390/ijms19051423
18. Saha SK, Yin Y, Chae HS, Cho SG. Opposing regulation of cancer properties via KRT19-mediated differential modulation of wnt/β-catenin/notch signaling in breast and colon cancers. Cancers. 2019;11(1):99. doi:10.3390/cancers11010099
19. Yan Z, Zhong Z, Shi C, Feng M, Feng X, Liu T. The prognostic marker KRT81 is involved in suppressing CD8 + T cells and predicts immunotherapy response for triple-negative breast cancer. Cancer Biol Ther. 2024;25(1). doi:10.1080/15384047.2024.2355705
20. Saghaeidehkordi A, Chen S, Yang S, Kaur K. Evaluation of a keratin 1 targeting peptide‐doxorubicin conjugate in a mouse model of triple‐negative breast cancer. Pharmaceutics. 2021;13(5):661. doi:10.3390/pharmaceutics13050661
21. Machado MCA, Ocarino NM, Serakides R, et al. Triple-negative mammary carcinoma in two male dogs. J Vet Diagn Invest. 2020;32(1):94–98. doi:10.1177/1040638719898686
22. Eivani D, Mortazavi P. The relationship between basal and luminal cytokeratins with histopathologic characteristics of canine mammary gland cancer. Pol J Vet Sci. 2016;19(2):261–269. doi:10.1515/pjvs-2016-0033
23. Bado I, Nikolos F, Rajapaksa G, et al. Somatic loss of estrogen receptor beta and p53 synergize to induce breast tumorigenesis. Breast Cancer Res. 2017;19(1). doi:10.1186/s13058-017-0872-z
24. WalyEldeen AA, Sabet S, Anis SE, Stein T, Ibrahim AM. FBLN2 is associated with basal cell markers Krt14 and ITGB1 in mouse mammary epithelial cells and has a preferential expression in molecular subtypes of human breast cancer. Breast Cancer Res Treat. 2024. doi:10.1007/s10549-024-07447-y
25. Dagher E, Simbault L, Abadie J, Loussouarn D, Campone M, Nguyen F. Identification of an immune-suppressed subtype of feline triple-negative basal-like invasive mammary carcinomas, spontaneous models of breast cancer. Tumor Biol. 2020;42(1). doi:10.1177/1010428319901052
26. Engstrøm MJ, Valla M, Bofin AM. Basal markers and prognosis in luminal breast cancer. Breast Cancer Res Treat. 2017;163(2):207–217. doi:10.1007/s10549-017-4182-z
27. Abdelrahman AE, Rashed HE, Abdelgawad M, Abdelhamid MI. Prognostic impact of EGFR and cytokeratin 5/6 immunohistochemical expression in triple-negative breast cancer. Ann Diagn Pathol. 2017;28:43–53. doi:10.1016/j.anndiagpath.2017.01.009
28. Maeda T, Nakanishi Y, Hirotani Y, et al. Immunohistochemical co-expression status of cytokeratin 5/6, androgen receptor, and p53 as prognostic factors of adjuvant chemotherapy for triple negative breast cancer. Med Mol Morphol. 2016;49(1):11–21. doi:10.1007/s00795-015-0109-0
29. Voutilainen S, Heikkilä P, Sampo M, Nevanlinna H, Blomqvist C, Mattson J. Expression of markers of stem cell characteristics, epithelial-mesenchymal transition, basal-like phenotype, proliferation, and androgen receptor in metaplastic breast cancer and their prognostic impact. Acta Oncol (Madr). 2021;60(9):1233–1239. doi:10.1080/0284186X.2021.1950927
30. Martin-Castillo B, Lopez-Bonet E, Buxó M, et al. Cytokeratin 5/6 fingerprinting in HER2-positive tumors identifies a poor prognosis and trastuzumab-resistant basal-HER2 subtype of breast cancer. 2015. Available from: www.impactjournals.com/oncotarget/.
