Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 6
Juvenile neuronal ceroid lipofuscinosis (Batten disease): current insights
Authors Ostergaard J
Received 4 May 2016
Accepted for publication 11 May 2016
Published 1 August 2016 Volume 2016:6 Pages 73—83
DOI https://doi.org/10.2147/DNND.S111967
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Müller
John R Ostergaard
Department of Paediatrics, Aarhus University Hospital, Centre for Rare Diseases, Aarhus, Denmark
Abstract: The present review is focused on juvenile neuronal ceroid lipofuscinosis (JNCL; Batten disease) due to a mutation in CLN3. Functional vision impairment occurring around 5–6 years of age is the first symptom in more than 80% of patients. Approximately 2 years later (though sometimes simultaneously), obvious signs of cognitive impairment appear. Behavior problems can occur in advance, especially in boys. These include anxious and depressed mood, aggressive behavior, and hallucinations, and even psychotic symptoms. Following the teens, severe dementia is present, including loss of memory, attention, and general reasoning abilities, as well as loss of independent adaptive skills such as mobility, feeding, and communicating. Sleep abnormalities, such as settling problems, nocturnal awakenings, and nightmares, are reported in more than half of patients. The vast majority, if not all, patients develop seizures, starting at approximately 10 years of age. Generalized tonic–clonic seizure occurs as the only type of seizure in approximately half of patients, and in combination with partial seizures in a third of patients. There seems to be no difference in seizure severity according to sex or genotype, and there is great variation in seizure activity among patients. Soon after diagnosis, patients begin to have slight ataxic symptoms, and at adolescence extrapyramidal symptoms (rigidity, bradykinesia, slow steps with flexion in hips and knees) occur with increasing frequency. Chewing and swallowing difficulties emerge as well, and food intake is hampered in the late teens. Disabling periodically involuntary movements may occur as well. A progressive cardiac involvement with repolarization disturbances, ventricular hypertrophy, and sinus-node dysfunction, ultimately leading to severe bradycardia and/or other conduction abnormalities, starts in the mid-teens. Patients are usually bedridden at 20 years of age, and death usually occurs in the third decade of life.
Keywords: Batten disease, neuronal ceroid lipofuscinosis, neurodegenerative disease, clinical course, dementia, diagnostic algorithm, juvenile
Introduction
The neuronal ceroid lipofuscinoses (NCLs) collectively constitute one of the most common forms of inherited childhood-onset neurodegenerative disorders. They form a heterogeneous group of incurable lysosomal storage diseases that lead to blindness, epilepsy, dementia, and motor deterioration. Traditionally, the NCL diseases were classified according to the age at disease onset, ie, in infantile, late-infantile, juvenile, and adult form.1 Although Batten disease was usually regarded as the juvenile form of NCL, some physicians used, and still use, the term “Batten disease” to describe all forms of NCL. Over the past 20 years, it has become apparent that the diseases are more heterogeneous and today, at least 14 different NCL forms have been described.2 In addition, mutations in the same gene may lead to different disease courses.3 Recently, an international NCL nomenclature has been developed that identifies each NCL disease both genetically and clinically.1,4 This nomenclature classifies both the defective gene and the age of onset (infantile, late-infantile, juvenile, and adult).2 The present review focuses on classic juvenile NCL (JNCL), which is due to mutations in the CLN3 gene. A juvenile onset has also been described due to mutations in the CLN1 gene, which most generally give rise to the classic infantile subtype of NCL. A strategy and algorithm of diagnostic investigations once NCL with juvenile onset is suspected is addressed in the latter part of the article.
Historical cases of JNCL
The first clinical description of patients who may have suffered from JNCL was published in 1826 by a Norwegian clinical practitioner – Otto Christian Stengel.5 He observed what he called a “singular illness” in four children of a local family. Following an unremarkable early development, their sight began to deteriorate. Within years, the disease led to blindness, progressive mental deterioration, loss of speech, and epileptic seizures. The two oldest siblings died at the ages of 20 and 21 years, respectively. No autopsies were performed, but in retrospect the clinical features were thought to be compatible with JNCL. This report remained unnoticed until the 1950s, after the English pediatrician and neurologist Frederick Batten (1865–1918), the German neuropathologist Walther Spielmeyer (1879–1935), the German neurologist Heinrich Vogt (1875–1936), and the Swedish psychiatrist Torsten Sjögren (1896–1974), in the first 3 decades of the 20th century separately had described further similar studies of familial cases with progressive loss of vision and psychomotor deterioration supplemented with neuropathological investigations, which showed intraneuronal accumulation of granular material with lipid-like staining qualities.6 It was not until the late 1960s that Zeman and Dyken7 proposed the new term “neuronal ceroid lipofuscinosis”, in order to distinguish these progressive diseases from Tay–Sachs disease and other gangliosidoses after they had demonstrated that the storage material was largely resistant to lipid solvent and bore a striking resemblance to the autofluorescent lipopigments ceroid and lipofuscin. In addition, the storage material showed curvilinear and fingerprint ultrastructural patterns. The juvenile type of NCL (NCL3) was linked to the haptoglobin locus on the long arm of chromosome 16 in 1989, and by positional cloning the gene CLN3 was isolated in 1995.8
The CLN3 gene and the CLN3 protein battenin
The CLN3 gene encodes a hydrophobic integral membrane protein (CLN3 – battenin) of 438 amino acids.8 The proposed functions of the CLN3 protein include lysosomal acidification, lysosomal arginine import, membrane fusion, vesicular transport, cytoskeletal-linked functions, autophagy, and apoptosis. The protein is expressed ubiquitously, and has been localized mostly in the endosomal/lysosomal compartment, in the plasma membrane, and in the synaptosomal fraction of neuronal cells. It is a highly conserved protein, and is present in small invertebrate animals such as Drosophila and in worms.
