Back to Journals » International Medical Case Reports Journal » Volume 11
Kartagener syndrome complicated by immunoglobulin A nephropathy
Authors Oka K, Sugase T ![]() , Akimoto T
, Akimoto T ![]() , Murakami T, Nagayama I
, Murakami T, Nagayama I ![]() , Kaneko M, Asakura M, Ohara K, Saito O, Nagata D
, Kaneko M, Asakura M, Ohara K, Saito O, Nagata D ![]()
Received 30 August 2018
Accepted for publication 24 October 2018
Published 10 December 2018 Volume 2018:11 Pages 359—362
DOI https://doi.org/10.2147/IMCRJ.S185887
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Kentaro Oka,1 Taro Sugase,1 Tetsu Akimoto,1,2 Takuya Murakami,1 Izumi Nagayama,1 Miwa Kaneko,1 Maki Asakura,1 Ken Ohara,1 Osamu Saito,1 Daisuke Nagata1
1Division of Nephrology, Department of Internal Medicine, Jichi Medical University, Tochigi, Japan; 2Department of Chronic Kidney Disease Pathophysiology, Jichi Medical University, Tochigi, Japan
Abstract: We herein report the case of a 36-year-old woman with Kartagener syndrome (KS), which is an autosomal recessive disorder defined by a triad of bronchiectasis, sinusitis, and situs inversus, with complications of asymptomatic microhematuria and proteinuria. She was finally diagnosed with biopsy-proven immunoglobulin (Ig) A nephropathy. KS constitutes a subgroup of primary ciliary dyskinesia (PCD) characterized by structural and/or functional ciliary abnormalities resulting in sinopulmonary involvement with varying severity. Our case does not allow us to corroborate the clinical impact of KS and/or PCD as a pathogenic basis for the IgA nephropathy, and each disease might develop independently. However, systematic studies on this topic are quite lacking, so we strongly recommend the accumulation of more cases similar to our own, which would allow us to clarify the nature of this disease state more precisely.
Keywords: primary ciliary dyskinesia, Kartagener syndrome, situs inversus, IgA nephropathy, glomerulonephritis
Introduction
Kartagener syndrome (KS) is an autosomal recessive disorder characterized by the triad of bronchiectasis, sinusitis, and situs inversus.1 It constitutes the subgroup of primary ciliary dyskinesia (PCD) characterized by structural and/or functional ciliary abnormalities resulting in sinopulmonary involvement with varying degrees of severity.2 Ciliary dysfunction results in clinically heterogeneous disorders that may lead to a single organ involvement, such as a congenital heart disease, or result in a syndromic phenotype.3,4 However, there are few reports describing patients with this disease state complicated by glomerulopathy.5–7 In this report, we describe one such case in a patient with complications of asymptomatic microhematuria and proteinuria, which were finally ascribed to immunoglobulin (Ig) A nephropathy.
Case report
A 36-year-old woman with a history of persistent cough and bronchiectasis was referred and admitted to our hospital for the evaluation of asymptomatic microhematuria with a urine protein-to-creatinine (Cr) ratio of 0.64 g/gCr, which had been found by her primary physician a few months previously. She was diagnosed with KS on a clinical basis in her childhood and experienced frequent respiratory tract infections, for which she received several courses of medical care, including the sporadic use of antibiotics and expectorants. A dipstick analysis performed 3 years previously revealed 1+ proteinuria and 1+ occult blood reaction when she was admitted to another hospital for bacterial pneumonia; however, her renal parameters were not regularly monitored after the analysis. She had never smoked and denied the use of any drugs. Her younger brother had also been diagnosed with KS.