31. Lee DHY, Tsang JY, Li JJ, et al. Cytokeratin 15 is a novel and independent predictor of poor outcome in luminal B HER2-negative breast carcinomas. Pathology. 2024;56(6):834–841. doi:10.1016/j.pathol.2024.03.009
32. Merkin RD, Vanner EA, Romeiser JL, et al. Keratin 17 is overexpressed and predicts poor survival in estrogen receptor–negative/human epidermal growth factor receptor-2–negative breast cancer. Hum Pathol. 2017;62:23–32. doi:10.1016/j.humpath.2016.10.006
33. Hendawy SR, Mansour M, Hamed M, Hossam A, Ramez AM. Serum cytokeratin 18 as a potential early marker for chemotherapy response in breast cancer patients: a prospective study. Asian Pac J Cancer Prev. 2023;24(3):969–975. doi:10.31557/APJCP.2023.24.3.969
34. Morales S, Velasco A, Gasol A, et al. Circulating tumor cells (CTCs) and cytokeratin 19 (CK19) mRNA as prognostic factors in heavily pretreated patients with metastatic breast cancer. Cancer Treat Res Commun. 2018;16:13–17. doi:10.1016/j.ctarc.2018.04.003
35. Wang XM, Zhang Z, Pan LH, Cao XC, Xiao C. KRT19 and CEACAM5 mRNA-marked circulated tumor cells indicate unfavorable prognosis of breast cancer patients. Breast Cancer Res Treat. 2019;174(2):375–385. doi:10.1007/s10549-018-05069-9
36. Tang S, Liu W, Yong L, et al. Reduced expression of KRT17 predicts poor prognosis in HER2high breast cancer. Biomolecules. 2022;12(9):1183. doi:10.3390/biom12091183
37. Zhao C, Lou Y, Wang Y, et al. A gene expression signature-based nomogram model in prediction of breast cancer bone metastases. Cancer Med. 2019;8(1):200–208. doi:10.1002/cam4.1932
38. Trask DK, Band V, Zajchowski DA, Yaswen P, Suh T, Sager R. Keratins as markers that distinguish normal and tumor-derived mammary epithelial cells (Intermediate filaments/immortalization/tumor and normal antigens/breast cancer). Proc Natl Acad Sci. 1990;87(6):2319–23.
39. Mohanty SK, Lai JP, Gordon OK, Pradhan D, Bose S, Dadmanesh F. BRCA-mutated invasive breast carcinomas: immunohistochemical analysis of insulin-like growth factor II mRNA-binding protein (IMP3), cytokeratin 8/18, and cytokeratin 14. Breast J. 2015;21(6):596–603. doi:10.1111/tbj.12494
40. McBrearty N, Bahal D, Platero S. Fast-tracking drug development with biomarkers and companion diagnostics. J Cancer Metastasis Treat. 2024;10. doi:10.20517/2394-4722.2023.134
41. Polioudaki H, Agelaki S, Chiotaki R, et al. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer. 2015;15(1). doi:10.1186/s12885-015-1386-7
42. Tanabe Y, Tsuda H, Yoshida M, et al. Pathological features of triple-negative breast cancers that showed progressive disease during neoadjuvant chemotherapy. Cancer Sci. 2017;108(7):1520–1529. doi:10.1111/cas.13274
43. Majumder A, Jagani R, Basu A. Double-positive in triple-negative? How significant is basal cytokeratin expression in breast cancer? Med J Armed Forces India. 2020;76(1):63–70. doi:10.1016/j.mjafi.2018.10.002
44. Limsakul P, Choochuen P, Jungrungrueang T, Charupanit K. Prognostic markers in tyrosine kinases specific to basal-like 2 subtype of triple-negative breast cancer. Int J Mol Sci. 2024;25(3):1405. doi:10.3390/ijms25031405
45. Rocca A, Braga L, Volpe MC, Maiocchi S, Generali D. The predictive and prognostic role of RAS–RAF–MEK–ERK pathway alterations in breast cancer: revision of the literature and comparison with the analysis of cancer genomic datasets. Cancers. 2022;14(21):5306. doi:10.3390/cancers14215306
46. An SJ, Anneken A, Xi Z, Choi C, Schlessinger J, Toomre D. Regulation of EGF-stimulated activation of the PI-3K/AKT pathway by exocyst-mediated exocytosis. Proc Natl Acad Sci. 2022;119(48). doi:10.1073/pnas.2208947119
47. Jiang W, Wang X, Zhang C, Xue L, Yang L. Expression and clinical significance of MAPK and EGFR in triple–negative breast cancer. Oncol Lett. 2020;19(3):1842–1848. doi:10.3892/ol.2020.11274
48. Li H, Liu ZY, Wu N, Chen YC, Cheng Q, Wang J. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer Bio. 2020;19(1). doi:10.1186/s12943-020-01227-0