Currently, there are more than 60 mutations (http://www.ucl.ac.uk/ncl/mutation.shtml) known to cause JNCL, the most common of which is a 1.02 kB deletion that removes exons 7 and 8 and which occurs in 85% of patients.3 Although not demonstrated in studies investigating the genotype–phenotype association among individuals that are homozygote or compound heterozygote for the CLN3 deletion, there is some evidence of a correlation between CLN3 genotype and clinical disease phenotype.9 In all cases of NCL caused by mutations in CLN3, visual failure has occurred by the age of 10 years, suggesting the importance of battenin in the retina. Most recently, certain mutations in CLN3 have been identified as a novel nonsyndromic retinal disease, with all patients who carry these mutations showing retinal degeneration phenotypes but without additional signs of JNCL.10
Human pathology
Pathological features of extracerebral tissues have been of diagnostic use for decades. In lymphocytes, vacuoles, which either look empty or contain solid material, can be observed. Sudan black-positive material and autofluorescence can be detected in skin biopsies (endothelial cells, sweat-gland epithelia, smooth-muscle cells), in skeletal muscle cells, and in ganglionic neurons following rectal biopsies.
Histological studies of eyes from patients with JNCL have shown that the retina is uniformly thinned, with severe loss of the photoreceptor and outer-nuclear and outer-plexiform layers in the macula and mid-periphery areas. In addition, there is atrophy of the nerve-fiber layer and ganglion cells and significant gliosis of the optic nerve.11 Autofluorescent cytoplasmic ceroid granules accumulate predominantly in the photoreceptor-cell layer and in the retinal ganglion cells. Autofluorescence is prominent in the epithelial cells of the conjunctiva and ciliary body as well, but not in the cornea. Though not well characterized, an element of inflammation is hypothesized to be associated with JNCL retinal changes, with observation of cell infiltration in the vitreous and of antiretinal antibody reactivity.12
Postmortem investigations of the brain have shown a diffuse symmetric atrophy of the cerebral hemispheres, cerebellum, and brain stem and a diffusely enlarged ventricular system.13 The cut surface revealed a narrow tawny cortex, reduced and firm-elastic white matter, small, grayish-white basal ganglia, and depigmentation of the substantia nigra.13 Microscopy revealed closely packed cytoplasmic lipopigment granules displacing the nucleus of the nerve cells. The changes were diffusely distributed throughout the layers in all gyri, in the brain stem, and in the spinal cord. In the cerebellum, the pigment was particularly prominent in the Purkinje cells. The ganglion cells of the spinal ganglia contained fine lipopigment granules, whereas the roots and peripheral nerves showed loss of axons and myelin sheaths, but no granular deposits.13 Cytoplasmic deposits with the same staining properties as that found in the central nervous system can also be seen in the dorsal root ganglion cells and widely distributed in visceral organs and lymph nodes.13,14 In the myocardium, the deposits are mainly localized in the perinuclear region of the muscle fibers. In the spleen, thymus, lymph nodes, and bone marrow, they are seen in large reticuloendothelial cells, and in the kidney the storage material is found in the epithelium of the distal part of the collecting tubules.14 In the liver, deposits have been demonstrated in both parenchymal cells and Kupffer cells. In the intestinal walls, autonomous nerve cells show large accumulations, and variable amounts of fluorescent granular deposits can be found in the anterior lobe of the pituitary gland, pancreas, adrenals, thyroid gland, and gonads.14 The skeletal muscles show varying degrees of neurogenic atrophy, but autofluorescent granules are not prominent.13 In the heart, extensive deposition has been demonstrated in the sinus node, atrioventricular node, and bundle of His.15
In accordance with postmortem and histologic investigations, magnetic resonance imaging reveals progressive cerebral and cerebellar atrophy.16 Cerebral atrophy is usually not found before the age of 9 years, whereas cerebellar atrophy is unusual before the age of 13 years.16 Cerebral atrophy appears first within the occipital cortex, whereas thinning of the frontal and parietal cortices occurs later. On magnetic resonance imaging, abnormally high intensity may be found in the white matter beside the lateral ventricle, and low-signal changes may be seen in the thalamus and basal ganglia.16
Clinical symptoms
The typical course relative to the time of onset (in average ranges) of specific clinical symptoms or milestones is outlined in Table 1.
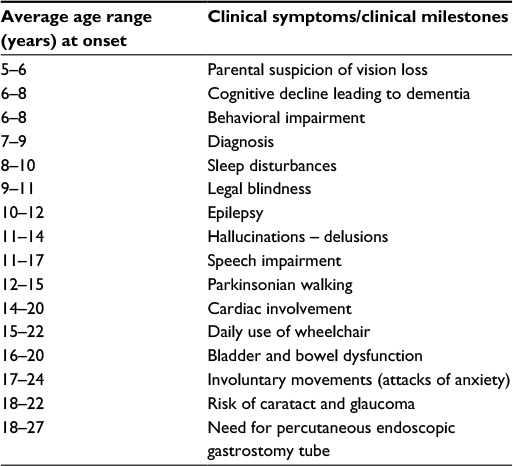 | Table 1 Typical time course relative to onset (years in average ranges) of specific clinical symptoms or milestones in patients with juvenile neuronal ceroid lipofuscinosis |
Vision loss
Functional vision impairment, including loss of visual acuity, is the first symptom that is most clearly linked to JNCL.17 It occurs as the first symptom in more than 80% of patients.17,18 The mean age at disease onset, as marked by a parental suspicion of vision loss, is around 5 years of age in males, and ∼6 years of age in female,18 with presentation to an ophthalmologist at 5.5–8.5 years.19 Accordingly, in a Danish study, females were diagnosed significantly later than males, ie, 7.9±1.2 years and 7±1.2 years (mean ±2 standard deviations), respectively.20 At that time, children often have eccentric viewing or “overlooking”: holding the eyes in a raised position when attempting to fixate.19 This feature is attributed to relative preservation of the superior peripheral retina, with loss of central and inferior visual fields. Rotary nystagmus may be present. At this point, there are no anterior segment abnormalities described.19 Fundus examination at presentation may range from normal to severe pigmentary retinopathy. Early in the disease, subtle granularity of the retinal pigment epithelium in the central macula may be present, although bull’s-eye maculopathy is classically described. Later in the disease, optic nerve atrophy, vascular attenuation, and pigment accumulation in the peripheral retina develops. The rate of progression is extremely rapid in comparison to other retinal degeneration, leading to legal blindness within 1–2 years of presentation.19
The electroretinogram shows significantly reduced rod- and cone-response amplitude, even at an early disease stage. At advanced-disease stages, the electroretinograms of JNCL patients show no recordable rod-mediated activity and significantly reduced cone-mediated activity.