At the time of admission, the patient was alert with a blood pressure of 120/71 mmHg. Her heart sounds were on the right side of the chest. Neither peripheral cyanosis nor clubbing of the digits was noted. Renal sonography showed that the right and left kidney long axis dimensions were 10.8 and 11.1 mm, respectively, with normal renal cortex echogenicity, while radiographic studies demonstrated bronchiectasis in the lower zone of both lungs, as well as dextrocardia (Figure 1). A full blood count demonstrated a white blood cell count of 4,600/µL with normal differentials, a hemoglobin level of 12.4 g/dL, and a platelet count of 32.5×104/µL. Further laboratory analyses revealed the following results: blood urea nitrogen, 8 mg/dL; serum, Cr 0.4 mg/dL; uric acid, 3.8 mg/dL; total protein, 6.4 g/dL; serum albumin, 3.3 g/dL; sodium, 140 mmol/L; potassium, 4.1 mmol/L; chloride, 104 mmol/L; Ca 9.5 mg/dL; phosphorus, 3.2 mg/dL; IgG, 1,793 mg/dL; IgA, 680 mg/dL; IgM, 90 mg/dL; C3, 99 mg/dL; C4, 24 mg/dL; and C-reactive protein, 0.02 mg/dL. A serological study was negative for antimyeloperoxidase antineutrophil cytoplasmic antibody (ANCA), antiproteinase 3-ANCA, and anti-glomerular basement membrane antibodies. A 24-hour urine specimen contained 0.16 g of protein, with a Cr clearance rate of 145.1 mL/min. Renal biopsy, which was performed on hospital day 2 (Figure 2), revealed two cores of renal parenchyma with nine glomeruli, one of which was globally sclerotic. The rest of the glomeruli demonstrated mild mesangial proliferation and moderate mesangial matrix accumulation. Predominant mesangial depositions of IgA and C3 were observed in an immunofluorescence analysis, while electron microscopy revealed paramesangial electron-dense deposits within a mildly expanded mesangium. Consequently, the patient’s renal abnormalities were ascribed to IgA nephropathy. In accordance with the Japanese therapeutic guidance,8 we prescribed a low protein diet and a periodic monitoring program, consisting of comprehensive renal care, was scheduled.
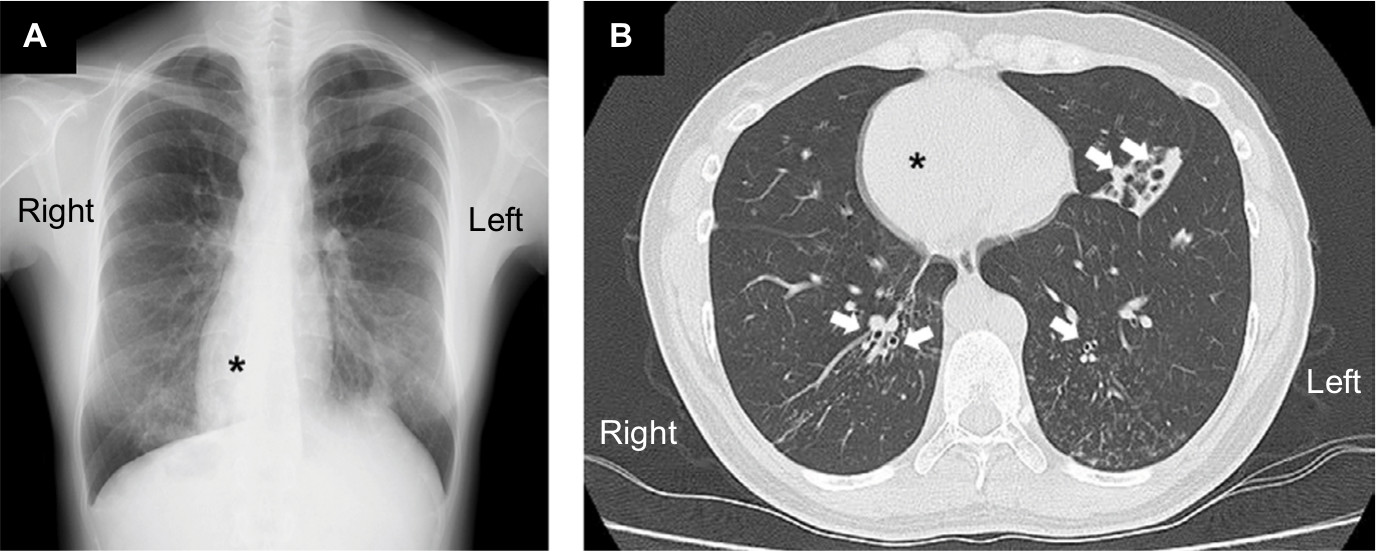 | Figure 1 The radiographic findings. Notes: A chest X-ray film (A) and a selected chest computed tomography image (B) demonstrate bronchial wall thickening and bronchial dilatation (arrows), which are suggestive of bronchiectasis, especially in the lower zone of both lungs, as well as dextrocardia (asterisks). |
 | Figure 2 The renal biopsy findings. Notes: A light micrograph (A) shows the glomerulus accompanied by the slight expansion of the mesangial matrices with mesangial cell proliferation (Periodic acid–Schiff staining). Immunofluorescence microscopy (B) shows granular staining for IgA limited to the mesangium. The scale bar is indicated in each panel. Abbreviation: Ig, immunoglobulin. |
Discussion
This report illustrates a rare case in which a renal biopsy performed as a diagnostic investigation for asymptomatic microhematuria and proteinuria led to the discovery of IgA nephropathy in a patient with KS. A spectrum of renal abnormalities, including several glomerular injuries as well as polycystic kidney disease, has been anecdotally shown in patients with the disease.4–7,9 Even patients with end-stage renal failure have been described.10,11 At present, whether or not KS and/or PCD has a pathogenic role in kidney disorders, and the chronic recurrent respiratory tract infections seen in this syndrome or medications for a comorbidity may be associated with certain types of glomerulopathy.5,9 Obviously, systematic studies on this topic are quite lacking, so we strongly recommend the accumulation of more cases similar to our own to clarify the nature of the combination between KS and IgA nephropathy more precisely. To the best of our knowledge, this is the first report of IgA nephropathy as a complication in a patient with KS.