49. Shi R, Wang C, Fu N, et al. Downregulation of cytokeratin 18 enhances BCRP-mediated multidrug resistance through induction of epithelial-mesenchymal transition and predicts poor prognosis in breast cancer. Oncol Rep. 2019;41(5):3015–3026. doi:10.3892/or.2019.7069
50. Abdelaziz LA, Ebian HF, Harb OA, Nosery Y, Taha HF, Nawar N. Clinical significance of cytokeratin 19 and OCT4 as survival markers in non-metastatic and metastatic breast cancer patients. Wspolczesna Onkologia. 2022;26(1):78–87. doi:10.5114/wo.2022.115678
51. Strati A, Nikolaou M, Georgoulias V, Lianidou ES. Rna-based ctc analysis provides prognostic information in metastatic breast cancer. Diagnostics. 2021;11(3):513. doi:10.3390/diagnostics11030513
52. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: update on effectors and inhibitors. Cancer Treat Rev. 2018;62:50–60. doi:10.1016/j.ctrv.2017.11.002
53. Zhang H, Hang W, Jing Z, et al. The role of notch signaling pathway in cancer: mechanistic insights, therapeutic potential, and clinical progress. Front Immunol. 2025;16. doi:10.3389/fimmu.2025.1567524
54. Zhang YU, Xie ZY, Guo XT, Xiao XH, Xiong LX. Notch and breast cancer metastasis: current knowledge, new sights and targeted therapy (review). Oncol Lett. 2019;18(3):2743–2755. doi:10.3892/ol.2019.10653
55. Xu X, Zhang M, Xu F, Jiang S. Wnt signaling in breast cancer: biological mechanisms, challenges and opportunities. Mol Cancer. 2020;19(1). doi:10.1186/s12943-020-01276-5
56. Song P, Gao Z, Bao Y, et al. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J Hematol Oncol. 2024;17(1). doi:10.1186/s13045-024-01563-4
57. Bonin S, Pracella D, Barbazza R, Sulfaro S, Stanta G. In stage II/III lymph node-positive breast cancer patients less than 55 years of age, keratin 8 expression in lymph node metastases but not in the primary tumour is an indicator of better survival. Virchows Archiv. 2015;466(5):571–580. doi:10.1007/s00428-015-1748-1
58. Aiad HA, Samaka RM, Asaad NY, Kandil MA, Shehata MA, Miligy IM. Relationship of CK8/18 expression pattern to breast cancer immunohistochemical subtyping in Egyptian patients. E Cancer Med Sci. 2014;8(1). doi:10.3332/ecancer.2014.404
59. Peng Y, Wang Y, Zhou C, Mei W, Zeng C. PI3K/Akt/mTOR pathway and its role in cancer therapeutics: are we making headway? Front Oncol. 2022;12. doi:10.3389/fonc.2022.819128
60. Hua H, Zhang H, Chen J, Wang J, Liu J, Jiang Y. Targeting Akt in cancer for precision therapy. J Hematol Oncol. 2021;14(1). doi:10.1186/s13045-021-01137-8
61. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. doi:10.1016/j.tibs.2011.03.006
62. Zhou H, Huang S. The complexes of mammalian target of rapamycin.
63. Lim Y, Kim S, Yoon HN, Ku NO. Keratin 8/18 regulate the akt signaling pathway. Int J Mol Sci. 2021;22(17):9227. doi:10.3390/ijms22179227
64. Akter H, Park M, Kwon OS, Song EJ, Park WS, Kang MJ. Activation of matrix metalloproteinase-9 (MMP-9) by neurotensin promotes cell invasion and migration through ERK pathway in gastric cancer. Tumor Biol. 2015;36(8):6053–6062. doi:10.1007/s13277-015-3282-9
65. Cheng JCH, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-κB signal transduction pathway. Oncogene. 2006;25(53):7009–7018. doi:10.1038/sj.onc.1209706
66. Nidai Ozes O, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine–threonine kinase. Nature. 1999;401(6748):82–85. doi:10.1038/43466
67. Wissink S, Van De Stolpe A, Caldenhoven E, Koenderman L, Van Der Saag PT. NF-κB/rel family members regulating the ICAM-1 promoter in monocytic THP-1 cells. Immunobiology. 1997;198(1–3):50–64. doi:10.1016/S0171-2985(97)80026-5
68. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–976. doi:10.1016/j.cell.2017.02.004
69. Bae GY, Choi SJ, Lee JS, et al. Loss of E-cadherin activates EGFR-MEK/ERK signaling, which promotes invasion via the ZEB1/MMP2 axis in non-small cell lung cancer. Oncotarget. 2013;4(12):2512–2522. doi:10.18632/oncotarget.1463
70. Dipankar P, Kumar P, Dash SP, Sarangi PP. Functional and therapeutic relevance of rho GTPases in innate immune cell migration and function during inflammation: an in silico perspective. Mediators Inflamm. 2021;2021(1). doi:10.1155/2021/6655412