Only very recently have pathological alterations beyond the retina and optic nerve been reported to be complications of JNCL.21 In their case series of 35 JNCL patients, Nielsen et al21 identified five patients with cataract, of which two developed acute glaucoma. The patients’ age at detection of cataract was 20.1±1.6 years (mean ±2 standard deviations), and even though cataract may not impact vision in JNCL due to loss of retinal function, the authors recommended that investigation of the anterior segment should be performed routinely in JNCL patients beyond 16 years of age.21 The authors suggested that the mechanism for cataract formation in JNCL was related to their severe retinitis pigmentosa.21 Of note is the recent identification of a nonsyndromic retinal disease in patients with certain mutations in CLN3 showing retinal degeneration phenotypes without additional signs of JNCL.10 This suggests that there likely are factors independent of neurodegeneration contributing to JNCL vision loss.
Cognitive decline
Approximately 2 years after the onset of visual impairment, though sometimes simultaneously, obvious signs of cognitive impairment appear. Behavior problems can, especially in boys, occur in advance.17,18 Comprehension and short-time memory seem to be impaired early in the disease, as is attention, whereas auditory perception remains quite stable.22,23 Cognitive decline was first quantitatively described in the 1990s, including cases with repeat assessments that provided information on change in cognition over time.24 Follow-up periods were for 4–8 years, and a significant decline was observed.24 However, the particular combination of symptoms, including loss of vision and speech intelligibility,23 leads to limits on assessment. Many intelligence tests evaluate both verbal and reasoning skills, and even some verbal tasks, such as picture naming, require visual identification. In children with JNCL, vision loss excludes tests of nonverbal/visual reasoning, but loss of speech intelligibility eventually also limits the ability to provide verbal responses to language based tasks. In addition, behavioral impairments (see later) limit compliance within the evaluation process. No doubt, however, disease duration is inversely correlated with a significant cognitive decline,23 and following the teens, severe dementia is present, including loss of memory, attention, and general reasoning abilities, as well as a loss of independent adaptive skills such as mobility, feeding, and communicating.9,23
The early mental impairment in JNCL indicates early habilitation. It is possible to teach Braille reading at an early age. After the age of 10 years, learning Braille becomes very difficult or even impossible, because of a short digit-memory span and impaired motor speed.22 If patients have learned to read Braille well at an early age, the ability may be preserved for several years.22
Behavioral impairment
Formal characterization of neurobehavioral features of JNCL started in the 1990s, and is primarily based on patient groups from Finland and North America.9,17,23–27 These studies provided details of behavioral and mood impairments, including anxious and depressed mood, aggressive behavior, and hallucinations, and even psychotic symptoms.23–27 The debut of these symptoms, along with cognitive impairment, appears to precede onset of motor impairment, and does not appear to be influenced by genotype.9,27 It has been hypothesized that behavioral symptoms may follow an inverse U-shaped function in which the problems initially worsen as disease advances, before dropping off in the later stages of the illness, as motor impairment limits the behavioral repertoire.27 However, there exists no formal characterization of behavioral features in patients with JNCL beyond 25 years of age. Behavioral and psychiatric symptoms have also been evaluated by using standardized questionnaires like the Child Behavior Checklist, Teacher’s Report Form, and Children’s Depression Inventory.26,27 These show increased-severity scores on internalizing, asocial, and externalizing domains like withdrawn or inattentive behavior, unusual or repetitive habits, uncooperative behavior, and disruptive behavior in the vast majority of patients. Many patients were described as routine-bound, ritualistic, and engaged in repetitive questioning or discussions. However, their need for routine seems not to be goal-directed, but may be a behavioral analog to echolalic speech and pervasive thoughts.27 Noteworthy is the fact that the patients themselves did not report depressive symptoms.26
The hallucinations reported are often visual, and are of a frightening nature: snakes, lizards, lots of ants, flies, mice, or other terrifying sights such as “eyes in the ceiling”, monsters, or threatening men. Some have delusions, also frightening, like “death of a family member or friend” or fear of being poisoned.28 The hallucinations can last for minutes, hours, or even several days, and the older the patient and the more cognitively they are affected, the more difficult is it for parental reassurance to have an impact. Females may exhibit greater anxiety and greater social difficulties.25 Pharmacotherapy for the behavioral symptoms can be necessary, but only limited research has been done29 and large controlled trials are very difficult or even impossible to conduct. In one Finnish pilot study,29 citalopram was the first-line drug used to treat depression and aggression, whereas risperidone was used to treat hallucinations, delusions, agitation, and restlessness. The clinical outcome was good or satisfactory in 70%, but adverse effects like fatigue and weight gain were seen in more than half the patients.29
Sleep disturbances
In JNCL, sleep disorders are reported in more than half of patients, and may be the most disturbing factor in the patient’s everyday life.30 The most typical problems are settling problems, nocturnal awakenings, and nightmares. In 2000, Kirveskari et al30 performed a very comprehensive investigation of 28 patients with JNCL (age range 6–27 years) using polysomnography, electrooculography, electromyography, electrocardiography, and electroencephalography (EEG). In the majority of patients, the total sleep time and sleep-efficiency index were significantly lower, and the average of non-rapid eye movement (REM) stage 1 and number of awakenings and stage shift were higher than in the control group. The percentage of REM sleep was significantly decreased. Sleep-structure abnormalities were evident throughout all age-groups, irrespective of whether the patients had shown sleep complaints or not, and irrespective of the degree of visual impairment. However, patients with complaints tended to have a more fragmented sleep. The younger patients with a less advanced-disease stage were also affected, but the percentages of non-REM stage 1 and the number of awakenings increased with age and progression of the disease. Half the patients had daytime sleepiness.