A genetic disturbance characterized by structural and functional abnormalities in the ciliary system leading to impaired mucociliary clearance have received focus under the term PCD.12 Patients with PCD may experience sinopulmonary infections many times and suffer from progressive bronchiectasis.12 Approximately 40%–50% of subjects with the disease may have mirror-image organ arrangement, a clinical variant that is categorized as KS.1,13,14 Since this condition was first described by Siewert and later as a triad by Kartagener in 1935,15,16 the cumulative number of publications on PCD has been slowly growing. In most patients, the diagnosis of this pathology is made in childhood, but there may be some patients who are diagnosed in late adulthood.17 No distinct gender predilection has been recognized,1,3,12 whereas the incidence of the disease remains to be clarified, as the current data are largely limited to retrospective studies and the number of patients with PCD or KS may be underdiagnosed.13,14,18 Nevertheless, Afzelius et al reported their experience, identifying 26 children with PCD among 286,409 children who were born in the study period; thus, the estimated prevalence of PCD, based on the population of the study, was as much as 1 in 11,000 births. Based on this figure, the estimated incidence of KS may be ~1 in 20,000–30,000.7,14,19 Given the rarity of this condition, the lack of reports describing glomerular diseases, including IgA nephropathy among patients with PCD or KS, may not be surprising. Otherwise, the paucity of information regarding glomerular diseases among PCD patients without situs inversus is interesting.5–7,9
IgA nephropathy is recognized as the most common form of primary chronic glomerulonephritis worldwide.8 It is diagnosed based on pathological findings and histological findings that require assessment by light microscopy and immunofluorescence microscopy; thus, there are considerable discrepancies in the incidence reported in previous studies, probably due to the different policies in nephrology centers regarding the selection of patients for renal biopsy.8,20 Asymptomatic renal abnormalities and macroscopic hematuria are known as common clinical manifestations of IgA nephropathy, while isolated microscopic hematuria with minimal proteinuria is regarded as having a favorable prognosis.21 Nevertheless, a proportion of patients will eventually develop hypertension and significant proteinuria, which are also independent predictors of progression to a chronic decline in the renal function.20 Thus, the long-term follow-up of these subjects should be mandatory.21,22 Our case does not allow us to corroborate the relationship between KS by PCD and IgA nephropathy, and there might not be any shared pathogenic properties for each disease. However, we must bear in mind that this comorbid condition can be seen in ordinary clinical practice and remember that the long-term prognostic implications may be mediated by the form of chronic kidney disease, which has been in focus as a major public health problem worldwide.23 Indeed, IgA nephropathy has been shown to be a significant morbidity. The 20-year cumulative rate of end-stage kidney disease in patients with this condition is reported to be ~40%.8,23
Consent for publication
Written informed consent was obtained from the patient to publish this case report and any accompanying images. The clinicians at our university are not required to obtain institutional review board approval for case reports of a single patient.
Acknowledgment
This study was supported in part by a Grant-in-Aid for Research on Advanced Chronic Kidney Disease, Practical Research Project for Renal Diseases from the Japan Agency for Medical Research and Development (AMED) and by a grant for Private University Research Branding Project from the Ministry of Education, Science and Culture, Japan.
Disclosure
TA is affiliated with an endowed department (Department of Chronic Kidney Disease Pathophysiology) supported by Terumo Co., Chugai Pharmaceutical Co., Ltd., and Kyowa Hakko Kirin Co., Ltd. The other authors report no conflicts of interest in this work.