71. Olson MF, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5(2):e29846. doi:10.4161/sgtp.29846
72. Calderwood DA, Shattil SJ, Ginsberg MH. Integrins and actin filaments: reciprocal regulation of cell adhesion and signaling. J Biol Chem. 2000;275(30):22607–22610. doi:10.1074/jbc.R900037199
73. Chen BJ, Tang Y, Tang YL, Liang X H. What makes cells move: requirements and obstacles for leader cells in collective invasion. Exp Cell Res. 2019;382(2). doi:10.1016/j.yexcr.2019.06.026
74. Clayton NS, Ridley AJ. Targeting rho GTPase signaling networks in cancer. Front Cell Dev Biol. 2020;8. doi:10.3389/fcell.2020.00222
75. Rodriguez-Hernandez I, Cantelli G, Bruce F, Sanz-Moreno V. Rho, ROCK and actomyosin contractility in metastasis as drug targets. F1000Res. 2016;5:783. doi:10.12688/f1000research.7909.1
76. Peláez R, Pariente A, Pérez-Sala Á, Larrayoz IM. Integrins: moonlighting proteins in invadosome formation. Cancers. 2019;11(5):615. doi:10.3390/cancers11050615
77. Augoff K, Hryniewicz-Jankowska A, Tabola R. Invadopodia: clearing the way for cancer cell invasion. Ann Transl Med. 2020;8(14):902. doi:10.21037/atm.2020.02.157
78. Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541(7637):321–330. doi:10.1038/nature21349
79. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22. doi:10.1038/nrc2748
80. Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155(7):1639–1651. doi:10.1016/j.cell.2013.11.029
81. Ying H, Li ZQ, Li MP, Liu WC. Metabolism and senescence in the immune microenvironment of osteosarcoma: focus on new therapeutic strategies. Front Endocrinol. 2023;14. doi:10.3389/fendo.2023.1217669
82. Salem B, Webb J, Jacobsohn D. An update on the clinical utility of extracorporeal photopheresis in the treatment of graft-versus-host disease. Int J Clin Transfusion Med. 2017;5:19–28. doi:10.2147/ijctm.s116657
83. Joshi RM, Telang B, Soni G, Khalife A. Overview of perspectives on cancer, newer therapies, and future directions. Endosc Ultrasound. 2024;10(3):105–109. doi:10.1097/ot9.0000000000000039
84. Hassan A, Aubel C. The PI3K/Akt/mTOR signaling pathway in triple-negative breast cancer: a resistance pathway and a prime target for targeted therapies. Cancers. 2025;17(13):2232. doi:10.3390/cancers17132232
85. Shibabaw T, Teferi B, Ayelign B. The role of Th-17 cells and IL-17 in the metastatic spread of breast cancer: as a means of prognosis and therapeutic target. Front Immunol. 2023;14. doi:10.3389/fimmu.2023.1094823
86. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9(8):556–567. doi:10.1038/nri2586
87. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007;7(1):41–51. doi:10.1038/nri1995
88. Wang J, Zhang Y, Yin K, et al. IL-17A weakens the antitumor immunity by inhibiting apoptosis of MDSCs in lewis lung carcinoma bearing mice. Vol 8. 2017. Available from: www.impactjournals.com/oncotarget/.
89. He D, Li H, Yusuf N, et al. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol. 2010;184(5):2281–2288. doi:10.4049/jimmunol.0902574
90. Zhang N, Zeng Y, Du W, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49(4):1360–1368. doi:10.3892/ijo.2016.3632
91. Lee KL, Lai TC, Lee WJ, et al. Sustaining the activation of EGFR signal by inflammatory cytokine IL17A prompts cell proliferation and EGFR-TKI resistance in lung cancer. Cancers. 2023;15(13):3288. doi:10.3390/cancers15133288
92. Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2–16. doi:10.1016/j.gene.2005.10.018
93. Yang PL, Liu LX, Li EM, Xu LY. STAT3, the challenge for chemotherapeutic and radiotherapeutic efficacy. Cancers. 2020;12(9):2459. doi:10.3390/cancers12092459
94. Wang Z, Wang C, Zuo D, et al. Attenuation of STAT3 phosphorylation promotes apoptosis and chemosensitivity in human osteosarcoma induced by raddeanin A. Int J Biol Sci. 2019;15(3):668–679. doi:10.7150/ijbs.30168
95. Zhai H, Du Y, He H, Shen X, Hu D. WDR54 enhances NF-κB signaling to promote progression of hepatocellular carcinoma. Cancer Genet. 2025;298-299:302–314. doi:10.1016/j.cancergen.2025.11.007
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.