Sleep disturbances are probably connected to the progressive encephalopathy seen in JNCL. Accompanying blindness, epilepsy, dementia, and psychological factors may further predispose to sleep abnormalities. It is worth noting that disturbances in circadian hormonal rhythms have been demonstrated only at an advanced stage of the disease,31 and that even in JNCL patients with no light perception, light can penetrate the visual system to the hypothalamic and pineal levels and regulate their neuroendocrine functions.32
Epilepsy
The vast majority of, if not all, patients with JNCL develop seizures. Collectively, the NCL diseases are often cited within the group of progressive myoclonus epilepsy, an epileptic encephalopathy characterized by the presence of myoclonus, epilepsy, and progressive neurological deterioration.33 Differentiation among the many NCL types, however, is not made in reference to association with progressive myoclonus epilepsy. In addition, the distinction between epileptic and nonepileptic myoclonus is not consistently delineated, and it is now generally accepted that myoclonic seizures are not a hallmark of JNCL.34,35 JNCL is most commonly characterized by generalized tonic–clonic seizures,34,35 starting at approximately 10 years of age.19,34,35 Generalized tonic–clonic seizure occurs as the only type of seizure in approximately half of the patients, and in combination with partial seizures in a third of patients.34,35 Myoclonic seizures occur in less than a third of patients.34,35 There seems to be no difference in seizure severity according to sex or genotype.34,35 Seizures occur on average less frequently than once every 3 months, but there is great variation among patients, with some having none for years, others having two to three seizures every month. Seizure frequency showed a weak positive correlation with age in one study35 but not in another study.34 In a Finnish study from 2000 comprising 60 genetically verified patients with JNCL, consecutive EEG recordings were performed.34 Abnormal background activity was found as early as age 7 years, and consisted of slightly irregular delta, alpha and beta-activity. In older children, δ-activity was seen diffusely or as short focal episodes. Paroxysmal, generalized, irregular slow-wave discharges were seen in 30% of the recordings and focal spikes in less than 20%, usually in those 10 years of age or older. In later recordings, background activity became slower, and paroxysmal activity tended to increase with age. Clinically, there was no obvious correlation between the EEG findings and the severity of seizures.34 It has recently been shown that progressive bilateral hippocampal atrophy occurs in patients with JNCL, and that the annual loss of hippocampal volume exceeds the annual loss of total gray-matter volume.36 However, further studies are needed in order to establish a possible causal relationship among hippocampal atrophy and occurrence of epilepsy in JNCL.
There are limited data specifically regarding seizure treatment, and there exist no placebo-controlled studies. In a single open-label study from Finland, lamotrigine, either as initial, add-on, or substitution therapy, was well tolerated and associated with reductions in seizure frequency and severity.37 In another Finnish series, 70% responded well to valproate as initial monotherapy.34 In a recently published observational study from the University of Rochester (Rochester, NY, USA), including 86 patients with JNCL from different parts of the US,35 valproate and levetiracetam were the most commonly used medications. However, as it was an observational study and not an interventional trial, the authors were unable to discern whether the specific treatment-regimen rationale was due to common prescribing preferences of some of the clinicians treating the JNCL patients or whether it was a treatment regimen to which individuals with JNCL were particularly responsive.
Valproate is generally well tolerated when serum levels are maintained within the therapeutic range, but serious side effects like valproate-induced hyperammonemic encephalopathy (VHE) may occur.38 Known risk factors for VHE include a high valproate dosage, need for polytherapy, and long duration of treatment, which are all incidents that occur in patients with JNCL.34 In addition, individuals with JNCL often have a reduced level of carnitine,39 which further increases the risk of VHE. In accordance, valproate-induced hyperammonemia and VHE have been described in JNCL, and attention to VHE should be given to JNCL patients treated with valproate, especially in case of polytherapy and long-lasting treatment.40 However, it is generally accepted that valproate and also lamotrigine, levetiracetam, and clonazepam are drugs that can be recommended as antiepileptic medicine in JNCL.34,35 Opposite this, carbamazepine and phenytoin have in case reports and case series been reported to be unfavorable in patients with JNCL.41,42 This has been further exacerbated by documentation that patients with JNCL develop disturbances of the cardiac conduction system, including the sinus node, the atrioventricular node, and the bundle of His.15,43
Motor impairment
Motor impairment can be divided into: 1) loss of function relative to voluntary movements, including loss of speech function and food intake; and 2) appearance of involuntary movements. In addition, 3) loss of control of micturition and defecation occur.