References
Chang WJ, Kung WC, Chiu WY. Kartagener syndrome. QJM. 2017;110(8):523. | ||
Pandey AK, Maithani T, Bhardwaj A. Kartagener’s syndrome: a clinical reappraisal with two case reports. Egyp J Ear Nose Throat Allied Sci. 2014;15(3):271–274. | ||
Kennedy MP, Omran H, Leigh MW, et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115(22):2814–2821. | ||
Ware SM, Aygun MG, Hildebrandt F. Spectrum of clinical diseases caused by disorders of primary cilia. Proc Am Thorac Soc. 2011;8(5):444–450. | ||
Egbert BM, Schwartz E, Kempson RL. Kartagener syndrome: report of a case with mesangiocapillary glomerulonephritis. Arch Pathol Lab Med. 1977;101(2):95–99. | ||
Krishnamurthy S, Choudhary B, Rajesh NG, Ramesh A, Srinivasan S. Kartagener syndrome associated with mesangioproliferative glomerulonephritis. Paediatr Int Child Health. 2012;32(2):116–118. | ||
Momeni A, Doroushi B, Taheri N. Kartagener syndrome with focal segmental glomerulosclerosis. Iran J Kidney Dis. 2013;7(6):499–501. | ||
Tomino Y. Diagnosis and treatment of patients with IgA nephropathy in Japan. Kidney Res Clin Pract. 2016;35(4):197–203. | ||
Kawasaki Y, Suzuki J, Sike T, et al. Bucillamine-induced nephropathy in a child with juvenile rheumatoid arthritis and Kartagener’s syndrome. Pediatr Int. 2000;42(3):316–318. | ||
Imafuku T, Ogihara T, Kudo H, Hayashi K, Ohtake K, Iyori S. Kartagener’s syndrome associated with infundibular pulmonic stenosis, chronic renal failure and azoospermia: a report of a case. Jpn J Med. 1986;25(2):195–198. | ||
Osman EM, Abboud OI, Sulaiman SM, Musa AR, Beleil OM, Sharfi AA. End-stage renal failure in Kartagener’s syndrome. Nephrol Dial Transplant. 1991;6(10):747. | ||
Noone PG, Leigh MW, Sannuti A, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med. 2004;169(4):459–467. | ||
Barbato A, Frischer T, Kuehni CE, et al. Primary ciliary dyskinesia: a consensus statement on diagnostic and treatment approaches in children. Eur Respir J. 2009;34(6):1264–1276. | ||
Ciancio N, de Santi MM, Campisi R, Amato L, di Martino G, di Maria G. Kartagener’s syndrome: review of a case series. Multidiscip Respir Med. 2015;10(1):18. | ||
Siewert A. Über einen Fall von Bronchiektasie bei einem Patienten mit Situs inversus viscerum. [A case of bronchiectasis in a patient with Situs inversus viscerum]. Berl Klin Wochenschr. 1904;41:139–141. German. | ||
Kartagener M. Zur Pathogenese der Bronchiektasen. I. Mitteilung: Bronchiektasien bei Situs viscerum inversus. [The pathogenesis of bronchiectasis. I. Report: bronchiectasis in Situs viscerum inversus]. Beitr Klin Tuberk. 1933;83:489–501. German. | ||
Mcmanus IC, Mitchison HM, Chung EM, Stubbings GF, Martin N. Primary ciliary dyskinesia (Siewert’s/Kartagener’s syndrome): respiratory symptoms and psycho-social impact. BMC Pulm Med. 2003;3:4. | ||
Katsuhara K, Kawamoto S, Wakabayashi T, Belsky JL. Situs inversus totalis and Kartagener’s syndrome in a Japanese population. Chest. 1972;61(1):56–61. | ||
Afzelius BA, Stenram U. Prevalence and genetics of immotile-cilia syndrome and left-handedness. Int J Dev Biol. 2006;50(6):571–573. | ||
Haas M. IgA nephropathy and Henoch-Schonlein purpura nephritis. In: Jennete JC, Olson JL, Schwartz MM, Silva FG, editors, Heptinstall’s Pathology of the Kidney. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2007:423–486. | ||
Rodrigues JC, Haas M, Reich HN. IgA nephropathy. Clin J Am Soc Nephrol. 2017;12(4):677–686. | ||
Szeto CC, Lai FM, To KF, et al. The natural history of immunoglobulin A nephropathy among patients with hematuria and minimal proteinuria. Am J Med. 2001;110(6):434–437. | ||
Tanaka S, Ninomiya T, Katafuchi R, et al. Secular trends in the incidence of end-stage renal disease and its risk factors in Japanese patients with immunoglobulin A nephropathy. Nephrol Dial Transplant. 2018;33(6):963–971. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.