At the time of diagnosis, children with JNCL can walk, jump, and run normally, and commonly they do not have any fine-motor problems. However, soon after, they begin to have slight ataxic symptoms, and at adolescence extrapyramidal symptoms (rigidity, bradykinesia, slow steps with flexion in hips and knees, and shuffling gait) occur with increasing frequency. Unlike Parkinson’s disease, tremor is uncommon. The extrapyramidal signs in JNCL lead to a severe impairment of spontaneous movement and gradually to a bedridden state. In a Finnish series comprising 53 patients with JNCL, the mean age at first sign of a parkinsonian type of walking was 13.7 years.41 Naturally, the ataxia and extrapyramidal dysfunction associated with amblyopia lead to a severe disturbance of locomotion, and use of a wheelchair becomes obligatory within a few years. In the Finnish study, the patients lost their ability to walk at a mean age of 17.3 years. In a recent Danish study, females were dependent upon daily use of a wheelchair at the age of 17 (range 13.8–20.6) years, which was significantly earlier than in males, in which the mean age at use of a wheelchair on a daily basis was 20.2 (range 14.3–22.1) years.20 From positron-emission tomography and single-photon-emission computed tomography studies, it seems evident that both thalamic dysfunction and nigrostriatal and striatal dysfunction contribute to the parkinsonian signs in JNCL.44,45 Only one controlled study has been published regarding antiparkinsonian treatment of patients with JNCL.46 This study included eight patients of which four received placebo. No effect was found. In an open study of 21 patients, of which ten received levodopa, six received selegiline, and five served as a control group, mean scores on the Unified Parkinson’s Disease Rating Scale (UPDRS) decreased significantly in the levodopa group when compared with the control group.47 However, not all levodopa-treated patients showed a decrease in UPDRS score, and larger controlled trials are needed to determine the long-term outcome of antiparkinsonian treatment in JNCL.47
Speech is an important indicator of motor function and movement coordination, and is thus an extremely sensitive marker of changes due to neurodegeneration. In accordance, in all individuals with JNCL, speech and communication skills will deteriorate. However, there are large individual differences, with some individuals showing their first signs of language disability early at school, while others may not show such difficulties until well into their teens. In some cases, speech disappears completely, but poor intelligibility due to articulatory dysfunction is a more common problem than a total lack of speech.48 Other changes that have been observed include increased dysfluency (stuttering or cluttering) and a greater prevalence of omissions of inflections at the end of words, splitting of words, word-finding problems, incorrect or idiosyncratic use of words without showing awareness of it, odd word constructions, syntactic errors, and problems initiating conversations.48 For some patients with JNCL, word-finding problems may be most apparent, while dysfluency of speech is the greatest problem for others. As the disease progresses, both word comprehension and speech production become increasingly dependent on being in a situation that is familiar and does not contain too many disturbing elements. As stuttered or cluttered speech progress, the speech may become unintelligible even to those who know the person well,48 and the deterioration in speech and communication skills is a major source of despair. Due to the development of extrapyramidal symptoms, it may be tempting to compare the speech disturbance in JNCL with that present in Parkinson’s disease, the so-called hypokinetic dysarthria, presenting with prosodic insufficiency, related to a monotony of pitch and intensity, a reduction of accentuation, variable speech rate, and possible phoneme imprecision. Unfortunately, there are very few descriptions of speech changes in individuals with JNCL, and a thorough and detailed investigation of the underlying neurological dysfunction is missing. Dysarthria is the most commonly used word in the description of the progressive speech abnormality in JNCL. However, it remains to be elucidated whether there is both a hypokinetic, spastic, or ataxic component.
In view of the oral motor dysfunctions, chewing and swallowing difficulties emerge as well, and food intake is hampered in the late teens. To ensure adequate fluidic and caloric intake, gastric feeding is required, which currently is done via percutaneous endoscopic gastrostomy-tube placement. In a recent Danish study comprising 35 patients with JNCL,20 a percutaneous endoscopic gastrostomy tube was found necessary in females 17.8–22.3 years of age (mean 19.8 years) and in males 19.8–27.1 years of age (mean 24 years).
Just as lesions of the basal ganglia in JNCL may lead to failure to facilitate desired movements (eg, the hypokinetic parkinsonian signs), failure to inhibit unwanted hyperkinetic movements may occur. Accordingly, involuntary movements have been described in JNCL,28,49,50 but only anecdotally. In their clinical study from 1979, Sørensen and Parnas28 reported that in some patients they observed “dystonic seizures lasting as long as 1 hour, with oculogyric crisis, at times accompanied by dystonia in the neck and throat muscles”. In certain cases, the dystonia was accompanied by anxiety. It “either stopped spontaneously or rather unpredictably following administration of choral hydrate or diazepam”. In one patient, an EEG was obtained during the attack, but there was no simultaneous paroxysmal activity seen. Also, in a case series from a Dutch residential institution for JNCL-affected children and young people,49 periodically involuntary movements were described. They occurred particularly with regard to intentional actions like lifting the patient from a bed to a wheelchair. These involuntary movements disappeared during sleep, but were otherwise unable to be suppressed totally by medication, and were perceived and described as myoclonias. The presence of a more or less continuous appearance of the involuntary movements, an urge to move constantly leading to sweating, increase in temperature, and exhaustion was also described.49 Elkay et al reported two sisters with JNCL who developed severe generalized dystonia culminating in dystonic storm.50 In addition to intubation and sedation, multiple medications, including botulinum toxin injections and intrathecal baclofen infusions, were tried in both patients without benefit. Ultimately, one sister was treated with bilateral pallidotomy, with a partial and transient effect, whereas the other sister received bilateral pallidotomy and deep-brain stimulation. Following the operation, she was free of abnormal movements for 7 months. Subsequently, adjusting the deep stimulations was able to maintain a reduction in her abnormal movements.50
There exists no systematic or thorough investigation of occurrence of involuntary movements in JNCL, and even less do we know concerning their etiology. In the cases described from Denmark and the Netherlands,28,49 it appears that involuntary movements occurred periodically, that the intensity and duration of involuntary movements may gradually increase, that patients seemed anxious and often demonstrated a fixed upward gaze, and that there were accompanying sweating and temperature increase. There was no simultaneous paroxysmal activity seen on EEG, although mental status seemed reduced. Sometimes, the movements were initiated by intentional actions like lifting the patient from bed to a wheelchair. Other times, the symptoms started for no apparent reason. The attacks seemed rather resistant to medical treatment, but seemed to disappear during sleep.49 In the two sisters reported by Elkay et al,50 severe generalized dystonia culminated in what was called a dystonic storm. Currently, there is no internationally agreed definition for dystonic storm or status dystonicus, but generally the mental status is described as normal, and there are no sweating or pupil alterations.51 This is in contrast to descriptions in patients with JNCL,28,49 which seem better suited to paroxysmal autonomic instability with dystonia, also called paroxysmal sympathetic storms.51,52 Until now, there have been no studies to specifically investigate the autonomic system in JNCL or sympathetic activity during periods of involuntary movements, but from postmortem studies we know that granular deposits are accumulated in the dorsal root ganglion cells and that peripheral nerves show loss of their axons and myelin sheaths.13,14 In addition, with advancing age, a significant reduction in parasympathetic activity on the heart has been demonstrated in patients with JNCL.43 If this reduction in parasympathetic activity is a more generalized phenomenon, it may lead to an autonomic disturbance with sympathetic hyperactivity in JNCL. A chronic and progressive generalized autonomic failure is a clinical hallmark in neurodegenerative disorders such as multiple-system atrophy and Parkinson’s disease,53 to which JNCL has some similarities. Therefore, future studies should focus on the possible component of autonomic dysfunction in patients with JNCL.
There exist no exploratory studies of bladder or bowel function in patients with JNCL, but like in other neurodegenerative disorders,53,54 patients with JNCL gradually lose control of micturition and defecation. Clinically, the bladder dysfunction seems to be a reduced detrusor activity leading to incomplete bladder emptying, increased postvoid residual volume, and eventually urinary retention, thus mimicking multiple-system atrophy more than Parkinson’s disease. Behavioral and emotional influences may affect the reflex of micturition as well, by integration via inputs from the frontal lobe.54
Cardiac involvement
Disease involvement of the heart has been described in early case series28,55 and in autopsy studies,15 but it was not until 2011 that a systematically or thoroughly clinical investigation of the cardiac involvement in JNCL was reported.43 In a Danish cross-sectional and follow-up study comprising 29 patients with JNCL, progressive cardiac involvement with repolarization disturbances, expressed by abnormally deeply inverted T waves, ventricular hypertrophy, and sinus-node dysfunction, ultimately leading to severe bradycardia and/or other conduction abnormalities, were demonstrated.43 The inverted T waves were present as early as 14 years of age, and were associated with an increased risk of death during the 7.5-year follow-up period. With increased age, heart rate and heart-rate variability, expressed as the vagal index, were significantly reduced, suggesting an age-dependent bidirectional effect of the heart rate: one through decreasing parasympathetic activity on the heart and the other through a direct negative influence on sinus-node automaticity.43 Four of seven patients beyond 20 years of age had hypertrophy of the left ventricular walls, but the ejection fractions were within normal limits. The demonstrated impact of the cardiac conduction system and the cardiac muscles are in accordance with postmortem studies, in which deposition of lipopigment occurs extensively in the sinus node, as well as in the atrioventricular node and in the bundle of His.15 In electrocardiography recordings, severe bradycardia, including sinus arrests for periods as long as 22 seconds, has been demonstrated55 and treatment with a pacemaker may be necessary and appropriate in order to increase quality of life. However, the general clinical condition of the patient should be taken into consideration. Due to the progressive cardiac symptoms, surveillance for heart involvement should be performed at least once every year beyond the age of 18 years.43
Hormonal changes
Hyperandrogenism, characterized by acne, hirsutism, and/or hyperandrogenemia, is a common finding in girls with JNCL.56 In addition, menarche occurs approximately 18 months earlier than in healthy controls.19,56 No single causative factor has been found to be responsible, but the neurodegenerative process and medication used in symptomatic treatment for epilepsy (ie, valproate) and psychosis (ie, risperidone) may also contribute to these findings. In boys, an increased incidence of acne is also present, and puberty starts and ends earlier than in healthy controls.
Disease course
Following progressive visual failure starting at 5–7 years of age, development of epilepsy and behavioral disturbances at 8–10 years of age, followed by dementia and motor dysfunction, patients are usually bedridden at 20 years of age, and death usually occurs in the third decade of life.18,20,41 Based on impressions from parental anecdotes, it has been suggested that females may have a more precipitous decline than males. This sex difference in disease course has been verified in both a North American study and a Danish study.18,20 Females demonstrated earlier loss of independent functions, had lower quality of life, and died approximately 18 months earlier. There is no obvious explanation for the demonstrated sex difference in JNCL, which is also in contrast to adult-onset neurodegenerative diseases like Parkinson’s, in which female sex is often associated with a milder disease course.57
Diagnostic strategy
The typical patient who should be suspected as having JNCL is a school-age child with visual loss and/or dementia and epilepsy. In cases of visual impairment, many parents send their child to an ophthalmologist, who by fundus examination will find a pigmentary retinopathy. Early in the disease, subtle granularity of the retinal pigment epithelium in the central macula may be present, although classical bull’s-eye maculopathy may also be seen. The testing strategy, including gene testing in order to confirm or establish the diagnosis of JNCL, appears in Table 2.
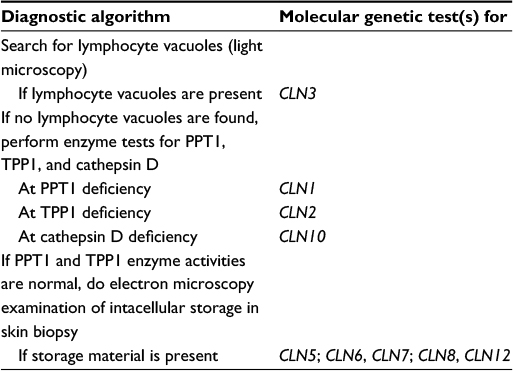 | Table 2 Testing strategy to confirm or establish the diagnosis of juvenile neuronal ceroid lipofuscinosis |
Treatment
Currently, there exists no curative treatment, and all treatment for JNCL is symptomatic and palliative. Animal models may point to new treatment strategies.58 An autoimmune component has been proposed.59 A 1-year trial with intermittent systemic prednisolone (dose 0.75 mg/kg/day, maximum 40 mg/day, for 10 days in each of 12 months) in eight children with JNCL was reported to increase IQ in two subjects. In two other children, the presence of anti-GAD65 antibodies was cleared during treatment, but there was no correlation between increase in IQ and the existence of GAD65 antibodies.60 One patient was withdrawn after 3 months due to behavioral side effects, and two patients experienced recurrent infections during prednisolone treatment. A 16-week double-crossover clinical trial of mycophenolate mofetil has been completed (NCT01399047), but so far no results have been published.
Symptomatic and palliative treatment represents a significant challenge, due to the wide range of symptoms from the different body systems affected. Recommendations in relation to individual organ manifestations have been provided under individual headings. The medications most often used in epilepsy and behavioral impairment are summarized in Table 3. Due to progressive brain degeneration, the effectiveness of medication may change over time, and due to the need for polypharmacy there is a great risk of unexpected toxicity. Besides anticonvulsive treatment, such symptoms as sleep disturbances, fear, aggressive behavior, depression, and hallucinations represent a certain challenge. Recognition of contexts and possible trigger factors are helpful, in order to manage possible environmental exacerbating factors. It is necessary to keep patients hydrated and nourished, the use of a percutaneous gastric tube should be considered, and one must be aware of urinary tract infections and pulmonary infections, which may exacerbate hallucinations, anxiety, and involuntary movements. Basic functions such as urination and defecation should be monitored, in order to avoid or to reduce the risk of urinary retention and constipation, which can further worsen quality of life. The disease leads relentlessly to death, and professionals should in due time inform and talk to the relatives of the progress and the likely course of the final period.
 | Table 3 Medications most often used in epilepsy and behavioral impairment in patients with juvenile neuronal ceroid lipofuscinosis (JNCL) |
Disclosure
The author reports no conflicts of interest in this work.
References
Mole SE, Williams R, Goebel HH. The Neuronal Ceroid Lipofuscinoses (Batten disease). Oxford: Oxford University Press; 2011. | ||
Schultz A, Kohlschütter A, Mink J, Simonati A, Willimas R. NCL diseases – clinical perspectives. Biochim Biophys Acta. 2013;1832:1801–1806. | ||
Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2011;33:42–63. | ||
Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79:183–191. | ||
Stengel C. [Report on a strange illness among four siblings near Røraas]. Beretning om et mærkeligt sygdomstilfælde hos fire søskende i nærheden af Røraas. Eyr Med Tidskr. 1826;1:347–352. Danish. | ||
Haltia M; Goebel HH. The neuronal ceroid lipofuscinoses: a historical introduction. Biochim Biophys Acta. 2013;1832:1795–1800. | ||
Zeman W, Dyken P. Neuronal ceroid lipofuscinosis (Batten’s disease): relationship to amaurotic family idiocy? Pediatrics. 1969;44:570–583. | ||
[No authors listed]. Isolation of a novel gene underlying Batten disease, CLN3. Cell. 1995;82:949–957. | ||
Adams HR, Beck CA, Levy E, et al. Genotype does not predict severity of behavioural phenotype in juvenile neuronal ceroid lipofuscinosis (Batten disease). Dev Med Child Neurol. 2010;52:637–643. | ||
Wang F, Wang H, Tuan HF, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133:331–345. | ||
Ouseph MM, Kleinman ME, Wang QJ. Vision loss in juvenile neuronal ceroid lipofuscinosis (CLN3 disease). Ann N Y Acad Sci. Epub 2016 Jan 8. | ||
Drack AV, Mullins RF, Pfeifer WL, Augustine EF, Stasheff SF, Hong SD. | ||
Bruun I, Reske-Nielsen E, Oster S. Juvenile ceroid-lipofuscinosis and calcifications in the CNS. Acta Neurol Scand. 1991;83:1–8. | ||
Kristensson K, Rayner S, Sourander P. Visceral involvement in juvenile amaurotic idiocy. Acta Neuropathol. 1963;4:421–424. | ||
Reske-Nielsen E, Baandrup U, Bjerregaard P, Bruun I. Cardiac involvement in juvenile amaurotic idiocy – a specific heart muscle disorder: histological findings in 13 autopsied patients. Acta Pathol Microbiol Scand A. 1981;89:357–365. | ||
Autti T, Raininko R, Vanhanen SL, Santavuori P. MRI of neuronal ceroid lipofuscinosis I: cranial MR of 30 patients with juvenile neuronal ceroid lipofuscinosis. Neuroradiology. 1995;38:476–482. | ||
Marshall FJ, de Blieck EA, Mink JW, et al. A clinical rating scale for Batten disease: reliable and relevant for clinical trials. Neurology. 2005;65:275–279. | ||
Cialone J, Adams H, Augustine EF, et al. Females experience a more severe disease course in Batten disease. J Inherit Metab Dis. 2012;35:549–555. | ||
Collins J, Holder GE, Herbert H, Adams GG. Batten disease: features to facilitate early diagnosis. Br J Ophthalmol. 2006;90:1119–1124. | ||
Nielsen AK, Ostergaard JR. Do females with juvenile ceroid lipofuscinosis (Batten disease) have a more severe disease course? The Danish experience. Eur J Paediatr Neurol. 2013;17:265–268. | ||
Nielsen AK, Drack AV, Ostergaard JR. Cataract and glaucoma development in juvenile neuronal lipofuscinosis (Batten disease). Ophthalmic Genet. 2015;36:39–42. | ||
Lamminranta S, Åberg LE, Autti, T, et al. Neuropsychological test battery in the follow-up of patients with juvenile neuronal ceroid lipofuscinosis. J Intellect Disabil Res. 2001;45:8–17. | ||
Adams HR, Kwon J, Marshall FJ, de Blieck EA, Pearce D, Mink JW. Neuropsychological symptoms of juvenile-onset batten disease: experiences from 2 studies. J Child Neurol. 2007;22:621–627. | ||
Lauranen L, Munroe PB, Järvelä I, et al. Delayed classical and protracted phenotypes of compound heterozygous juvenile neuronal ceroid lipofuscinosis. Neurology. 1999;52:360–365. | ||
Santavuori P, Linnankivi T, Jaeken J, Vanhanen SL, Telakivi T, Haiskala H. | ||
Bäckman ML, Santavuoiri P, Åberg LE, Aronen ET. Psychiatric symptoms of children and adolescents with juvenile neuronal ceroid lipofuscinosis. J Intellect Disabil Res. 2005;49:25–32. | ||
Adams H, de Blieck EA, Mink JW, et al. Standardized assessment of behavior and adaptive skills in juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2006;48:259–264. | ||
Sørensen JB, Parnas J. A clinical study of 44 patients with juvenile amaurotic family idiocy. Acta Psychiatr Scand. 1979;59:449–461. | ||
Bäckman ML, Åberg LE, Aronen ET, Santavuori PR. New antidepressive and antipsychotic drugs in juvenile neuronal ceroid lipofuscinoses – a pilot study. Eur J Paediatr Neurol. 2001;5 Suppl A: | ||
Kirveskari E, Partinen M, Salmi T, et al. Sleep alterations in juvenile neuronal ceroid lipofuscinosis. Pediatr Neurol. 2000;22:347–354. | ||
Heikkilä E, Hàtònen T, Telakivi T, et al. Circadian rhythm studies in neuronal ceroid-lipofuscinosis (NCL). Am J Med Genet. 1995;57: | ||
Hätönen T, Laasko ML, Heiskala H, Alila A, Saino K, Santavuori P. Bright light suppresses melatonin in blind patients with neuronal ceroid-lipofuscinosis (NCL). Neurology. 1998;50:1445–1450. | ||
Ramachandran CN, Girard JM, Turnbull J, Minassian BA. The autosomal recessively inherited progressive myoclonus epilepsies and their genes. Epilepsia. 2009;50:29–36. | ||
Åberg LE, Bäckman M, Kirveskari E, Santavuori P. Epilepsy and antiepileptic drug therapy in juvenile neuronal ceroid lipofuscinosis. Epilepsia. 2000;41:1296–1302. | ||
Augustine EF, Adams HE, Beck CA, et al. Standardized assessment of seizures in patients with juvenile neuronal ceroid lipofuscinosis. Dev Med Child Neurol. 2015;57:366–371. | ||
Tokola AM, Salli EK, Åberg LE, Autti TH. Hippocampal volumes in juvenile neuronal ceroid lipofuscinosis: a longitudinal magnetic resonance imaging study. Pediatr Neurol. 2014;50:158–163. | ||
Åberg L, Kirveskari E, Santavuori P. Lamotrigine therapy in juvenile neuronal ceroid lipofuscinosis. Epilepsia. 1999;40:796–799. | ||
Lewis C, Deshpande A, Tesar GE, Dale R. Valproate-induced hyperammonemic encephalopathy: a brief review. Curr Med Res Opin. 2012;28:1039–1042. | ||
Katz ML. Decreased plasma carnitine and trimethyl-L-lysine levels associated with lysosomal accumulation of a trimethyl-L-lysine containing protein in Batten disease. Biochim Biophys Acta. 1996;1317:192–198. | ||
Larsen E, Østergaard JR. Valproate-induced hyperammonemia in juvenile ceroid lipofuscinosis (Batten disease). Seizure. 2014;23: | ||
Santavouri P, Heiskala H, Westermarck T, Saino K, Moren R. Experience over 17 years with antioxidant therapy in Spielmeyer-Sjögren disease. Am J Med Genet. 1988;5:265–274. | ||
Viukari NM. Intolerance to diphenylhydatoin in five cases of Vogt-Spielmeyer disease. J Ment Defic Res. 1969;13:245–248. | ||
Østergaard JR, Rasmussen T, Mølgaard H. Cardiac involvement in juvenile neuronal ceroid lipofuscinosis (Batten disease). Neurology. 2011;76:1245–1251. | ||
Routtinen HM, Rinne JO, Haaparanta M, et al. [18F]Fluorodopa PET shows striatal dopaminergic dysfunction in juvenile neuronal ceroid lipofuscinosis. J Neurol Neurosurg Psychiatry. 1997;62:622–625. | ||
Åberg L, Liewendahl K, Nikkinen P, Autti T, Rinne JO, Santavuori P. Decreased striatal dopamine transporter density in JNCL patients with parkinsonian symptoms. Neurology. 2000;54:1069–1074. | ||
Zweije-Hofman IL, van der Zee HJ, van Nieuwenhuizen O. Anti-Parkinson drugs in the Batten-Speimeyer-Vogt syndrome; a pilot study. Clin Neurol Neurosurg 1982;84:101–105. | ||
Åberg LE, Rinne JO, Rojantie I, Santavuori P. A favorable response to antiparkinsonian treatment in juvenile neuronal ceroid lipofuscinosis. Neurology. 2001;56:1236–1239. | ||
von Tetzchner S, Fosse P, Elmerskog B. Juvenile neuronal ceroid lipofuscinosis and education. Biochim Biophys Acta. 2013;1832:1894–1905. | ||
Hofman IL. Observations in institutionalized neuronal ceroid-lipofuscinosis patients with special reference to involuntary movements. J Inherit Metab Dis. 1993;16:249–251. | ||
Elkay M, Silver K, Penn RD, Dalvi A. Dystonic storm due to Batten’s disease treated with pallidotomy and deep brain stimulation. Mov Disord. 2009;27:1048–1053. | ||
Allen NH, Lin JP, Lynch T, King MD. Status dystonicus: a practical guide. Dev Med Child Neurol. 2014;56:105–112. | ||
Blackman JA, Patrick PD, Buck ML, Rust RS. Paroxysmal autonomic instability with dystonia after brain injury. Arch Neurol. 2004;61: | ||
Benarroch EE. The clinical approach to autonomic failure in neurological disorders. Nat Rev Neurol. 2014;10:396–407. | ||
Benarroch EE. Neural control of the bladder: recent advances and neurologic implications. Neurology. 2010;75:1839–1846. | ||
Hofman IL, van der Wal AC, Dingemans KP, Becker AE. Cardiac pathology in neuronal ceroid lipofuscinosis – a clinicopathologic correlation in three patients. Eur J Paediatr Neurol 2001;5(Suppl A): | ||
Åberg LE, Tiitinen A, Autti TH, Kivisaari L, Santavuori PR. Hyperandrogenism in girls with juvenile neuronal ceroid lipofusinosis. Eur J Paediatr Neurol 2002;6:199–205. | ||
Gillies GE, McArthur S. Estrogen actions in the brain and the basis for differential action in men and women: a case for sex-specific medicine. Pharmacol Rev. 2010;62:155–198. | ||
Sondhi D, Scott EC, Chen A, et al. Partial correction of the CNS lysosomal storage defect in a mouse model of juvenile neuronal ceroid lipofuscinosis by neonatal CNS administration of an adeno-associated virus serotype rh.10 vector expressing the human CLN3 gene. Hum Gene Ther. 2014;25:223–239. | ||
Lim MJ, Beake J, Bible E, et al. Distinct patterns of serum immunoreactivity as evidence for multiple brain-directed autoantibodies in juvenile neuronal ceroid lipofuscinosis. Neuropathol Appl Neurobiol. 2006;32:469–482. | ||
Aberg L, Talling M, Härkönen T, et al. Intermittent prednisolone and autoantibodies to GAD65 in juvenile neuronal ceroid lipofuscinosis. Neurology. 2008;70:1218–1220. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.